 Audrey Hemadou1
Audrey Hemadou1 Véronique Giudicelli2
Véronique Giudicelli2 Melissa Laird Smith3†
Melissa Laird Smith3† Marie-Paule Lefranc2*
Marie-Paule Lefranc2* Patrice Duroux2
Patrice Duroux2 Sofia Kossida2
Sofia Kossida2 Cheryl Heiner3
Cheryl Heiner3 N. Lance Hepler3
N. Lance Hepler3 John Kuijpers3Alexis Groppi4Jonas Korlach3
John Kuijpers3Alexis Groppi4Jonas Korlach3 Philippe Mondon5Florence Ottones1
Philippe Mondon5Florence Ottones1 Marie-Josée Jacobin-Valat1
Marie-Josée Jacobin-Valat1 Jeanny Laroche-Traineau1‡
Jeanny Laroche-Traineau1‡ Gisèle Clofent-Sanchez1*‡
Gisèle Clofent-Sanchez1*‡
- 1CRMSB, UMR 5536, CNRS, Bordeaux, France
- 2IMGT®, The International ImMunoGeneTics Information System®, Laboratoire d’ImmunoGénétique Moléculaire, LIGM, Institut de Génétique Humaine, IGH, UMR 9002, CNRS, Montpellier University, Montpellier, France
- 3Pacific Biosciences, Menlo Park, CA, United States
- 4Université de Bordeaux, Centre de Bioinformatique de Bordeaux (CBiB), Bordeaux, France
- 5LFB Biotechnologies, Lille, France
Phage-display selection of immunoglobulin (IG) or antibody single chain Fragment variable (scFv) from combinatorial libraries is widely used for identifying new antibodies for novel targets. Next-generation sequencing (NGS) has recently emerged as a new method for the high throughput characterization of IG and T cell receptor (TR) immune repertoires both in vivo and in vitro. However, challenges remain for the NGS sequencing of scFv from combinatorial libraries owing to the scFv length (>800 bp) and the presence of two variable domains [variable heavy (VH) and variable light (VL) for IG] associated by a peptide linker in a single chain. Here, we show that single-molecule real-time (SMRT) sequencing with the Pacific Biosciences RS II platform allows for the generation of full-length scFv reads obtained from an in vivo selection of scFv-phages in an animal model of atherosclerosis. We first amplified the DNA of the phagemid inserts from scFv-phages eluted from an aortic section at the third round of the in vivo selection. From this amplified DNA, 450,558 reads were obtained from 15 SMRT cells. Highly accurate circular consensus sequences from these reads were generated, filtered by quality and then analyzed by IMGT/HighV-QUEST with the functionality for scFv. Full-length scFv were identified and characterized in 348,659 reads. Full-length scFv sequencing is an absolute requirement for analyzing the associated VH and VL domains enriched during the in vivo panning rounds. In order to further validate the ability of SMRT sequencing to provide high quality, full-length scFv sequences, we tracked the reads of an scFv-phage clone P3 previously identified by biological assays and Sanger sequencing. Sixty P3 reads showed 100% identity with the full-length scFv of 767 bp, 53 of them covering the whole insert of 977 bp, which encompassed the primer sequences. The remaining seven reads were identical over a shortened length of 939 bp that excludes the vicinity of primers at both ends. Interestingly these reads were obtained from each of the 15 SMRT cells. Thus, the SMRT sequencing method and the IMGT/HighV-QUEST functionality for scFv provides a straightforward protocol for characterization of full-length scFv from combinatorial phage libraries.
Introduction
Immunoglobulin (IG) or antibody fragments displayed as single chain Fragment variable (scFv) on filamentous phages (scFv-phages) are classically selected from scFv-phage combinatorial libraries to obtain human antibodies specific for a given target (1–3). This selection from scFv-phage display libraries is widely used for the discovery of novel specificities for therapeutic antibodies in cancer, cardiovascular, autoimmune, infectious or neurodegenerative pathologies, with many of them at various stages of clinical or research development (4–10). Classical in vitro phage display approaches involve multiple rounds of selection (or panning) for the enrichment of scFv-phages that demonstrate the desired specificity against a target followed, at the last selection round, by functional screening and characterization of selected candidates using appropriate assays. At this very last step, analysis of the selected scFv via Sanger sequencing is commonly used to identify sequences of interest, taking advantage of the genotype–phenotype linkage inherent to the display system. A critical limitation of using biological assays followed by Sanger sequencing is that only a minute fraction of the selected library is actually sampled, a few hundred at best, whereas the selected library may usually contain up to 105 to 106 variants. This limitation is further enhanced when scFv-phage selection is performed in vivo (biopanning) in different pathological models in which scFv-phages can encounter a very large panel of unknown biomarkers (11–13). Currently available next-generation sequencing (NGS) platforms allow the simultaneous sequencing of millions of reads. However, a main challenge for the NGS sequencing of scFv from combinatorial libraries remains the scFv length >800 bp, which is too long for most NGS platforms. Up to now, NGS methods have only provided reads encompassing one variable (V) domain (400 bp), therefore losing a critical piece of information found in scFv sequences, that of the association of two specific V domains [variable heavy (VH) and variable light (VL) for the IG] by the peptide linker. Although a few approaches have been proposed, retrieving information regarding V domain association has still not been solved (14–16).
The analysis of antibody scFv sequences is a difficult exercise because not only are scFv composed of two V domains, but these two V domains are different from each other and each can potentially be extremely diverse. Indeed, the huge diversity of IG or antibodies results from complex in vivo mechanisms that occur during the synthesis of the VH and VL domains, which include the molecular rearrangements at the DNA level of the variable (V), diversity (D) (only for VH), and joining (J) genes with nucleotide deletions and insertions (N-diversity) at the V-(D)-J junctions in the bone marrow pre-B and immature B cells (17, 18). In spleen and lymph nodes, somatic hypermutations accumulate in the mature B cell VH and VL, creating a huge diversity of the B cell membrane IG for the recognition of foreign antigens. Following a specific antibody-antigen interaction the B cell proliferates and generates clones engaged in in vivo selection and affinity maturation. The specificity of the V domains is conferred by the complementarity determining regions (CDR) and more particularly the CDR3 (19–21). The same features are observed in in vitro combinatorial libraries, which mimic the natural in vivo diversity, selection and affinity maturation (1–3).
In order to manage, analyze and compare the extraordinary diversity of the immune repertoires, IMGT®, the international ImMunoGeneTics information system® 1 (22), was created in 1989 in Montpellier by Marie-Paule Lefranc (Montpellier University, CNRS), which marked the birth of immunoinformatics (18), a new science at the interface between immunogenetics and bioinformatics. IMGT® has developed online tools that provide a detailed and accurate analysis of the V domains and which, in the case of nucleotide sequences, include IMGT/V-QUEST (23–25) for the analysis of the rearranged V-(D)-J sequences of the IG or antibodies and T cell receptors (TR), and IMGT/JunctionAnalysis (26, 27) for the analysis of the V-(D)-J junctions and of the included CDR3. The algorithms and IMGT reference directories of these tools have been implemented in IMGT/HighV-QUEST (28–31), the first and only web portal for NGS sequence analysis of IG and TR, begun in 2010. IMGT/HighV-QUEST analyses up to 500,000 NGS reads per batch and includes a statistical module for IMGT clonotype identification and comparison (analyses are performed on the results of up to one million reads, from one or several batches) (30). IMGT/StatClonotype (32, 33), a stand-alone tool and R package, allows for the comparison of IMGT clonotype diversity and expression between two NGS data sets, using the IMGT/HighV-QUEST statistical results output. In order to overcome the analysis challenge of the NGS scFv, the IMGT/V-QUEST functionality “Analysis of single chain Fragment variable (scFv)” which includes the search and characterization of two V domains in a single sequence [IMGT/V-QUEST Documentation2 (34)] has recently been integrated in IMGT/HighV-QUEST (IMGT/HighV-QUEST Documentation3).
The main challenge addressed in this study was to obtain high quality NGS reads of the scFv long enough to encompass the two VH and VL domains and to analyze and characterize the association of the two V domains in the NGS scFv reads using the scFv functionality in IMGT/HighV-QUEST. Full-length scFv reads are expected to have a length of around 800 bp with a whole insert of around 1,000 bp (including the 5′ and 3′ sequences of the vector and primer sequences at both ends). We used the Pacific Biosciences (PacBio) third-generation NGS technology, which provides long sequencing reads and the highest consensus accuracy available today (35–38). For this project, the high accuracy is a result of the generation of circular consensus sequencing (CCS) reads, by which the long sequencing reads allow for multiple passes of the same insert and thus removal of random sequencing errors upon consensus construction. Practically, one single molecule, real-time (SMRT) cell has 150,000 zero-mode waveguides (ZMWs), of which 50,000–75,000 are loaded with single molecules during sequencing, resulting in the production of ~50,000–75,000 unique consensus reads per run and per SMRT cell. PacBio NGS sequencing was performed from amplified DNA of scFv-phages isolated from the third round of an in vivo biopanning in an animal model of atherosclerosis (12, 13). The PacBio scFv CCS (version 2) reads were first analyzed using IMGT/HighV-QUEST with the scFv functionality. In a second step, the sequence quality was evaluated by tracking the NGS reads of a scFv-phage clone P3, identified by biological assays and Sanger sequencing.
Materials and Methods
In Vivo Selection of scFv-Phages Specific to Atheroma Plaque
A fully human-recombinant scFv antibody library (scFv cloned in the pMG72 phagemid vector, containing the ampicillin-resistant gene for the selection and maintenance of the phagemid) with a diversity of 3.4 × 109 clones (full description in patent WO2007137616) was expressed by phage display and selected in vivo, as previously described (13). The scFv-phages were obtained from the scFv-phagemid combinatorial library by expression of the scFv on the phage surface, following the addition of a helper filamentous phage to recombinant phagemid infected bacteria in the exponential phase (39). Three rounds of biopanning were performed in atheromatous injured rabbits (12). All animal experiments were performed in conformity with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and were accredited by the local ethical committee (Animal Care and Use Committee of Bordeaux, France under the No. 50120192). Briefly, the procedure was the following (Figure 1): 2.4 × 1012 colony-forming units (cfu) of scFv-phages were injected into an atheromatous rabbit. After 1 h in circulation, the animal was sacrificed, the aorta was retrieved and scFv-phages binding to the aorta were eluted in different fractions. The eluted scFv-phages were reamplified in XL1-Blue bacteria and following scFv expression at the phage surface as above (39), the amplified scFv-phages were reinjected in another atheromatous animal. Rounds 2 and 3 were conducted following the same procedure. The number of reinjected colony-forming units were 4.8 × 1011 in round 2 and 3.9 × 1011 in round 3. The total number of eluted scFv-phages from the third round was 1.5 × 107 cfu (total for seven fractions corresponding to different areas of the aorta).
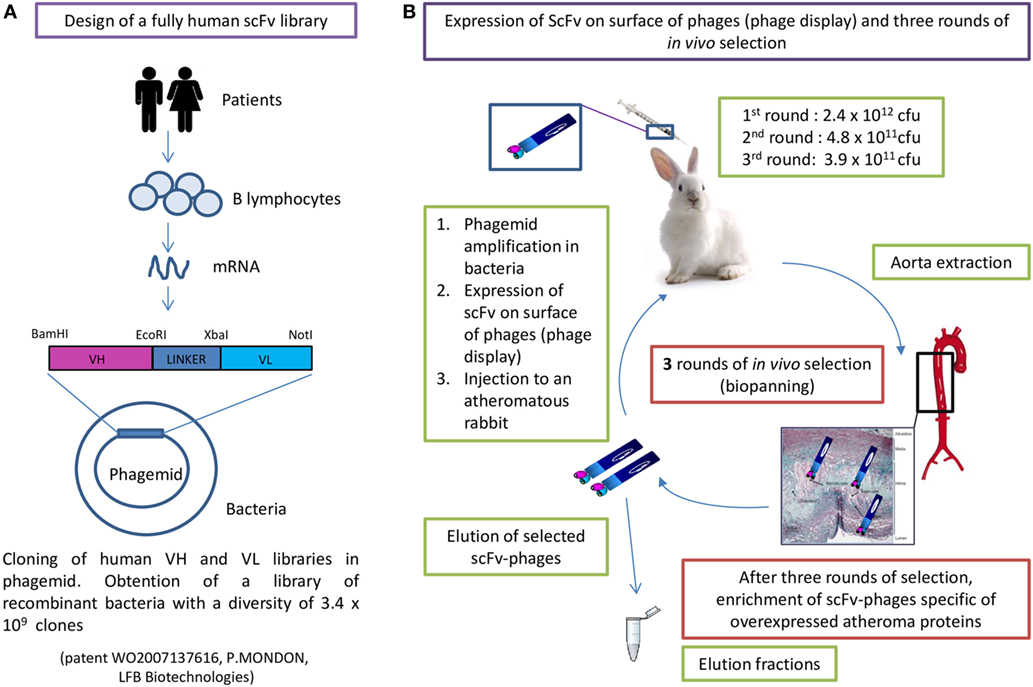
Figure 1. Human single chain fragment variable-phagemid combinatorial library construction (A) and in vivo phage display selection (B).
In this study, the analyzed fraction is the one recovered after the third round of selection from the endothelial cells of the damaged abdominal aorta vessel wall (named AAR3 for abdominal aorta round 3) (Figure 2). The scFv-phages were amplified in XL1-Blue bacteria and plated on 145 mm Petri dishes for storage of the whole AAR3 fraction before sequencing, and on 80 mm Petri dishes for limiting dilution (quantification of recombinant bacteria and picking of individual clones) (Figure 2). The recombinant bacteria plated on 145 mm Petri dishes were scratched and stored at −80°C in 50% v/v glycerol. Around 3.5 × 105 clones issuing from the whole AAR3 fraction were counted by limiting dilution of recombinant bacteria on 80 mm Petri dishes. Ninety-six recombinant bacteria clones were individually picked and grown in selective medium on a 96-well MASTERBLOCK® polypropylene storage plate (Greiner Bio-One, France) and stored for in vitro bioreactivity assays (in vitro screening of scFv-phages on atheromatous proteins) and for Sanger sequencing.
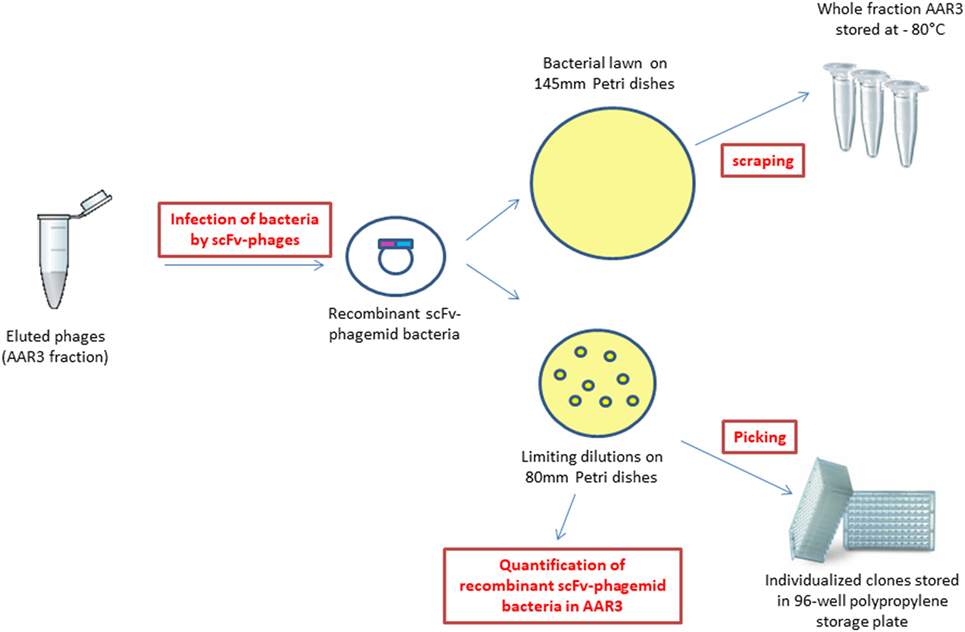
Figure 2. Obtaining recombinant single chain fragment variable-phagemid bacteria from the AAR3 fraction, storage for next-generation sequencing, quantification, and picking of individual clones.
Each of the 96 individual recombinant bacteria clones from the MASTERBLOCK® was repicked on an agarose 96-Well plate (Beckman Coulter, France). After incubation at 37°C overnight, plasmid DNA extraction, PCR amplification and sequencing were then performed by Beckman Coulter (France). One of these sequenced clones, P3 (767 bp), was used in this study for tracking the PacBio reads that were identical or related to it.
PacBio RS II Sequencing of the Whole AAR3 Fraction
Generating High Quality PCR Products
Single-molecule real-time sequencing requires high-quality, doubled-stranded DNA as input. To ensure this, plagemid DNA of recombinant bacteria from the whole AAR3 fraction was extracted directly from the frozen extract just before PCR amplification, using the QIAprep spin miniprep kit (Qiagen, France) according to manufacturer’s instructions. To generate clean, undamaged and non-chimeric amplicons, the highest fidelity polymerase was used. All PCR reactions were performed in volumes of 50 µL using 25 µL of the KAPA HiFi™ HotStart ready Mix (Kapa Biosystems, France) and 20 ng of DNA template. Each primer was used at a final concentration of 0.3 µM. PCR reactions were performed with the forward primer (Primer 1, 23-mer FWD) 5′-TGCAAATTCTATTTCAAGGAGAC-3′ and the reverse primer (Primer 2, 20-mer REV) 5′-TCACGTGCAAAAGCAGCGGC-3′. These primers were designed based on the phagemid vector; for primer 1 from position −96 to −74 upstream of the BamHI site and for primer 2 from positions 95 to 114 downstream of the NotI site (Figure 3A).
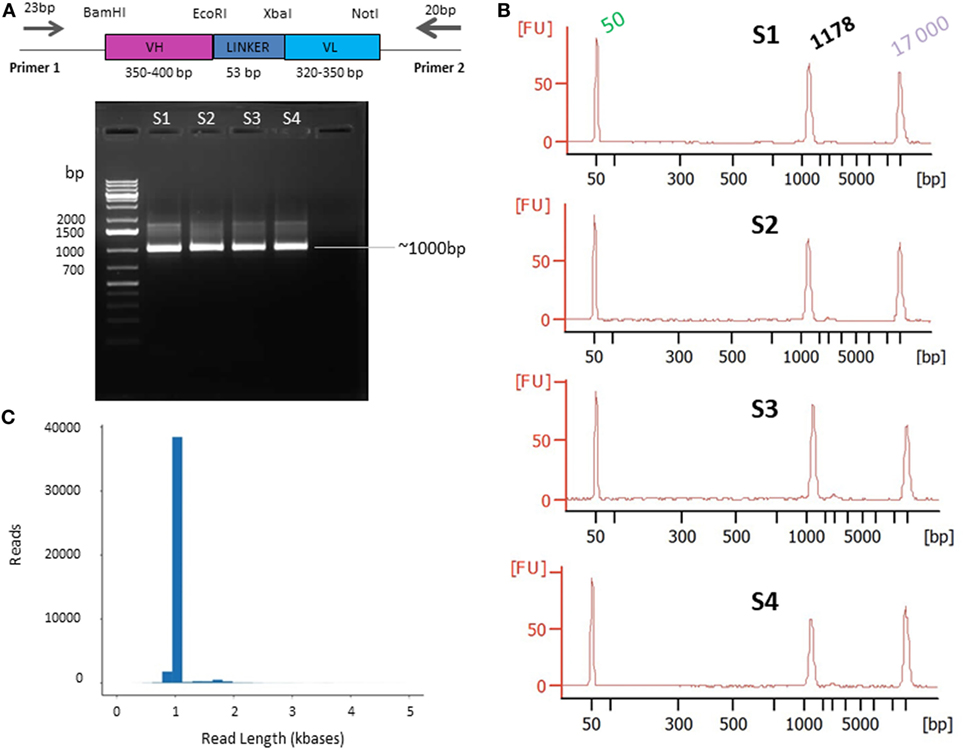
Figure 3. Primer design (A) and quality control presequencing (B) and postsequencing (C). (A) Primers designed on the phagemid vector and used for single chain fragment variable (scFv) PCR amplification. The scFv (VH-LINKER-VL) length range is between ~720 and ~800 bp [variable heavy (VH) between ~350 and ~400 bp and variable light (VL) between ~320 and ~350 bp]. The linker is 53 bp including the EcoRI and XbaI sites. The PCR products are expected to be ~1,000 bp on average, including the 5′ and 3′ region and the primers. (B) Agarose gel electrophoresis of PCR products. The DNA was amplified from the AAR3 fraction and PCR products were analyzed on 1.2% (w/v) agarose gel. The band at ~1,000 bp corresponds to the expected size for scFv amplicons. S1, S2, S3, and S4 correspond to the samples 1, 2, 3, and 4, respectively. The Bioanalyzer trace of the four samples shows the purity of amplicons with a high-quality single peak. (C) Pacific Biosciences RS II CCS2 read length distribution using P6-C4 chemistry for 1 SMRT cell (similar results were obtained for the 15 SMRT cells). Data are based on a 1-kb size-selected scFv library using a 6 h movie.
The PCR cycling protocol was chosen according to the manufacturer’s instructions and consisted of 95°C for 3 min; 15 cycles of 98°C 20 s, 65°C 15 s, 72°C 30 s; one 72°C 1 min followed by one cycle at 4°C. The reduction of the number of PCR cycles minimizes the PCR bias, however when further decreases in the number of PCR cycles were attempted, it led to several contaminating, off-target bands on an agarose gel.
The required quantity in PacBio guidelines for insert sizes superior to 750 bp is 500 ng of DNA. Four PCR reactions were performed on the same AAR3 fraction for reproducibility purposes. Agarose gel electrophoresis was used to confirm amplification, correct fragment size, and to check for non-specific product contamination. A sizing marker was included to confirm size specificity (GeneRuler 1 kb Plus DNA Ladder, Thermo Fisher Scientific, France). Amplicons were cleaned using 1× ratio of AMPure PB Beads (Pacific Biosciences). DNA purity and quantification (sample volume, yields and size distributions) were evaluated and measured using the Agilent 2100 Bioanalyzer DNA12000 kit (Agilent technologies).
SMRTbell Template Preparation
SMRTbell templates (PacBio, CA, USA) were constructed following the standard Amplicon Sequencing Protocol.4 The full procedure is explained in Figure S1 in Supplementary Material.
Briefly, PCR products were treated to repair DNA damage and then hairpin adapters were added via blunt end ligation to produce SMRTbell templates using the SMRTbell template prep kit 1.0. Exonucleases III and VII were used to remove failed ligation products and SMRTbell templates were purified with AMPure PB Beads. The ratio of sequencing primer and polymerase was determined by a PacBio calculator to correlate with SMRTbell concentrations and the 1,100-bp insert size. The sequencing primer was annealed to the single-stranded loop region of the SMRTbell template, and primer-annealed templates were then bound to DNA polymerase P6 using the DNA/polymerase binding kit P6v2. The DNA–polymerase complexes were loaded on 15 SMRT cells using MagBeads onto the PacBio RS II system and sequenced using the C4 chemistry and 6-h movies.
Initial loading titrations were performed to identify the optimal loading concentration, identifying 0.03–0.035 nM as the best loading conditions for scFv PCR products. Each SMRT cell generates ~50,000 reads on average (considering that 50,000–75,000 ZMW can be optimally loaded with a single molecule of DNA). A total of 15 SMRT cells were loaded for the four different PCR products of the same AAR3 fraction (three SMRT for sample 1 and four SMRT for samples 2, 3, and 4) to cover, per PCR sample, the diversity of the AAR3 fraction (Figure S2 in Supplementary Material). The use of 15 SMRT cells was chosen to provide a thorough and sound proof-of-concept and read comparison from four different PCR amplicons generated from the same fraction (AAR3).
PacBio CCS Read Generation
The PacBio RSII instrument produces sequencing reads with an average read length of ~15 kb, which would theoretically pass over a 1,100 bp molecule more than 10 times, producing CCS2 sequences with an accuracy ~99.9%. Longer read lengths can be achieved by increasing the instrument run time, so these data were collected using 15 SMRT cells and 6 h movies, to maximize read length and, thus, number of passes. Following sequencing, the raw data were processed using the CCS2 pipeline (CCS code used available at https://github.com/PacificBiosciences/unanimity). All CCS2 reads that were 99.9% accurate or greater were exported for further analysis. The raw NGS data of the 85-related P3 sequences can be found in the NCBI Sequence Read Archive with the accession number SRP124616.
IMGT/HighV-QUEST Analysis of the PacBio CCS2 Reads
Characterizing IMGT/HighV-QUEST scFv Reads from AAR3
The FASTQ files of the PacBio CCS2 reads were imported and converted to FASTA sequences for submission to IMGT/HighV-QUEST5 (29, 30), which implements IMGT/V-QUEST program version 3.4.2 (August 4, 2016) and IMGT/V-QUEST reference directory release 201631-4. The analysis was performed with the advanced functionality “Analysis of single chain Fragment variable (scFv)” (IMGT/HighV-QUEST Documentation, see text footnote 3).
Data filtering was applied with the following criteria to be fulfilled for each of the two V domains: (i) >85% of identity of the V-REGION of the V domain with the V-REGION of the closest germline IMGT gene and allele and (ii) in-frame V-(D)-J junction. Filtered sequences were then analyzed to identify the closest V, D (for VH) and J IMGT genes and alleles, to characterize the amino acid (AA) junction, to evaluate the mutations and to give a complete description of the scFv with IMGT labels (IMGT Index > scFv6).
Tracking and Analysis of Identical and Related PacBio Reads of the AAR3 scFv-Phage Clone P3
In order to evaluate the PacBio scFv read sequencing quality, reads identical or closely related to the sequence of a scFv-phage clone P3 (previously isolated from the same fraction AAR3 and Sanger sequenced) were tracked among the total reads generated in the 15 SMRT cells by two approaches. First, reads potentially related to P3 were searched for based on the expected VH-VL characteristics determined by IMGT/HighV-QUEST (same V and J genes and alleles, and same AA junctions). Second, a Blast search was performed to check whether P3-related reads could have been missed by the IMGT/HighV-QUEST filtering. The fasta headers of the sequences were modified to include the sequence set identifier and a blast database was built (formatdb 2.2.26) from the accumulated reads from the 15 SMRT cells. The database search was performed using the blastn program (2.2.26), with the combined following criteria: longest alignments on the P3 Sanger (767 bp) sequence, highest identity percentage, and maximum number of 20 mismatches or indels. Among the extracted reads only those fulfilling the P3 characteristics (in terms of V and J genes, allele names and AA junctions) were retained.
Results
Generation and Analysis of PCR Products from the Whole AAR3 Fraction
The unbiased characterization of scFv from phage-display combinatorial libraries, in conjunction with sequencing on the PacBio RS II system, requires high-quality PCR products with undamaged, clean and non-chimeric amplicons. Creating PCR protocols to generate products that are truly representative of the starting cell population is a major challenge.
To achieve this aim: (1) the complete frozen AAR3 fraction was directly used, without amplification, as the source of recombinant bacteria; (2) efforts were made to limit the number of amplification cycles (the four samples subject to 15 cycles) in order to reduce quantitative distortions as well as error rates due to PCR artifact (data not shown); and (3) different high-fidelity Taq polymerases were tested so as to fit with our amplification system. The required quantity (500 ng) and the correct size of amplicons (~1,000 bp) were obtained with the KAPA HiFi™ HotStart polymerase (Figure 3A). The quality and quantity of amplicons was confirmed using an Agilent 2100 Bioanalyzer. After confirmation of the purity (Figure 3B), PCR samples were sequenced on the PacBio platform.
PacBio Sequencing and CCS2 Analysis
Sequencing was done on the PacBio RS II system using P6-C4 sequencing chemistry and SMRTbell libraries generated from DNA amplified from the AAR3 fraction (corresponding to 3.5 × 105 scFv-phagemids). For each of the four PCR samples, 3 or 4 SMRT cells were run to ensure adequate sampling of the scFv as described in Section “Materials and Methods” (Figure S2 in Supplementary Material). A total of 15 SMRT cells were used.
Circular consensus sequencing (CCS2) analysis of the 15 SMRT cells produced 450,558 reads. These reads were filtered to remove any double-loaded wells or other artifactual/lower accuracy reads. The data obtained from filtered, post-CCS2 analysis represent the reads achieving 99.9% accuracy or above, derived only from wells loaded with a single molecule. Any reads that did not reach consensus coverage of QV30 (99.9% accurate) or above were filtered out. Those settings and filters are built into the CCS2 pipeline that is available via the PacBio web-based analysis software (SMRT Link). Thus, these 450,558 reads passed initial filtering with an average pass number of 24 and a quality score of minimum 99.94% accuracy (Figure 3C). Another contributing factor to data quality and throughput was the use of the longest movie lengths possible at the time (6 h) to ensure the longest read lengths for analysis.
IMGT/HighV-QUEST Analysis of the scFv PacBio Reads
The 450,558 FASTQ PacBio CCS2 reads were converted in FASTA sequences and analyzed using IMGT/HighV-QUEST, as described in Section “Materials and Methods.” After analysis, a total of 391,655 scFv “candidates” (i.e., sequences with two V regions, IMGT label V-REGION) (86.93% of the submitted PacBio reads) were identified (Table 1). The scFv sequences were then filtered according to the criteria described in Section “Materials and Methods” [>85% of identity of the V-REGION of the V domains with the V-REGION of the closest germline IMGT genes and alleles and in-frame V-(D)-J junction, determined for both V domains]. The threshold of 85% of identity is the standard filter for classical IG repertoire analysis in IMGT/HighV-QUEST (29). Following this filtering, 348,659 full-length scFv representing 89.02% of the filtered sequences were identified (Table 1). These scFv reads include 346,934 VH-VL or VL-VH expected scFv sequences (Table S1 in Supplementary Material). The other 1,725 scFv reads comprise 171 VH-VH (5–22 found per SMRT cell) and 1,554 VL-VL (68–158 found per SMRT cell). These combinations most probably occurred during the construction of the original scFv-phage combinatorial library. Thus these results provide a useful and detailed overview of the content of the scFv combinatorial library.
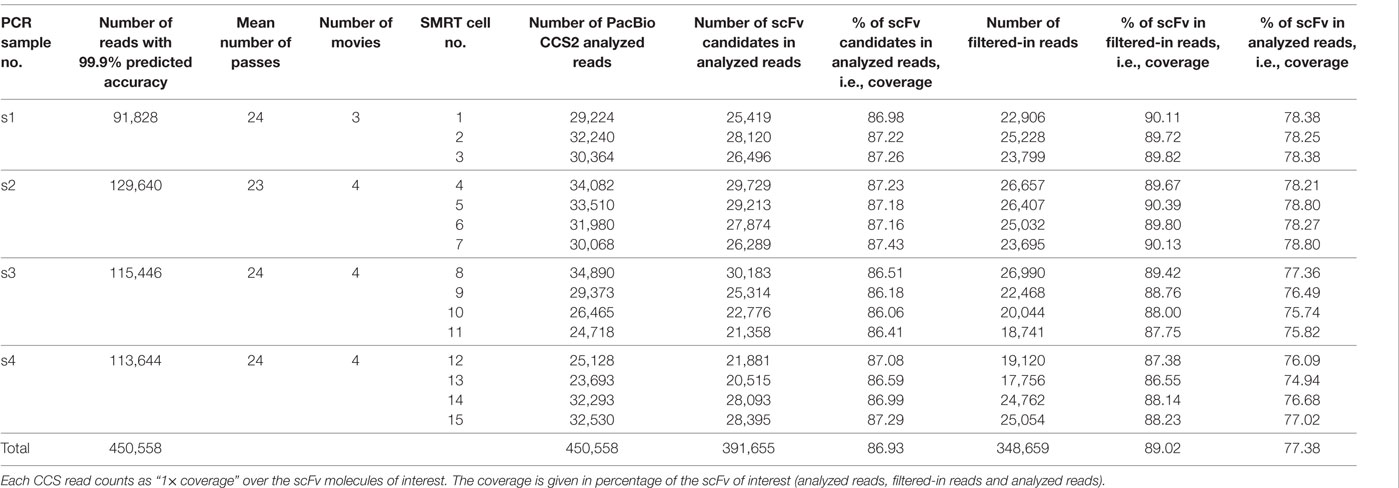
Table 1. IMGT/HighV-QUEST analysis of the scFv Pacific Biosciences (PacBio) reads.
The IMGT/HighV-QUEST analysis of the scFv reads included identification of the closest V, D (if VH), and J IMGT genes and alleles, characterization of the junction, evaluation of the mutations, and complete description of both V domains with IMGT labels (see text footnotes 2 and 5). These results demonstrate that both domains of the scFv reads sequenced by PacBio could be fully characterized with the functionality for scFv.
Tracking and Analysis of PacBio Reads Identical or Related to the P3 Clone across 15 SMRT Cells
Single chain fragment variable-phages issuing from the in vivo AAR3 selected fraction have been screened by a high-throughput flow cytometry method against atherosclerotic rabbit proteins. Some of the selected clones were then processed into scFv-Fc format in HEK293 cells and validated by immunohistochemistry on atheromatous aorta sections of two animal models of atherosclerosis (ApoE−/− mouse and New Zealand White rabbit submitted to hypercholesterolemic diet) and on human endarteriectomy biopsies. A scFv-phage clone, named P3, selected for its high accuracy in biological assays in the two animal models and human biopsies was Sanger sequenced and patented (EP 17306337.1). The whole scFv P3 sequence (Figure S3 in Supplementary Material) was then tracked among the 346,934 VH-VL and VL-VH reads (Table S1 in Supplementary Material) from the 15 SMRT cells, by the two approaches described in Section “Materials and Methods,” in order to evaluate the PacBio scFv read sequencing quality.
Analysis of PacBio Reads Identical to the P3 Clone
Sixty PacBio reads with 100% identity to the region aligned with the P3 Sanger sequence (767 bp) were obtained by both approaches. Interestingly, the P3 scFv was identified in the top 100 of the most abundant VH-VL associations in all the 15 data sets. These PacBio reads have a length of 977 bp, including the 5′ and 3′ regions and the primers. Fifty-three of the 60 reads have a 100% identity on their full length of 977 bp. As these PacBio reads were obtained from the 15 SMRT cells (Table 2), we can confidently say that no sequencing error was observed in the 51,781 bp of these 53 reads.
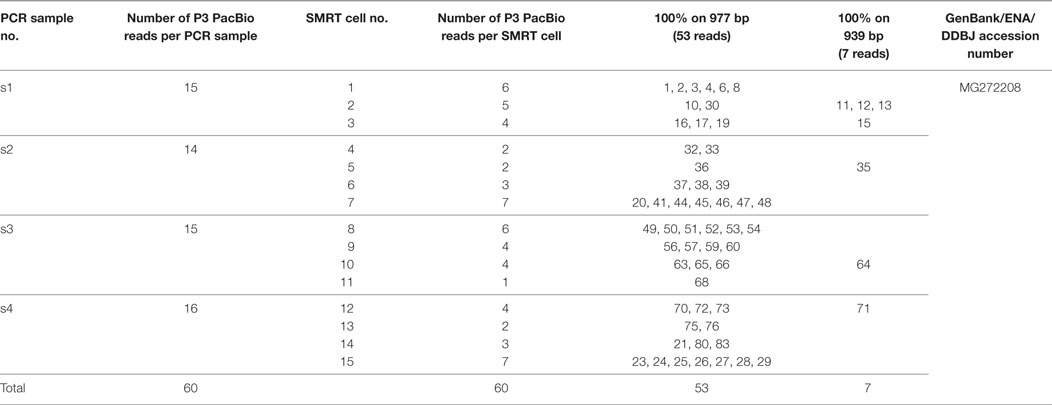
Table 2. Pacific Biosciences (PacBio) reads 100% identical to the aligned P3 Sanger sequence and 100% identical between them on 977 bp (53 reads) or 939 bp (7 reads).
Seven reads showed 100% identity on a 939 bp length (Table 2). Interestingly their mutations are all localized at the ends of the primers and/or in the immediate vicinity of the 3′ primer (Table 3). A most probable explanation is that they occur during the sequencing polymerization (the priming step could be excluded as different mutations were observed in different SMRT cells and no degenerate bases were used in the primers). Ignoring the mutations in or next to the primers, no sequencing error was observed in 6,573 nucleotides of the 7 reads (100% identity on 939 bp). Combining these results with those of the 53 reads (100% identity on the full length of 977 bp), no sequencing error was detected on 58,354 nucleotides for the 60 scFv reads.
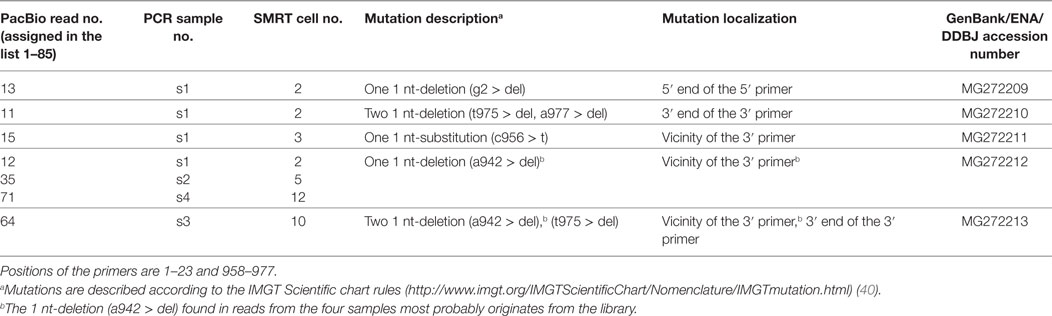
Table 3. Mutations observed at the 5′ and 3′ end of the seven Pacific Biosciences (PacBio) reads with 100% identity on 939 bp (positions 3–941).
Analysis of PacBio CCS Reads Related to the P3 Clone
In addition to the 60 PacBio reads with a 100% identity (53 reads on 977 bp and seven reads on 939 bp) (Table 2), 25 “related” P3 reads were identified (Figure S4 in Supplementary Material). These 25 P3-related reads showed 15 different mutation types in the insert and, for each type, a limited number of reads (1–6) (Table 4). Altogether, the 15 mutation types are described by 28 different individual mutations. This heterogeneity is in sharp contrast with the 60 reads with a 100% identical insert.
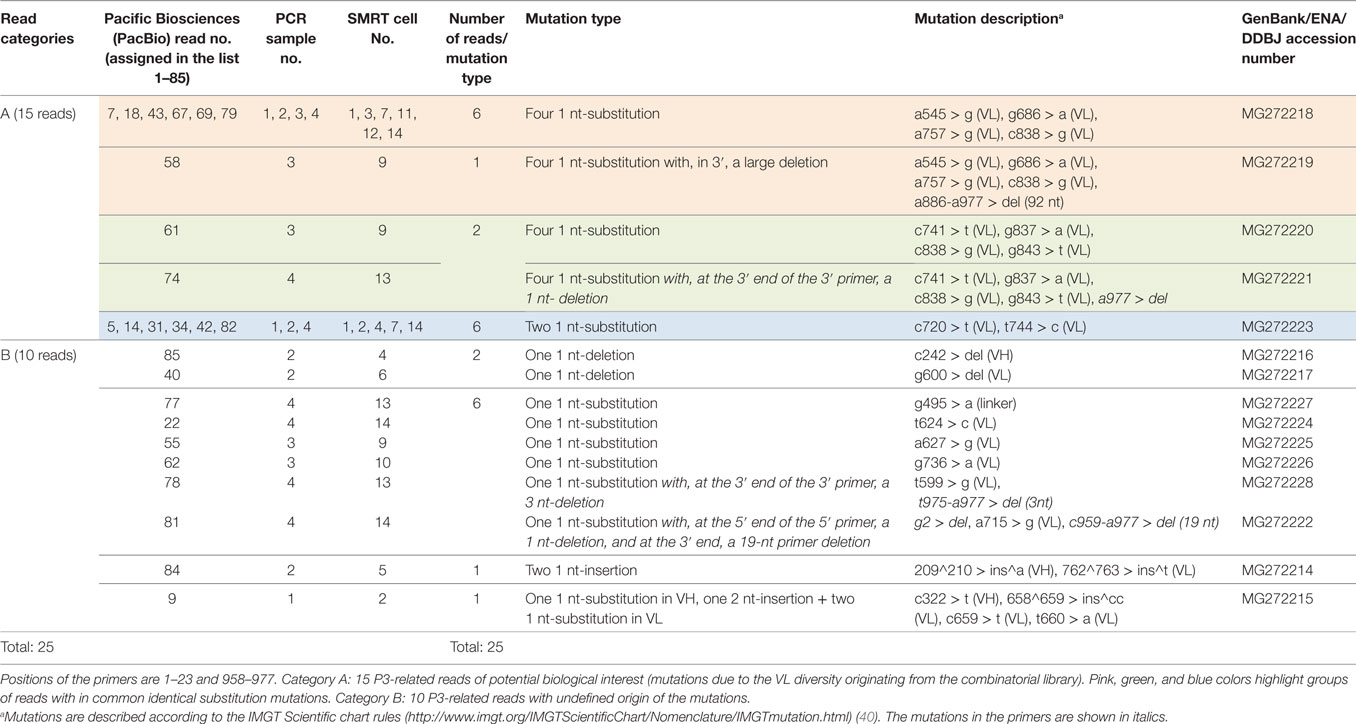
Table 4. Mutation heterogeneity observed in 25 P3-related PacBio reads, in contrast with the 60 P3 identical PacBio reads with 100% identity on the complete scFv.
The observation of related reads was expected as the scFv are from a phagemid combinatorial library in which point mutations were initially introduced to mimic the IG somatic hypermutations. The analysis of the 25 reads was therefore performed in an attempt to distinguish among them related clones with potential biological interest (as reflecting the diversity of the original library) from reads with artifactual differences.
For 15 of these PacBio CCS reads, the origin of the mutations (mostly substitutions) was clearly from the scFv phagemid combinatorial library (and therefore of potential interest given their relatedness to P3). This was supported by the fact that identical mutated reads were obtained from different PCR and from different SMRT cells (category A in Table 4). These included 7 reads (highlighted in pink) with four identical individual mutations, 2 reads (highlighted in green) with three identical individual mutations different from those described above and one shared by both groups [c838 > g (VL)], 6 reads (highlighted in blue) with two identical individual mutations (and still different from those of the previous mutation types). All the substitution mutations are localized in the VL, in agreement with mutations of the VL and VH domains being targeted differently before the assembly into the scFv and confirming that the differences observed are intrinsic to the VL domain, and not due to PCR or sequencing errors. The IMGT/V-QUEST alignment of the 15 PacBio NGS sequences with the initial P3 Sanger sequence is provided as an additional PDF file in supplementary data (Figure S5 in Supplementary Material).
For the other 10 PacBio reads, present in one copy (category B in Table 4), the origin of the differences could not be determined with certainty: and any of the possible explanations: putative sequencing errors, PCR amplification errors or diversity of the combinatorial library could not be formally excluded at this stage. The 1 nt-deletion observed in VH and VL for reads 85 and 40, respectively, the large nt deletion at the 3′ end in read 78, the nt deletion at both 5′ and 3′ ends for read 81 and the one or two nt insertions in VH or VL for reads 84 and 9 could be considered as sequencing errors. The six reads with different single substitution and the one with 3 mutations could be attributed to PCR amplification errors or diversity of the combinatorial library. Moreover, and even if these differences are due to diversity of the combinatorial library, their single copy number suggests that they are from unselected (or poorly selected) scFv-phages from the library background and could be ignored in the library screening.
Discussion
Antibody libraries are important resources to derive antibodies to be used for a wide range of diagnostic and therapeutic applications. In vivo or ex vivo phage-display selections have emerged as interesting ways to identify accurate antibodies in the context of the pathologic microenvironment (11). Although advancements in automation of biological assays have greatly improved screening strategies, high-throughput campaigns are still severely limited in the number of antibody fragments that can be interrogated, providing only a tiny fraction of the enriched phage library. Access to the genetic information encoded in antibody repertoires by NGS should allow more in depth analysis of the diversity of the selected library. However, the currently available NGS platforms that were capable of providing several million reads per run, generated only short reads of up to 300–700 bp (41). Therefore, only synthetic combinatorial scFv libraries, in which diversity was confined to CDR3 regions of the heavy and light chains, have benefited from NGS potential in the extensive in silico analysis of complex collections of selected antibodies (16).
While Mi-Seq, 454, or Ion-Torrent technologies will completely sequence heavy and light variable domains, they are currently insufficient to cover the full-length scFv, which are comprised of both VH and VL domains, connected by a peptide linker. These technologies often necessitate consensus building of multiple reads originating from the scFv fragment to obtain whole sequence information (42–44). Therefore, NGS sequencing of scFv fragments longer than 800 bp is still hampered by technical limitations in the length of reads. These limitations could be circumvented by the third generation PacBio sequencing platform. Capitalizing on PacBio SMRT DNA sequencing for high-resolution and high-throughput HLA typing (36, 37), we develop here a PacBio SMRT/CCS2 approach, combined with IMGT/HighV-QUEST analysis of full-length scFv reads to provide a straightforward protocol for characterization of the complete VH and VL domains of scFv from fully human combinatorial libraries.
Pacific Biosciences SMRT sequencing generated 450,558 reads of about 1,000 bp across 15 SMRT cells, following DNA amplification of the scFv insert from 3.5 × 105 in vivo selected scFv-phages. IMGT/HighV-QUEST with its scFv functionality allowed for further filtration and characterization of the 348,659 PacBio CCS reads as containing full-length scFv, which represent 89.02% of the overall filtered sequences from the 15 SMRT cells run. Among them, 346,934 identified expected VH-VL or VL-VH full-length scFv reads.
In order to evaluate the PacBio scFv read sequencing quality, a selected scFv-phage clone P3, previously identified from the AAR3 fraction by biological screenings and sequenced by the Sanger methodology, was tracked within the 346,934 VH-VL and VL-VH scFv reads characterized by IMGT/HighV-QUEST. Sixty PacBio CCS2 reads were identified from the 15 SMRT cells that demonstrated 100% identity on a length of 939 bp (including the complete scFv of 767 bp and the 5′ and 3′ regions) and for 53 of them on the full length of 977 bp (including the primers at both ends). Thus no sequencing error was observed on a total of 51,728 bp for these 53 reads, obtained from the 15 SMRT cells (and 58,354 bp if the seven reads with 100% on 939 bp are included), validating the capacity of the PacBio SMRT-CCS2 method to produce reads with an accuracy of >99.9% for complete sequences of inserts approaching 1,000 bp. It is of interest to note that these PacBio reads were obtained from the 15 SMRT cells.
In addition to the 60 PacBio reads with a 100% identity, 25 “related” P3 reads were identified and for 15 of them, mutation analysis (category A in Table 4) clearly showed that they are related to P3. This was supported by the fact that identical mutated reads in the VL (in agreement with the diversity generated during the library construction) were obtained from different PCR samples and from different SMRT cells. P3 (60 reads) with its 15 related reads are among the top 100 most represented associated VH-VL domains in all the 15 data sets.
This study provides the first proof-of-concept that similar sequences could be tracked in phage-display selected scFv samples and their frequency determined by the number of reads. The favorite candidates chosen for their high frequency of enrichment could be rescued from these in silico data for implementation of downstream biological assays. This could be easily done by custom gene chemical synthesis, which offers the utmost flexibility and efficiency with high production yields (45).
Thus the large number of sequenced reads delivered, following the enrichment process, could be ideally suited for a more extensive evaluation of antibody candidates by biological assays. In that way, bringing full sequence data from NGS will accelerate search for identification of both the antibodies and their targeted biomarkers. We thus aimed to combine the sensitivity of the sequencing approach with the functional information provided by the immune assays. This in silico approach could be applied to any other pathology and phage-display screening methodology. Resolving the issue of complete scFv sequencing has the potential to profoundly impact the selection process of antibodies with desired properties after phage display biopanning with a special focus of in vivo selections. It is expected that this will contribute to therapeutic antibody discovery, selection and development.
Limitations
Scientists working on IGs are very concerned by the problems of sequencing errors versus mutations and of short read assembly. This study demonstrates that the PacBio SMRT-CCS2 method is able to produce reads with an accuracy of >99.9% for complete sequences of scFv inserts without the need of in silico VH and VL assembly as usually necessary using MiSeq or Ion Torrent technologies (43, 44). To assess the read quality of PacBio sequencing, we performed a pilot study using different PCR samples and 3 or 4 SMRT for each PCR, starting from the same AAR3 fraction. Using the biologically validated P3 clone as a reference, we demonstrated that P3 clone is among the top 100 in vivo selected clones with a representativity of 0.025%. Moreover, among the 85 P3 related sequences, 60 were 100% identical and 15 clearly originated from the scFv phagemid combinatorial library. From the IMGT/V-QUEST alignment of P3 pink, blue, and green groups of reads with the initial P3 Sanger sequence, we can confidently say that 88% of reads were free of sequencing errors. However, a doubt remains for 10 of them, present in just one copy. The nt-deletions or nt insertions observed in VH and VL for six of the 10 reads could be definitively considered as sequencing errors. For the single substitutions observed in seven reads, it is obviously impossible to determine their origin. To circumvent this limitation, other technologies could be considered, such as inverse PCR method based on VH CDR3 or VL CDR3 sequences (44) to synthesize these clones from the AAR3 fraction. Nonetheless, what is of upmost importance in our study is to identify over-represented clones (from the top 100 candidates) and to proceed to the rescue of highly enriched scFv and not isolated clones. Indeed, during phage-display selections, the reads of greatest interest will have the greatest depth of coverage, having expanded in the pool and thus receiving greater proportional read depth.
Ethics Statement
All animal experiments were performed in conformity with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996) and were accredited by the local ethical committee (Animal Care and Use Committee of Bordeaux, France under the No. 50120192).
Author Contributions
AH, FO, M-JJ-V, JL-T, MS, SK, M-PL, and GC-S conceived and designed the experiments. PM provided the library. MS sequenced the data. NH processed the raw data. VG designed the analysis algorithms and PD implemented the tool for NGS. CH, JKuijpers, AG, and JKorlach supervised the sequencing procedure. GC-S and M-PL supervised the project. AH, MS, GC-S and M-PL wrote the article. All the authors have read and approved the final manuscript.
Conflict of Interest Statement
CH, NH, JKuijpers, and JKorlach are full-time employees at Pacific Biosciences, a company developing single-molecule sequencing technologies. MS was a full-time employee at Pacific Biosciences at the time of this work. PM is full-time employee at LFB, a pharmaceutical group specializing in biological medicinal products. The other authors declare that they have no conflict of interest.
Acknowledgments
We would like to thank Franck Salin for his helpful advice on DNA amplification and analysis using the Agilent 2100 Bioanalyzer. We are really grateful to Alexandre Fontayne for his help in the P3 patenting process. We thank Roberto Lleras for helpful information. We are grateful to Gérard Lefranc for helpful comments, Arthur Lavoie and Karthik Kalyan for IMGT/HighV-QUEST and to all IMGT team members for their constant motivation. IMGT® is Academic Institutional Member of the International Medical Informatics Association (IMIA) and of the Global Alliance for the Genomics and Health (GA4GH).
Funding
IMGT® is currently supported by the Centre National de la Recherche Scientifique (CNRS); the Ministère de l′Enseignement Supérieur et de la Recherche (MESR); the Montpellier University, France; the Agence Nationale de la Recherche (ANR) Labex MabImprove [ANR-10-LABX-5301]; BioCampus Montpellier; Région Languedoc-Roussillon [Grand Plateau Technique pour la Recherche (GPTR)]. This work was granted access to the HPC@LR and to the High Performance Computing (HPC) resources of the Centre Informatique National de l′Enseignement Supérieur (CINES) and to Très Grand Centre de Calcul (TGCC) of the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA) under the allocation [036029] (2010–2017) made by GENCI (Grand Equipement National de Calcul Intensif). This study was achieved within the context of the Laboratory of Excellence TRAIL ANR-10-LABX-57, referenced ANR-10-LABX-0057 and named TRAIL. A public grant from the French National Agency within the context of the Investments for the Future Program (ANR-13-BSV5-0018 SVSE5 Program), named ATHERANOS supported this work.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01796/full#supplementary-material.
Abbreviations
CCS, circular consensus sequencing; IG, immunoglobulin; IMGT, IMGT®, the International ImMunoGeneTics Information System®; NGS, next-generation sequencing; scFv, single chain Fragment variable; SMRT, single-molecule real-time; TR, T cell receptor.
Footnotes
- ^http://www.imgt.org.
- ^http://www.imgt.org/IMGT_vquest/share/textes/imgtvquest.html.
- ^http://www.imgt.org/HighV-QUEST/doc.action.
- ^http://www.pacb.com/wp-content/uploads/Procedure-Checklist-Amplicon-Template-Preparation-and-Sequencing.pdf.
- ^http://www.imgt.org/HighV-QUEST/login.action.
- ^http://www.imgt.org/IMGTindex/scFv.php.
References
1. McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature (1990) 348(6301):552–4. doi:10.10138/348552a0
2. Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol (1991) 222(3):581–97. doi:10.1016/0022-2836(91)90498-U
3. Griffiths AD, Malmqvist M, Marks JD, Bye JM, Embleton MJ, McCafferty J, et al. Human anti-self antibodies with high specificity from phage display libraries. EMBO J (1993) 12(2):725–34.
4. Peneff C, Lefranc M-P, Dariavach P. Characterisation and specificity of two single-chain Fv antibodies directed to the protein tyrosine kinase Syk. J Immunol Methods (2000) 236:105–15. doi:10.1016/S0022-1759(99)00228-8
5. Pelat T, Hust M, Laffly E, Condemine F, Bottex C, Vidal D, et al. A high affinity, human-like antibody fragment (scFv) neutralising the lethal factor (LF) of Bacillus anthracis by inhibiting PA-LF complex formation. Antimicrob Agents Chemother (2007) 51:2758–64. doi:10.1128/AAC.01528-06
6. Thie H, Meyer T, Schirrmann T, Hust M, Dübel S. Phage display derived therapeutic antibodies. Curr Pharm Biotechnol (2008) 9:439–46. doi:10.2174/138920108786786349
7. Marcus WD, Wang H, Lindsay SM, Sierks MR. Characterization of an antibody scFv that recognizes fibrillar insulin and beta-amyloid using atomic force microscopy. Nanomedicine (2008) 4(1):1–7. doi:10.1016/j.nano.2007.11.003
8. Pelat T, Hust M, Hale M, Lefranc M-P, Dübel S, Thullier P. Isolation of a human-like antibody fragment (scFv) that neutralizes ricin biological activity. BMC Biotechnol (2009) 9:60. doi:10.1186/1472-6750-9-60
9. Tian H, Davidowitz E, Lopez P, He P, Schulz P, Moe J, et al. Isolation and characterization of antibody fragments selective for toxic oligomeric tau. Neurobiol Aging (2015) 36(3):1342–55. doi:10.1016/j.neurobiolaging.2014.12.002
10. Williams SM, Venkataraman L, Tian H, Khan G, Harris BT, Sierks MR. Novel atomic force microscopy based biopanning for isolation of morphology specific reagents against TDP-43 variants in amyotrophic lateral sclerosis. J Vis Exp (2015) 96:1–13. doi:10.3791/52584
11. Krag DN, Shukla GS, Shen GP, Pero S, Ashikaga T, Fuller S, et al. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res (2006) 66:7724–33. doi:10.1158/0008-5472.CAN-05-4441
12. Deramchia K, Jacobin-Valat MJ, Laroche-Traineau J, Bonetto S, Sanchez S, Dos Santos P, et al. By-passing large screening experiments using sequencing as a tool to identify scFv fragments targeting atherosclerotic lesions in a novel in vivo phage display selection. Int J Mol Sci (2012) 13:6902–23. doi:10.3390/ijms13066902
13. Deramchia K, Jacobin-Valat MJ, Vallet A, Bazin H, Santarelli X, Sanchez S, et al. In vivo phage display to identify new human antibody fragments homing to atherosclerotic endothelial and subendothelial tissues. Am J Pathol (2012) 180:2576–89; Erratum in: Am J Pathol (2012) 181(5):1889. doi:10.1016/j.ajpath.2012.02.013
14. Ravn U, Gueneau F, Baerlocher L, Osteras M, Desmurs M, Malinge P, et al. By-passing in vitro screening – next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res (2010) 38(21):e193. doi:10.1093/nar/gkq789
15. Larman HB, Xu GJ, Pavlova NN, Elledge SJ. Construction of a rationally designed antibody platform for sequencing-assisted selection. Proc Natl Acad Sci U S A (2012) 109:18523–8. doi:10.1073/pnas.1215549109
16. Ravn U, Didelot G, Venet S, Ng KT, Gueneau F, Rousseau F, et al. Deep sequencing of phage display libraries to support antibody discovery. Methods (2013) 60(1):99–110. doi:10.1016/j.ymeth.2013.03.001
18. Lefranc M-P. Immunoglobulin and T cell receptor genes: IMGT(®) and the birth and rise of immunoinformatics. Front Immunol (2014) 5:22. doi:10.3389/fimmu.2014.0002223
19. Martinez O, Gangi E, Mordi D, Gupta S, Dorevitch S, Lefranc M-P, et al. Diversity in the complementarity determining region 3 (CDR3) of antibodies from mice with evolving anti-TSHR antibody responses. Endocrinology (2007) 148:752–61. doi:10.1210/en.2006-1096
20. Li L, Wang XH, Williams C, Volsky B, Steczko O, Seaman MS, et al. A broad range of mutations in HIV-1 neutralizing human monoclonal antibodies specific for V2, V3, and the CD4 binding site. Mol Immunol (2015) 66(2):364–74. doi:10.1016/j.molimm.2015.04.011
21. Marillet S, Lefranc M-P, Boudinot P, Cazals F. Novel structural parameters of Ig-Ag complexes yield a quantitative description of interaction specificity and binding affinity. Front Immunol (2017) 8:34. doi:10.3389/fimmu.2017.00034
22. Lefranc M-P, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, et al. IMGT®, the international ImMunoGeneTics information system® 25 years on. Nucleic Acids Res (2015) 43:D413–22. doi:10.1093/nar/gku1056
23. Giudicelli V, Chaume D, Lefranc M-P. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res (2004) 32:W435–40. doi:10.1093/nar/gkh412
24. Brochet X, Lefranc M-P, Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res (2008) 36:W503–8. doi:10.1093/nar/gkn316
25. Giudicelli V, Brochet X, Lefranc M-P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb Protoc (2011) 6:695–715. doi:10.1101/pdb.prot5633
26. Yousfi Monod M, Giudicelli V, Chaume D, Lefranc M-P. IMGT/JunctionAnalysis: the first tool for the analysis of the immunoglobulin and T cell receptor complex V-J and V-D-J JUNCTIONs. Bioinformatics (2004) 20:i379–85. doi:10.1093/bioinformatics/bth945
27. Giudicelli V, Lefranc M-P. IMGT/JunctionAnalysis: IMGT standardized analysis of the V-J and V-D-J junctions of the rearranged immunoglobulins (IG) and T cell receptors (TR). Cold Spring Harb Protoc (2011) 6:716–25. doi:10.1101/pdb.prot5634
28. Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc M-P. IMGT/HighV-QUEST: the IMGT® web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res (2012) 8(1):2. doi:10.4172/1745-7580.1000056
29. Alamyar E, Duroux P, Lefranc M-P, Giudicelli V. IMGT® tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol (2012) 882:569–604. doi:10.1007/978-1-61779-842-9_32
30. Li S, Lefranc M-P, Miles JJ, Alamyar E, Giudicelli V, Duroux P, et al. IMGT/HighV-QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat Commun (2013) 4:2333. doi:10.1038/ncomms3333
31. Giudicelli V, Duroux P, Lavoie A, Aouinti S, Lefranc M-P, Kossida S. From IMGT-ONTOLOGY to IMGT/HighV-QUEST for NGS immunoglobulin (IG) and T cell receptor (TR) repertoires in autoimmune and infectious diseases. Autoimmun Infec Dis (2015) 1:1. doi:10.16966/aidoa.103
32. Aouinti S, Malouche D, Giudicelli V, Kossida S, Lefranc M-P. IMGT/HighV-QUEST statistical significance of IMGT clonotype (AA) diversity per gene for standardized comparisons of next generation sequencing immunoprofiles of immunoglobulins and T cell receptors. PLoS One (2015) 10(11):e0142353. doi:10.1371/journal.pone.0142353
33. Aouinti S, Giudicelli V, Duroux P, Malouche D, Kossida S, Lefranc M-P. IMGT/StatClonotype for pairwise evaluation and visualization of NGS IG and TR IMGT clonotype (AA) diversity or expression from IMGT/HighV-QUEST. Front Immunol (2016) 7:339. doi:10.3389/fimmu.2016.00339
34. Giudicelli V, Duroux P, Kossida S, Lefranc M-P. IG and TR single chain fragment variable (scFv) sequence analysis: a new advanced functionality of IMGT/V-QUEST and IMGT/HighV-QUEST. BMC Immunol (2017) 18(1):35. doi:10.1186/s12865-017-0218-8
35. Rhoads A, Au KF. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics (2015) 13:278–89. doi:10.1016/j.gpb.2015.08.002
36. Mayor NP, Robinson J, McWhinnie AJ, Ranade S, Eng K, Midwinter W, et al. HLA typing for the next generation. PLoS One (2015) 10:e0127153. doi:10.1371/journal.pone.0127153
37. Westbrook CJ, Karl JA, Wiseman RW, Mate S, Koroleva G, Garcia K, et al. No assembly required: full-length MHC class I allele discovery by PacBio circular consensus sequencing. Hum Immunol (2015) 76:891–6. doi:10.1016/j.humimm.2015.03.022
38. Carapito R, Radosavljevic M, Bahram S. Next-generation sequencing of the HLA locus: methods and impacts on HLA typing, population genetics and disease association studies. Hum Immunol (2016) 77:1016–23. doi:10.1016/j.humimm.2016.04.002
39. Hua TD, Souriau C, Marin M, Lefranc M-P, Weill M. Construction d’un répertoire de fragments d’anticorps scFv et son expression à la surface de phages filamenteux [French]. In: Lefranc and Lefranc, editor. Ingénierie des anticorps. Banques combinatoires. Techniques en Immunologie. (Chap. 5), Paris: Les Editions INSERM (1997). p. 39–54.
40. Lefranc M-P. IMGT locus on focus: a new section of experimental and clinical immunogenetics. Exp Clin Immunogenet (1998) 15:1–7. doi:10.1159/000019049
41. Glanville J, D’Angelo S, Khan TA, Reddy ST, Naranjo L, Ferrara F, et al. Deep sequencing in library selection projects: what insight does it bring? Curr Opin Struct Biol (2015) 33:146–60. doi:10.1016/j.sbi.2015.09.001
42. Yang W, Yoon A, Lee S, Kim S, Han J, Chung J. Next-generation sequencing enables the discovery of more diverse positive clones from a phage-displayed antibody library. Exp Mol Med (2017) 49:e308. doi:10.1038/emm.2017.22
43. DeKosky BJ, Kojima T, Rodin A, Charab W, Ippolito GC, Ellington AD, et al. In-depth determination and analysis of the human paired heavy- and light-chain antibody repertoire. Nat Med (2015) 21:86–91,1. doi:10.1038/nm.3743
44. D’Angelo S, Kumar S, Naranjo L, Ferrara F, Kiss C, Bradbury AR. From deep sequencing to actual clones. Protein Eng Des Sel (2014) 27:301–7. doi:10.1093/protein/gzu032
Keywords: human antibody, IMGT/HighV-QUEST, immunoinformatics, immunoglobulin, Pacific Biosciences sequencing, phage combinatorial library, single chain fragment variable, next-generation sequencing
Citation: Hemadou A, Giudicelli V, Smith ML, Lefranc M-P, Duroux P, Kossida S, Heiner C, Hepler NL, Kuijpers J, Groppi A, Korlach J, Mondon P, Ottones F, Jacobin-Valat M-J, Laroche-Traineau J and Clofent-Sanchez G (2017) Pacific Biosciences Sequencing and IMGT/HighV-QUEST Analysis of Full-Length Single Chain Fragment Variable from an In Vivo Selected Phage-Display Combinatorial Library. Front. Immunol. 8:1796. doi: 10.3389/fimmu.2017.01796
Received: 30 October 2017; Accepted: 30 November 2017;
Published: 20 December 2017
Edited by:
Prabakaran Ponraj, Intrexon, United StatesReviewed by:
Lei Xu, National Cancer Institute (NIH), United StatesKwan-Ki Hwang, Duke University, United States
Copyright: © 2017 Hemadou, Giudicelli, Smith, Lefranc, Duroux, Kossida, Heiner, Hepler, Kuijpers, Groppi, Korlach, Mondon, Ottones, Jacobin-Valat, Laroche-Traineau and Clofent-Sanchez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie-Paule Lefranc, bWFyaWUtcGF1bGUubGVmcmFuY0BpZ2guY25ycy5mcg==;
Gisèle Clofent-Sanchez, Z2lzZWxlLmNsb2ZlbnQtc2FuY2hlekBybXNiLnUtYm9yZGVhdXgyLmZy
†Present address: Melissa Laird Smith, Department of Genetics and Genomic Sciences, Icahn School of Medicine Mount Sinai, New York, United States
‡Equivalent position of authors.