 Young-Mee Moon1†Seon-Yeong Lee1†Seung-Ki Kwok2†Seung Hoon Lee1†
Young-Mee Moon1†Seon-Yeong Lee1†Seung-Ki Kwok2†Seung Hoon Lee1† Deokhoon Kim3,4Woo Kyung Kim5Yang-Mi Her1Hea-Jin Son1Eun-Kyung Kim1Jun-Geol Ryu1Hyeon-Beom Seo1Jeong-Eun Kwon1Sue-Yun Hwang6
Deokhoon Kim3,4Woo Kyung Kim5Yang-Mi Her1Hea-Jin Son1Eun-Kyung Kim1Jun-Geol Ryu1Hyeon-Beom Seo1Jeong-Eun Kwon1Sue-Yun Hwang6 Jeehee Youn7Rho H. Seong8Dae-Myung Jue9Sung-Hwan Park2Ho-Youn Kim2Sung-Min Ahn10*†
Jeehee Youn7Rho H. Seong8Dae-Myung Jue9Sung-Hwan Park2Ho-Youn Kim2Sung-Min Ahn10*† Mi-La Cho1*†
Mi-La Cho1*†
- 1Laboratory of Immune Network, The Rheumatism Research Center, College of Medicine, Catholic Research Institute of Medical Science, The Catholic University of Korea, Seoul, South Korea
- 2Center for Rheumatic Disease, Division of Rheumatology, Department of Internal Medicine, Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 3Asan Institute for Life Sciences, University of Ulsan College of Medicine, Asan Medical Center, Seoul, South Korea
- 4Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, South Korea
- 5Department of Developmental Biology, Washington University School of Medicine, St. Louis, MO, United States
- 6Department of Chemical Engineering, Hankyong National University, Anseong, South Korea
- 7Department of Biomedical Sciences, College of Medicine, Hanyang University, Seoul, South Korea
- 8Department of Biological Sciences, Institute of Molecular Biology and Genetics, Research Center for Functional Cellulomics, Seoul National University, Seoul, South Korea
- 9Department of Biochemistry, College of Medicine, The Catholic University of Korea, Seoul, South Korea
- 10Department of Hemato-oncology, Bioinformatics, Cancer Research, Systems Biology, Gachon University, Seongnam, South Korea
Dysfunction of T helper 17 (Th17) cells leads to chronic inflammatory disorders. Signal transducer and activator of transcription 3 (STAT3) orchestrates the expression of proinflammatory cytokines and pathogenic cell differentiation from interleukin (IL)-17-producing Th17 cells. However, the pathways mediated by STAT3 signaling are not fully understood. Here, we observed that Fos-related antigen 1 (FRA1) and JUNB are directly involved in STAT3 binding to sites in the promoters of Fosl1 and Junb. Promoter binding increased expression of IL-17 and the development of Th17 cells. Overexpression of Fra1 and Junb in mice resulted in susceptibility to collagen-induced arthritis and an increase in Th17 cell numbers and inflammatory cytokine production. In patients with rheumatoid arthritis, FRA1 and JUNB were colocalized with STAT3 in the inflamed synovium. These observations suggest that FRA1 and JUNB are associated closely with STAT3 activation, and that this activation leads to Th17 cell differentiation in autoimmune diseases and inflammation.
Introduction
T helper 17 (Th17) cells are a pathogenic subset of T helper lymphocytes that play a key role in inflammatory disorders leading to a severe chronic immune inflammatory response. Th17 cells release several proinflammatory cytokines including interleukin (IL)-17A, IL-21, and IL-22 (1). IL-17A is an inflammatory cytokine produced predominantly by Th17 cells with strong effects on stromal cells. IL-17A induces inflammatory cytokine production and leukocyte recruitment, which act to link the innate and adaptive branches of immunity (2). Both IL-17A and Th17 cells are highly involved in the pathogenesis of several autoimmune diseases, including rheumatoid arthritis (RA) (3, 4), while playing a significant role in autoimmune disorders mediated by excessive inflammation.
Signal transducer and activator of transcription 3 (STAT3) is an important transcription factor with DNA-binding properties whose activity is paramount to the inflammatory response. It has been suggested that STAT3 activation upregulates IL-17 production via Th17 cell proliferation (5, 6). Several transcription factors including STAT3 regulate Th17 cell differentiation (7–12); however, STAT3 also plays a key role in the immune inflammatory response. There is a general consensus that STAT3 is essential for Th17 cell differentiation (13). Moreover, STAT3 modulates the production of several cytokines including IL-17A and activates downstream transcription factors, such as RAR-related orphan receptor gamma isoform 2 (RORγt), which is responsible for the Th17 phenotype (14, 15).
The activator protein 1 (AP-1) family is a group of structurally and functionally related JUN (c-JUN, JUNB, and JUND) and FOS [c-FOS, FOSB, Fos-related antigen 1 (FRA1), and FRA2] transcription factors. AP-1 heterodimers are involved in a variety of biological processes including cell proliferation, differentiation, apoptosis, and inflammation (16, 17). It has been suggested that AP-1 proteins are involved in several pathological conditions (18–21), while JUN and FOS proteins are also associated with the immune inflammatory response. Modulation of c-FOS and c-JUN expression is critical for inhibition of IL-17 production (22) and the maintenance of suppressive regulatory T-cell function (23). Additionally, production of FRA1, a member of the FOS protein family, is increased by B cell stimulation (24). Furthermore, JUNB modulates the proliferation of B cells (25). This evidence suggests that FRA1 and JUNB may be involved in regulating the inflammatory immune response.
We hypothesized that FRA1 and JUNB modulate the Th17 cell-mediated inflammatory response. The aim of this study was to elucidate whether FRA1 and JUNB regulate autoimmune arthritis via Th17 cell differentiation and factors downstream of STAT3. We used in vitro models, in vivo animal models, and clinical specimens from patients with RA to investigate the biological importance of this pathway.
Materials and Methods
Mice
Collagen-induced arthritis (CIA) was induced in 6–8-week-old male DBA/1J, BALB/c, and C57BL/6 mice (Orient, Korea). To generate Fra1/Junb Tg mice, a pcDNA3.1+HA (Invitrogen, CA, USA) vector containing the FRA1 and JUNB proteins coupled to a linker peptide (3 × GGGGS) was constructed. The Fra1/Junb fragment was synthesized by GenScript Corporation (NJ, USA), with codon optimization for expression in mammalian cells. Fra1/Junb Tg mice were bred from the C57BL/6 line and maintained in facilities at Macrogen Laboratories (Seoul, Korea). All mice were maintained under specific-pathogen-free conditions at the Institute of Medical Science, The Catholic University of Korea. The presence of the transgene in the founders was confirmed by PCR of genomic DNA extracted from the tail samples. Genotyping was performed by PCR analysis of genomic DNA obtained from mice at 3 weeks of age. All experimental procedures were examined and approved by the Animal Research Ethics Committee at the Catholic University of Korea.
Accession Codes
Raw RNA-seq data have been deposited in the NCBI Sequence Read Archive (SRR6320798 and SRR6320799).
A detailed description of all other experimental procedures and the statistical analysis is provided in the Section “Supplementary Materials and Methods” in Data Sheet S1 in Supplementary Material.
Results
STAT3 Target Genes Are Differentially Expressed in Mouse Th17 Cells
Potential STAT3-binding sites were identified using publicly available chromatin immunoprecipitation sequencing (ChIP-Seq) data cross-referenced with differentially expressed genes in Th17 cells (14). We sequenced mRNA obtained from Th17 cells and naïve T cells and compared the results with potential STAT3-binding targets identified by ChIP-Seq to identify STAT3-regulated genes involved in Th17 cell differentiation. The literature was systematically reviewed for downstream STAT3 targets in humans and mice in multiple biological contexts. By combining these three datasets, we searched for genes with STAT3-binding sites that are upregulated during Th17 cell differentiation (Figure 1A; Table S1 in Supplementary Material). RNA-Seq was performed to identify genes differentially expressed between Th17 cells and naïve T cells. This integrated approach suggested that Fosl1 (the gene locus of Fra1) and Junb are Th17 cell differentiation factors downstream of STAT3. As shown in Figures 1B–F and Table S2 in Supplementary Material, Fos and Jun were the predominant AP-1 subtypes expressed in naïve (CD4+CD62L+) T cells, whereas Fosl1 and Junb were the predominant subtypes in Th17 cells. Other AP-1 subtypes were downregulated, suggesting that Fosl1 and Junb AP-1 subtypes play an important role in Th17 cell differentiation.
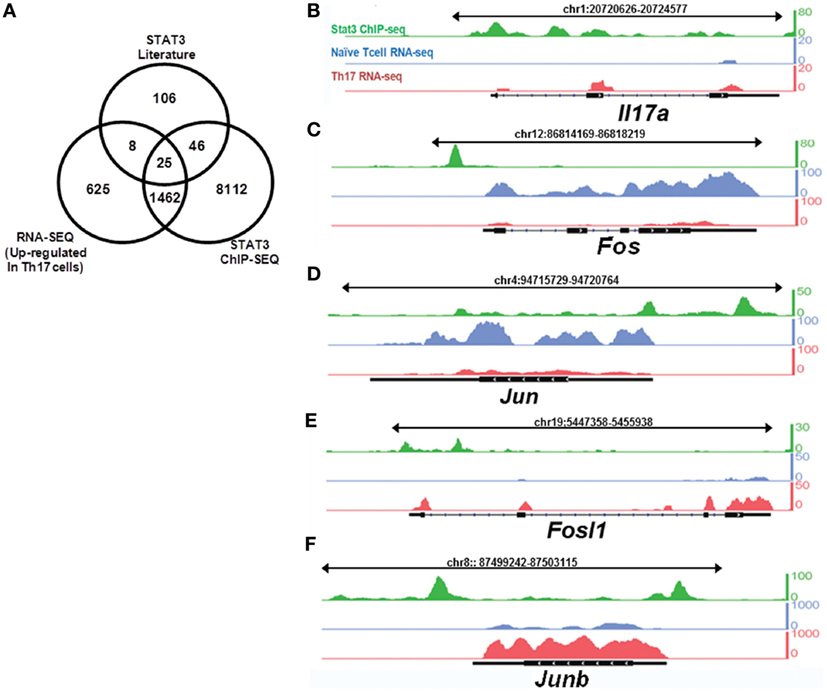
Figure 1. Signal transducer and activator of transcription 3 (STAT3) target genes are differentially expressed in naïve and T helper 17 (Th17) cells. Naïve T cells (CD4+CD62L+) were isolated from 6-week-old mice (n = 3), stimulated under Th17-polarizing conditions for 3 days, and then sorted for CD4+CD62L− expression. RNA was sequenced using the Illumina GA IIx. STAT3 chromatin immunoprecipitation sequencing (ChIP-Seq) data for CD4+ T cells were downloaded from the NCBI Sequence Read Archive (SRP002451). (A) A comparison of differentially expressed genes identified by RNA-Seq, STAT3-binding target genes identified by ChIP-Seq, and the downstream targets of STAT3. The expression of (B) IL17a and (C–F) activator protein 1 transcription factor target genes [(C) Fos, (D) Jun, (E) Fosl1, and (F) Junb] was compared between naïve T cells and Th17 cells, with reference to STAT3-binding target genes (obtained from STAT3 ChIP-Seq data in Th17 cells). The peak profiles are color-coded as follows: naïve T cells (RNA-Seq: blue), Th17 cells (RNA-Seq: red), and STAT3-binding genes (ChIP-Seq: green).
Fra1 and Junb Are Highly Expressed in Th17 Cells, and Their Expression Is Regulated Directly by STAT3 in Mice
Quantitative real-time reverse transcription PCR (qRT-PCR) and immunoblot analyzes were used to confirm Fra1 and Junb expression. Among the various AP-1 subtypes, only Fra1 and Junb mRNA levels were significantly increased in Th17 cells compared with naive T cells (Figures 2A,B). These findings supported the RNA-Seq results. Immunoblot analysis also showed high levels of FRA1 and JUNB expression in Th17 cells (Figure 2C). Co-immunoprecipitation studies were performed using antibodies against Jun family proteins (i.e., JUNB and c-JUN), followed by immunoblotting using antibodies against FOS family proteins (i.e., FRA1, c-FOS, and FOSB) to determine whether FRA1 and JUNB form a heterodimeric AP-1 complex. It was found that FRA1 forms a complex with JUNB, but not with c-JUN (Figure 2D).
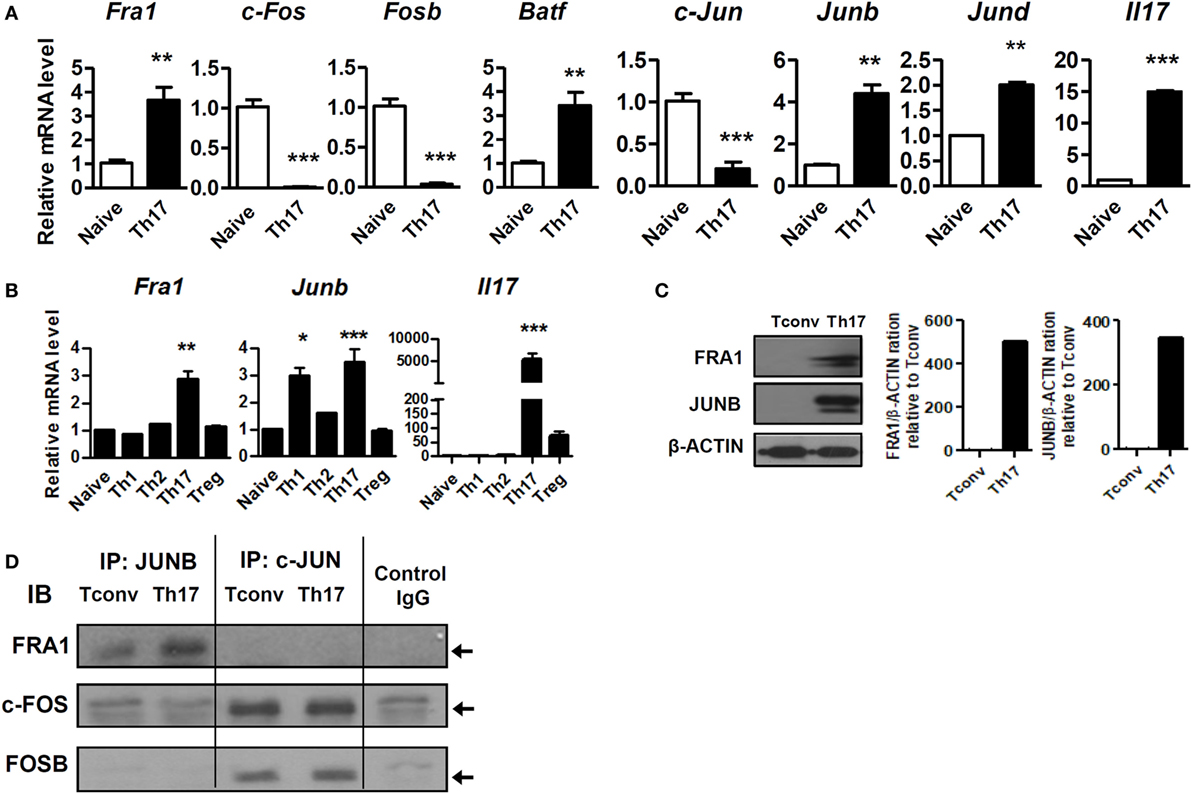
Figure 2. Both Fos-related antigen 1 (FRA1) and JUNB are highly expressed in T helper 17 (Th17) cells. (A,B) Naïve T cells (CD4+CD62L+) were isolated from 6-week-old C57BL/6 mice (n = 3) and cultured for 3 days under the indicated promoters. Relative mRNA levels were measured by qRT-PCR. (C) Protein extracts were prepared from CD4+ T cells (Tconv) and CD4+ T cells cultured for 3 days under Th17-polarizing conditions (Th17). FRA1 and JUNB protein levels were evaluated by immunoblotting. (D) CD4+ T cells cultured under Th17-polarizing conditions were lysed, and the cell lysates were subjected to co-immunoprecipitation with anti-JUNB and anti-c-JUN antibodies and immunoblotting with anti-FRA1, anti-c-FOS, and anti-FOSB antibodies. Data are representative of three (A–C) or two (D) independent experiments, each performed in triplicate (A,B). The data were analyzed using an unpaired t-test or one-way ANOVA with Bonferroni’s post hoc test. Error bars show the SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
Next, we investigated the functional association between the FRA1–JUNB complex and STAT3. The STAT3 signaling pathway was inhibited using genetically engineered STAT3-deficient CD4+ T cells and STA-21, which blocks the DNA-binding activity of STAT3 (26). As shown in Figure 3A, CD4+ T cells harboring Stat3fl/flCd4-Cre had markedly decreased levels of Fra1 and Junb mRNA under both neutral and Th17-polarizing conditions. Treatment with STA-21 profoundly decreased the expression of FRA1, JUNB, and STAT3 (Figure 3B). Moreover, STA-21 decreased the levels of STAT3 phosphorylation under Th17-polarizing conditions (Figure 3C). STAT3 overexpression increased the relative levels of Fra1 and Junb mRNA (Figure 3D), while in vitro treatment with IL-6 upregulated Fra1 and Junb mRNA (Figure 3E).
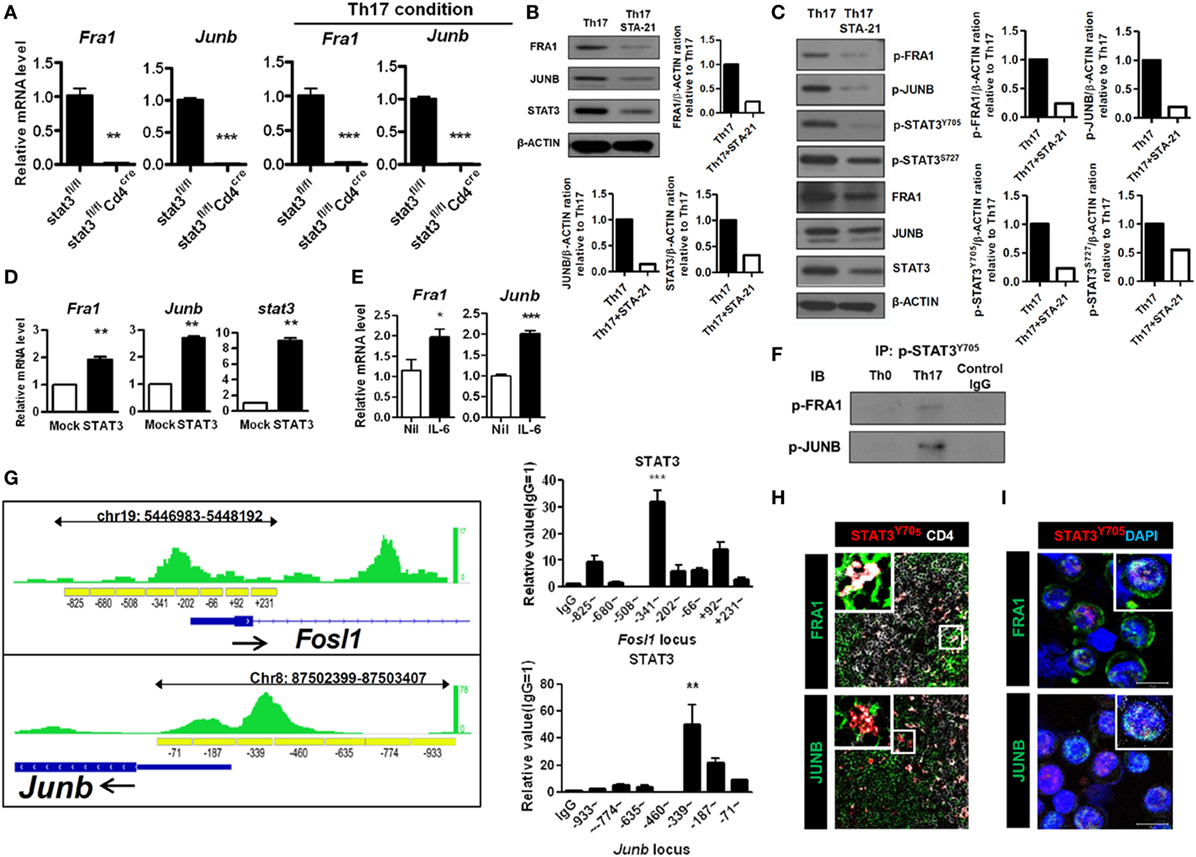
Figure 3. Signal transducer and activator of transcription 3 (STAT3)-mediated activation of Fos-related antigen 1 (FRA1) and JUNB in T helper 17 (Th17) cells. (A) CD4+ T cells from Stat3fl/fl and Stat3fl/flCd4-Cre were isolated (left panel) or cultured under Th17-polarizing conditions for 3 days (right panel). (B,C) CD4+ T cells were pretreated with STA-21 for 1 day, cultured under Th17-polarizing conditions for a further 3 days, and then treated with interleukin (IL)-6 for 2 h (C) before harvesting and lysing. (D) LBRM cells were transfected with a STAT3 expression vector, and the cells were cultured for 3 weeks in the presence of neomycin (10 µg/ml). (E) CD4+ T cells were cultured in the presence of IL-6 for 16 h. (F) CD4+ T cells were cultured under Th0- or Th17-polarizing conditions for 3 days and the lysed. The cell lysates were subjected to immunoprecipitation with an anti-phospho-STAT3Y705 antibody and immunoblotting with anti-phospho-FRA1 or anti-phospho-JUNB antibodies. (G) The STAT3-binding region (green) detected by STAT3 chromatin immunoprecipitation (ChIP) sequencing in Th17 cells is shown along with the Fosl1 and Junb loci, including the primer sites (yellow boxes) (left panel). The negative control cyclophilin showed no enrichment (data not shown). CD4+ T cells were cultured under Th17-polarizing conditions for 3 days. ChIP-qPCR analysis was performed using an anti-STAT3 antibody and primers specific for Fosl1 and Junb (right panel). (H,I) Splenic tissue was collected from collagen-induced arthritis mice. CD4+ splenic T cells and Th17 cells from arthritic mice were stained for CD4 (white), FRA1, JUNB (green), and phospho-STAT3Y705 (H) or DAPI (I) (blue). Data are representative of three (B–I) and two (A) independent experiments, each performed in triplicate (A,D,E,G). The data were analyzed using an unpaired t-test; error bars show the SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
Co-immunoprecipitation studies were performed using anti-FRA1 and anti-JUNB antibodies followed by immunoblotting with antibodies against STAT3 to determine whether FRA1 and JUNB physically interact with STAT3. Both FRA1 and JUNB were found to form a complex with STAT3 (Figure 3F). We performed ChIP and qPCR to examine whether STAT3 binds directly to the promoter regions of Fosl1 and Junb. The ChIP-qPCR results were compared with publicly available ChIP-Seq data from Th17 cells. We found that STAT3 binds to the predicted recognition sites upstream of Fosl1 and Junb, suggesting that it regulates their expression by binding directly to their promoters (Figure 3G). Using confocal microscopy, we found that FRA1 and JUNB were colocalized with phosphorylated STAT3 in CD4+ splenic T cells and Th17 cells from arthritic mice (Figures 3H,I). These findings suggest that STAT3 signaling is critical for the expression and activation of FRA1 and JUNB during Th17 cell differentiation.
Expression of IL-17 Is Directly Regulated by the FRA1–JUNB Complex
The AP-1 family plays a versatile role in T-cell development (27). Levels of IL-17 were measured in Fra1- and Junb-overexpressing and silenced cells. The relative levels of IL17 mRNA were significantly increased in cells overexpressing Fra1 and Junb. The relative mRNA levels of other genes associated with Th17 cells, such as IL21, IL22, and Rorc (the RORγt gene locus), were also increased (Figures 4A,B). CD4+IL-17+ cells and IL17 mRNA levels were significantly decreased when Fra1 and Junb were silenced under Th17-polarizing conditions (Figures 4C,D). By contrast, when c-Fos and c-Jun, the dominant AP-1 subtypes in naïve T cells, were overexpressed, IL17 mRNA levels were decreased significantly (Figures 4E,F). These findings support the specificity of the role of the FRA1–JUNB complex in Th17 cell differentiation.
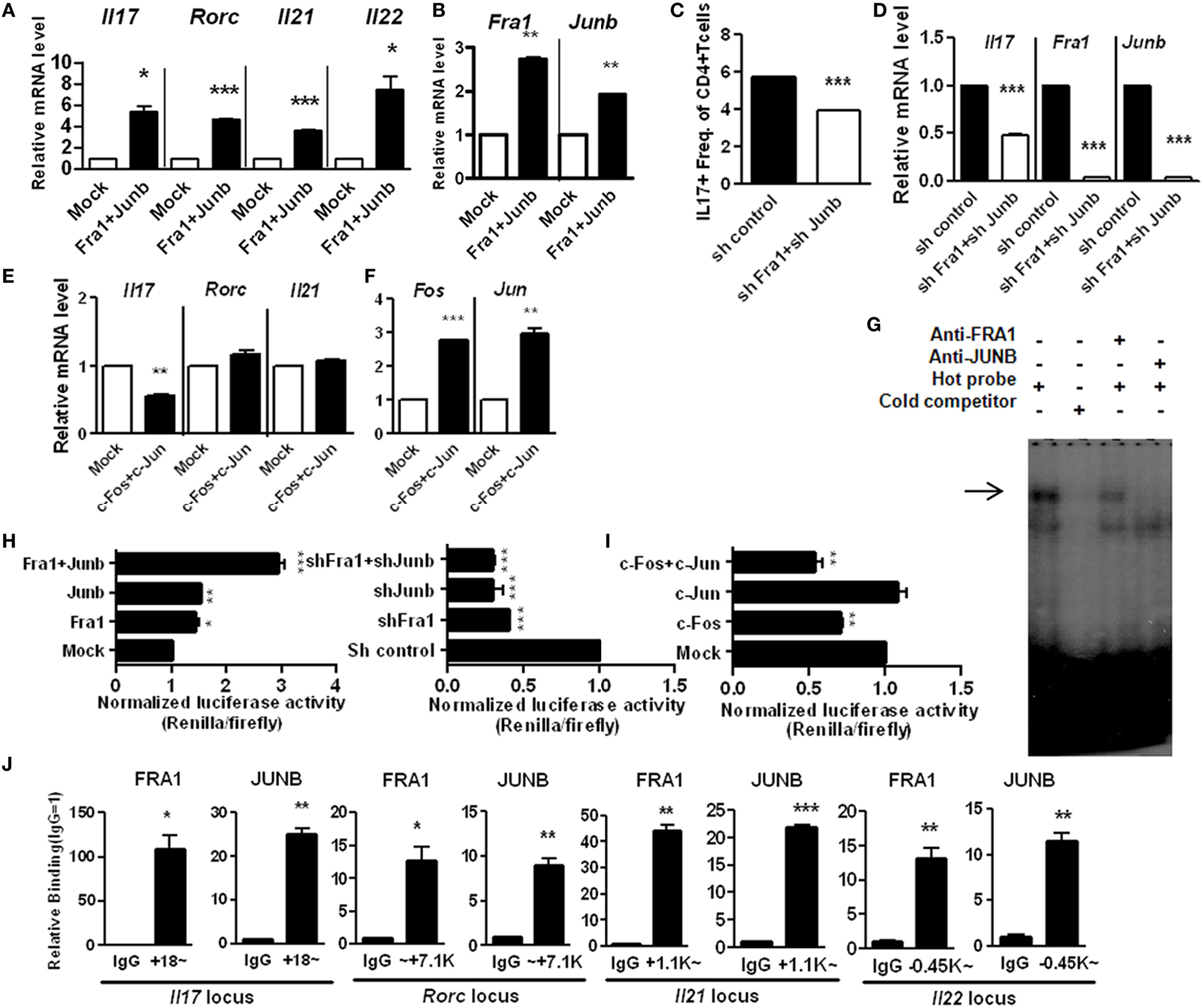
Figure 4. Expression of interleukin (IL)-17 is directly mediated by the Fos-related antigen 1 (FRA1)–JUNB complex in T helper 17 (Th17) cells. LBRM cells were transfected with vectors and cultured for 3 weeks with neomycin (10 µg/ml) (A,B,E,F). (A,B) The relative mRNA levels of each gene were measured by qRT-PCR. (C,D) CD4+ T cells were infected with lentiviruses harboring constructs designed to knockdown Fra1 and Junb. Cells were then cultured under Th17-polarizing conditions for 3 days. CD4+IL-17+ T cells were identified by flow cytometry, and relative mRNA levels were measured by qRT-PCR. (E,F) The relative mRNA level of each gene was measured by qRT-PCR. (G) Nuclear extracts from Th17 cells were analyzed by electrophoretic mobility shift assay incorporating a radiolabeled probe derived from the activator protein 1-binding site of the IL17a locus and antibodies specific for FRA1 or JUNB. (H,I) EL4 cells were transiently transfected with expression vectors and IL17a promoter constructs with a CNS2 enhancer region. Luciferase activity was measured using a dual-luciferase reporter assay system. (J) CD4+ T cells were cultured under Th17-polarizing conditions. Chromatin immunoprecipitation-qPCR was performed using anti-FRA1 or anti-JUNB antibodies with primers specific for each gene locus. Data are representative of at least three independent experiments, performed in triplicate. Data were analyzed using an unpaired t-test; error bars show the SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
We next investigated whether the FRA1–JUNB complex directly regulates IL-17 expression by binding to the IL17a promoter. The interaction between the FRA1–JUNB complex and the AP-1-binding motif within the IL17a promoter region was examined by electrophoretic mobility shift assay. The anti-FRA1 and anti-JUNB antibodies caused a super shift of the labeled DNA probe, indicating that the AP-1-binding motif interacts specifically with FRA1 and JUNB (Figure 4G). This finding was confirmed by an analysis of the IL17a promoter using a luciferase reporter system. Overexpression of both FRA1 and JUNB increased luciferase activity in EL4 cells. Simultaneous overexpression of FRA1 and JUNB caused luciferase activity to increase twofold (Figure 4H). Silencing of Fra1 and Junb caused a greater than twofold reduction in luciferase activity. Simultaneous overexpression of c-FOS and c-JUN caused a significant decrease in the luciferase activity induced by the IL17a promoter (Figure 4I). ChIP-qPCR analyses showed that FRA1 and JUNB bind directly to the IL17a gene loci and to gene loci encoding other Th17 cell differentiation activators, such as IL21, IL22, and Rorc (Figure 4J). These findings indicate that the FRA1–JUNB complex binds to the IL17a promoter region, and that this binding activates IL17 gene transcription.
FRA1 and JUNB Are Critical for the Pathogenesis of CIA
T helper 17 cells and IL-17 contribute significantly to the development of RA (28, 29). To examine the in vivo role of the FRA1–JUNB complex in rheumatoid inflammation, Fra1 and Junb were silenced in CIA mice by injecting Fra1 and Junb short hairpin RNA vectors. This markedly reduced the incidence of arthritis and decreased the arthritis score (Figure 5A). The numbers of FRA1+, JUNB+, and IL-17+ cells were significantly lower within the joint tissues (Figure 5B). Histological examination showed that the ankle joints of Fra1/Junb-silenced mice had less inflammation and cartilage damage (Figure 5C). Inhibition of Fra1/Junb also reduced the expression of Th17 cytokines such as IL-6, IL-17, IL-1β, and tumor necrosis factor-α in joint tissue. Other mediators of joint destruction such as vascular endothelial growth factor and receptor activator of nuclear factor κB ligand (RANKL) were also downregulated (Figure S1A in Supplementary Material). In CIA mice, disease severity was correlated with the level of type II collagen (CII)-specific IgG antibodies (30). Silencing of Fra1 and Junb reduced T-cell proliferation and CII-specific antibody production (Figures S1B,C in Supplementary Material). It also substantially reduced the population of IL-17-producing CD4+ T cells, considered to be Th17 cells, in the spleen and draining lymph nodes (Figure S1D in Supplementary Material).
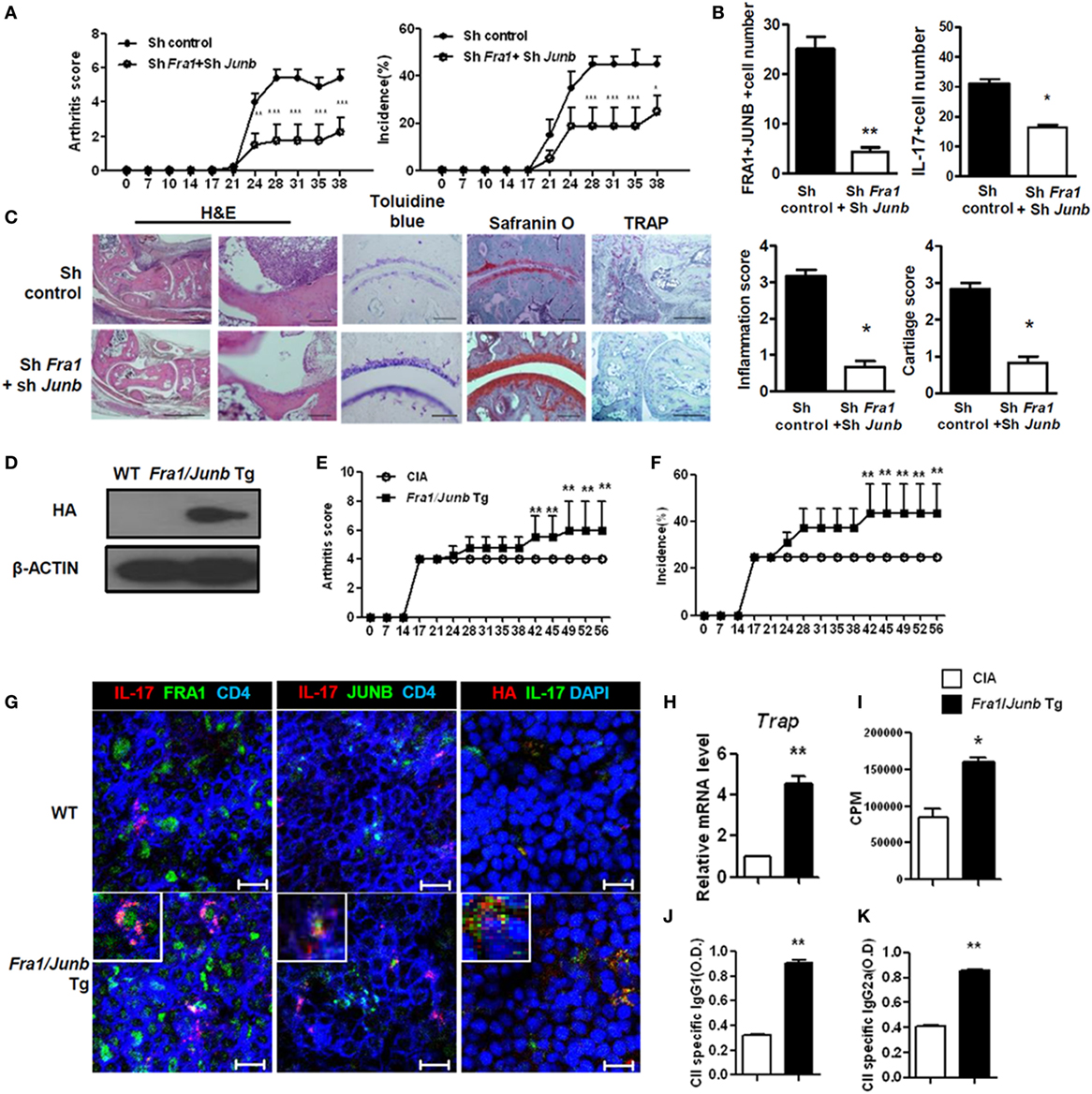
Figure 5. Fos-related antigen 1 (FRA1) and JUNB are involved in the pathogenesis of collagen-induced arthritis, T helper 17 cell differentiation, and rheumatoid inflammation in mice. Arthritis was induced in mice, and shRNA control or shRNAFra1/shRNAJunb vectors were injected on days 7 and 14 (n = 5–7). (A) Arthritis severity was measured using the incidence and mean arthritis score. (B) Splenic tissue from each mouse was stained. Positively stained cells were counted visually by four individuals from high-magnification images projected onto a screen, and the mean values are presented as a histogram. (C) On day 30 after immunization, tissues were sampled from the ankle joints and stained with toluidine blue, Safranin O (scale bar = 100 µm), H&E [scale bar = 500 µm (left); 100 µm (right)], or tartrate-resistant acid phosphatase (TRAP) (scale bar = 200 µm). The inflammation and cartilage scores are shown in bar graphs (right panel). (D) Splenocytes were examined for the HA-tagged FRA1/JUNB transgene by immunoblotting. (E,F) Disease severity was recorded using the incidence and mean arthritis score. (G) Splenic tissue from each mouse was stained for FRA1, JUNB, and interleukin-17 in CD4+ T cells and examined by laser confocal microscopy. (H) The relative levels of TRAP mRNA in cells from the ankle joints were measured by qRT-PCR. (I) T-cell proliferation was analyzed using a mixed-lymphocyte reaction. (J,K) The levels of IgG1 and IgG2a antibodies specific to type II collagen were measured in serum. Data are representative of more than two independent experiments. Data were analyzed using an unpaired t-test or two-way ANOVA with Bonferroni’s post hoc test; error bars show the SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
CD4+ T cells in Fra1/Junb Tg mice had different proportions of Th1 and Th17 cells (Figure S2 in Supplementary Material). CIA was induced in Fra1/Junb transgenic mice to investigate the in vivo effects of Fra1 and Junb overexpression. Splenocytes were examined for HA-tagged FRA1/JUNB transgene by immunoblotting (Figure 5D). The incidence of arthritis and the arthritis score were significantly increased (Figures 5E,F). There was a marked increase in the levels of IL-17, FRA1, and JUNB in the spleen (Figure 5G). The relative mRNA level of the osteoclastogenic marker tartrate-resistant acid phosphatase was significantly higher (Figure 5H), while CII-specific T-cell proliferation and IgG production were also increased (Figures 5I–K). Overexpression of FRA1/JUNB profoundly increased CIA joint pathology (Figure S3A in Supplementary Material). Immunostaining showed that the expression of various proinflammatory cytokines and RANKL, a mediator of osteoclastogenesis, was significantly greater in the joint tissues of transgenic mice with CIA (Figure S3B in Supplementary Material). IL-17 mRNA levels and Th17 cell numbers were increased markedly (Figures S3C,D in Supplementary Material). These findings suggest that the FRA1–JUNB complex plays a major role in the development of CIA, likely through the induction of Th17 cell differentiation.
Expression and Potential Functions of FRA1 and JUNB in RA Patients
We demonstrated in vivo roles of FRA1 and JUNB in mice with CIA, an animal disease model for RA. To confirm the roles of FRA1 and JUNB in patients with RA, FRA1 was overexpressed in CD4+ T cells isolated from healthy controls. FRA1 overexpression increased the proportion of IL-17-producing CD4+ T cells during Th17 polarization (Figure 6A). These cells produced significantly higher levels of IL17a mRNA and cytokines (Figure 6B). Fra1 silencing decreased IL17a mRNA levels and IL-17 secretion (Figure 6C). There were significantly increased the transcription levels of Fra1 and Junb among peripheral blood mononuclear cells (PBMCs) and synovial fluid mononuclear cells (SFMCs) from RA patients compared with PBMCs from healthy controls (Figure 6D). Junb was also upregulated in RA patients. When Fra1 was silenced in CD4+ T cells isolated from SFMCs of RA patients, IL-17 secretion was decreased during Th17 polarization (Figure 6E). The expression of both FRA1 and JUNB was particularly high in the inflamed synovium of RA patients, a region enriched with inflammatory immune cells (Figure 6F). Of note, FRA1, JUNB, and phosphorylated STAT3 were colocalized in the inflamed synovium, which further supported our in vitro experimental results. A schematic illustration of the inflammatory signaling pathway induced by FRA1/JUNB is shown in Figure 6G. Th17 conditioning induces an inflammatory signal by promoting FRA1/JUNB. The inflammatory response is mediated by activation of STAT3, which regulates genes involved in Th17 cell differentiation. This cascade induces excessive inflammation and autoimmunity resulting in RA.
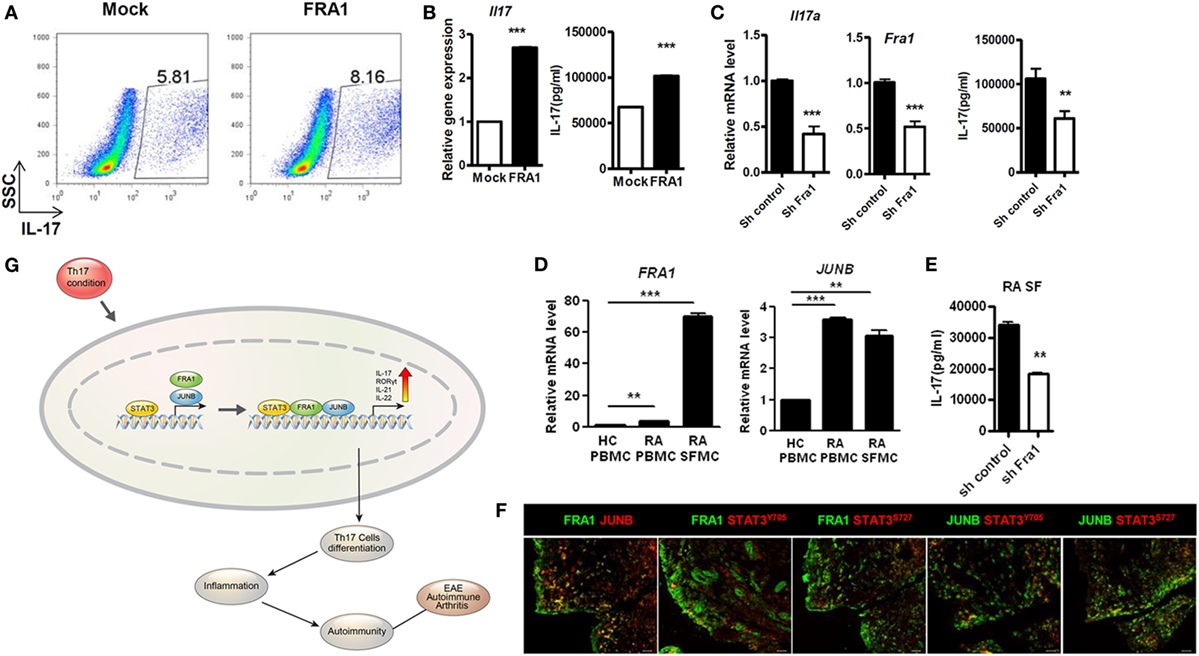
Figure 6. Expression and potential function of Fos-related antigen 1 (FRA1) and JUNB in rheumatoid arthritis (RA) patients. (A,B) Flow cytometry, qRT-PCR, and ELISA. CD4+ T cells were isolated from healthy peripheral blood mononuclear cells (PBMCs) and infected with retroviruses containing FRA1 on day 1. The infected cells were then cultured under T helper 17 (Th17)-polarizing conditions for 3 days and analyzed. (C) qRT-PCR and ELISA. CD4+ T cells from healthy controls were infected with lentiviruses harboring constructs designed to knockdown Fra1. Cells were then cultured under Th17-polarizing conditions for 3 days. (D) qRT-PCR analysis. Total RNA was extracted from PBMCs isolated from healthy controls and from PBMCs and synovial fluid mononuclear cells (SFMCs) from RA patients. (E) ELISA. CD4+ T cells in SFMCs from RA patients were infected with lentiviruses harboring constructs designed to knockdown Fra1. Cells were then cultured under Th17-polarizing conditions for 3 days. (F) Synovial tissue obtained from RA patients. Sections were immunostained. Data are representative of three (A–C,F) and two (D,E) independent experiments, each performed in triplicate. (G) A schematic illustration of the inflammatory signaling pathway mediated by FRA1/JUNB. (B–E) Data are representative of more than three independent experiments. Data were analyzed using an unpaired t-test; error bars show the SEM. *p < 0.05, **p < 0.005, and ***p < 0.0005.
Discussion
Although FRA1 and JUNB may have modulatory activity in the immune inflammatory response (18, 19), there is currently no evidence in the literature to support this notion in autoimmune disorders mediated by STAT3 and Th17 cells. In this study, we found evidence of previously unidentified functions of FRA1 and JUNB in Th17 cell differentiation and autoimmune disease. We investigated the modulatory function and underlying mechanisms of FRA1 and JUNB in a murine model of inflammatory arthritis. The predominant conclusion from this study was that FRA1 and JUNB exacerbate inflammation via the induction of Th17 cells. To our knowledge, this is the first study to implicate FRA1 and JUNB directly in STAT3 activation and regulation of Th17 cell proliferation. These observations suggest a previously unidentified function of these factors in autoimmune disease.
Activator protein 1 is comprised of dimers of FOS and JUN protein family members (31, 32). The diverse functions of AP-1 family members result from heterodimer formation between the FOS and JUN proteins (33). AP-1 is also involved in cell functions including proliferation, differentiation, and apoptosis. For example, the FRA2–JUND complex is involved in the terminal differentiation of granulosa cells into luteal cells (34) and T-cell activation (35). Additionally, AP-1 factors may play important roles in autoimmune disease and Th17 cell differentiation. It was suggested that basic leucine zipper transcription factor, ATF-like (BATF), an AP-1 protein, leads to increased Th17 cell differentiation, whereas BATF deficiency results in IL-17 downregulation (12). Loss of the AP-1 family member Fosl2 in T cells improved EAE severity via suppression of Th17 cell plasticity (36). We performed an integrative analysis using RNA-Seq and ChIP-Seq data to identify key factors downstream of STAT3 involved in Th17 cell differentiation. This integrative analysis suggested that upregulation of Fra1 and Junb drives and promotes Th17 cell differentiation.
Signal transducer and activator of transcription 3-mediated IL-17 expression and Th17 cell differentiation involve AP-1 family proteins. Previous studies have shown a biological link between the STAT3 signaling pathway and the AP-1 family of transcription factors (12, 37, 38). In particular, STAT3-mediated expression of BATF, an AP-1 subfamily member, is associated with Th17 cell differentiation (12). In this study, RNA-Seq analysis showed that the expression of cFos and cJun was higher in naïve T cells. This suggests that AP-1 subtype switching from c-FOS–c-JUN to FRA1/JUNB is essential for Th17 cell differentiation. It can also be postulated that activation of c-FOS–c-JUN must be suppressed in parallel with activation of FRA1/JUNB by STAT3 during polarization toward the Th17 subtype. The present study identified an AP-1 complex comprising FRA1 and JUNB, which appears to function as a key factor downstream of STAT3 in Th17 cell differentiation. It appears that AP-1 subtype switching, particularly from c-FOS, FOSB, and c-JUN to FRA1 and JUNB, is required for STAT3-mediated Th17 cell differentiation.
The AP-1 family includes the c-JUN and c-FOS proteins, which play important roles in IL-17 expression and Th17 cell functions. It has been demonstrated that BATF binds to intergenic components in the IL17a locus and to IL17 promoters, both of which are essential for Th17 differentiation (12). Additionally, the BATF–JUNB and BATF–JUND complexes have been shown to cooperate with IRF4 in Th17 cells to promote transcription of IL17 (39). The c-JUN transcription factor that heterodimerizes with c-FOS to generate the AP-1 transcription factor complex is involved in suppression of IL-17 production in developing Th17 cells (16, 22). FRA2 is also a negative regulator of IL-17 (36). We observed that overexpression of FRA2 decreased the expression of IL17a in a promoter-fused luciferase reporter assay (data not shown). We also found that expression levels of c-Fos and c-Jun were significantly reduced, whereas Batf expression was increased in Th17 cells compared with naïve T cells. Overexpression of c-Fos and c-Jun significantly decreased IL17 mRNA levels. The FRA1–JUNB complex directly activates expression of IL-17, a cytokine produced by Th17 cells. This complex plays a role in promoting the expression of other cytokines associated with Th17 cells, such as IL-21 and IL-22, by directly binding to their promoter regions. These results suggest that FRA1 and JUNB are involved in IL-17 production and Th17 cell differentiation.
Although FRA1/JUNB and c-FOS/c-JUN compete for the same AP-1-binding sites within the IL17a promoter, we found in this study that IL17a promoter activity was increased by FRA1 and JUNB overexpression but decreased by c-FOS and c-JUN overexpression. We observed an interaction between p-STAT3 and p-FRA1 or p-JUNB in Th17 cells; therefore, FRA1/JUNB and c-FOS/c-JUN may be involved in AP-1 subtype switching. Additionally, p-FRA1 binds to p-JUNB in Th17 cells. Further study is needed to confirm the AP-1 subtype switching factors involved and the mechanism of interaction between p-FRA1 and p-JUNB in Th17 cells.
This study has certain limitations. One is that the animal studies were conducted using FRA1/JUNB-1 overexpression and knockdown vectors. Adoptive transfer studies in conditional overexpression and knockout mice are required to confirm that FRA1/JUNB-1 regulates CD4+ T cells, leading to autoimmune arthritis. Nonetheless, this study is the first to demonstrate functions of FRA1/JUNB-1 in Th17 cell differentiation and autoimmune arthritis. Future studies using adoptive transfer models with CD4+ T cells differentially expressing FRA1/JUNB-1 are required to validate our data more precisely.
Uncontrolled Th17 cell activation is responsible for the onset of several autoimmune diseases. Th17 cells cause tissue injury via production of IL-17, as observed in CIA mouse models of disease. The present study showed that the FRA1–JUNB complex plays a central role in the development of RA, which is a Th17-mediated autoimmune disease. The FRA1–JUNB complex may be clinically relevant as it functions as a Th17 cell-specific regulator of proinflammatory cytokine production in CIA mouse models and hence RA. In the RA mouse model used in this study, FRA1/JUNB overexpression resulted in the development of severe inflammatory symptoms and augmented expression of IL-17 in mice. Notably, this has fueled interest in the genetic importance of FRA1/JUNB in human RA in the context of promoted STAT3 activity. FRA1 overexpression activated Th17 cell differentiation in PBMCs from healthy controls; however, FRA1 silencing downregulated IL-17 production in SFMCs from RA patients. This may indicate the potential of the FRA1–JUNB complex as a therapeutic target in human autoimmune disease. In conclusion, our results suggest a previously unidentified function of FRA1/JUNB in T-cell-mediated autoimmune diseases. FRA1/JUNB directly modulates STAT3 activation and Th17 cell differentiation. Our identification of the involvement of FRA1/JUNB in T-cell development provides a potential therapeutic target for the treatment of autoimmune diseases.
Ethics Statement
The Animal Care Committee of The Catholic University of Korea approved the experimental protocol. All experimental procedures were evaluated and carried out in accordance with the protocols approved by the Animal Research Ethics Committee at the Catholic University of Korea (CMCU-2012-0156-01). All procedures performed followed the ethical guidelines on animal use. Approval by the ethics committee of Seoul St. Mary’s Hospital (Seoul, Republic of Korea) was obtained for all procedures. All human experimental procedures were approved by the Ethics Committee of Seoul St. Mary’s Hospital (Seoul, Republic of Korea, KC13TISE0032).
Author Contributions
Y-MM, S-YL, S-KK, SL, S-MA, and M-LC designed the experiments and analyzed the data. Y-MM, S-YL, S-KK, and SL wrote the manuscript along with input from DK, WK, Y-MH, H-JS, E-KK, J-GR, H-BS, J-EK, S-YH, and JY. Y-MM and S-YL performed all in vitro assays with help from S-KK, SL, DK, WK, and H-BS. S-YL, H-JS, and J-GR performed the animal experiments. E-KK conducted all immunohistochemistry experiments. Y-MM, S-YL, S-KK, SL, RS, D-MJ, H-YK, S-HP, S-MA, and M-LC discussed and developed the study concept. All authors critically reviewed and approved the final form of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry for Health & Welfare, Republic of Korea (grant no. HI14C3417), by the Basic Science Research Program from the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (grant no. 2012-0006135), and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant no. HI15C3062).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01793/full#supplementary-material.
Figure S1. Knockdown of FRA1/JUNB Decreases Th17 Cell Differentiation and Attenuates Rheumatoid Inflammation in Mice.
Figure S2. T cell proportion of CD4+Tcells in Fra1/Junb Tg mice.
Figure S3. Overexpression of FRA1/JUNB Causes Th17 Cell Differentiation and Increases CIA Disease Severity.
References
1. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol (2005) 6:1123–32. doi:10.1038/ni1254
2. Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev (2008) 226:57–79. doi:10.1111/j.1600-065X.2008.00699.x
3. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol (2006) 177:566–73. doi:10.4049/jimmunol.177.1.566
4. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. doi:10.1172/JCI5703
5. Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, et al. STAT3 and STAT4 direct development of IL-17-secreting Th cells. J Immunol (2007) 178:4901–7. doi:10.4049/jimmunol.178.8.4901
6. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327:1098–102. doi:10.1126/science.1178334
7. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
8. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control Th17 responses in a STAT3-dependent manner. Science (2009) 326:986–91. doi:10.1126/science.1172702
9. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem (2007) 282:9358–63. doi:10.1074/jbc.C600321200
10. Pan F, Ye Z, Cheng L, Liu JO. Myocyte enhancer factor 2 mediates calcium-dependent transcription of the interleukin-2 gene in T lymphocytes: a calcium signaling module that is distinct from but collaborates with the nuclear factor of activated T cells (NFAT). J Biol Chem (2004) 279:14477–80. doi:10.1074/jbc.C300487200
11. Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol (2008) 9:1297–306. doi:10.1038/ni.1663
12. Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, et al. The AP-1 transcription factor BATF controls T(H)17 differentiation. Nature (2009) 460:405–9. doi:10.1038/nature08114
13. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A (2006) 103:8137–42. doi:10.1073/pnas.0600666103
14. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity (2010) 32:605–15. doi:10.1016/j.immuni.2010.05.003
15. Bi Y, Liu G, Yang R. Th17 cell induction and immune regulatory effects. J Cell Physiol (2007) 211:273–8. doi:10.1002/jcp.20973
16. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci (2004) 117:5965–73. doi:10.1242/jcs.01589
17. Thomsen MK, Bakiri L, Hasenfuss SC, Hamacher R, Martinez L, Wagner EF. JUNB/AP-1 controls IFN-gamma during inflammatory liver disease. J Clin Invest (2013) 123:5258–68. doi:10.1172/JCI70405
18. Pirusian ES, Nikol’skaia TA, Abdeev RM, Bruskin SA. Transcription factor AP-1 components as a psoriasis candidate genes. Mol Biol (Mosk) (2007) 41:1069–80.
19. Sabatakos G, Rowe GC, Kveiborg M, Wu M, Neff L, Chiusaroli R, et al. Doubly truncated FosB isoform (Delta2DeltaFosB) induces osteosclerosis in transgenic mice and modulates expression and phosphorylation of Smads in osteoblasts independent of intrinsic AP-1 activity. J Bone Miner Res (2008) 23:584–95. doi:10.1359/jbmr.080110
20. Eferl R, Hasselblatt P, Rath M, Popper H, Zenz R, Komnenovic V, et al. Development of pulmonary fibrosis through a pathway involving the transcription factor FRA-2/AP-1. Proc Natl Acad Sci U S A (2008) 105:10525–30. doi:10.1073/pnas.0801414105
21. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol (2012) 14:1203–11. doi:10.1038/ncb2590
22. Coquet JM, Middendorp S, van der Horst G, Kind J, Veraar EA, Xiao Y, et al. The CD27 and CD70 costimulatory pathway inhibits effector function of T helper 17 cells and attenuates associated autoimmunity. Immunity (2013) 38:53–65. doi:10.1016/j.immuni.2012.09.009
23. Lee SM, Gao B, Fang D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood (2008) 111:3599–606. doi:10.1182/blood-2007-09-115014
24. Huo L, Rothstein TL. Isolation and characterization of murine FRA-1: induction mediated by CD40 and surface Ig is protein kinase C dependent. J Immunol (1996) 157:3812–8.
25. Szremska AP, Kenner L, Weisz E, Ott RG, Passegue E, Artwohl M, et al. JUNB inhibits proliferation and transformation in B-lymphoid cells. Blood (2003) 102:4159–65. doi:10.1182/blood-2003-03-0915
26. Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits STAT3 function in breast cancer cells. Proc Natl Acad Sci U S A (2005) 102:4700–5. doi:10.1073/pnas.0409894102
27. Jochum W, Passegue E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene (2001) 20:2401–12. doi:10.1038/sj.onc.1204389
28. Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther (2007) 9:202. doi:10.1186/ar2107
29. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev (2008) 223:87–113. doi:10.1111/j.1600-065X.2008.00628.x
30. Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M. Complement deficiency ameliorates collagen-induced arthritis in mice. J Immunol (2002) 169:454–9. doi:10.4049/jimmunol.169.1.454
31. Jariel-Encontre I, Salvat C, Steff AM, Pariat M, Acquaviva C, Furstoss O, et al. Complex mechanisms for c-FOS and c-JUN degradation. Mol Biol Rep (1997) 24:51–6. doi:10.1023/A:1006804723722
32. Stancovski I, Gonen H, Orian A, Schwartz AL, Ciechanover A. Degradation of the proto-oncogene product c-FOS by the ubiquitin proteolytic system in vivo and in vitro: identification and characterization of the conjugating enzymes. Mol Cell Biol (1995) 15:7106–16. doi:10.1128/MCB.15.12.7106
33. Bakiri L, Matsuo K, Wisniewska M, Wagner EF, Yaniv M. Promoter specificity and biological activity of tethered AP-1 dimers. Mol Cell Biol (2002) 22:4952–64. doi:10.1128/MCB.22.13.4952-4964.2002
34. Sharma SC, Richards JS. Regulation of AP1 (JUN/FOS) factor expression and activation in ovarian granulosa cells. Relation of JUND and FRA2 to terminal differentiation. J Biol Chem (2000) 275:33718–28. doi:10.1074/jbc.M003555200
35. Boise LH, Petryniak B, Mao X, June CH, Wang CY, Lindsten T, et al. The NFAT-1 DNA binding complex in activated T cells contains FRA-1 and JUNB. Mol Cell Biol (1993) 13:1911–9. doi:10.1128/MCB.13.3.1911
36. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell (2012) 151:289–303. doi:10.1016/j.cell.2012.09.016
37. Leu JI, Crissey MA, Leu JP, Ciliberto G, Taub R. Interleukin-6-induced STAT3 and AP-1 amplify hepatocyte nuclear factor 1-mediated transactivation of hepatic genes, an adaptive response to liver injury. Mol Cell Biol (2001) 21:414–24. doi:10.1128/MCB.21.2.414-424.2001
38. Song Y, Qian L, Song S, Chen L, Zhang Y, Yuan G, et al. FRA-1 and STAT3 synergistically regulate activation of human MMP-9 gene. Mol Immunol (2008) 45:137–43. doi:10.1016/j.molimm.2007.04.031
Keywords: Fos-related antigen 1-JUNB, signal transducer and activator of transcription 3, T helper 17, autoimmune arthritis, inflammation
Citation: Moon Y-M, Lee S-Y, Kwok S-K, Lee SH, Kim D, Kim WK, Her Y-M, Son H-J, Kim E-K, Ryu J-G, Seo H-B, Kwon J-E, Hwang S-Y, Youn J, Seong RH, Jue D-M, Park S-H, Kim H-Y, Ahn S-M and Cho M-L (2017) The Fos-Related Antigen 1–JUNB/Activator Protein 1 Transcription Complex, a Downstream Target of Signal Transducer and Activator of Transcription 3, Induces T Helper 17 Differentiation and Promotes Experimental Autoimmune Arthritis. Front. Immunol. 8:1793. doi: 10.3389/fimmu.2017.01793
Received: 11 September 2017; Accepted: 30 November 2017;
Published: 18 December 2017
Edited by:
Guixiu Shi, Xiamen University, ChinaCopyright: © 2017 Moon, Lee, Kwok, Lee, Kim, Kim, Her, Son, Kim, Ryu, Seo, Kwon, Hwang, Youn, Seong, Jue, Park, Kim, Ahn and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sung-Min Ahn, c21haG5AZ2FjaG9uLmFjLmty;
Mi-La Cho, aWFtbWlsYUBjYXRob2xpYy5hYy5rcg==
†These authors have contributed equally to this work.