 Federico Nicolás Penas1,2
Federico Nicolás Penas1,2 Davide Carta3
Davide Carta3 Ganna Dmytrenko4
Ganna Dmytrenko4 Gerado A. Mirkin1,2
Gerado A. Mirkin1,2 Carlos Pablo Modenutti5
Carlos Pablo Modenutti5 Ágata Carolina Cevey1,2
Ágata Carolina Cevey1,2 Maria Jimena Rada1,2
Maria Jimena Rada1,2 Maria Grazia Ferlin3
Maria Grazia Ferlin3 María Elena Sales4
María Elena Sales4 Nora Beatriz Goren1,2,6*
Nora Beatriz Goren1,2,6*
- 1Departamento de Microbiología, Parasitología e Inmunología, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina
- 2Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM)-CONICET, Buenos Aires, Argentina
- 3Department of Pharmaceutical and Pharmacological Sciences, University of Padova, Padova, Italy
- 4Centro de Estudios Farmacológicos y Botánicos (CEFyBO)-CONICET, Facultad de Medicina, Universidad de Buenos Aires, Buenos Aires, Argentina
- 5Instituto de Química Biológica, Facultad de Ciencias Exactas y Naturales (IQUIBICEN)-CONICET, Universidad de Buenos Aires, Buenos Aires, Argentina
- 6Universidad de Buenos Aires, Consejo Nacional de Investigaciones Científicas y Técnicas, Instituto de Investigaciones Biomédicas en Retrovirus y Sida (INBIRS), Facultad de Medicina, Buenos Aires, Argentina
Trypanosoma cruzi infection induces an intense inflammatory response in diverse host tissues. The immune response and the microvascular abnormalities associated with infection are crucial aspects in the generation of heart damage in Chagas disease. Upon parasite uptake, macrophages, which are involved in the clearance of infection, increase inflammatory mediators, leading to parasite killing. The exacerbation of the inflammatory response may lead to tissue damage. Peroxisome proliferator-activated receptor gamma (PPARγ) is a ligand-dependent nuclear transcription factor that exerts important anti-inflammatory effects and is involved in improving endothelial functions and proangiogenic capacities. In this study, we evaluated the intermolecular interaction between PPARγ and a new synthetic PPARγ ligand, HP24, using virtual docking. Also, we showed that early treatment with HP24, decreases the expression of NOS2, a pro-inflammatory mediator, and stimulates proangiogenic mediators (vascular endothelial growth factor A, CD31, and Arginase I) both in macrophages and in the heart of T. cruzi-infected mice. Moreover, HP24 reduces the inflammatory response, cardiac fibrosis and the levels of inflammatory cytokines (TNF-α, interleukin 6) released by macrophages of T. cruzi-infected mice. We consider that PPARγ agonists might be useful as coadjuvants of the antiparasitic treatment of Chagas disease, to delay, reverse, or preclude the onset of heart damage.
Introduction
Chagas disease (American trypanosomiasis) is caused by the protozoan parasite Trypanosoma cruzi. The acute phase of infection is characterized by the presence of parasites in the host bloodstream and diverse tissues.
Acute T. cruzi infection is characterized by parasite invasion of the heart and other organs. Monocytes that are recruited from blood to the heart differentiate into macrophages that mediate the control of the parasite load. Macrophages produce pro-inflammatory mediators in situ, such as nitric oxide (NO), TNF-α, and interleukin 6 (IL-6), that inhibit T. cruzi multiplication and differentiation, precluding the spread of the infection within the host (1). In this regard, some studies have shown that mice administered iNOS inhibitors exhibit higher parasite levels and greater mortality than untreated mice (2, 3). Also, other authors have shown the relevant role of TNF-α in protection of mice during the acute T. cruzi infection (4–6).
On the other hand, a pro-inflammatory response may precipitate pathological conditions. In Chagas disease, the antigenic stimuli persist for years. The expression of pro-inflammatory mediators such as tumor necrosis factor-alpha (TNF-α), IL-6, interleukin 1 beta (IL-1β), and nitric oxide synthase 2 (NOS2) is associated with progressive tissue damage, leading to cardiac pathological conditions (7–11).
The infection-associated immunopathology and microvascular abnormalities are crucial aspects in the generation of heart disease, which is characterized by myocytolysis, thromboembolism, dysrhythmia, and cardiac hypertrophy (12). The progression of the disease is favored by these features, which lead to a gradual reduction in coronary flow. In this microenvironment M2 macrophages might play a crucial role, because they contribute to a range of physiological processes, including homeostasis, repair, metabolic functions, and angiogenesis, by secreting a plethora of proangiogenic factors like vascular endothelial growth factor A (VEGF-A), CD31, NOS3, and other cytokines, which increase the proliferation of endothelial and epithelial cells to induce neovascularization (13–15).
Peroxisome proliferator-activated receptor gamma (PPARγ) is a ligand-dependent transcription factor of the nuclear receptor superfamily, involved in the regulation of lipid metabolism, insulin sensitivity, and inflammatory response (16). It has been suggested that PPARγ is involved in the molecular mechanisms that regulate neoangiogenesis, through the action of growth factors and cytokines that stimulate migration, proliferation, and survival of endothelial cells (17). PPARγ ligands enhance VEGF-A expression in human vascular smooth muscle cells and upregulate NOS3 expression in myocardial infarction (18, 19). Particularly, it has been demonstrated that troglitazone enhances the expression of VEGFR-2 in HUVEC cells through PPARγ activation (20). However, some studies have shown that PPARγ and PPARα activation inhibits angiogenesis in vitro and in vivo, affecting vascular remodeling and leading to reduction of tumor cell growth (21). Thus, it is important to undertake studies to increase the knowledge about the possible role of PPARγ receptor and its ligands in heart angiogenesis, particularly in Chagas disease.
The new PPARγ synthetic ligand 3-hydroxy-4-pyridinecarboxylic acid derivative 24 (HP24), an aza-analog of salicylic acid and structurally close to other potent anti-inflammatory pyridine compounds, has been tested in dextran sulfate sodium-induced colitis in mice, where it showed a significant decrease in colonic myeloperoxidase activity and IL-1β tissue levels, exhibiting its anti-inflammatory activity without cytotoxic activity (22).
Pharmacological interventions leading to enhanced vascular development and reduction of inflammation and fibrosis might be useful to prevent heart functional abnormalities. The aim of this study was to determine the effect of the new PPARγ ligand HP24 in angiogenesis and in the levels of inflammatory mediators and to analyze the participation of macrophages in these processes.
Materials and Methods
Ethics Statement
The BALB/c mice used in this study were bred and maintained in the animal facility at the Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM), Universidad de Buenos Aires–CONICET. All the procedures were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, Facultad de Medicina, Universidad de Buenos Aires, CD No. 2271/2014) and are in accordance with the guidelines of the Argentinean National Administration of Medicines, Food and Medical Technology (ANMAT), Argentinean National Service of Sanity and Agrifoods Quality (SENASA) and also based on the US NIH Guide for the Care and Use of Laboratory Animals.
Mice and Infection
All mice were provided with a 12-h day/night cycle and water and food ad libitum with a standard diet. Seven male mice per group were infected intraperitoneally with 1 × 105 bloodstream trypomastigotes of a lethal RA (pantropic/reticulotropic) subpopulation of T. cruzi (23) and sacrificed by CO2 inhalation at 10 days postinfection (dpi). Each experiment was carried out at least three times.
Synthesis of 1-Methyl-3-Hydroxy-4-Pyridinecarboxylic Acid Derivative 24 (HP24)
1-Methyl-3-hydroxy-4-pyridinecarboxylic acid derivative was resynthesized following the previously reported pathway by Brun et al. (22) with some modifications in the reaction conditions for the final steps of the synthesis and the purification step that led to the desired compound HP24 in the zwitterion form (Figure 1A) instead of the previously described chloride compound. 3-Hydroxy-isonicotinic acid (1 g, 7.18 mmol) was suspended in 5 ml of DMF in a 25-ml round bottomed flask. The resulting suspension was stirred at room temperature, and 10% NaOH (7.5 ml) was added dropwise until complete dissolution of the solid (pH 9–10). Methyl iodide (2.06 g, 14.46 mmol, d = 2.28 g/ml, 0.9 ml) was added under stirring and the solution was then refluxed, monitoring the reaction progress by thin layer chromatography (n-butanol:H2O:AcOH, 1:1:1). Once the starting material disappeared, the solvent was removed under reduced pressure, obtaining a deep orange colored solid, which was dissolved in boiling water (50 ml). The solution was acidified with 37% HCl (3.5 ml), and 10% H2O2 (1 ml) was added. Then, the iodine was exhaustively extracted with CHCl3 (5× 15 ml) in a separating funnel. The organic phase was concentrated under pressure to dryness, obtaining an orange crude raw powdery solid (1.662 g), which was purified by reversed-phase chromatography in a Biotage Isolera Spektra Flash Chromatography apparatus equipped with prepacked C18 cartridges. The fractions containing the product were pooled and concentrated to dryness by means of a rotary evaporator, yielding a white powdery product (0.956 g, 6.21 mmol).
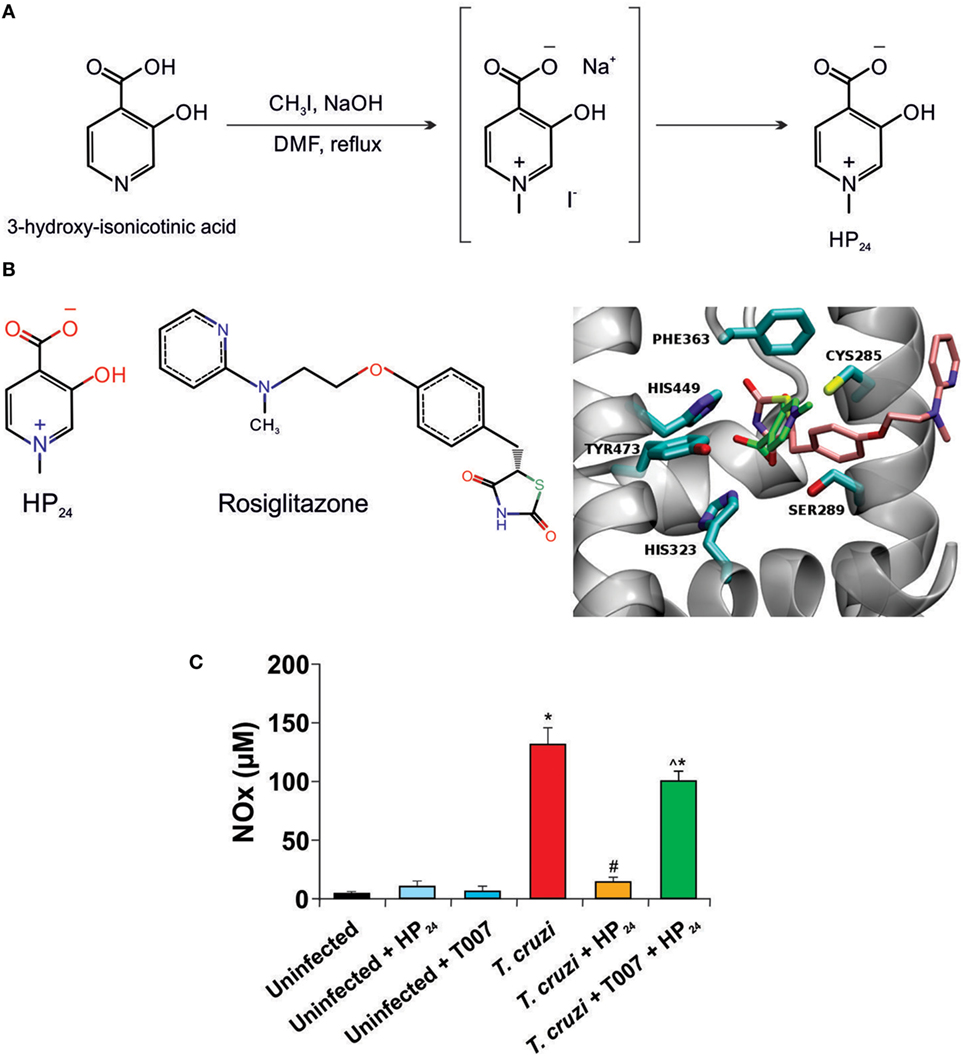
Figure 1. HP24 interacts with peroxisome proliferator-activated receptor gamma (PPARγ). (A) Route for HP24 synthesis. (B) Predicted binding mode for HP24 to PPARγ. On the right panel, we show rosiglitazone (RSG) from complex crystallographic structure (PDB id 2PRG) superimposed to most probably conformation of HP24 compound from docking calculations. RSG, HP24, and receptor binding site amino acids are shown in stick representation, colored by atom type (oxygen, nitrogen, sulfur in red, blue, and yellow, respectively) except carbons, which are colored in pink, green, and cyan respectively. Receptor backbone is shown in gray with New Ribbon representation. On the left panel, we show chemical formula of RSG and HP24. The images were generated using VMD software (24). (C) Peritoneal macrophages from Trypanosoma cruzi-infected mice were preincubated with T0070907 (50 µM), a PPARγ specific antagonist, for 30 min. Then, incubated with HP24 (100 µM). After 48 h, nitrite accumulation was measured in the cultured medium. For panel (C), results represent the mean ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. uninfected cells, #P < 0.05 vs. T. cruzi-infected cells. ^P < 0.05 vs. T. cruzi-infected HP24-treated cells.
3-Hydroxy-1-Methylpyridin-1-Ium-4-Carboxylate (HP24)
Yield: 86.4%; mp: 236°C (decomposition); Rf: 0.13 (n-butanol:H2O:AcOH, 1:1:1); IR (KBr): ν (cm−1) = 3,432 (OH), 3,079 (=C–H), 2,850 (CH3), 1,654 (COO−), 1,480 (C=C), 1,381 (C=N), 1,300 (C–N) cm−1; 1H NMR (300 MHz, [D6] DMSO) δ = 8.43 (s, 1H, H-2), 8.01 (d, J = 6.00 Hz, 1H, H-6), 7.98 (d, J = 6.03 Hz, 1H, H-5), 4.19 ppm (s, 3H, N-CH3); 13C NMR (75 MHz, [D6] DMSO) δ = 47.98 (N-CH3), 126.85 (C-5), 129.73 (C-4), 130.72 (C-6), 137.54 (C-2), 164.99 (C-3), 166.95 ppm (COO−); HRMS (ESI-MS, 140 eV): m/z [M + H+] calculated for , 154.0504; found, 154.0545; RT-HPLC, C18: tR = 5.40 min, 97.61 A%; elemental analyses: calculated for C7H7NO3, C 54.9%, H 4.61%, N 9.15%; found: C 54.47%, H 4.39%, N 8.98%.
Treatment of Mice with HP24
Mice were treated by oral gavage with HP24 (400 mg/kg/day) suspended in phosphate-buffered saline (PBS), according to Brun et al. (25), for 9 days, since day 1 postinfection (pi), and sacrificed at day 10 pi. Euthanasia was carried out at this time for humanitarian reasons due to the unhealthy conditions of the mice infected with the RA parasite strain.
Parasitemia, Body Weight, and Survival of Mice
Parasitemia of T. cruzi-infected mice was analyzed through a small incision at the end of the tail. Blood from T. cruzi-infected mice and T. cruzi-infected HP24-treated mice was obtained at 3, 5, 8, and 10 dpi. Fivefold dilutions were obtained in red blood cell lysis buffer (150 mM NH4Cl, 0.1 mM EDTA, and 10 mM KHCO3, pH 7.4). Parasitemia was measured in a Neubauer chamber. Body weight gain or loss was monitored on the days described previously. For survival studies, two independent groups (T. cruzi-infected and T. cruzi-infected HP24-treated mice) of seven animals each were followed up daily until different days postinfection and analyzed by the Kaplan–Meyer method.
Molecular Modeling
To elucidate the binding mode of the HP24 molecule, docking calculations were performed against a murine PPARγ receptor. First, we built an homology model with MODELLER software (26) using human PPARγ structures as template from Protein Data Bank (http://www.pdb.org), PDB id 5DV6 and 2PRG (27). Then, we built and optimized HP24 ligand structure with Molefacture module of VMD software (24) employing default settings for convergence criteria. Finally, AutoDock4 software (28) with Water Site Biased Method (29) was used for docking experiments. The PPARγ binding site (BS) was defined as all atoms within a radius of 10 Å from the co-crystallized ligand rosiglitazone (RSG, PDB id 2PRG). The receptor BS amino acids were treated as rigid structures during the calculations. A total of 100 different docking runs were performed, and the results were clustered according to the ligand-heavy atom RMSD using a cutoff of 2 Å. The genetic algorithm parameters for each conformational search run were kept at their default values. The AutoDock4.2 energy function (OA atom type map) was modified adding an additional energy term for each crystallographic water (CWS) placed on ABS BS in the apo form (PDB id 1PRG) to the original function (29).
Isolation of Peritoneal Macrophages
Macrophages were obtained by washing the peritoneal cavity of mice with 8 ml of RPMI-1640 culture medium (Invitrogen Life Technologies, Grand Island, NY, USA), supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (Internegocios S.A., Argentina) and antibiotics (50 µg/ml of penicillin, streptomycin, and gentamicin). Cells were left to adhere to the plastic surface of cell culture dishes, 35 mm × 10 mm (Greiner Bio One International AG) for 3 h at 37°C under a 5% CO2 atmosphere (30).
Treatment with HP24 and T0070907 In Vitro
Peritoneal macrophages were isolated from T. cruzi-infected mice as indicated above. The cells were preincubated for 30 min with specific PPARγ antagonist, T0070907 (50 µM in DMSO) (Sigma-Aldrich Co., St. Louis, MO, USA), and treated afterward with HP24 (100 µM in PBS) for 48 h. Cell viability was examined by Trypan Blue dye exclusion test, for each treatment.
Measurement of NO Production by Peritoneal Macrophages In Vitro
Release of NO by peritoneal macrophages from T. cruzi-infected mice was assessed by the Griess reaction, as previously described (31). The absorbance at 540 nm was compared with a standard curve of NaNO2.
Macrophage-Induced Angiogenesis
Macrophage-induced angiogenesis was quantified with an in vivo bioassay. After detaching, the concentration of macrophages was adjusted to 2 × 105 cells/ml in culture medium without FBS. Seven normal syngeneic male mice per group were inoculated intradermally in both flanks with 0.1 ml of cell suspension. Five days after inoculation, mice were sacrificed, and the internal layer of skin was separated from the underlying tissues, and the vascular response was observed with a dissecting microscope (Konus USA Corporation, Miami, FL, USA) at a 7.5× magnification and photographed with an incorporated digital camera (Canon Power Shot A45, Canon USA, Inc., Lake Success, NY, USA). Photos were projected on a reticular screen to count the number of vessels per square millimeters of skin. Angiogenesis was quantified as vessel density, calculated as the total number of vessels divided by the total number of squares (32).
Coculture of Macrophages with Heart Explants
Macrophages (1.5 × 106) from uninfected, T. cruzi-infected and T. cruzi-infected HP24-treated mice were obtained and cultured with heart slices (100 mg/sample) from uninfected and T. cruzi-infected mice in 4 ml RPMI-1640 culture medium (Invitrogen Life Technologies, Grand Island, NY, USA). After 48 h, the culture supernatants were collected, and hearts were homogenized to obtain total proteins as previously described (33).
Histopathological Studies
Hearts of all experimental groups were fixed in 4% paraformaldehyde in PBS, dehydrated and embedded in paraffin. Six non-contiguous sections (5 µm) were stained with hematoxylin–eosin or Masson trichrome stain. Cellular infiltrates, presence of amastigote nests and collagen deposition were examined in using a Nikon Eclipse E600 microscope (Nikon Inc.). Images were captured with a Spot RT digital camera. At least 30 random microscopic fields (400×) were analyzed in each microscopic section, using the open source Image J software (NIH, USA) (34).
Quantitative Real-time Reverse-Transcriptase Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from frozen cells by using a QuickZol reagent (Kalium Technologies, Buenos Aires-Argentina). Total RNA was reverse-transcribed using Expand Reverse Transcriptase (Invitrogen Corp., MA, USA). RT-qPCR was performed using a 5× HOT FIREPol® EvaGreen® qPCR Mix Plus (ROX) (Solis BioDyneCorp., Estonia) in an Applied Biosystems 7,500 sequence detector. Primer sequences were: 18S: Fw 5′AACACGGGAAACCTCACCC 3′, Rv 5′ CCACCAACTAAGAACGGCCA 3′; connective tissue growth factor (CTGF): Fw 5′ CCTAAAATCGCCAAGCCTGT 3′, Rv 5′ CACCCCGCAGAACTTAGCC 3′, and PPARγ: Fw 5′ ATCTACACGATGCTGGC 3′, Rv 5′ GGATGTCCTCGATGGG 3′; PCR parameters were 52°C for 2 min, 95°C for 15 min, and 40 cycles of 95°C for 30 s and 60°C (for 18S), 63°C (for CTGF) or 54°C (for PPARγ). Quantification was calculated using the comparative threshold cycle (Ct) method and the efficiency of the RT reaction The replicates were then averaged, and fold induction was determined, considering the value at time 0 as 1 (35).
Determination of Cytokine Levels
TNF-α and IL-6 levels in culture supernatants were quantified by enzyme-linked immunosorbent assays using DuoSet antibody pairs (R&D Systems, Minneapolis, MN, USA).
Preparation of Total Protein Extracts for Western Blot
Total protein extracts were obtained after washing the hearts with PBS and adding 300 ml of RIPA modified lysis buffer (50 mM NaCl, 50 mM Tris–HCl (pH 7.40), 1% Triton X-100, 1 mM EDTA, 1 mM PMSF; 2.5 g/l Protease Inhibitor Cocktail (Sigma-Aldrich Co., St. Louis, MO, USA), 1 mM Na3VO4, 1 mM NaF), or washing the cultured cells and scraped off the dishes with 50 µl of the same buffer. Then, the tubes were kept on ice for 30 min with swirling, and the samples were centrifuged at 7,000 g at 4°C for 10 min. The supernatants were stored at −20°C. Protein concentrations were determined by the Bradford method using the Bio-Rad Protein Assay (Bio-Rad, USA) and bovine serum albumin (Sigma-Aldrich Co., St. Louis, MO, USA) as a standard (36). For Western blot analysis, total proteins were boiled in Laemmli sample buffer, and equal amounts of protein (40–50 µg) were separated by 10–12% SDS-PAGE. The gels were blotted onto a Hybond-P membrane (GE Healthcare, Madrid, Spain) and incubated with the following antibodies: anti-NOS2, anti-NOS3, anti-Arginase I (Arg-I), anti-CD31, anti-VEGF-A, and anti-α-actin (Santa Cruz Biotechnology, CA, USA). The blots were revealed by enhanced chemiluminescence in an Image Quant 300 cabinet (GE Healthcare Biosciences, PA, USA) following the manufacturer’s instructions. Band intensity was analyzed using the NIH-ImageJ software (37).
Statistical Analysis
Data are expressed as the mean of three independent experiments ± SEM for each treatment (seven mice/group). The Kaplan–Meier test was used to compare survival curves between groups. One-way ANOVA was used to analyze the statistical significance of the differences observed between the uninfected, uninfected HP24-treated, T. cruzi-infected, and T. cruzi-infected HP24-treated mice. The Tukey post hoc test was performed to compare all pairs of groups. Kruskal–Wallis test and Dunn’s post hoc test was used to analyze the differences in collagen deposition between uninfected, T. cruzi-infected, and T. cruzi-infected HP24-treated mice. Differences were considered statistically significant when P < 0.05. All analyses were performed using the Prism 5.01 software (GraphPad, USA).
Results
The New Pyridinecarboxylic Acid Derivative 24 (HP24) Interacts with PPARγ
Selection of the 3,4-pyridinecarboxylic derivative HP24 was based on its properties as PPARγ ligand and on its ability to reduce the pro-inflammatory response in a Dextran-induced colitis mouse model, as previously reported (22). A simplified synthesis route is shown in Figure 1A.
To elucidate how HP24 interacts with PPARγ, we compared its binding against that of co-crystallized structure of synthetic PPARγ ligand, rosiglitazone (RSG). Figure 1B illustrates the superimposition of the best energy docking result for HP24 (estimated free energy of binding −4.05 kcal/mol), as well as the experimental position of the co-crystallized RSG (estimated free energy of binding −8.95 kcal/mol), present in the 2PRG PDB structure. The most important interaction between RSG and PPARγ is a hydrogen bond network with residues GLN286, SER289, HIS323, and TYR473. Hydrophobic contacts with LEU330, ILE341, MET364, and CYS285 are considered as secondary interactions (27). Besides, a detailed visual inspection of the binding modes for HP24 reveals the same hydrogen bond network, adding an extra-polar contact with HIS449 in the hydrophilic region of PPARγ BS (Figure 1B, panel right).
Since ligand binding can be inhibited by specific antagonists, we designed an in vitro assay to confirm that HP24 binds to PPARγ as modeled by the in silico binding analysis. This involved the use of NO release by T. cruzi-infected peritoneal macrophages, as an indicator system of the inhibitory effect of HP24 on pro-inflammatory mediators release. T. cruzi-infected macrophages release significantly higher amounts of NO than uninfected macrophages. Moreover, HP24 significantly reduced the release of this pro-inflammatory mediator. Notably, preincubation of T. cruzi-infected macrophages with the specific PPARγ antagonist, T0070907 impeded the effect of HP24 on NO release, thus confirming the specific binding and activation of PPARγ by HP24 (Figure 1C).
As previously reported by our group, infection of mice with T. cruzi increases PPARγ expression in the heart and in peritoneal macrophages (30, 38). In this study, we tested the effect of HP24 treatment on PPARγ mRNA levels in macrophages and hearts of uninfected and T. cruzi-infected mice. Figure 2 shows that HP24 treatment increased the PPARγ mRNA expression in uninfected mice, but did not modify the already increased expression in T. cruzi-infected mice.
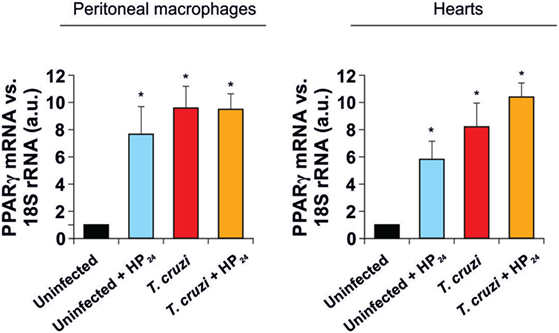
Figure 2. Peroxisome proliferator-activated receptor gamma (PPARγ) expression. PPARγ mRNA expression was analyzed by quantitative real-time reverse-transcriptase polymerase chain reaction in peritoneal macrophages and heart homogenates from uninfected and Trypanosoma cruzi-infected mice, treated or not with HP24. Results were normalized against 18S rRNA. Results represent the mean ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice.
HP24 Treatment Does Not Affect the Course of Infection
We evaluated whether the treatment with HP24 modified the parasitemia levels, weight and survival of T. cruzi-infected mice. These parameters were evaluated at 3, 5, 8, 10, and 13 dpi. We found no differences in parasitemia levels, weight or survival between T. cruzi-infected and T. cruzi-infected HP24-treated mice (Table 1). These results are consistent with those previously reported by our group with other PPARγ ligands (34).
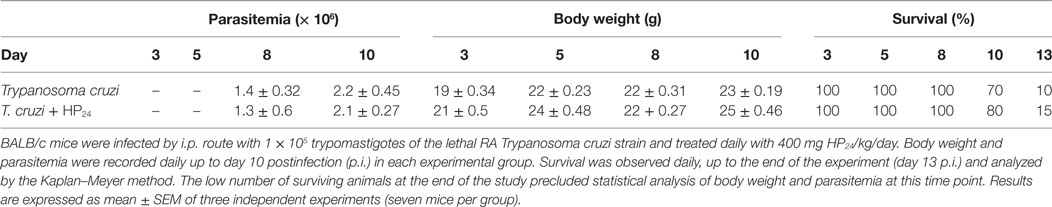
Table 1. Effect of HP24 on the course of infection.
Effect of HP24 on T. cruzi-Infected Macrophages
We have previously demonstrated that PPARα and PPARγ ligands promote the polarization of macrophages isolated from T. cruzi-infected mice toward an M2 profile (30).
Here, we evaluated the role of the new PPARγ ligand HP24 as an anti-inflammatory ligand. We determined its effect on the expression of NOS2 and pro-inflammatory cytokines. The treatment with HP24 significantly inhibited NOS2 expression as well as TNF-α and IL-6 secretion in macrophages from T. cruzi-infected mice (Figure 3A). The PPARγ agonist did not affect the secretion of pro-inflammatory cytokines in macrophages from uninfected mice (Figure 3A).
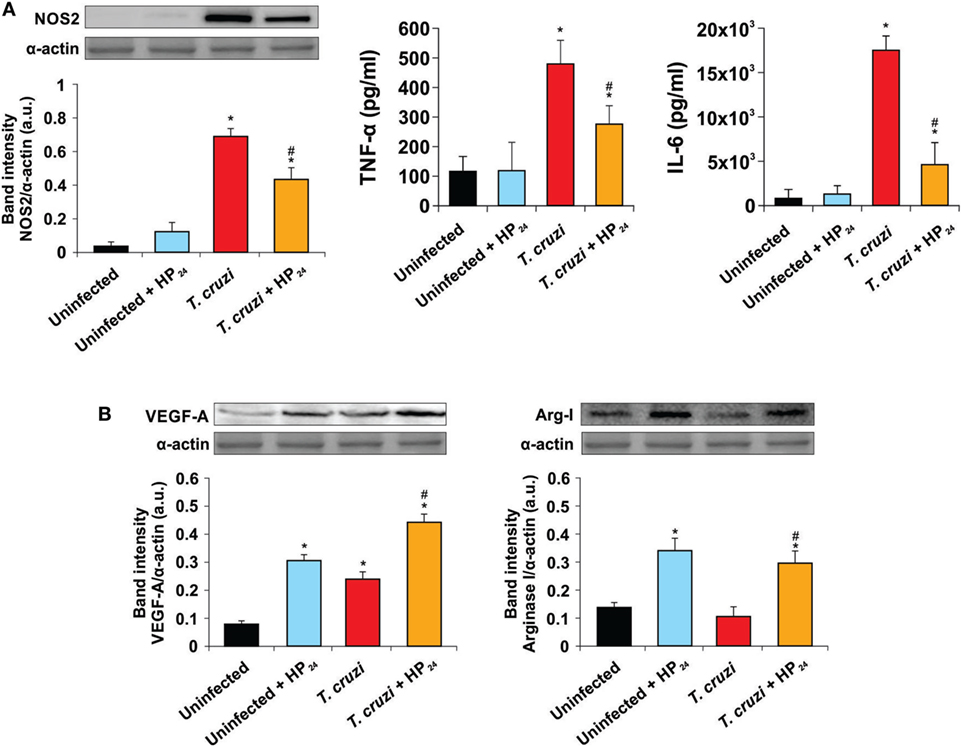
Figure 3. Effect of HP24 on Trypanosoma cruzi-infected macrophages. (A) Nitric oxide synthase 2 (NOS2) expression was determined by Western blot with a specific antibody. The release of TNF-α and interleukin 6 (IL-6) to the culture medium was quantitated by capture enzyme-linked immunosorbent assays. (B) Peritoneal macrophages were obtained from uninfected, uninfected HP24-treated, T. cruzi-infected, and T. cruzi-infected HP24-treated mice at 10 dpi. Vascular endothelial growth factor A (VEGF-A) and Arginase I expressions were determined by Western blot with a specific antibody. Protein levels were normalized against α-actin. Results represent the mean ± SEM band intensity of three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice, #P < 0.05 vs. T. cruzi-infected mice.
We also analyzed whether HP24 was able to promote the participation of macrophages in tissue repair and neovascularization processes. To this end, we analyzed the expression of representative proangiogenic markers like VEGF-A and Arg-I in peritoneal macrophages of T. cruzi-infected mice. Both T. cruzi infection and HP24 treatment significantly increased VEGF-A expression in peritoneal macrophages with respect to uninfected and untreated mice. This effect was potentiated in macrophages from T. cruzi-infected HP24-treated mice (Figure 3B). However, HP24 was able to significantly increase the expression of Arg-I, both in T. cruzi-infected macrophages and in uninfected control cells (Figure 3B).
Participation of HP24 in Macrophage-Induced Angiogenesis
The formation of new blood vessels requires the sprouting of preexisting ones and their subsequent fusion with others (39). It has been previously reported that peritoneal macrophages from tumor-bearing mice are able to induce a strong neovascular response in the skin of syngeneic normal mice (32). Based on these results, we investigated whether HP24 treatment modulated the ability of peritoneal macrophages to induce neovascularization in the skin of normal syngeneic mice. Peritoneal macrophages from T. cruzi-infected donor mice increased vessel density in comparison with macrophages from uninfected donors. Furthermore, macrophages from T. cruzi-infected mice-treated in vitro with HP24 induced a higher increase in neovascularization than macrophages from T. cruzi-infected donor mice, upon passive transfer in the skin of normal syngeneic mice (Figure 4A). To assess the participation of PPARγ in the effects of macrophage-induced neovascularization, the cells were pretreated with the PPARγ antagonist T0070907 and then treated with HP24. As shown in Figure 4A the PPARγ antagonist reduced the effect of HP24 on new vessel formation in the skin of syngeneic normal mice.
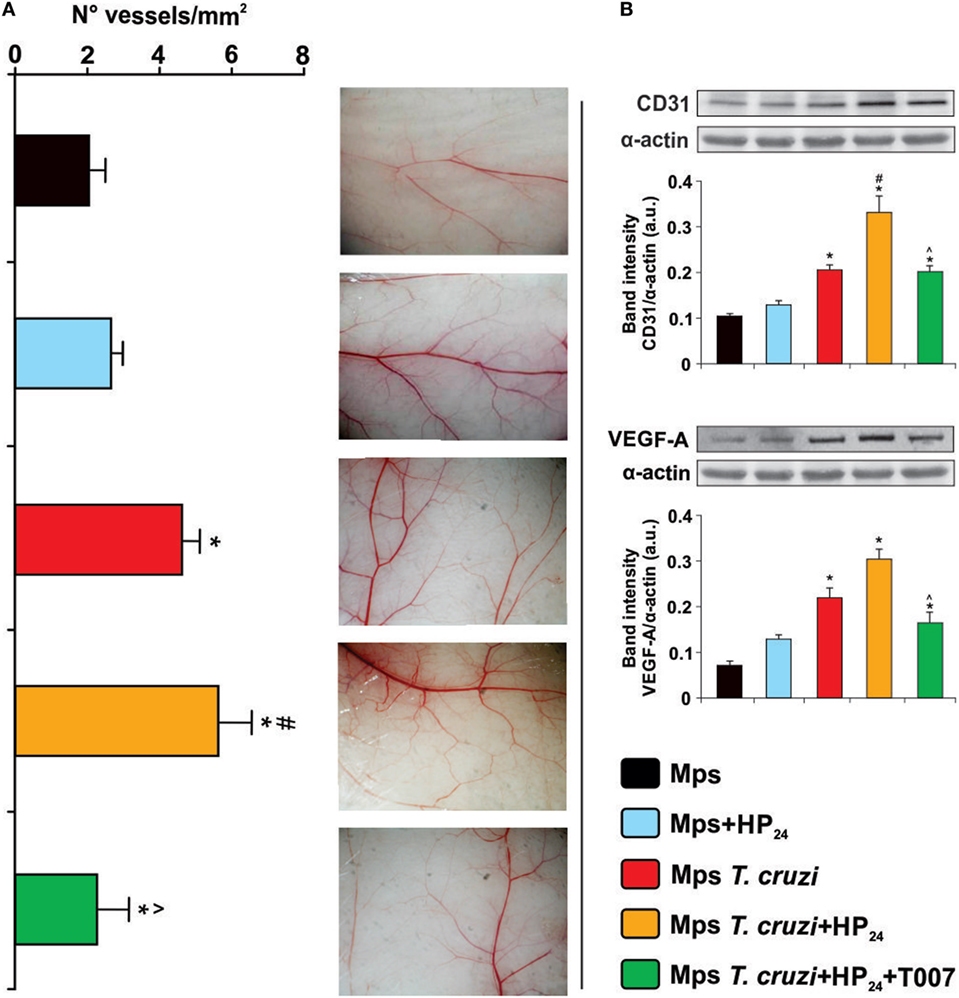
Figure 4. Participation of HP24 in macrophage-induced angiogenesis. (A) Macrophage-induced angiogenesis in mice was determined in vivo by quantification of newly formed vessels in the skin. Peritoneal macrophages from different groups were pretreated with the peroxisome proliferator-activated receptor gamma antagonist T0070907 and treated with HP24. Then, the cells were harvested and adjusted to 2 × 105 cells/ml. One-tenth milliliters of the cell suspension was inoculated by intradermal route in both flanks of syngeneic normal mice. Five days after inoculation, recipient mice were sacrificed, the inner layer of the skin exposed, and photographs were acquired. Images were projected on a reticular screen to count the number of vessels per square millimeters. Angiogenesis was quantified as the vessel density, calculated as the total number of vessels divided by the total number of squares. (B) CD31 and Vascular endothelial growth factor A (VEGF-A) expression were determined by Western blot with a specific antibody. Anti-CD31 and anti-VEGF-A-specific antibody were used. Protein levels were normalized against α-actin. Results are mean band intensity ± SEM for three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice macrophages, #P < 0.05 vs. Trypanosoma cruzi-infected mice macrophages. ^P < 0.05 vs. HP24-treated macrophages from T. cruzi-infected mice. Mps, macrophages.
Blood vessel density positively correlated with CD31 and VEGF-A expression. Western blot analysis showed higher expression of CD31 and VEGF-A in skin extracts of normal syngeneic recipient mice, upon transfer of macrophages from T. cruzi-infected mice treated in vitro with HP24, than upon transfer of macrophages from T. cruzi-infected mice. The PPARγ antagonist T0070907 significantly reduced the effects of HP24 on the expression of CD31 and VEGF-A in the skin of normal syngeneic recipient mice upon passive transfer of macrophages from T. cruzi-infected mice (Figure 4B).
As expected, CD31 and VEGF-A expression was higher in the skin of recipient mice transferred with macrophages from T. cruzi-infected mice than in mice transferred with macrophages from uninfected controls (Figure 4B).
Effect of Macrophages from T. cruzi-Infected HP24-Treated Mice on the Expression of Proangiogenic Proteins in Heart Explants
To investigate whether macrophages from T. cruzi-infected HP24-treated mice participate in heart angiogenesis, we cocultured macrophages from these mice with heart explants from T. cruzi-infected mice. Then, we evaluated the expression of proangiogenic markers in those explants. Macrophages from T. cruzi-infected mice induced an increase in VEGF-A expression in heart explants, whereas macrophages from control mice were unable to modify VEGF-A expression in the same explants (Figure 5). When heart explants were cocultured with macrophages from T. cruzi-infected mice treated in vitro with HP24, the expression of VEGF-A, CD31 and Arg-I was further increased (Figure 5).
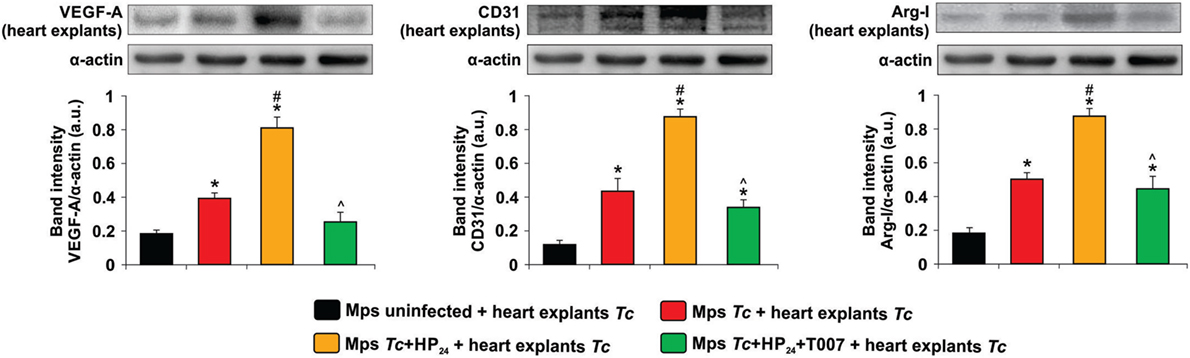
Figure 5. Effect of Trypanosoma cruzi-infected macrophages from untreated or HP24-treated mice on the expression of proangiogenic proteins in T. cruzi-infected heart explants. Peritoneal macrophages were obtained from uninfected and T. cruzi-infected mice at 10 dpi. Macrophages were either pretreated or not with T0070907 and then treated with HP24. Then, the cells were cocultured with heart explants (100 mg/sample) from T. cruzi-infected mice. After 48 h, heart explants were collected and homogenized. Vascular endothelial growth factor A (VEGF-A), CD31, and Arginase I (Arg-I) expressions were determined by Western blot with specific antibodies. Protein levels were normalized against α-actin. Results represent the mean band intensity ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. macrophages from uninfected mice + heart explants from T. cruzi-infected mice, #P < 0.05 vs. macrophages from T. cruzi-infected mice + heart explants from T. cruzi-infected mice. ^P < 0.05 vs. HP24-treated macrophages from T. cruzi-infected mice + heart explants from T. cruzi-infected mice. Mps, macrophages; Tc, T. cruzi.
Moreover, the effect of HP24 on the expression of VEGF-A, CD31 and Arg-I in heart explants was reverted when the macrophages from T. cruzi-infected mice were pretreated with the PPARγ antagonist T0070907 (Figure 5).
The same pattern was observed when macrophages were cocultured with heart explants from uninfected mice although the expression levels were lower (Figure S1 in Supplementary Material). Besides, the expression of CD31, VEGF-A and Arg-I did not differ in heart explants from T. cruzi-infected mice cultured alone or in the presence of macrophages from uninfected control mice (Figure S2 in Supplementary Material).
HP24 Treatment Reduces Inflammatory Response and Heart Fibrosis in T. cruzi-Infected Mice
We investigated the effects of HP24 treatment on the inflammatory response in the heart of T. cruzi-infected mice. Mice infected with T. cruzi showed intense inflammatory reaction, consisting of mononuclear cell infiltrates. Treatment with HP24 significantly reduced heart inflammation (number of inflammatory foci/field, T. cruzi vs. T. cruzi-HP24, 0.84 ± 0.41 vs. 0.21 ± 0.05, N = 5, P < 0.05, Figure 6). We did not find significant differences in the number of amastigote nests per field between the T. cruzi-infected and T. cruzi-infected HP24-treated groups (Figure 6).
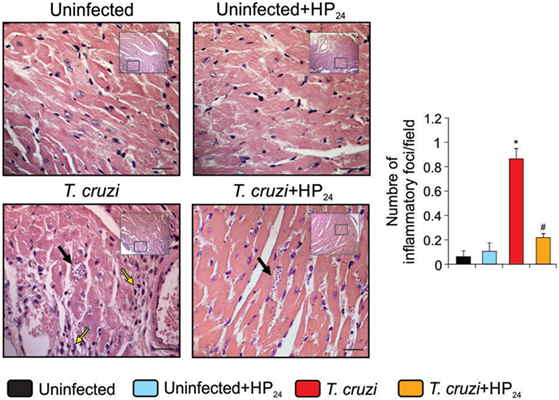
Figure 6. HP24 reduces heart inflammatory reaction in Trypanosoma cruzi-infected mice. Heart inflammatory reaction and parasite nests were analyzed at 10 dpi in histological sections of T. cruzi-infected, T. cruzi-infected HP24-treated, and uninfected mice, stained with hematoxilin–eosin (bar: 100 µm, yellow arrows, inflammatory infiltrates, black arrows, amastigote nests). Results represent the mean ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice, #P < 0.05 vs. T. cruzi-infected mice.
Fibrosis was observed in heart sections of T. cruzi-infected mice using Masson’s trichrome staining (Figure 7A). The heart area compromised by collagen deposits was significantly reduced in T. cruzi-infected HP24-treated mice (Figure 7B). In addition, we analyzed the mRNA expression of CTGF as a profibrotic marker. RT-qPCR showed that CTGF expression levels were higher in the hearts from T. cruzi-infected mice than in those from uninfected mice. Interestingly, treatment with HP24 significantly inhibited CTGF mRNA in infected mice (Figure 7C).
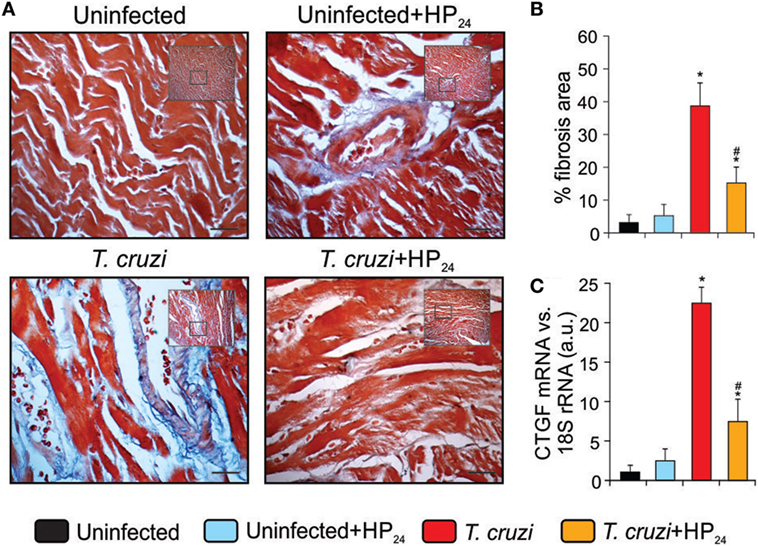
Figure 7. Reduction of heart fibrosis by HP24 in Trypanosoma cruzi-infected mice (A).Collagen deposits in heart sections of the experimental groups were assessed using Masson’s trichrome (bar: 100 µm). (B) The bar graph shows the percentage of area with fibrosis. (C) Connective tissue growth factor (CTGF) mRNA expression was analyzed by quantitative real-time reverse-transcriptase polymerase chain reaction in heart homogenates from all experimental groups. Results were normalized against 18S rRNA. All studies were performed at 10 dpi. Results represent the mean ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice, #P < 0.05 vs. T. cruzi-infected mice.
HP24 Treatment Increases the Expression of Proangiogenic Markers in the Heart of T. cruzi-Infected Mice
To demonstrate the proangiogenic role of the HP24 treatment in the hearts of T. cruzi-infected mice, we evaluated the cardiac expression of CD31, VEGF-A, NOS3, and Arg-I. Western blot analysis showed increased expression of CD31 and VEGF-A in hearts upon infection while that of NOS3 and Arg-I remained unchanged. Treatment with HP24 promoted the increase of all proangiogenic markers in the heart of T. cruzi-infected mice. (Figure 8A). Moreover, HP24 treatment in vivo reduced the expression of NOS2 in the hearts of T. cruzi-infected mice in comparison with untreated T. cruzi-infected mice (Figure 8B).
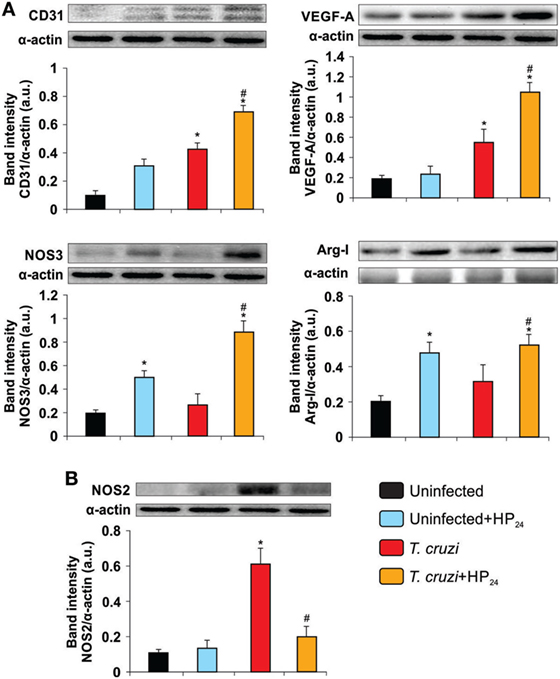
Figure 8. HP24 increases the expression of proangiogenic markers and decreases nitric oxide synthase 2 (NOS2) in the heart of mice infected with Trypanosoma cruzi. Expression of CD31, vascular endothelial growth factor A (VEGF-A), NOS3, Arginase I (Arg-I) (A), and NOS2 (B) was determined in the heart by Western blot. Anti-CD31, anti-VEGF-A, anti-NOS3, anti-Arg-I, and anti-NOS2 specific antibodies were used, and protein levels were normalized against α-actin. Results show a representative experiment out of three performed. Results represent the mean band intensity ± SEM of three independent experiments (five mice per group). *P < 0.05 vs. uninfected mice, #P < 0.05 vs. T. cruzi-infected mice.
Discussion
In this study, we analyzed the role of the new synthetic PPARγ ligand HP24 (22) in cardiac damage and neovascularization, in an experimental model of Chagas disease, considering the participation of macrophages. We demonstrated that HP24 treatment increased the expression of proangiogenic factors, inhibited pro-inflammatory mediators, and reduced fibrosis in the heart of infected mice. Moreover, we evidenced that HP24 potentiates the ability of macrophages to stimulate angiogenesis in our experimental model. PPARγ and its ligands have a wide spectrum of functions, regulating metabolism, attenuating inflammation, maintaining the balance of immune cells, inhibiting apoptosis and oxidative stress, and improving endothelial function (40). Results from different experimental models have shown that PPARγ and its ligands play a critical role in the regulation of various biological processes in the cardiovascular system under pathological conditions: they attenuate cardiac fibrosis in diabetic rats (41, 42), and alleviate ischemia–reperfusion injury through the inhibition of inflammation, improve endothelial function, reduce oxidative, stress, and calcium overload in rabbits (43).
Molecular modeling used in this work predicted that the main interactions between the PPARγ and HP24 differ little from another synthetic agonist, RSG. Our data suggest that HP24 has a lower number of non-polar contacts because its volume is lower than that of RSG. Only PHE363 and CYS285 are at a suitable distance for hydrophobic interaction. In addition, the carbon 2 of the pyridine ring is at an optimal distance for a possible nucleophilic attack by the CYS285. This may be determinant for the PPARγ agonist function, since such a covalent bond is widely described for the positive regulators of this receptor (44, 45). Thus, while the interaction free energy of HP24 with PPARγ is higher than that of RSG, the covalent binding that might be formed between CYS285 and the pyridine ring of HP24 would make the binding stable.
To evaluate the implication of PPARγ in HP24 effects, mouse peritoneal macrophages were pretreated with the specific PPARγ antagonist T0070907 and NO production was evaluated. Reversion of the inhibitory effects of HP24 on the release of NO, together with the docking analysis, strongly suggests the binding of HP24 to PPARγ.
In the present work, we demonstrated that HP24 treatment increases VEGF-A expression in macrophages from T. cruzi-infected HP24-treated mice. Our results are in agreement with those of Kotlinowski et al. (16), who showed that the activation of PPARγ by rosiglitazone increases the proangiogenic potential of endothelial cells and of bone marrow-derived proangiogenic cells (16). In this regard, Biscetti et al. found that the activation of PPARγ and PPARα receptors stimulates neoangiogenesis through a VEGF-dependent mechanism (17).
We have previously demonstrated that the treatment with Wy14643 and 15dPGJ2 (PPARα and PPARγ ligands, respectively) drives peritoneal macrophages toward an M2 profile in T. cruzi-infected mice (30). In view of these results, we analyzed whether the treatment with HP24 could influence the profiling of macrophages. We showed that macrophages from T. cruzi-infected HP24-treated mice upregulated Arg-I expression and downregulated NOS2 expression, processes that strongly suggest M2 differentiation. Recently, Assunção et al. demonstrated that M2 polarization of macrophages triggered by Schistosomal-derived lipids occurs through a PPARγ-dependent mechanism (46). Interestingly, Odegaard et al. showed that the promoter region of the Arg-I gene has PPARγ response elements (47).
We previously shown that 15dPGJ2 is a potent modulator of the inflammatory processes through PPARγ-dependent and -independent pathways, in cultures of T. cruzi-infected neonatal cardiomyocytes and T. cruzi-infected mice (37, 38). Here, we determined the ability of HP24 to exert anti-inflammatory actions by inhibiting the expression of a pro-inflammatory enzyme as NOS2 and releasing inflammatory cytokines such as TNF-α and IL-6 in macrophages from T. cruzi-infected mice. In the same line of evidence, Brun et al. (22) showed that the synthetic PPARγ agonist HP24 improves the outcome of dextran-induced colitis, by reducing colonic myeloperoxidase activity and IL-1β levels in the gut of mice. These authors also demonstrated that HP24 prevents LPS-induced TNF-α and IL-8 release in the same model (22).
During acute Chagas disease, oxidative stress associated with inflammation of the heart contributes to the tissue damage triggered by T. cruzi infection. This condition stimulates the release of a new set of proangiogenic mediators such as VEGF, CD31, angiopoietin (Ang)-1 and Ang-2 (48). We cannot discard that macrophages participate in this angiogenic process. Regarding the latter, our group reported that peritoneal macrophages from tumor-bearing mice are able to induce a strong neovascular response (32). In this study, we showed that passive transfer of macrophages from T. cruzi-infected mice treated in vitro with HP24 enhances angiogenesis, as shown by the increase of VEGF-A and CD31 expression in the skin of normal syngeneic recipient mice. This effect was dependent on PPARγ signaling, since pretreatment of macrophages with the PPARγ antagonist precluded the HP24-induced increase of CD31 and VEGF-A in skin homogenates of syngeneic normal recipient mice.
Recently, Guedes-da-Silva et al. (49) showed that antigens from the Y strain of T. cruzi are able to promote inflammatory neovascularization, probably induced by angiogenic mediators produced by macrophages (49). Furthermore, Shrestha et al. (50) demonstrated that T. cruzi infection increases the inflammatory and angiogenic mediators in the heart of infected mice (50). Taking into account that the evolution of Chagas disease is linked to microvascular lesions, including obstruction due to thrombosis, perivascular inflammation, and lesions in the coronary arterioles (33), we believe that the increased neovascularization observed during T. cruzi infection may act as a delaying factor of the physiopathological mechanisms leading to the cardiac symptoms during the course of Chagas disease.
Peritoneal macrophages from T. cruzi-infected mice treated in vitro with HP24 induced higher expression of VEGF-A, CD31, and Arg-I than macrophages from T. cruzi-infected mice in heart explants. As observed in the skin model, these findings suggest that HP24 plays a role in inducing macrophages from infected mice to promote cardiac angiogenesis. In fact, similarly to what was found in the case of passive transfer to the skin, the effect of HP24 on the expression of VEGF-A, CD31, and Arg-I depended on PPARγ, since pretreatment of macrophages with the PPARγ antagonist T0070907 significantly reduced the expression of proangiogenic markers in heart explant homogenates. Ashoff et al. demonstrated that treatment of diabetic rats with pioglitazone, a synthetic PPARγ agonist, increases the density of capillaries in heart and skeletal muscle (51). Besides, de la Torre et al. (33) demonstrated that LPS-treated macrophages from septic mice show increased CD31 and VEGF-A expression in the heart (33).
However, some studies have shown that PPARγ has antiangiogenic activity. Pigment epithelial-derived factor and its functional peptides inhibit angiogenesis by means of upregulation of PPARγ in a rat model of acute myocardial infarction and in an in vitro model of myocardial angiogenesis (52). Funovics et al. (53) and Kim et al. (54) have shown that PPARγ activation results in reduced expression of VEGF-R2 and VEGF-R1 in endothelial cells (53, 54). These seemingly contradictory findings highlight the need for additional studies to determine not only the mechanisms through which different PPAR ligands promote angiogenesis but also the physiopathological significance of the increase of the proangiogenic markers and mediators in the case of Chagas disease.
The myocardial infection caused by T. cruzi elicits an intense inflammatory response that may act as a “double-edged” sword. Although necessary for the control of parasite proliferation, inflammation results in tissue damage, leading to myocardial fibrosis and remodeling. CTGF, a fibrogenic cytokine, has hypertrophic effects, modulating the myocardial phenotype (55). In this regard, Koitabashi et al. (56) demonstrated that a disproportionate increase in CTGF expression in cardiac myocytes plays a central role in the induction of excessive myocardial fibrosis and diastolic heart failure (56). In this study, we showed that treatment of T. cruzi-infected mice with HP24 significantly reduces inflammatory response and heart fibrosis, and inhibits CTGF expression. We have recently reported that 15dPGJ2, a natural PPARγ ligand, is able to reduce the inflammatory response, fibrosis, and enzyme markers of liver damage in T. cruzi-infected mice (34). It has been shown that activation of PPARγ by pioglitazone can attenuate cardiac fibrosis and partly ameliorate cardiac remodeling and function by suppressing activity of RAS, in a rat model of diabetes (42). Here, we demonstrated that, besides reducing heart fibrosis, HP24 treatment induces the expression of proangiogenic markers (VEGF-A, CD31, Arg-I, and NOS3) and reduces that of NOS2, in the heart of T. cruzi-infected mice.
In conclusion, taking into account that during T. cruzi infection microvascular lesions contribute to ischemia and necrosis, the early treatment with drugs that reduce inflammation and fibrosis, and simultaneously increase microvascular development, might be useful as a coadjuvant of the antiparasitic treatment to delay, reverse, or preclude the onset of heart damage during the course of Chagas disease.
Ethics Statement
All the procedures were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, Facultad de Medicina, Universidad de Buenos Aires, CD No. 2271/2014) and are in accordance with the guidelines of the Argentinean National Administration of Medicines, Food and Medical Technology (ANMAT), Argentinean National Service of Sanity and Agrifoods Quality (SENASA) and also based on the US NIH Guide for the Care and Use of Laboratory Animals.
Author Contributions
NG, MS, and FP designed experiment; FP, NG, and GM contributed to the writing of the manuscript; FP, GD, GM, ÁC, and MR did experiments; FP, NG, MS, and GM analyzed data; MF and DC provided the 3-hydroxy-4-pyridinecarboxylic acid derivative (HP24); NG, MS, GM, and FP contributed to final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Mr. Ricardo Chung and Mr. Eduardo Alejandro Giménez and for their technical assistance. The authors would also like to thank Ms. Victoria González Eusevi for her assistance in English grammar and spelling corrections.
Funding
This work was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas [Grant number PIP 0672]; Universidad de Buenos Aires [Grant number 20020130100774BA]; Agencia Nacional de Promoción Científica y Tecnológica [Grant number 2014-1049]; and Fundación Bunge y Born [Grant number 2015/16].
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01738/full#supplementary-material.
References
1. Melo RCN. Acute heart inflammation: ultrastructural and functional aspects of macrophages elicited by Trypanosoma cruzi infection. J Cell Mol Med (2009) 13:279–94. doi:10.1111/j.1582-4934.2008.00388
2. Petray P, Castaños-Velez E, Grinstein S, Orn A, Rottenberg ME. Role of nitric oxide in resistance and histopathology during experimental infection with Trypanosoma cruzi. Immunol Lett (1995) 47:121–6. doi:10.1016/0165-2478(95)00083-H
3. Petray P, Rottenberg ME, Grinstein S, Orn A. Release of nitric oxide during the experimental infection with Trypanosoma cruzi. Parasite Immunol (1994) 16:193–9. doi:10.1111/j.1365-3024.1994.tb00340.x
4. Muñoz-Fernández MA, Fernández MA, Fresno M. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur J Immunol (1992) 22:301–7. doi:10.1002/eji.1830220203
5. Silva JS, Vespa GNR, Cardoso MAG, Aliberti JCS, Cunha FQ. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun (1995) 63:4862–7.
6. Abrahamsohn IA, Coffman RL. Trypanosoma cruzi: IL-10, TNF, IFN-γ, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol (1996) 84:231–44. doi:10.1006/expr.1996.0109
7. Chandrasekar B, Melby PC, Troyer DA, Colston JT, Freeman GL. Temporal expression of pro-inflammatory cytokines and inducible nitric oxide synthase in experimental acute chagasic cardiomyopathy. Am J Pathol (1998) 152:925–34.
8. Souza PEA, Rocha MOC, Rocha-Vieira E, Menezes CAS, Chaves ACL, Gollob KJ, et al. Monocytes from patients with indeterminate and cardiac forms of Chagas’ disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun (2004) 72:5283–91. doi:10.1128/IAI.72.9.5283-5291.2004
9. Keating SM, Deng X, Fernandes F, Cunha-Neto E, Ribeiro AL, Adesina B, et al. Inflammatory and cardiac biomarkers are differentially expressed in clinical stages of Chagas disease. Int J Cardiol (2015) 199:451–9. doi:10.1016/j.ijcard.2015.07.040
10. Gomes JAS, Bahia-Oliveira LMG, Rocha MOC, Martins-Filho OA, Gazzinelli G, Correa-Oliveira R. Evidence that development of severe cardiomyopathy in human Chagas’ disease is due to a Th1-specific immune response. Infect Immun (2003) 71:1185–93. doi:10.1128/IAI.71.3.1185-1193.2003
11. Cunha-Neto E, Chevillard C. Chagas disease cardiomyopathy: immunopathology and genetics. Mediators Inflamm (2014) 2014:11. doi:10.1155/2014/683230
12. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Mem Inst Oswaldo Cruz (2009) 104(Suppl):152–8. doi:10.1590/S0074-02762009000900021
13. Eltzschig HK, Carmeliet P, Carmeliet P. Hypoxia and inflammation. N Engl J Med (2011) 364:656–65. doi:10.1056/NEJMra0910283
14. Konisti S, Kiriakidis S, Paleolog EM. Hypoxia – a key regulator of angiogenesis and inflammation in rheumatoid arthritis. Nat Rev Rheumatol (2012) 8:153–62. doi:10.1038/nrrheum.2011.205
15. Costa C, Incio J, Soares R. Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis (2007) 10:149–66. doi:10.1007/s10456-007-9074-0
16. Kotlinowski J, Grochot-przeczek A, Taha H, Kozakowska M, Pilecki B, Skrzypek K, et al. PPAR γ activation but not PPAR γ haplodeficiency affects proangiogenic potential of endothelial cells and bone marrow-derived progenitors. Cardiovasc Diabetol (2014) 13:150. doi:10.1186/s12933-014-0150-7
17. Biscetti F, Gaetani E, Flex A, Aprahamian T, Hopkins T, Straface G, et al. Selective activation of peroxisome induces neoangiogenesis through a vascular endothelial growth factor – dependent mechanism. Diabetes (2008) 57:1394–404. doi:10.2337/db07-0765
18. Yue TL, Chen J, Bao W, Narayanan PK, Bril A, Jiang W, et al. In vivo myocardial protection from ischemia/reperfusion injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. Circulation (2001) 104:2588–94. doi:10.1161/hc4601.099403
19. Yamakawa K, Hosoi M, Koyama H, Tanaka S, Fukumoto S, Morii H, et al. Peroxisome proliferator-activated receptor-γ agonists increase vascular endothelial growth factor expression in human vascular smooth muscle cells. Biochem Biophys Res Commun (2000) 271:571–4. doi:10.1006/bbrc.2000.2665
20. Fujii M, Inoki I, Saga M, Morikawa N, Arakawa K, Inaba S, et al. Aldosterone inhibits endothelial morphogenesis and angiogenesis through the downregulation of vascular endothelial growth factor receptor-2 expression subsequent to peroxisome proliferator-activated receptor gamma. J Steroid Biochem Mol Biol (2012) 129:145–52. doi:10.1016/j.jsbmb.2011.12.014
21. Bishop-bailey D. PPARs and angiogenesis. Biochem Soc Trans (2011) 39:1601–5. doi:10.1042/BST20110643
22. Brun P, Dean A, Di Marco V, Surajit P, Castagliuolo I, Carta D, et al. Peroxisome proliferator-activated receptor-γ mediates the anti-inflammatory effect of 3-hydroxy-4-pyridinecarboxylic acid derivatives: synthesis and biological evaluation. Eur J Med Chem (2013) 62:486–97. doi:10.1016/j.ejmech.2013.01.024
23. Mirkin GA, Jones M, Sanz OP, Rey R, Sica RE, González Cappa SM. Experimental Chagas’ disease: electrophysiology and cell composition of the neuromyopathic inflammatory lesions in mice infected with a myotropic and a pantropic strain of Trypanosoma cruzi. Clin Immunol Immunopathol (1994) 73:69–79. doi:10.1006/clin.1994.1171
24. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph (1996) 14(33–8):27–8. doi:10.1016/0263-7855(96)00018-5
25. Brun P, Dean A, Di V, Surajit P, Castagliuolo I, Carta D, et al. Peroxisome proliferator-activated receptor-g mediates the anti-inflammatory effect of 3-hydroxy-4-pyridinecarboxylic acid derivatives: synthesis and biological evaluation. Eur J Med Chem (2013) 62:486–97. doi:10.1016/j.ejmech.2013.01.024
26. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol (1993) 234:779–815. doi:10.1006/jmbi.1993.1626
27. Milburn MV, Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature (1998) 395:137–43. doi:10.1038/25931
28. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem (2009) 30:2785–91. doi:10.1002/jcc.21256
29. Modenutti C, Gauto D, Radusky L, Blanco J, Turjanski A, Hajos S, et al. Using crystallographic water properties for the analysis and prediction of lectin-carbohydrate complex structures. Glycobiology (2015) 25:181–96. doi:10.1093/glycob/cwu102
30. Penas F, Mirkin GA, Vera M, Cevey Á, González CD, Gómez MI, et al. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta (2015) 1852:893–904. doi:10.1016/j.bbadis.2014.12.019
31. Goren N, Cuenca J, Martin Sanz P, Bosca L. Attenuation of NF-kappaB signalling in rat cardiomyocytes at birth restricts the induction of inflammatory genes. Cardiovasc Res (2004) 64:289–97. doi:10.1016/j.cardiores.2004.06.029
32. de la Torre E, Davel L, Jasnis MA, Gotoh T, de Lustig ES, Sales ME. Muscarinic receptors participation in angiogenic response induced by macrophages from mammary adenocarcinoma-bearing mice. Breast Cancer Res (2005) 7:345–52. doi:10.1186/bcr1005
33. de la Torre E, Hovsepian E, Penas FN, Dmytrenko G, Castro ME, Goren NB, et al. Macrophages derived from septic mice modulate nitric oxide synthase and angiogenic mediators in the heart. J Cell Physiol (2013) 228:1584–93. doi:10.1002/jcp.24320
34. Penas FN, Cevey ÁC, Siffo S, Mirkin GA, Goren NB. Hepatic injury associated with Trypanosoma cruzi infection is attenuated by treatment with 15-deoxy-Δ12,14 prostaglandin J2. Exp Parasitol (2016) 170:100–8. doi:10.1016/j.exppara.2016.09.015
35. Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren NB. IL-10 inhibits the NF-κB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in Trypanosoma cruzi-infected cardiomyocytes. PLoS One (2013) 8:e79445. doi:10.1371/journal.pone.0079445
36. Kruger NJ. The Bradford method for protein quantitation. Methods Mol Biol (1994) 32:9–15. doi:10.1385/0-89603-268-X:9
37. Hovsepian E, Mirkin GA, Penas F, Manzano A, Bartrons R, Goren NB. Modulation of inflammatory response and parasitism by 15-Deoxy-Δ(12,14) prostaglandin J(2) in Trypanosoma cruzi-infected cardiomyocytes. Int J Parasitol (2011) 41:553–62. doi:10.1016/j.ijpara.2010.12.002
38. Penas F, Mirkin GA, Hovsepian E, Cevey Á, Caccuri R, Sales ME, et al. PPARγ ligand treatment inhibits cardiac inflammatory mediators induced by infection with different lethality strains of Trypanosoma cruzi. Biochim Biophys Acta Mol Basis Dis (2013) 1832:239–48. doi:10.1016/j.bbadis.2012.08.007
39. Escudero CA, Herlitz K, Troncoso F, Guevara K, Acurio J, Aguayo C, et al. Pro-angiogenic role of insulin: from physiology to pathology. Front Physiol (2017) 8:204. doi:10.3389/fphys.2017.00204
40. Liao HH, Jia XH, Liu HJ, Yang Z, Tang QZ. The role of PPARs in pathological cardiac hypertrophy and heart failure. Curr Pharm Des (2017) 23:1677–86. doi:10.2174/1381612822666160928150040
41. Abou Daya K, Abu Daya H, Nasser Eddine M, Nahhas G, Nuwayri-Salti N. Effects of rosiglitazone (PPARγ agonist) on the myocardium in non-hypertensive diabetic rats. J Diabetes (2015) 7:85–94. doi:10.1111/1753-0407.12140
42. Zhao S-M, Li H-W, Guo C-Y, Shen L-H. Cardiac fibrosis in diabetic rats: regulation and mechanism of activation of the PPARgamma signal pathway. Chin J Physiol (2010) 53:262–7. doi:10.4077/CJP.2010.AMK076
43. Hu Q, Chen J, Jiang C, Liu H-F. Effect of peroxisome proliferator-activated receptor gamma agonist on heart of rabbits with acute myocardial ischemia/reperfusion injury. Asian Pac J Trop Med (2014) 7:271–5. doi:10.1016/S1995-7645(14)60036-5
44. Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, Balint BL, et al. Structural basis for the activation of PPARgamma by oxidized fatty acids. Nat Struct Mol Biol (2008) 15:924–31. doi:10.1038/nsmb.1474
45. Egawa D, Itoh T, Yamamoto K. Characterization of covalent bond formation between PPARγ and oxo-fatty acids. Bioconjug Chem (2015) 26:690–8. doi:10.1021/acs.bioconjchem.5b00021
46. Assunção LS, Magalhães KG, Carneiro AB, Molinaro R, Almeida PE, Atella GC, et al. Schistosomal-derived lysophosphatidylcholine triggers M2 polarization of macrophages through PPARγ dependent mechanisms. Biochim Biophys Acta (2017) 1862:246–54. doi:10.1016/j.bbalip.2016.11.006
47. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature (2007) 447:1116–20. doi:10.1038/nature05894
48. Scholz A, Plate KH, Reiss Y. Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci (2015) 1347:45–51. doi:10.1111/nyas.12726
49. Guedes-da-Silva FH, Shrestha D, Salles BC, Figueiredo VP, Lopes LR, Dias L, et al. Trypanosoma cruzi antigens induce inflammatory angiogenesis in a mouse subcutaneous sponge model. Microvasc Res (2015) 97:130–6. doi:10.1016/j.mvr.2014.10.007
50. Shrestha D, Bajracharya B, Paula-Costa G, Salles BC, Leite ALJ, Menezes APJ, et al. Expression and production of cardiac angiogenic mediators depend on the Trypanosoma cruzi-genetic population in experimental C57BL/6 mice infection. Microvasc Res (2017) 110:56–63. doi:10.1016/j.mvr.2016.12.002
51. Ashoff A, Qadri F, Eggers R, Jöhren O, Raasch W, Dendorfer A. Pioglitazone prevents capillary rarefaction in streptozotocin-diabetic rats independently of glucose control and vascular endothelial growth factor expression. J Vasc Res (2012) 49:260–6. doi:10.1159/000335214
52. Zhang H, Wei T, Jiang X, Li Z, Cui H. PEDF and 34-mer inhibit angiogenesis in the heart by inducing tip cells apoptosis via up-regulating PPAR- c to increase surface FasL. Apoptosis (2016) 21:60–8. doi:10.1007/s10495-015-1186-1
53. Funovics P, Brostjan C, Nigisch A, Fila A, Grochot A, Mleczko K, et al. Effects of 15d-PGJ(2) on VEGF-induced angiogenic activities and expression of VEGF receptors in endothelial cells. Prostaglandins Other Lipid Mediat (2006) 79:230–44. doi:10.1016/j.prostaglandins.2006.02.002
54. Kim KY, Ahn JH, Cheon HG. Anti-angiogenic action of PPARγ ligand in human umbilical vein endothelial cells is mediated by PTEN upregulation and VEGFR-2 downregulation. Mol Cell Biochem (2011) 358:375–85. doi:10.1007/s11010-011-0989-9
55. Tanowitz HB, Machado FS, Spray DC, Friedman JM, Oren S, Lora JN, et al. Developments in the management of Chagas cardiomyopathy. Expert Rev Cardiovasc Ther (2016) 13:1393–409. doi:10.1586/14779072.2015.1103648.Developments
Keywords: Trypanosoma cruzi, angiogenesis, new peroxisome proliferator-activated receptor gamma agonist, macrophages, inflammatory mediators, heart fibrosis
Citation: Penas FN, Carta D, Dmytrenko G, Mirkin GA, Modenutti CP, Cevey ÁC, Rada MJ, Ferlin MG, Sales ME and Goren NB (2017) Treatment with a New Peroxisome Proliferator-Activated Receptor Gamma Agonist, Pyridinecarboxylic Acid Derivative, Increases Angiogenesis and Reduces Inflammatory Mediators in the Heart of Trypanosoma cruzi-Infected Mice. Front. Immunol. 8:1738. doi: 10.3389/fimmu.2017.01738
Received: 29 June 2017; Accepted: 23 November 2017;
Published: 11 December 2017
Edited by:
Jixin Zhong, Case Western Reserve University, United StatesReviewed by:
Xiaoxuan Lyu, California Institute for Biomedical Research, United StatesZhibin Zhang, Harvard University, United States
Martin Rottenberg, Karolinska Institute (KI), Sweden
Copyright: © 2017 Penas, Carta, Dmytrenko, Mirkin, Modenutti, Cevey, Rada, Ferlin, Sales and Goren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nora Beatriz Goren, bmdvcmVuQGZtZWQudWJhLmFy