 Kevin A. Henry1
Kevin A. Henry1 Dae Young Kim1Hiba Kandalaft1Michael J. Lowden1Qingling Yang1Joseph D. Schrag2
Dae Young Kim1Hiba Kandalaft1Michael J. Lowden1Qingling Yang1Joseph D. Schrag2 Greg Hussack1
Greg Hussack1 C. Roger MacKenzie1,3
C. Roger MacKenzie1,3 Jamshid Tanha1,3,4*
Jamshid Tanha1,3,4*
- 1Human Health Therapeutics Research Centre, National Research Council Canada, Ottawa, ON, Canada
- 2Human Health Therapeutics Research Centre, National Research Council Canada, Montréal, QC, Canada
- 3School of Environmental Sciences, University of Guelph, Guelph, ON, Canada
- 4Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada
Human autonomous VH/VL single-domain antibodies (sdAbs) are attractive therapeutic molecules, but often suffer from suboptimal stability, solubility and affinity for cognate antigens. Most commonly, human sdAbs have been isolated from in vitro display libraries constructed via synthetic randomization of rearranged VH/VL domains. Here, we describe the design and characterization of three novel human VH/VL sdAb libraries through a process of: (i) exhaustive biophysical characterization of 20 potential VH/VL sdAb library scaffolds, including assessment of expression yield, aggregation resistance, thermostability and tolerance to complementarity-determining region (CDR) substitutions; (ii) in vitro randomization of the CDRs of three VH/VL sdAb scaffolds, with tailored amino acid representation designed to promote solubility and expressibility; and (iii) systematic benchmarking of the three VH/VL libraries by panning against five model antigens. We isolated ≥1 antigen-specific human sdAb against four of five targets (13 VHs and 7 VLs in total); these were predominantly monomeric, had antigen-binding affinities ranging from 5 nM to 12 µM (average: 2–3 µM), but had highly variable expression yields (range: 0.1–19 mg/L). Despite our efforts to identify the most stable VH/VL scaffolds, selection of antigen-specific binders from these libraries was unpredictable (overall success rate for all library-target screens: ~53%) with a high attrition rate of sdAbs exhibiting false positive binding by ELISA. By analyzing VH/VL sdAb library sequence composition following selection for monomeric antibody expression (binding to protein A/L followed by amplification in bacterial cells), we found that some VH/VL sdAbs had marked growth advantages over others, and that the amino acid composition of the CDRs of this set of sdAbs was dramatically restricted (bias toward Asp and His and away from aromatic and hydrophobic residues). Thus, CDR sequence clearly dramatically impacts the stability of human autonomous VH/VL immunoglobulin domain folds, and sequence-stability tradeoffs must be taken into account during the design of such libraries.
Introduction
The concept of an autonomous single immunoglobulin variable domain (single-domain antibodies or sdAbs) as the smallest representation of an antigen-binding-competent antibody was first described by Ward et al. in the mouse (1). With the discovery of naturally occurring heavy chain-only antibodies in Camelidae (2) and in cartilaginous sharks (3) several years later (the single variable domains of which can recognize antigen autonomously), it became clear that sdAbs represented not only a theoretical possibility but a viable immunological solution to the problem of antigen recognition. Although the human humoral immune system produces only conventional antibodies with paired heavy and light chains and not sdAbs, the question of whether human sdAbs (autonomous variable heavy- or light-chain domains, VHs or VLs) could be isolated and/or molecularly engineered in vitro was brought to light.
The identification, engineering and biophysical characterization of a handful of non-antigen-specific human VH/VL sdAbs has been extensively reported and discussed (4). The first efforts to produce human VH/VL sdAbs with novel antigen-binding specificities used “camelized” scaffolds that incorporated the solubilizing framework region (FR) substitutions found in camelid sdAbs (5–9). Although this approach yielded antigen-specific sdAbs with excellent solubility and biophysical properties, it relied on undesirable sequence deviation from the human IGHV germline. Later, rare fully human rearranged VH and VL variable domains were discovered that were autonomously stable and monomeric and large phage display libraries were constructed by randomizing their complementarity-determining regions (CDRs), although it was clear from the mid-2000s that certain CDR sequences (potentially low in hydrophobic content and rich in negative charge) were better compatible with solubility and stability of these molecules (9–11). There are now many examples of fully human antibodies (primarily VHs) isolated from such libraries against a variety of targets, including α-amylase (12), β-galactosidase (13, 14), Candida albicans MP65 and SAP-2 (15), carbonic anhydrase (12), CD154 (16), CD28 (17), CD40 (18, 19), CD40L (20), Clostridium difficile toxin B (21), EGFR (22), glypican-2 (23), glypican-3 (24), human serum albumin (HSA) (25–27), lysozyme (28–30), maltose-binding protein (31), MDM4 (32), mesothelin (33), TNF-α (34), TNFR1 (35), and VEGF (22). These fully human VH/VL sdAbs exhibit a variety of antigen-binding modes and functional activities and several have entered clinical development, where they have been generally well-tolerated albeit unexpectedly immunogenic (36, 37).
Here, we report the design, construction and characterization of three novel phage-displayed, synthetically randomized human VH/VL sdAb libraries. We attempted to circumvent the unfavorable biophysical properties of many human VH/VL sdAbs by (i) selecting ultra-stable VH/VL sdAbs tolerant to CDR modification as library scaffolds, (ii) maximizing randomized sequence diversity in CDRs using trinucleotide mutagenesis, and (iii) spiking the library with negatively charged residues to encourage solubility. Similarly to the experiences of others, we were able to isolate monomeric, high-affinity VH/VL sdAbs from the libraries against some antigens but not against others. The stochastic process of selecting binders from human VH/VL sdAb libraries is likely a consequence of fundamental tradeoffs between CDR sequence and human VH/VL sdAb stability and aggregation resistance.
Materials and Methods
Identification of Human Autonomous VH/VL sdAb Scaffolds
The human autonomous VH and VL sdAb scaffolds used in this study (Table 1; Figure S1 in Supplementary Material) were isolated as previously described by To et al. (38) and Kim et al. (39). Disulfide-stabilized versions of each VH/VL sdAb (bearing an intradomain disulfide linkage formed between Cys residues at IMGT positions 54 and 78) were produced by overlap extension PCR as described in Kim et al. (40).
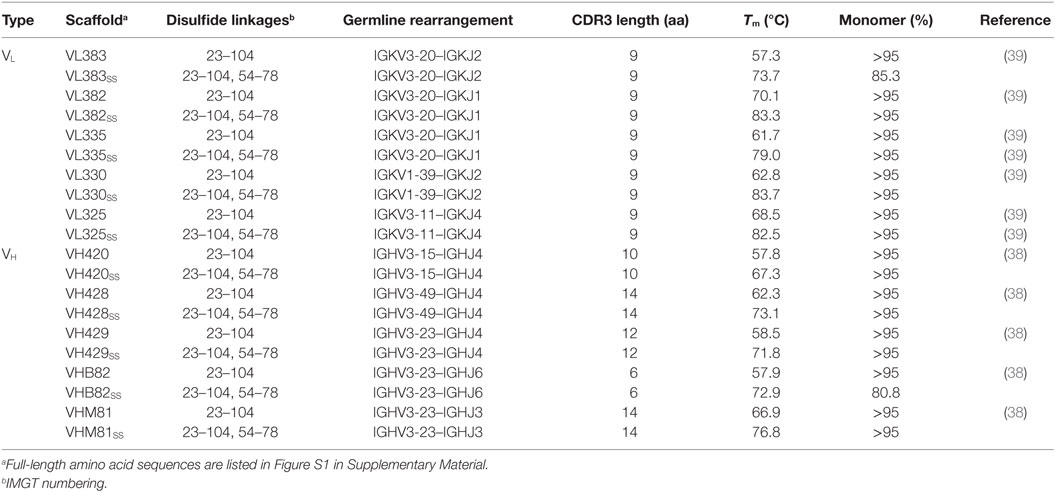
Table 1. Properties of human VH and VL single-domain antibody scaffolds used in this study.
CDR Shuffling
All three CDRs of each VH/VL scaffold were simultaneously exchanged for those listed in Table 2 (20 VH/VL scaffolds × 12 CDR sets = 240 CDR-shuffled variants). DNA constructs encoding the CDR-shuffled variants were synthesized commercially (GeneArt/Life Technologies, Regensberg, Germany) and subcloned into the pSJF2H bacterial expression vector (12).
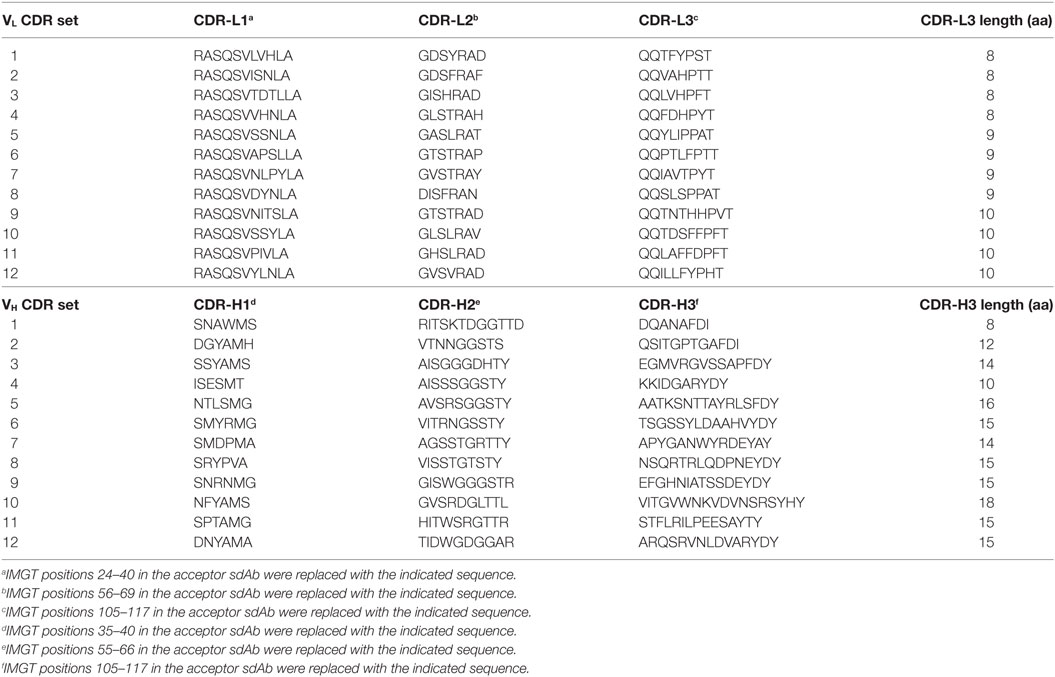
Table 2. Amino acid sequences of complementarity-determining region (CDR) sets introduced into human VH/VL single-domain antibody (sdAb) scaffolds for biophysical stability assessment.
Design and Construction of Synthetic Human VH/VL sdAb Phage Display Libraries
Three phage-displayed sdAb libraries were constructed by in vitro randomization of the sdAb scaffolds VH428, VHB82SS and VL383SS. Briefly, nondegenerate oligonucleotides spanning each sdAb were chemically synthesized using the phosphoramidite method (GeneArt/Life Technologies) and purified by HPLC. CDRs were randomized via incorporation of defined mixtures of trinucleotide phosphoramidite building blocks (41) during oligonucleotide synthesis. Oligonucleotides were assembled without amplification by Klenow fragment extension, gel purified using a GeneJET™ gel extraction kit (Thermo-Fisher, Waltham, MA, USA) according to the manufacturer’s instructions, and resuspended in a total volume of 1 mL TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Non-amplified library DNA was quantified by real-time PCR using Fast SYBR™ Green master mix (Thermo-Fisher), and a StepOnePlus™ real-time PCR system (Thermo-Fisher), then approximately 232–1,069 ng (4.3 × 1011 to 1.5 × 1012 molecules) of DNA was PCR-amplified with Phusion® high-fidelity DNA polymerase (Thermo-Fisher) using M13F and M13R primers (sequences in Table S1 in Supplementary Material), gel purified using a GeneJET™ gel extraction kit, then digested and ligated between the NcoI and NotI sites of the phagemid vector pMED1 (42). Escherichia coli TG1 cells were transformed with the ligation products by electroporation, yielding final library sizes between 1.0 × 1010 and 2.3 × 1010 independent transformants, ≥95% of which carried VH/VL sdAb inserts as shown by colony PCR.
Antibodies, Proteins and Reagents
Recombinant human CEACAM6 (residues 145–232) and mouse GITR (residues 20–153) ectodomains were produced by transient transfection of HEK293-6E cells as previously described (43). HSA was from Sigma-Aldrich (St. Louis, MO, USA; Cat. No. A9511). Recombinant human CTLA4 ectodomain (residues 1–162) was from Sino Biological (Beijing, China; Cat. No. 11159-H08H). E. coli 0157:H7 intimin (residues 658–934) fused C-terminally to maltose-binding protein (MBP-intimin) was from GenScript (Piscataway, NJ, USA). Horseradish peroxidase-conjugated antibodies used in ELISA (mouse anti-M13, Cat. No. 27942101; mouse anti-c-Myc, Cat. No. 11814150001; rabbit anti-6 × His, Cat. No. A190-114P) were from GE Healthcare (Piscataway, NJ, USA), Bethyl Laboratories (Montgomery, TX, USA) and Roche Diagnostics (Basel, Switzerland), respectively. M13K07 helper phage was from New England BioLabs (Ipswich, MA, USA) and M13K07ΔpIII hyper phage was from Progen Biotechnik (Heidelberg, Germany).
Isolation of Antigen-Specific Human VH/VL sdAbs by Panning
Phage particles displaying monovalent VH/VL sdAbs were prepared by rescue of the three synthetic human VH/VL sdAb phagemid libraries with M13K07 helper phage as previously described (42). Briefly, 2 × YT broth (4 L) containing 1% (w/v) glucose and 100 µg/mL ampicillin was inoculated with 1 mL phagemid-bearing E. coli TG1 cells (7.3 × 1010 to 1.6 × 1011) and grown at 37°C with 250 rpm shaking to an OD600 of 0.5. Phagemid-bearing cells were superinfected with M13K07 helper phage at a multiplicity of infection of 20:1 at 37°C for 30 min with no shaking, then pelleted and resuspended in 6 L of 2 × YT broth containing 100 µg/mL ampicillin and 50 µg/mL kanamycin. The next day, phage particles were purified from bacterial supernatants using two rounds of polyethylene glycol precipitation; neither heat treatment nor filtration steps were used to remove residual bacterial cells. For panning, 5 µg of each protein in 35 µL phosphate-buffered saline (PBS), pH 7.4, was adsorbed overnight at 4°C in wells of NUNC MaxiSorp™ 96-well microtiter plates (Thermo-Fisher). The next day, wells were blocked with 200 µL PBS containing either 2% (w/v) bovine serum albumin (BSA) or 2% (w/v) skim milk for 1 h at 37°C, then rinsed 3 × with PBS. Approximately 5 × 1011 infective library phage particles (5 × 1012 virions) were added to wells in 100 µL of PBS containing 1% BSA or skim milk and 0.1% (v/v) Tween-20 for 2 h at room temperature. The blocking protein (BSA and skim milk) was switched in alternate rounds of panning, except for panning on HSA in which only skim milk was used. Wells were washed 5 × with PBS containing 0.1% Tween-20, 2 × with PBS and then bound phage were eluted for 10 min with 50 µL of 100 mM triethylamine, neutralized with 50 µL 1 M Tris-HCl, pH 7.5, and used to reinfect exponentially growing E. coli TG1 cells. The cultures were superinfected with M13K07 helper phage. The next day, amplified phage were purified by polyethylene glycol precipitation from 10 mL overnight cultures and used in subsequent panning rounds. After four or five rounds of selection, antigen-specific VH- or VL-displaying phage clones were identified by their binding in polyclonal and monoclonal phage ELISA as previously described (42).
Selection for Monomeric and Expressible Human VH/VL sdAbs by Panning
Phage particles displaying monovalent VH/VL sdAbs were prepared by rescue of phagemid libraries with M13K07 helper phage as described above. Phage particles displaying multivalent VH/VL sdAbs on all copies of pIII were prepared as described above, except that: (i) smaller volumes (100 mL) of 2 × YT broth were inoculated with 1 mL phagemid-bearing E. coli TG1 cells, (ii) phagemid libraries were rescued with M13K07ΔpIII hyper phage at a multiplicity of infection of 10:1, and (iii) the final overnight culture volume was 150 mL of 2 × YT broth.
For panning, 5 µg of either protein A (for the VHB82SS library; Thermo-Fisher) or protein L (for the VL383SS library; Thermo-Fisher) in 50 µL PBS, pH 7.4, was adsorbed overnight at 4°C in wells of NUNC MaxiSorp™ 96-well microtiter plates. The next day, wells were blocked with 300 µL PBS containing either 2% BSA or skim milk for 1 h at 37°C, then rinsed 1 × with PBS. Approximately 1011 infective library phage particles (1012 virions) were added to wells in 100 µL of PBS containing 1% skim milk and 0.1% Tween-20 for 2 h at room temperature. Wells were washed 5 × with PBS containing 0.1% Tween-20, 2 × with PBS and then bound phage were eluted for 10 min with 50 µL of 100 mM triethylamine, neutralized with 50 µL 1 M Tris-HCl, pH 7.5, and used to reinfect exponentially growing E. coli TG1 cells. The cultures were superinfected with either M13K07 helper phage or M13K07ΔpIII hyper phage. The next day, amplified phage were purified by polyethylene glycol precipitation from 10 mL overnight cultures and used in subsequent panning rounds. After three rounds of selection, pools of sdAb-phage (either phage bound by and eluted from protein A/L, or phage amplified from overnight cultures) were interrogated using next-generation DNA sequencing (NGS) as described below.
Soluble VH/VL Protein Expression
Monomeric VH/VL sdAbs bearing C-terminal 6 × His and c-Myc tags were expressed from overnight cultures of E. coli TG1 cells grown in 250 mL to 1 L of 2 × YT broth under IPTG (isopropyl β-d-1-thiogalactopyranoside) induction, then extracted from periplasmic space by osmotic sucrose shock and purified by immobilized metal affinity chromatography as previously described (42). For small-scale expression screening, 5 mL overnight cultures were grown as above, lysed using FastBreak™ reagent (Promega, Madison, WI, USA) and sdAbs purified using PureProteome™ nickel magnetic beads (EMD Millipore, Billerica, MA, USA). Expression yields from 5 mL cultures were determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA) as per manufacturer’s instructions with a VHH sdAb of known concentration as the protein standard. Titration ELISAs using soluble VH/VL sdAbs were performed as previously described (42, 44, 45), using either anti-6 × His or anti-c-Myc secondary antibodies to detect binding.
Size Exclusion Chromatography (SEC) and SEC with Multiangle Light Scattering (MALS)
Size exclusion chromatography analyses of monomeric VH/VL sdAbs were conducted using a Superdex™ 75 GL column (GE Healthcare) connected to an ÄKTA FPLC protein purification system (GE Healthcare) as previously described (42). UPLC-SEC-MALS analyses of VH/VL sdAbs were conducted essentially as previously described (39) using an Acquity BEH-125 column (Waters, Milford, MA) connected to an Acquity UPLC H-Class Bio system (Waters) with miniDAWN™ MALS detector and Optilab® UT-rEX™ refractometer (Wyatt Technology, Santa Barbara, CA, USA). VH/VL sdAbs (10–20 µg) were injected at 30°C in a mobile phase consisting of calcium- and magnesium-free DPBS (GE Healthcare) at a flow rate of 0.4 mL/min. Weighted average molecular mass (MMALS) was calculated using a protein concentration determined using A280 from the PDA detector with extinction coefficients calculated from amino acid sequences. Data were processed using ASTRA 6.1 software (Wyatt).
Surface Plasmon Resonance (SPR)
All monomeric human VH/VL sdAbs were SEC-purified and buffer-exchanged into HBS-EP buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% (v/v) surfactant P20, pH 7.4) prior to SPR analyses. All SPR analyses were performed on a Biacore™ 3000 instrument (GE Healthcare) at a temperature of 25°C. Briefly, proteins (CEACAM6, 370 resonance units (RUs); HSA, 1614 RUs; GITR, 796 RUs; MBP-intimin, 1536 RUs) were immobilized via amine coupling on research-grade CM5 sensor chips (GE Healthcare) in 10 mM acetate buffer, pH 4.0, according to the manufacturer’s instructions. VH/VL sdAbs were injected at concentrations ranging from 1 nM to 10 µM, at flow rates of 20–50 µL/min and with contact time between 120 s and 300 s, then allowed to dissociate for 7–10 min. All surfaces were regenerated using 10 mM glycine, pH 1.5. Ethanolamine-blocked flow cells served as reference surfaces. The data were fitted to a 1:1 interaction model and binding affinities (KDs) and/or kinetic parameters were determined either by steady-state analysis or by multicycle kinetic analysis using BIAevaluation 4.1 software (GE Healthcare).
Surface plasmon resonance analyses of two VL sdAbs (VLSS-2 and VLSS-5) against MBP-intimin were conducted as described above, except that 234 RUs of MBP-intimin were immobilized on a research-grade C1 sensor chip (GE Healthcare) via amine coupling in 10 mM acetate buffer, pH 4.5, according to the manufacturer’s instructions. These two VL sdAbs were injected in HBS-EP buffer containing either 150 or 500 mM NaCl with an extended contact time (600 s).
Protein Turbidity Assays
VH/VL sdAb samples (0.2 mL) in 1.5 mL microcentrifuge tubes were heated to 85°C in a heating block for 10 min, then allowed to cool to room temperature for 30 min. Absorbance at 360 nm was measured pre- and post-heat treatment in a quartz NanoQuant Plate™ (Tecan, Männedorf, Switzerland) using an Infinite® M200 PRO microplate reader (Tecan). VH/VL sdAb concentrations were adjusted to 0.25 mg/mL in PBS prior to analysis.
Thermal Shift Assays
VH/VL sdAb samples (45 µL) in 96-well thin-wall optical plates (Bio-Rad) were mixed with 5 µL SYPRO® Orange (diluted 1:100 from 5,000 × stock; Life Technologies, Carlsbad, CA, USA) and sealed with optical quality sealing tape (Bio-Rad). Using an iQ™ 5 real-time PCR system (Bio-Rad), a temperature ramp of 1°C/min was applied and thermal unfolding was monitored by measuring fluorescence at 0.5°C intervals as previously described (46, 47). The wavelengths for excitation and emission were 490 and 575 nm, respectively. Melting temperatures (Tms) were calculated as the temperature at which the maximum rate of change in fluorescent signal (d(RFU)/dt) was achieved. VH/VL sdAb concentrations were adjusted to 1 mg/mL in PBS prior to analysis.
Circular Dichroism
Tms were also determined by circular dichroism as previously described (39, 40). Ellipticity of VH/VL sdAbs (100 µg/mL) was measured at wavelengths between 205 and 210 nm in 100 mM sodium phosphate buffer, pH 7.4. Ellipticity measurements were normalized to a percentage scale and then Tms were determined by plotting percent folded versus temperature and fitting the data to a Boltzmann distribution.
Next-Generation DNA Sequencing
VH/VL sdAb libraries were interrogated using an Illumina MiSeq instrument as described previously (44, 45, 48). Amplicons for NGS were prepared using FR1- and FR4-specific barcoded primers (sequences in Table S1 in Supplementary Material) using as template either phagemid DNA (~10 ng) or phage virions (~106 particles).
Statistical Analyses
Descriptive statistics (mean, median) were used to describe datasets as described in figure legends. No inferential statistical tests were used.
Results
Antigen-specific human autonomous VH/VL sdAbs do not exist in nature, and are most commonly isolated from synthetically randomized in vitro display libraries. These molecules are notoriously unstable and aggregation prone (4), which probably negatively impacts the selection of antigen-specific binders from synthetic VH/VL sdAb libraries. In an effort to mitigate these factors, we attempted to identify ultra-stable VH/VL sdAb scaffolds upon which we could construct highly diverse, stability-enhanced phage-displayed VH/VL sdAb libraries designed to yield soluble and thermostable antigen-specific human sdAbs.
Biophysical Stability Assessment of Wild-Type Human VH/VL sdAb Scaffolds
The Tms and aggregation tendencies of 22 potential VH sdAb scaffolds and 18 potential VL sdAb scaffolds were determined by circular dichroism and SEC (Tables S2 and S3 in Supplementary Material). Five VH and five VL sdAb scaffolds as well as their disulfide-stabilized equivalents (Table 1) were selected as the most promising candidates for library construction based on: (i) primarily monomeric folding, (ii) high thermostability with reversible thermal unfolding, and (iii) reasonable expression yields (>1 mg/L) from overnight E. coli TG1 cultures. As reported previously (39, 40), incorporation of a disulfide linkage spanning IMGT positions 54–78 increased the Tm of each scaffold by ~5–20°C but also had unpredictable effects on expression yields.
Tolerance of Human VH/VL sdAb Scaffolds to CDR Modification
As a preliminary investigation into which of the 20 VH/VL sdAb scaffolds might best tolerate library randomization, we grafted 12 sets of exogenous CDRs (Table 2) into each scaffold and assessed the resulting molecules’ expression level (Bradford assay), aggregation tendency (SEC-MALS), Tm (thermal shift assay), and ability to refold after thermal denaturation (turbidity assay). For VL sdAb scaffolds, the 12 sets of exogenous CDRs were derived from non-antigen-specific human VL sdAbs isolated from previously constructed libraries with expression yields ≥10 mg/L and for VH sdAb scaffolds, the 12 sets of exogenous CDRs were selected from either human VH sdAbs of unknown antigen specificity isolated from previously constructed libraries or camelid VHH sdAbs with expression yields ≥3 mg/L.
The VL sdAb scaffolds yielding the best-expressing CDR-shuffled variants were VL383SS, VL325SS and VL335SS (Figure 1A). CDR-shuffled variants of VL383SS and VL325SS were primarily monomeric by SEC-MALS, while CDR-shuffled variants of VL335SS showed some tendency to aggregate (Figure 1C). All three of these VL sdAb scaffolds, especially VL325SS, yielded CDR-shuffled variants with high Tms (Figure 1E); however, many CDR-shuffled variants of VL325SS showed significant turbidity upon heat denaturation (Figure 1G), reflecting their inability to refold as soluble monomers. The VH sdAb scaffolds yielding the best-expressing CDR-shuffled variants were VH420, VH428, VHB82, VHB82SS, and VHM81SS (Figure 1B). CDR-shuffled variants of all five of these VH sdAb scaffolds were similarly monomeric (Figure 1D) but as expected, the two disulfide-stabilized sdAb scaffolds (VHB82SS and VHM81SS) yielded CDR-shuffled variants with higher Tms (Figure 1F). Similar proportions of the CDR-shuffled variants of all five VH sdAb scaffolds showed minor turbidity upon heat denaturation (Figure 1H). On the basis of these data, we selected one VL sdAb scaffold (VL383SS) and two VH sdAb scaffolds (VH428 and VHB82SS) for library construction, reflecting a balance of their tolerance to CDR modification as well as their differing CDR3 lengths and germline gene rearrangements.
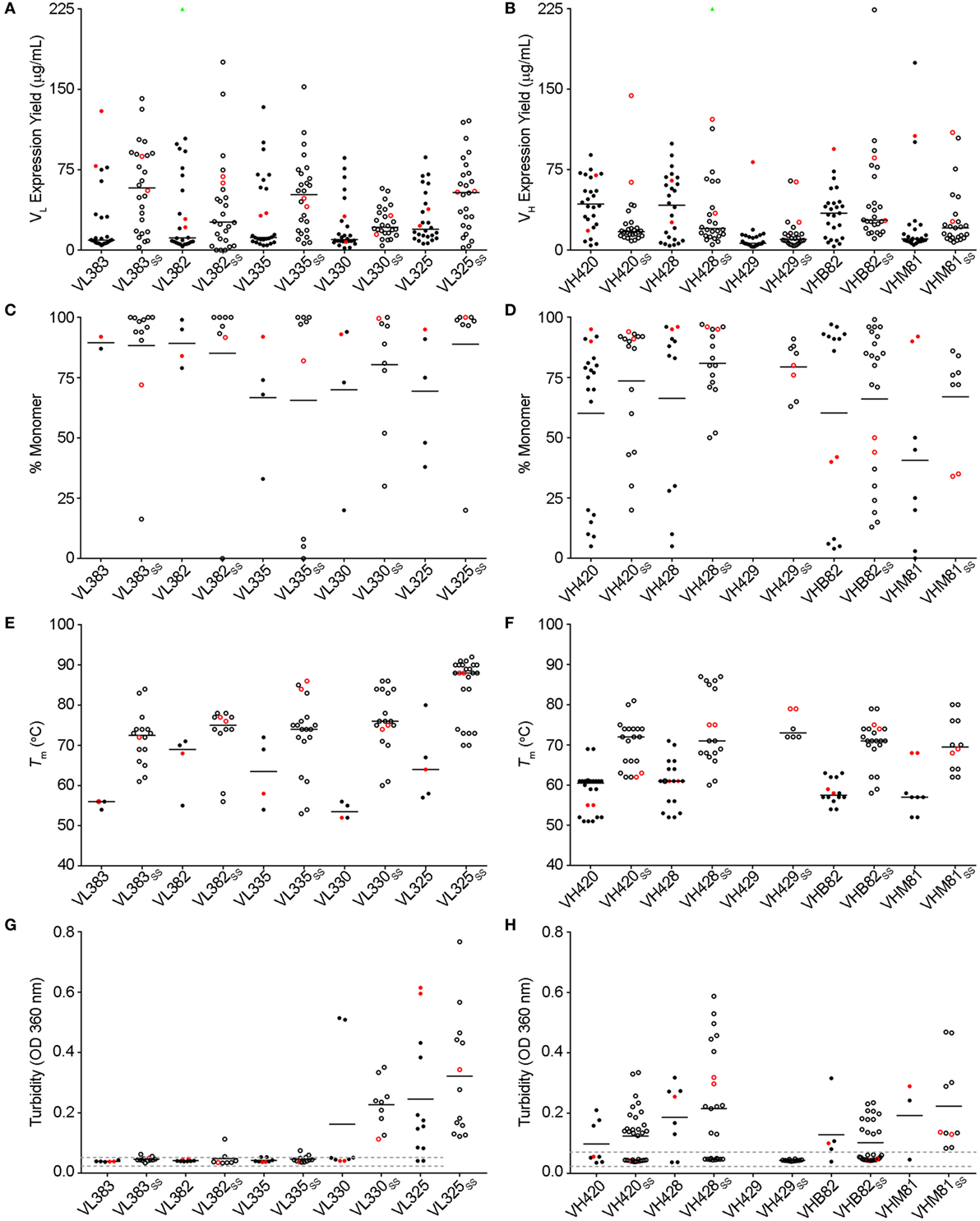
Figure 1. Biophysical stability assessment of complementarity-determining region (CDR)-shuffled human VH/VL single-domain antibody (sdAb) variants. (A,B) Expression yields of CDR-shuffled VL (A) and VH (B) sdAbs from 5 mL overnight cultures, as determined by Bradford assay. Two outliers (VL382 + CDR set 9, expression yield 467.7 µg/mL and VH428SS + CDR set 2, 356.4 µg/mL) are indicated by green triangles. (C,D) Aggregation tendencies of CDR-shuffled VL (C) and VH (D) sdAbs, as determined by SEC-MALS. (E,F) Melting temperatures (Tms) of CDR-shuffled VL (E) and VH (F) sdAbs, as determined by thermal shift assay. (G,H) Turbidity upon thermal denaturation of CDR-shuffled VL (G) and VH (H) sdAbs, as measured by spectrophotometry at 360 nm wavelength. Dashed lines represent the range of turbidity measurements for unheated VH/VL sdAbs. The aggregation tendencies, Tms and turbidities of CDR-shuffled variants of VH429 were not characterized due to inadequate expression yields. Horizontal lines represent mean (C,D,G,H) or median (A,B,E,F) values and wild-type VH/VL sdAbs with unmodified CDRs are shown in red. Open and solid circles represent VH/VL sdAbs with, or without, a stabilizing exogenous disulfide linkage. The number of data points in each panel reflects whether the experiment was conducted in singlicate (C,E) or duplicate (A,B,D,F–H) and for panels (C–H), whether the CDR-shuffled variant expressed sufficiently for analysis.
Design and Construction of Phage-Displayed Human VH/VL sdAb Libraries
Three synthetic sdAb libraries (VL383SS, VH428, and VHB82SS) were constructed by limited in vitro randomization of the CDRs of these VH/VL sdAb scaffolds and cloning of the resulting DNA into the pMED1 phagemid vector (Figures 2A,B). CDR3 length was varied in each library (Figures 2A,D; VL383SS library: 8, 9, or 10 residues; VH428 and VHB82SS libraries: 10, 12, 14, 16, 18, 20, or 22 residues) while CDR1 and CDR2 lengths were constant. The major technical advances of these over our previously described phage-displayed synthetic human VH/VL sdAb libraries (12, 21, 31) were (i) trinucleotide mutagenesis allowed for incorporation of defined mixtures of amino acids at each randomization position, based on alignments of human and llama antibodies with additional bias toward solubility-promoting residues (Asp, Glu) and against undesirable residues (Asn, Cys, and Met) and stop codons (Figures 2B,C; Figure S2 in Supplementary Material) and (ii) near-complete randomization of all three CDRs of each VH/VL sdAb scaffold resulted in much higher sequence diversity for the vast majority of library molecules (Figure 2E). All three VH/VL sdAb libraries were transformed into E. coli TG1 cells, yielding final library sizes (1.0–2.3 × 1010 independent transformants) that, as expected, vastly under-sampled theoretical library diversity (VL383SS: 2.9 × 1013 unique sequences; VHB82SS: 2.3 × 1033 unique sequences; VH428: 2.3 × 1035 unique sequences). At this stage, prior to helper phage rescue, no major clonality biases or deviations from library design were observed (data not shown).
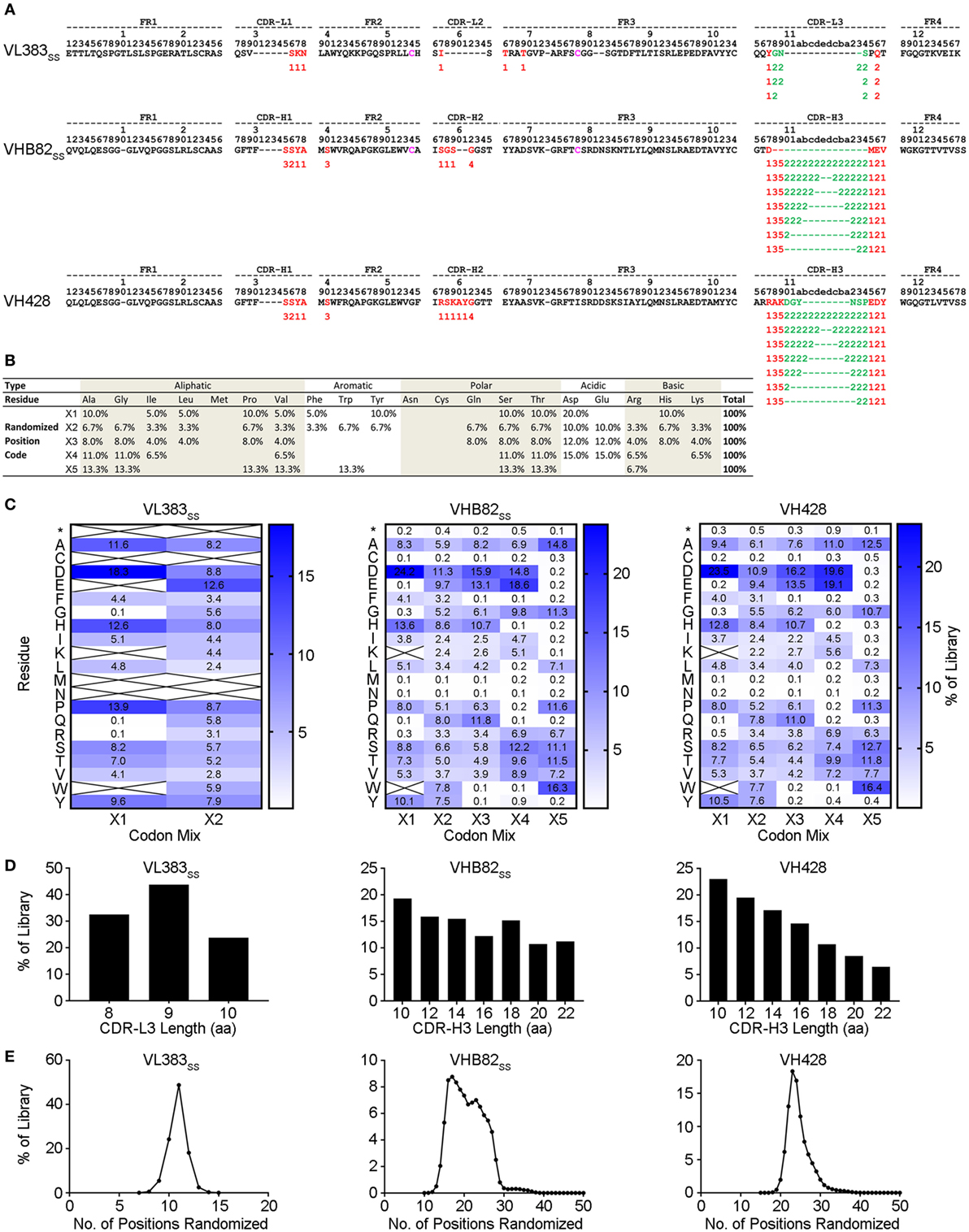
Figure 2. Design and construction of the VL383SS, VH428, and VHB82SS synthetic human VH/VL single-domain antibody (sdAb) libraries. (A) Design of the human VH/VL sdAb libraries. The parental VH/VL sdAb sequence is shown at top with IMGT numbering. Intradomain disulfide linkage-forming cysteine residues at positions 54 and 78 are shown in magenta. Randomization positions are highlighted in either red (no length polymorphism) or green (length polymorphism). Arabic numerals indicate the codon mixture (X1, X2, X3, X4, or X5) incorporated at each randomization position. (B) Expected amino acid frequencies at VH/VL sdAb library randomization positions. (C) Observed amino acid frequencies at VH/VL sdAb library randomization positions, measured from phagemid DNA isolated from Escherichia coli TG1 cells. Crossed-out cells indicate a frequency of <0.1%. (D) CDR3 length distributions of the VH/VL sdAb libraries. (E) Degree of randomized sequence diversity (Levenshtein distance, or number of amino acid changes with respect to the parental scaffold) in the VH/VL sdAb libraries. Analyses shown in (C–E) are representative of 7.7 × 104 to 7.9 × 105 sequences per library.
Isolation and Characterization of Antigen-Specific Human VH/VL sdAbs
All three VH/VL sdAb libraries were rescued with M13K07 helper phage and panned for four or five rounds against five model antigens (CEACAM6, CTLA4, GITR, HSA, and MBP-intimin). All library pannings were performed in duplicate by two independent operators at the same time and using the same materials, except for panning against MBP-intimin, in which panning was done in triplicate. High attrition rates of VH/VL sdAbs showing false positive binding either as sdAb-phage or as soluble sdAb proteins (VL383SS: 53% attrition; VH428: 69% attrition; VHB82ss: 71% attrition; Table 3) resulted in a final yield of 20 unique antigen-specific VH/VL sdAbs against four targets. Of the 15 library-target screens we conducted, eight (53%) yielded at least one antigen-specific VH/VL sdAb (four screens yielded one sdAb, two screens yielded two sdAbs, one screen yielded three sdAbs, and two screens yielded five sdAbs). Most of the recovered sdAbs were monomeric (Figure 3A; Table 4; Figure S3 in Supplementary Material), thermostable (Figure 3B; Table 4), and had affinities for antigen ranging from 5 nM to ~12 μM (Figures 3C,D; Table 4). One notable exception was the α-GITR VH sdAb VHB82SS-6, whose elution volume by SEC suggested it existed in solution as a strict dimer (Figure 3A). Stable homodimerization has previously been reported for several human VH sdAbs and was critical for antigen binding (49–52); this phenomenon is distinct from the general tendency of some VH domains to forms soluble aggregates. Antigen-specific VH/VL sdAbs isolated from all three libraries had aggregation tendencies and thermostabilities reflective of the parental VH/VL sdAb scaffold from which they were derived, but generally poorer expression yields (Figure 4A). Reproducibility in the selection of specific sdAb sequences from the libraries was modest, with only 45% (9/20) of antigen-specific VH/VL sdAbs recovered by both operators (Figure 4B) and no apparent connection between antigen-binding affinity and consistency of isolation.
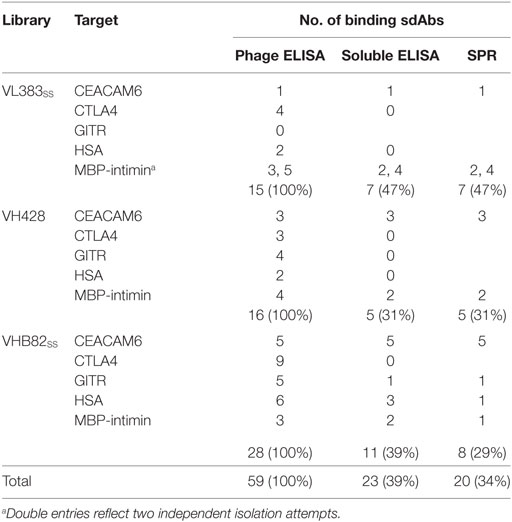
Table 3. Attrition rates of human VH/VL single-domain antibodies (sdAbs) through isolation and characterization.
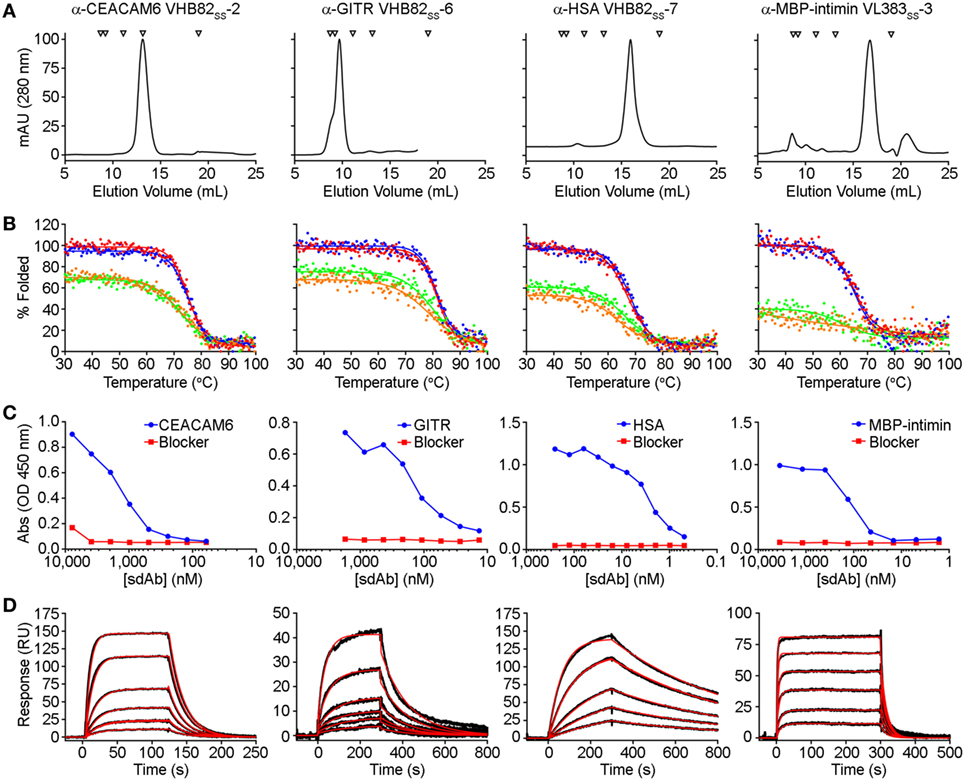
Figure 3. Properties of selected antigen-specific single-domain antibodies (sdAbs) isolated from the VL383SS, VH428, and VHB82SS synthetic human VH/VL sdAb libraries. The single highest affinity sdAb against each target is shown, except for MBP-intimin, where VLSS-3 is shown instead of VLSS-4 or VLSS-5 because of better fit to a 1:1 binding model. (A) Size exclusion chromatography (SEC) profiles of representative antigen-specific human VH/VL sdAbs. Arrows show molecular mass standards, from left to right: thyroglobulin (670 kDa), gamma globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.35 kDa). (B) Thermal unfolding of representative antigen-specific VH/VL sdAbs as determined by circular dichroism. Replicate unfolding curves are shown in red and blue; the sdAbs were then cooled to room temperature and remelted (shown in orange and green). (C) Titration ELISA of representative antigen-specific VH/VL sdAbs. Horseradish peroxidase mouse anti-c-Myc secondary antibody (clone 9E10, diluted 1:3,000) was used for detection. (D) Binding of antigen-specific VH/VL sdAbs to cognate antigen by SPR. Each antigen was immobilized on a CM5 sensor chip using amine coupling, then the indicated VH or VL sdAb was flowed over the surface at concentrations ranging from 25 to 1,000 nM (α-CEACAM6 VHB82SS-2), 25 to 500 nM (α-GITR VHB82SS-6), 2.5 to 50 nM (α-HSA VHB82SS-7), or 62.5 to 2,500 nM (α-MBP-intimin VL383SS-3). Black lines show data and red lines show fits.
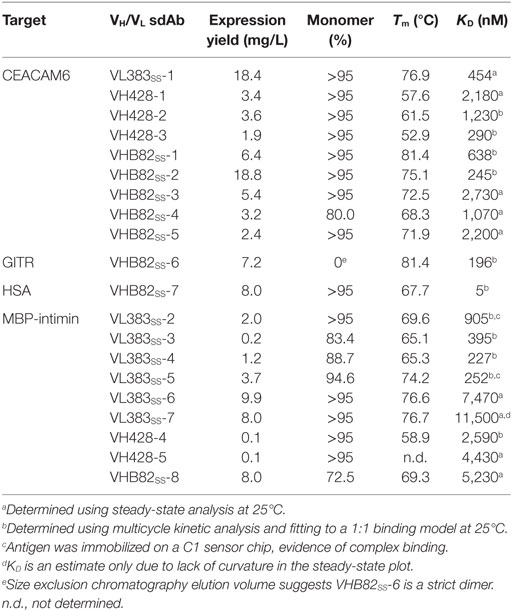
Table 4. Properties of antigen-specific single-domain antibodies (sdAbs) isolated from the phage-displayed synthetic human VH/VL libraries.
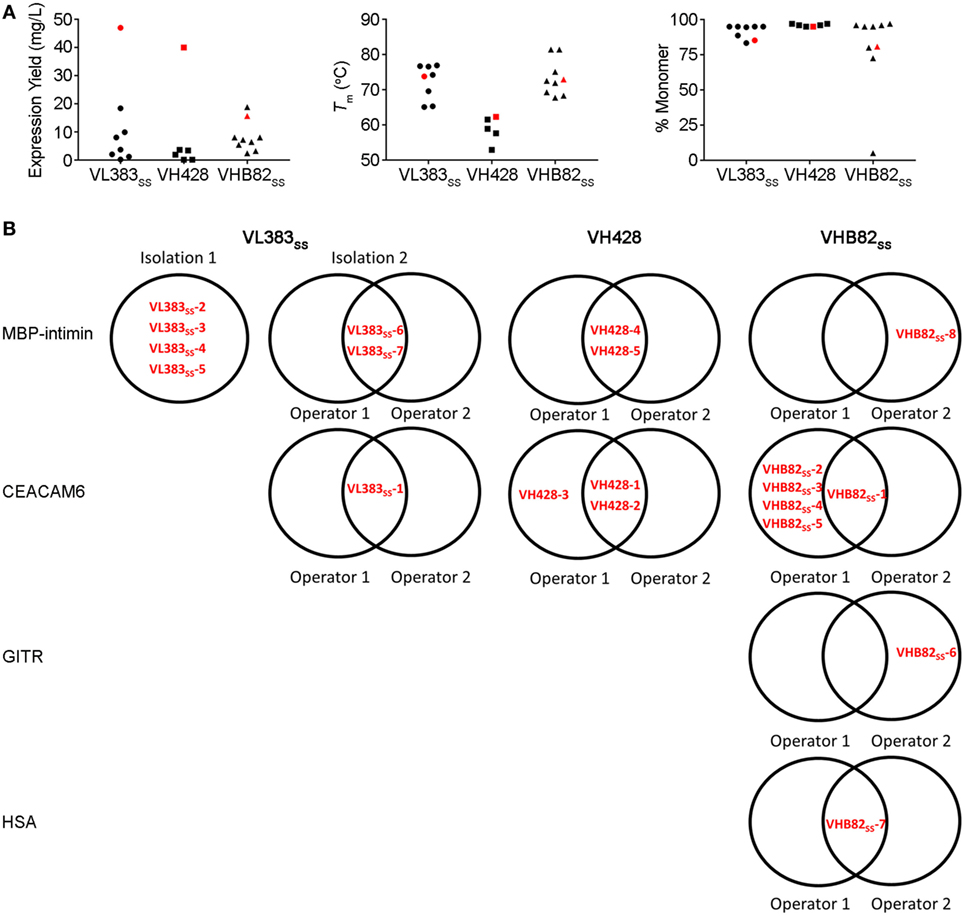
Figure 4. Properties of all antigen-specific single-domain antibodies (sdAbs) isolated from the VL383SS, VH428 and VHB82SS synthetic human VH/VL sdAb libraries. (A) Expression yields (as measured by spectrophotometry at 280 nm), Tms (as determined by circular dichroism), and aggregation tendencies (as determined by size exclusion chromatography) of antigen-specific VH/VL sdAbs isolated from the libraries in comparison to the parental scaffold from which they were derived (shown in red). (B) Interoperator reproducibility in isolating individual VH/VL sdAb sequences.
Evaluation of Stability-Sequence Diversity Tradeoffs in Human VH/VL sdAb Libraries
To better understand potential constraints on the CDR sequences of monomeric and stable human VH/VL sdAbs, we examined the effects of stability selection (three rounds of protein A or L selection followed by amplification of the eluted phage overnight in E. coli TG1 cells; Figure 5) on randomized sequence diversity of two VH/VL sdAb libraries (VL383SS and VHB82SS). We performed this experiment in duplicate using either phage particles displaying monovalent sdAb (rescued with M13K07 helper phage; simultaneous expression of pIII from helper phage and pIII-sdAb from phagemid) or phage particles displaying multivalent sdAb (rescued with M13K07ΔpIII hyper phage; only pIII-sdAb expressed from phagemid). Out-of-frame and/or stop-codon-encoding VH/VL sdAb clones were rare in both libraries (“Library Cells”), but rose substantially in frequency upon phage rescue (“Library Phage”) and upon amplification in E. coli (“Amplification in E. coli”), suggesting a growth advantage for non-sdAb expressing cells over sdAb-expressing ones (Figures 5A,B); this phenomenon was mitigated somewhat using hyper phage rescue. After three rounds of protein A/L selection and overnight amplification in E. coli with helper phage rescue, there were clear clonal biases observed in the resulting populations of sdAb-phage (Figures 5C,D). Using an arbitrary frequency cutoff (0.00006% for VL383SS and 0.00004% for VHB82SS; sdAbs at frequencies greater than these cutoffs were not present in any other dataset) to identify unique sdAb sequences enriched by stability selection, we found that different sdAb clones were selected in the two replicate pannings, although both sets of sdAbs were heavily biased toward the parental VH/VL sdAb scaffold’s CDR3 length (9 residues for the VL383SS library and 10 residues for the VHB82SS library, which is the shortest CDR3 length possible in the library design and the nearest to the VHB82SS scaffold’s CDR3 length of 6 residues; Figure S4 in Supplementary Material). However, stability selection reproducibly yielded VH/VL sdAbs with biased CDR amino acid composition (Figures 5E,F; Figures S5 and S6 in Supplementary Material). In order of magnitude, stability biases in the VL383SS library favored Asp, His, Pro, Ser and Thr and disfavored aromatic (Phe, Trp, Tyr) and hydrophobic (Ile, Leu, Val) residues. Similarly, stability biases in the VHB82SS library favored Asp, His, Pro, and Gln and disfavored aromatic (Phe, Trp, Tyr) and hydrophobic (Ile, Leu, Val) residues. These biases were especially acute at the C-terminus of CDR1 and N-terminus of CDR2, both of which flank FR2. Identical molecular signatures were observed to a lesser degree in the pools of all sdAb-phage (irrespective of frequency or mono- vs. multivalent display format) subjected to three rounds of stability selection (Figures S7 and S8 in Supplementary Material) as well as in the pools of sdAb-phage after phage rescue and eluted from protein A/L (data not shown), suggesting a common ontogeny.
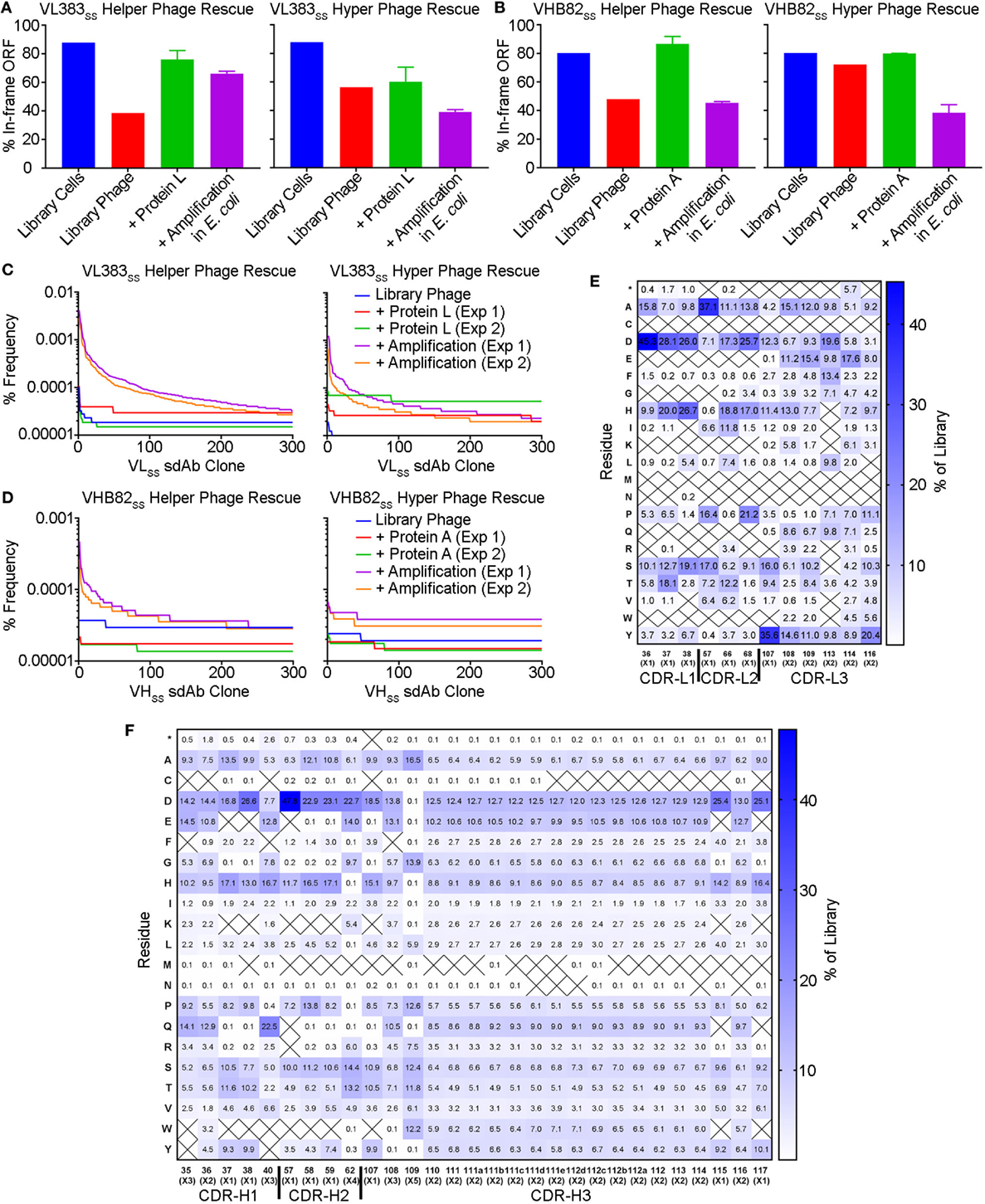
Figure 5. Impact of stability selection on human VH/VL single-domain antibody (sdAb) library sequence diversity. After transformation of Escherichia coli TG1 cells with phagemid DNA, phage were rescued by superinfection of overnight cultures with M13K07 helper phage or M13K07ΔpIII hyper phage. The purified sdAb-displaying phage were bound and eluted from protein A (VHB82SS library) or protein L (VL383SS library), then amplified by reinfection of E. coli TG1 cells. Three rounds of panning were performed, and VH/VL sdAb sequences were interrogated by NGS at the stage of rescued phage, phage eluted after a single round of protein A/L selection, and phage amplified after the final round of panning. (A,B) Proportion of functional sdAb sequences (in-frame ORF, no stop codons) observed in VL (A) and VH (B) library phage and panning outputs. (C,D) Clonality of VL (C) and VH (D) library phage and panning outputs. (E,F) Complementarity-determining region (CDR) amino acid composition of enriched VL (E) and VH (F) sdAb clones after three rounds of stability selection. Crossed-out cells indicate a frequency of <0.1%. Analyses in (A–F) are representative of 7.7 × 104 to 7.9 × 105 sequences per sample.
Discussion
The starting point for this investigation was the observation that several of our previously described synthetic human VH/VL sdAb libraries performed unpredictably (12, 21, 31), yielding monomeric antigen-specific binders against some targets but not others. Thus, we tested the hypothesis that these issues might be solved by: (i) building libraries on ultrastable VH/VL sdAb scaffolds that maintain their biophysical properties upon CDR modification and (ii) randomizing the CDRs of the VH/VL sdAb libraries more extensively using trinucleotide mutagenesis. A few general lessons became apparent during the course of these experiments. First, although VH/VL sdAb scaffolds clearly vary in their tolerance to CDR modification, no “perfect” scaffold exists (i.e., some CDR-shuffled variants of every scaffold showed poor biophysical properties). Second, the trinucleotide mutagenesis approach used here was dramatically more effective at incorporating randomized sequence diversity in VH/VL sdAb CDRs compared to the Kunkel mutagenesis (21, 31) and splicing by overlap extension PCR mutagenesis approaches using degenerate oligonucleotides (12) that we have used previously. Third, as we showed previously (31), incorporating a stabilizing exogenous disulfide linkage spanning IMGT positions 54–78 into VH/VL sdAb libraries is clearly compatible with selection of antigen-specific binders, as 15/20 of the binders shown here bear this linkage. Finally, it is clearly possible, at least under some circumstance, to select monomeric, high-affinity binders from synthetic human VH/VL sdAbs libraries, as evidenced by the α-HSA VHSS-7 sdAb (KD: 5 nM) reported here.
However, the more general issues afflicting synthetic human VH/VL sdAb libraries (high attrition rates due to false positive binding, possibly caused by sdAb aggregation; inconsistency of isolation; variable expression yield and aggregation of isolated sdAbs) were not completely solved by using more stable VH/VL scaffolds or building more diverse and non-degenerate libraries. The additional randomized sequence diversity we achieved by the use of trinucleotide mutagenesis is almost certainly negligible when compared to the theoretical size of the libraries, and massive undersampling may account at least in part for inconsistent isolation of sdAbs. Other potential solutions to the problem of undersampling include increasing throughput (53), use of VH/VL sdAb-transgenic mice (54, 55) and use of in vitro VDJ recombination systems (56). Nonetheless, modest inter-operator reproducibility in isolating the same binders clearly demonstrates that antigen-specific human VH/VL sdAbs are not always isolated even when they were present in the library. We currently have no understanding of the factors influencing the number of VH/VL sdAb binders isolated, their affinities or biophysical properties, nor if these depend on the library, the target antigen quality or composition, or the panning methodology. One possibility, for example, is that the numbers of input library sdAb-phage used here, the target antigen surface size and density, and/or the numbers of eluted phage amplified from each round of selection were inadequate to consistently recover very rare sdAb specificities that may be present in the libraries. These factors could be investigated empirically in future studies.
It is virtually certain that some of the challenges of human synthetic VH/VL sdAbs relate to fundamental tradeoffs between stability and sequence diversity. There is no necessary reason why rare autonomous rearranged human VH/VL sdAbs should be compatible with any CDR sequence; rather, it should be expected that these molecules rely chiefly on particular CDR sequences for their solubility and stability, given that human VH and VL domains have evolved to be paired with one another, occluding a hydrophobic surface between the two domains. We expect that such challenges are much less of a problem for protein domains that naturally exist as soluble monomers such as camelid and shark sdAbs and non-antibody scaffolds (e.g., FN3 and SH3 domains). Some camelid VHHs rely on CDR residues for solubility as well (57), albeit likely to a lesser extent due to the presence of solubility-enhancing FR2 residues. One obvious explanation for the previously described bias toward negatively charged residues resulting in acidic overall pIs of VH/VL sdAbs (4, 12), especially in CDR1 and CDR2 (10, 11), is that this may enhance solubility and aggregation resistance. The role of charge and negative charge in particular in solubilizing VH/VL sdAbs with hydrophobic CDRs has been previously demonstrated (58, 59), and appears to be highly position and scaffold dependent (4). Bias in favor of His residues is less easy to explain, although its imidazole side chain may also have stabilizing and solubilizing effects near physiological pH (60). Notably, “camelized” human VH sdAbs and human VH/VL sdAbs selected in vitro to bear solubility-promoting FR substitutions (9, 61), may not be subject to the same stability-sequence diversity tradeoffs. We caution that although the growth advantages conferred by VH/VL sdAbs with the most stable and soluble CDR sequences were most apparent using helper phage rescue of phagemid libraries, forcing pIII-sdAb expression using hyper phage rescue or by using phage (not phagemid) or yeast display systems would not be expected to circumvent the issue of bias; instead, it might be expected to result in immediate loss of a large population of VH/VL sdAbs with poorly stable and soluble CDR sequences.
In conclusion, we have described three novel synthetic human VH/VL sdAb libraries as well as antigen-specific binders against a variety of target antigens selected from these libraries. Future work will seek to better understand the constraints imposed on human VH/VL sdAbs by stability-sequence diversity tradeoffs, and whether it is possible to circumvent them by in vitro engineering of human autonomous immunoglobulin variable domain folds. One possibility would be to identify and fix a minimal set of stability-enhancing CDR residues in human VH/VL sdAb libraries, which might allow for more effective randomization of the remaining CDR positions. However, doing so without significant divergence from human germline IGHV sequences, increasing the risk of immunogenicity, may be very challenging.
Author Contributions
DK, HK, and JT isolated and characterized human VH/VL sdAb scaffolds and conducted CDR-shuffling experiments and designed the libraries. JS conducted SEC-MALS experiments. ML, HK, and KH isolated and characterized antigen-specific sdAbs. ML and KH conceived and carried out stability selections and next-generation DNA sequencing analyses. QY, GH, and CRM designed, carried out, and analyzed surface plasmon resonance experiments. KH and GH made the figures and KH wrote the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
JT and DK are inventors of US patents 8293233B2 and 9371371B2 as well as other patents and patent applications governing human VH/VL sdAb scaffolds.
Acknowledgments
We gratefully acknowledge the excellent technical help of Camille Hebert-Martineau, Sonia Leclerc, Hongtao Qi, Ken Chan, Shannon Ryan, and Julie Champagne.
Funding
This work was supported by funding from the National Research Council Canada.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01759/full#supplementary-material.
References
1. Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature (1989) 341:544–6. doi:10.1038/341544a0
2. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature (1993) 363:446–8. doi:10.1038/363446a0
3. Greenberg AS, Avila D, Hughes M, Hughes A, McKinney EC, Flajnik MF. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature (1995) 374:168–73. doi:10.1038/374168a0
4. Kim DY, Hussack G, Kandalaft H, Tanha J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim Biophys Acta (2014) 1844:1983–2001. doi:10.1016/j.bbapap.2014.07.008
5. Tanha J, Nguyen TD, Ng A, Ryan S, Ni F, MacKenzie R. Improving solubility and refolding efficiency of human VHs by a novel mutational approach. Protein Eng Des Sel (2006) 19:503–9. doi:10.1093/protein/gzl037
6. Tanha J, Xu P, Chen Z, Ni F, Kaplan H, Narang SA, et al. Optimal design features of camelized human single-domain antibody libraries. J Biol Chem (2001) 276:24774–80. doi:10.1074/jbc.M100770200
7. Davies J, Riechmann L. Single antibody domains as small recognition units: design and in vitro antigen selection of camelized, human VH domains with improved protein stability. Protein Eng (1996) 9:531–7. doi:10.1093/protein/9.6.531
8. Davies J, Riechmann L. Antibody VH domains as small recognition units. Biotechnology (N Y) (1995) 13:475–9. doi:10.1038/nbt0595-475
9. Ma X, Barthelemy PA, Rouge L, Wiesmann C, Sidhu SS. Design of synthetic autonomous VH domain libraries and structural analysis of a VH domain bound to vascular endothelial growth factor. J Mol Biol (2013) 425:2247–59. doi:10.1016/j.jmb.2013.03.020
10. Dudgeon K, Famm K, Christ D. Sequence determinants of protein aggregation in human VH domains. Protein Eng Des Sel (2009) 22:217–20. doi:10.1093/protein/gzn059
11. Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A (2012) 109:10879–84. doi:10.1073/pnas.1202866109
12. Arbabi-Ghahroudi M, To R, Gaudette N, Hirama T, Ding W, MacKenzie R, et al. Aggregation-resistant VHs selected by in vitro evolution tend to have disulfide-bonded loops and acidic isoelectric points. Protein Eng Des Sel (2009) 22:59–66. doi:10.1093/protein/gzn071
13. Christ D, Famm K, Winter G. Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel (2007) 20:413–6. doi:10.1093/protein/gzm037
14. Christ D, Famm K, Winter G. Tapping diversity lost in transformations – in vitro amplification of ligation reactions. Nucleic Acids Res (2006) 34:e108. doi:10.1093/nar/gkl605
15. De Bernardis F, Liu H, O’Mahony R, La Valle R, Bartollino S, Sandini S, et al. Human domain antibodies against virulence traits of Candida albicans inhibit fungus adherence to vaginal epithelium and protect against experimental vaginal candidiasis. J Infect Dis (2007) 195:149–57. doi:10.1086/509891
16. Pinelli DF, Wagener ME, Liu D, Yamniuk A, Tamura J, Grant S, et al. An anti-CD154 domain antibody prolongs graft survival and induces Foxp3(+) iTreg in the absence and presence of CTLA-4 Ig. Am J Transplant (2013) 13:3021–30. doi:10.1111/ajt.12417
17. Suchard SJ, Davis PM, Kansal S, Stetsko DK, Brosius R, Tamura J, et al. A monovalent anti-human CD28 domain antibody antagonist: preclinical efficacy and safety. J Immunol (2013) 191:4599–610. doi:10.4049/jimmunol.1300470
18. Yamniuk AP, Suri A, Krystek SR, Tamura J, Ramamurthy V, Kuhn R, et al. Functional antagonism of human CD40 achieved by targeting a unique species-specific epitope. J Mol Biol (2016) 428:2860–79. doi:10.1016/j.jmb.2016.05.014
19. Walker A, Chung CW, Neu M, Burman M, Batuwangala T, Jones G, et al. Novel interaction mechanism of a domain antibody-based inhibitor of human vascular endothelial growth factor with greater potency than ranibizumab and bevacizumab and improved capacity over aflibercept. J Biol Chem (2016) 291:5500–11. doi:10.1074/jbc.M115.691162
20. Xie JH, Yamniuk AP, Borowski V, Kuhn R, Susulic V, Rex-Rabe S, et al. Engineering of a novel anti-CD40L domain antibody for treatment of autoimmune diseases. J Immunol (2014) 192:4083–92. doi:10.4049/jimmunol.1303239
21. Hussack G, Keklikian A, Alsughayyir J, Hanifi-Moghaddam P, Arbabi-Ghahroudi M, van Faassen H, et al. A VL single-domain antibody library shows a high-propensity to yield non-aggregating binders. Protein Eng Des Sel (2012) 25:313–8. doi:10.1093/protein/gzs014
22. Zhang H, Yun S, Batuwangala TD, Steward M, Holmes SD, Pan L, et al. A dual-targeting antibody against EGFR-VEGF for lung and head and neck cancer treatment. Int J Cancer (2012) 131:956–69. doi:10.1002/ijc.26427
23. Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci U S A (2017) 114:E6623–31. doi:10.1073/pnas.1706055114
24. Feng M, Gao W, Wang R, Chen W, Man YG, Figg WD, et al. Therapeutically targeting glypican-3 via a conformation-specific single-domain antibody in hepatocellular carcinoma. Proc Natl Acad Sci U S A (2013) 110:E1083–91. doi:10.1073/pnas.1217868110
25. Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol (2004) 22:1161–5. doi:10.1038/nbt1000
26. Walker A, Dunlevy G, Rycroft D, Topley P, Holt LJ, Herbert T, et al. Anti-serum albumin domain antibodies in the development of highly potent, efficacious and long-acting interferon. Protein Eng Des Sel (2010) 23:271–8. doi:10.1093/protein/gzp091
27. Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, et al. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel (2008) 21:283–8. doi:10.1093/protein/gzm067
28. Rouet R, Dudgeon K, Christie M, Langley D, Christ D. Fully human VH single domains that rival the stability and cleft recognition of camelid antibodies. J Biol Chem (2015) 290:11905–17. doi:10.1074/jbc.M114.614842
29. Mandrup OA, Friis NA, Lykkemark S, Just J, Kristensen P. A novel heavy domain antibody library with functionally optimized complementarity determining regions. PLoS One (2013) 8:e76834. doi:10.1371/journal.pone.0076834
30. Jespers L, Schon O, James LC, Veprintsev D, Winter G. Crystal structure of HEL4, a soluble, refoldable human VH single domain with a germ-line scaffold. J Mol Biol (2004) 337:893–903. doi:10.1016/j.jmb.2004.02.013
31. Henry KA, Kandalaft H, Lowden MJ, Rossotti MA, van Faassen H, Hussack G, et al. A disulfide-stabilized human VL single-domain antibody library is a source of soluble and highly thermostable binders. Mol Immunol (2017) 90:190–6. doi:10.1016/j.molimm.2017.07.006
32. Yu GW, Vaysburd M, Allen MD, Settanni G, Fersht AR. Structure of human MDM4 N-terminal domain bound to a single-domain antibody. J Mol Biol (2009) 385:1578–89. doi:10.1016/j.jmb.2008.11.043
33. Tang Z, Feng M, Gao W, Phung Y, Chen W, Chaudhary A, et al. A human single-domain antibody elicits potent antitumor activity by targeting an epitope in mesothelin close to the cancer cell surface. Mol Cancer Ther (2013) 12:416–26. doi:10.1158/1535-7163.MCT-12-0731
34. Gay RD, Clarke AW, Elgundi Z, Domagala T, Simpson RJ, Le NB, et al. Anti-TNF-α domain antibody construct CEP-37247: full antibody functionality at half the size. MAbs (2010) 2:625–38. doi:10.4161/mabs.2.6.13493
35. Bertok S, Wilson MR, Morley PJ, de Wildt R, Bayliffe A, Takata M. Selective inhibition of intra-alveolar p55 TNF receptor attenuates ventilator-induced lung injury. Thorax (2012) 67:244–51. doi:10.1136/thoraxjnl-2011-200590
36. Holland MC, Wurthner JU, Morley PJ, Birchler MA, Lambert J, Albayaty M, et al. Autoantibodies to variable heavy (VH) chain Ig sequences in humans impact the safety and clinical pharmacology of a VH domain antibody antagonist of TNF-α receptor 1. J Clin Immunol (2013) 33:1192–203. doi:10.1007/s10875-013-9915-0
37. Cordy JC, Morley PJ, Wright TJ, Birchler MA, Lewis AP, Emmins R, et al. Specificity of human anti-variable heavy (VH) chain autoantibodies and impact on the design and clinical testing of a VH domain antibody antagonist of tumour necrosis factor-α receptor 1. Clin Exp Immunol (2015) 182:139–48. doi:10.1111/cei.12680
38. To R, Hirama T, Arbabi-Ghahroudi M, MacKenzie R, Wang P, Xu P, et al. Isolation of monomeric human VHs by a phage selection. J Biol Chem (2005) 280:41395–403. doi:10.1074/jbc.M509900200
39. Kim DY, To R, Kandalaft H, Ding W, van Faassen H, Luo Y, et al. Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. MAbs (2014) 6:219–35. doi:10.4161/mabs.26844
40. Kim DY, Kandalaft H, Ding W, Ryan S, van Faassen H, Hirama T, et al. Disulfide linkage engineering for improving biophysical properties of human VH domains. Protein Eng Des Sel (2012) 25:581–9. doi:10.1093/protein/gzs055
41. Virnekas B, Ge L, Pluckthun A, Schneider KC, Wellnhofer G, Moroney SE. Trinucleotide phosphoramidites: ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res (1994) 22:5600–7. doi:10.1093/nar/22.25.5600
42. Baral TN, MacKenzie R, Arbabi Ghahroudi M. Single-domain antibodies and their utility. Curr Protoc Immunol (2013) 103:Unit 2.17. doi:10.1002/0471142735.im0217s103
43. Dorion-Thibaudeau J, St-Laurent G, Raymond C, De Crescenzo G, Durocher Y. Biotinylation of the Fcγ receptor ectodomains by mammalian cell co-transfection: application to the development of a surface plasmon resonance-based assay. J Mol Recognit (2016) 29:60–9. doi:10.1002/jmr.2495
44. Henry KA, Hussack G, Collins C, Zwaagstra JC, Tanha J, MacKenzie CR. Isolation of TGF-β-neutralizing single-domain antibodies of predetermined epitope specificity using next-generation DNA sequencing. Protein Eng Des Sel (2016) 29:439–43. doi:10.1093/protein/gzw043
45. Henry KA, Tanha J, Hussack G. Identification of cross-reactive single-domain antibodies against serum albumin using next-generation DNA sequencing. Protein Eng Des Sel (2015) 28:379–83. doi:10.1093/protein/gzv039
46. Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem (2004) 332:153–9. doi:10.1016/j.ab.2004.04.031
47. Ericsson UB, Hallberg BM, Detitta GT, Dekker N, Nordlund P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem (2006) 357:289–98. doi:10.1016/j.ab.2006.07.027
48. Henry KA, Sulea T, van Faassen H, Hussack G, Purisima EO, MacKenzie CR, et al. A rational engineering strategy for designing protein A-binding camelid single-domain antibodies. PLoS One (2016) 11:e0163113. doi:10.1371/journal.pone.0163113
49. Baral TN, Chao SY, Li S, Tanha J, Arbabi-Ghahroudi M, Zhang J, et al. Crystal structure of a human single domain antibody dimer formed through VH-VH non-covalent interactions. PLoS One (2012) 7:e30149. doi:10.1371/journal.pone.0030149
50. Sepulveda J, Jin H, Sblattero D, Bradbury A, Burrone OR. Binders based on dimerised immunoglobulin VH domains. J Mol Biol (2003) 333:355–65. doi:10.1016/j.jmb.2003.08.033
51. Jin H, Sepulveda J, Burrone OR. Selection and characterisation of binders based on homodimerisation of immunoglobulin VH domains. FEBS Lett (2003) 554:323–9. doi:10.1016/S0014-5793(03)01182-7
52. Jin H, Sepulveda J, Burrone OR. Specific recognition of a dsDNA sequence motif by an immunoglobulin VH homodimer. Protein Sci (2004) 13:3222–9. doi:10.1110/ps.04921704
53. Wu Y, Batyuk A, Honegger A, Brandl F, Mittl PRE, Plückthun A. Rigidly connected multispecific artificial binders with adjustable geometries. Sci Rep (2017) 7:11217. doi:10.1038/s41598-017-11472-x
54. Drabek D, Janssens R, de Boer E, Rademaker R, Kloess J, Skehel J, et al. Expression cloning and production of human heavy-chain-only antibodies from murine transgenic plasma cells. Front Immunol (2016) 7:619. doi:10.3389/fimmu.2016.00619
55. Brüggemann M, Zou X, Inventors; Crescendo Biologics Ltd., Assignee. Mouse λ Light Chain Locus. United States patent US 9439405 B2 (2016).
56. Gallo M, Kang JS, Pigott CR, Inventors; Innovative Targeting Solutions, Inc., Assignee. Sequence Diversity Generation in Immunoglobulins. United States patent US 8012714 B2 (2011).
57. Conrath K, Vincke C, Stijlemans B, Schymkowitz J, Decanniere K, Wyns L, et al. Antigen binding and solubility effects upon the veneering of a camel VHH in framework-2 to mimic a VH. J Mol Biol (2005) 350:112–25. doi:10.1016/j.jmb.2005.04.050
58. Perchiacca JM, Ladiwala AR, Bhattacharya M, Tessier PM. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng Des Sel (2012) 25:591–601. doi:10.1093/protein/gzs042
59. Perchiacca JM, Lee CC, Tessier PM. Optimal charged mutations in the complementarity-determining regions that prevent domain antibody aggregation are dependent on the antibody scaffold. Protein Eng Des Sel (2014) 27:29–39. doi:10.1093/protein/gzt058
60. Chan P, Warwicker J. Evidence for the adaptation of protein pH-dependence to subcellular pH. BMC Biol (2009) 7:69. doi:10.1186/1741-7007-7-69
Keywords: single-domain antibody, synthetic antibody, human VH/VL, phage display, protein engineering
Citation: Henry KA, Kim DY, Kandalaft H, Lowden MJ, Yang Q, Schrag JD, Hussack G, MacKenzie CR and Tanha J (2017) Stability-Diversity Tradeoffs Impose Fundamental Constraints on Selection of Synthetic Human VH/VL Single-Domain Antibodies from In Vitro Display Libraries. Front. Immunol. 8:1759. doi: 10.3389/fimmu.2017.01759
Received: 30 September 2017; Accepted: 27 November 2017;
Published: 12 December 2017
Edited by:
Abdul Qader Abbady, Atomic Energy Commission of Syria, SyriaReviewed by:
Serge Muyldermans, Vrije Universiteit Brussel, BelgiumOscar R. Burrone, International Centre for Genetic Engineering and Biotechnology, Italy
Copyright: © 2015 Her Majesty the Queen in Right of Canada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamshid Tanha, amFtc2hpZC50YW5oYUBucmMtY25yYy5nYy5jYQ==
This is NRC-HHT publication number: 53364.