 Shengjie Xu
Shengjie Xu Mari L. Shinohara
Mari L. Shinohara- 1Department of Immunology, Duke University School of Medicine, Durham, NC, United States
- 2Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC, United States
Invasive fungal infections result in high morbidity and mortality. Host organs targeted by fungal pathogens vary depending on the route of infection and fungal species encountered. Cryptococcus neoformans infects the respiratory tract and disseminates throughout the central nervous system. Candida albicans infects mucosal tissues and the skin, and systemic Candida infection in rodents has a tropism to the kidney. Aspergillus fumigatus reaches distal areas of the lung once inhaled by the host. Across different tissues in naïve hosts, tissue-resident macrophages (TRMs) are one of the most populous cells of the innate immune system. Although they function to maintain homeostasis in a tissue-specific manner during steady state, TRMs may function as the first line of defense against invading pathogens and may regulate host immune responses. Thus, in any organs, TRMs are uniquely positioned and specifically programmed to function. This article reviews the current understanding of the roles of TRMs during major fungal infections.
Introduction
Macrophages were initially discovered in the late nineteenth century by Metchnikoff and named for its phagocytic activity as “devouring cells” in Greek (1, 2). They are capable of engulfing and digesting cellular debris, foreign substances, and microorganisms, which are critical for tissue remodeling and immune defense against pathogens. Based on the morphology, function, origin, and kinetics of these phagocytes, macrophages were categorized into the “mononuclear phagocytes system (MPS)” (3). Even after a century since the discovery of macrophages, research efforts have continuously focused on the origins and functions of macrophages for their significant impact on tissue homeostasis and disease pathogenesis.
Tissue-resident macrophages (TRMs) consist of heterogeneous subsets of macrophages distributed in tissues across the body and contribute to tissue homeostasis and immunosurveillance (4, 5). Depending on which organs they reside, some TRMs have specific names, such as alveolar macrophages (AMs) (lung), microglia (brain), Kupffer cells (liver), renal macrophages (kidney), and osteoclasts (skeletal system). As such specific names indicate, TRMs are considered to have specific functions due to various tissue microenvironments (6, 7). This mini-review provides an outline of several major TRMs in fungal infections, mainly focusing on murine studies, by which a majority of mechanistic insights about TRMs have been obtained.
Origins of TRMs
Developmental Origins of TRMs
Tissue-resident macrophages used to be considered as cells derived from circulating monocytes during the early establishment of the MPS (3). However, a series of recent studies drastically changed this notion, particularly through the technical advancement of in vivo cellular lineage-tracing by employing the “fate-mapping” technique using the mouse Cre-lox genetic system. Such in vivo lineage-tracing approaches have shown, for example, that microglia arise early in mouse development and are derived from primitive macrophages in the yolk sac (YS) (8). These studies suggested that microglia are ontogenically distinct from monocyte-derived macrophages (MDMs), which are of the hematopoietic origin. In addition to microglia, F4/80hi Kupffer cells and epidermal Langerhans cells were demonstrated to be YS-derived and do not require Myb, a transcription factor required for the development of hematopoietic stem cells (HSCs) (9). By employing the conditional CX3CR1 fate-mapping system, another study showed that origins of Kupffer cells, AMs, splenic, and peritoneal macrophages, are also embryonic, at least in part (10). Introduction of fate-mapping markers other than CX3CR1 further clarified that TRMs in many tissues consist of mixed populations of the embryonic (YS and/or fetal liver) and the BM hematopoietic origins, except for microglia that are exclusively of the YS-origin (11–13).
A majority of TRMs are self-maintained throughout adult life with minimal contribution from circulating monocytes (14). However, populations of TRMs can also be replaced. For example, intestinal macrophages in mouse neonates are derived from YS and fetal liver, but do not persist into adulthood and are replaced by MDMs around the time of weaning (15). Cardiac macrophages are established from YS and fetal monocyte progenitors, but disruption of homeostasis replaces the population with MDMs (11). These murine studies strongly suggested that TRMs, in general, are derived from diverse precursors including YS macrophages, fetal liver monocytes, and even circulating HSC-derived monocytes; and ontogenic origins of TRMs greatly vary depending on tissues.
TRMs Reflecting Organ-Specific Microenvironments
Tissue-resident macrophages develop locally and adapt to tissue microenvironments during embryogenesis and beyond. Distinct gene expression patterns were identified among local TRMs from various tissues (6, 7, 16, 17), and are often reflected at the epigenetic level, particularly indicated by differential histone marks on the enhancer landscape (6, 7). Multiple pieces of evidence have suggested that such tissue-specific patterns of gene expression in TRMs are influenced by tissue-specific environmental factors, including heme (18), retinoic acid (6, 17), and TGF-β (6, 19). Interestingly, macrophage “precursors” derived from YS, fetal liver, and adult monocytes appear to have the plasticity to become certain TRMs, based on tissue-specific gene expression profiles. For example, macrophages precursors from various origins develop into functional and self-maintaining AMs, when transplanted to an empty alveolar niche (20). However, once differentiated into organ-specific macrophages, TRMs, except for Kupffer cells, cannot efficiently colonize the empty AM niche (20), suggesting that the plasticity would be lost after the precursor stage. Thus, functions of TRMs are actively shaped by their local tissue microenvironment.
TRMs in Antifungal Responses
Critical steps to protect hosts from infections include; early recognition of the fungi, activation of host immunity, and killing of the spores and vegetative fungal cells to contain fungal dissemination (21–24). During early stages of fungal infections, infected hosts rely on tissue-resident “cells,” not necessarily TRMs alone, to function as the first line of defense. Here, despite the tissue-specific functions of TRMs from various organs, a general expectation for TRMs is to function as immune sentinels to detect infections at the front line. In fact, TRMs express a wide array of cell surface receptors that sense intruding microbes and produce chemokines and cytokines to recruit and activate other cell subsets for further help (25, 26). However, do TRMs always work to protect hosts? We will visit this topic in the following subsections. As some backgrounds for this section, we would like to mention that TRMs are not considered to play a role in T cell priming with microbe-derived antigens in draining lymph nodes because they are not migratory cells (27). It is also of note that CCR2+ inflammatory MDMs play critical role in fungal clearance (28–32). Here, CCR2+ MDMs are recruited from circulation by chemoattractants secreted by sentinel cells. In the following subsections and Table 1, we focus on the early interaction of TRMs with fungi.
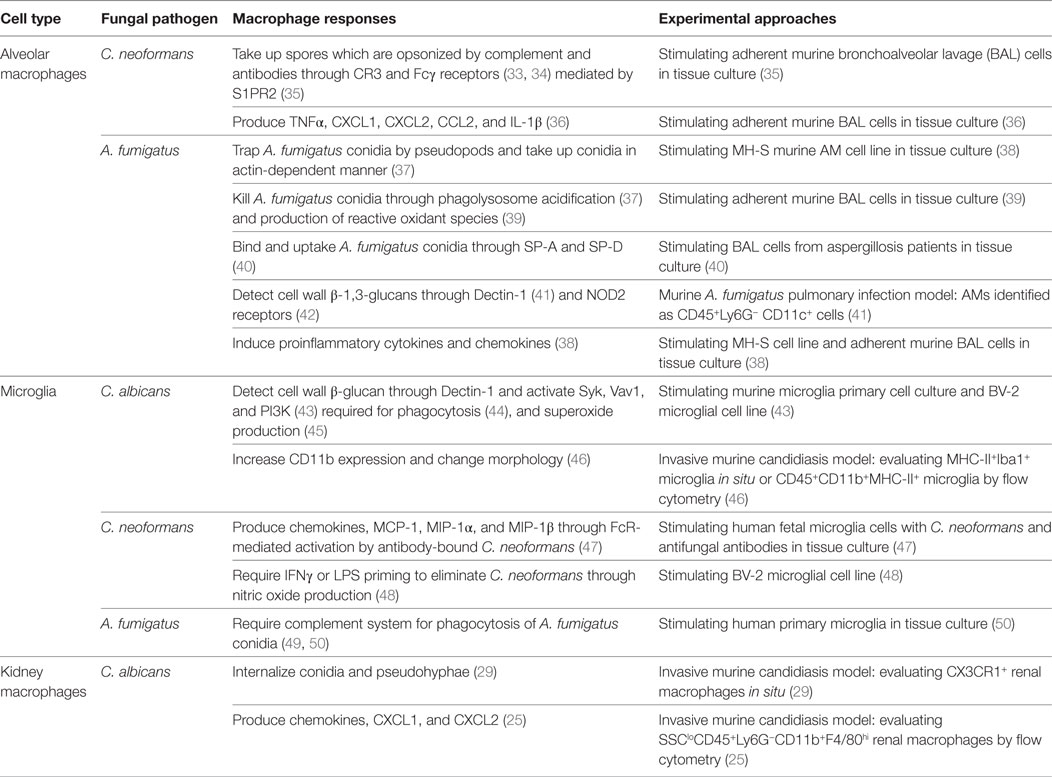
Table 1. Tissue-resident macrophage antifungal response.
Lung—AMs
Because lungs are exposed to the outer environment, they constantly inhale microbes, which enter the distal airway to bronchioles and alveoli. AMs are lung-resident macrophages considered to be largely derived from fetal liver monocytes (10, 13, 51) and represent more than 90% of leukocytes in a bronchoalveolar lavage in healthy animals (52). Since fungal infections through the pulmonary route have been intensively studied, AMs may be the best-documented TRMs in fungal infections. Here, we discuss AMs and two major pulmonary fungal pathogens, Cryptococcus neoformans and Aspergillus fumigatus, which can cause serious invasive cryptococcosis and aspergillosis, respectively (21, 53–55).
C. neoformans spores and A. fumigatus conidia enter into the lungs by inhalation and encounter lung-resident cells first, including AMs. Although AMs are not effective in antigen presentation to T cells due to their low level of costimulatory molecules (56), AMs are considered to be at the first line of immune defense against pulmonary pathogens (57). AMs express complement receptor 3 (CR3) and Fcγ receptors (FcγR) to opsonize and phagocytose C. neoformans spores (33, 34, 58, 59). Phagocytosis of C. neoformans spores is enhanced by extracellular sphingosine-1-phosphate, which upregulates FcγR expression on AMs (35). In A. fumigatus infection, AMs can trap dormant A. fumigatus conidia with pseudopods and endocytose conidia in an actin-dependent manner (37, 60). Although neutrophils are the main population involved in complement-dependent opsonization, phagocytosis, and killing of the fungi (61, 62), AMs can also kill internalized A. fumigatus conidia by detection of conidia swelling and the endosome–phagosome fusion, resulting in acidification of the organelles (37). Activation of NADPH oxidase in AMs was also reported (39), suggesting AMs to gain an “M1” phenotype. Alternatively, another study showed that A. fumigatus infection promotes AMs to gain an alternative activated macrophage phenotype, or also known as the M2 phenotype, based on upregulation of M2 macrophage markers, such as gene transcripts encoding arginase-1 (Arg1), Ym1, and CD206 (63). Interestingly, the study did not observe the induction of Nos2, a major M1 macrophage marker (63). It was suggested that Arg1-expressing AMs potentially deprive L-arginine, a substrate of arginase. Since L-arginine is an essential nutrient source of fungi, the expression of Arg1 may result in inhibiting fungal growth through arginine deprivation (63). These studies suggested the presence of multiple mechanisms by which AMs protects hosts from fungal infections.
Failure in the initial clearance of invaded fungi allows them to take advantage of the humid and nutrient-rich milieu in the lung to disseminate. As the next layer to contain fungal dissemination, inflammatory neutrophils and monocytes need to be recruited in the lung. Here, it is possible that AMs play a sentinel role to recruit such inflammatory cells by secreting cytokines and chemokines to fight against fungi. For example, dectin-1 on AMs detects β-glucans on the fungal cell surface (41, 64) and stimulates the production of proinflammatory cytokines TNFα, IL-6, and IL-18 (65). Intracellular receptor NOD2 in AMs can also induce the synthesis of cytokines, such as IL-12, IFN-γ, GM-CSF, CCL2/MCP-1, CXCL2/MIP-2, and CXCL1/KC (38, 42). It is of note that the majority of these studies on cytokine and chemokine expression were performed with isolated AMs or cell lines in tissue culture. Thus, in vivo protein expression patterns of AMs to A. fumigatus and C. neoformans infections need to be studied.
Neutrophil chemoattractants, such as CXCL1 and CXCL2, have a great impact on the host protection from A. fumigatus infection (66, 67), and the main source of the chemoattractants in A. fumigatus infection was reported to be epithelial cells, rather than AMs (68). Indeed, AM depletion by clodronate does not alter neutrophil recruitment and host mortality in pulmonary A. fumigatus infection (69). Thus, a role of AMs in A. fumigatus may be minor. In contrast in C. neoformans infection, AMs highly express CXCL1 and CXCL2, as well as TNFα (36), but C. neoformans can survive in AMs and contribute to latent infection (70). However, it is puzzling that depletion of AMs and DCs “together,” by using CD11c-DTR mice (AMs and DCs are CD11c positive), resulted in more neutrophil infiltration in the lung 4 days after C. neoformans infection and enhanced mortality with severe lung inflammation (71). Although it is not clear which cell type, DCs, or AMs, is dominant in inhibiting neutrophil recruitment in the lung, questions that can be brought up are how DCs and/or AMs inhibit neutrophil recruitment and whether the inhibition occurs only under some conditions. Since it is technically difficult to deplete AMs alone, we still need to wait to understand if and how AMs are detrimental or protective in fungal infections.
Central Nervous System (CNS)—Microglia
Fungal infections in the CNS are usually secondary to infections in peripheral tissues. Yet, once fungal pathogens reach to the CNS, it can be fatal to hosts. Some species of Candida, Cryptococcus, and Aspergillus can cause life-threatening CNS infections in immunocompromised patients (72–74). Microglia reside in the CNS parenchyma and are poised to provide the first line of defense against invading pathogens. Through the expression of various pattern-recognition receptors, microglia can recognize a wide range of pathogens that colonize the CNS (75, 76). In this section, we discuss responses of microglia during CNS infection by these fungi.
Candida albicans commonly colonizes the mucocutaneous locations in the host, and can also invade the bloodstream to cause systemic candidiasis. Innate immunity is the dominant protective mechanism against disseminated candidiasis. Microglia detect β-glucans through dectin-1, resulting in phosphorylation of Syk (43), and activation of Vav1 and PI3K, which are required for phagocytosis and superoxide production (45). However, dectin-1 stimulation alone is not sufficient for microglia to induce cytokines or chemokine production (43). This suggests a unique mechanism of dectin-1 signaling in microglia distinct from other types of TRMs and MDMs, in which dectin-1 signaling is sufficient for production of cytokines and chemokines. Microglia are also found in the retina and activated by invasive candidiasis, resulting in enhanced expression of cell surface CD11b, and morphological change (46), as well as phagocytosis of C. albicans conidia through dectin-1 activation (44).
In contrast to Candida, C. neoformans spores are not effectively cleared by microglia. Thus, microglia require other immune cells and mechanisms to effectively combat C. neoformans infection in the CNS (77, 78). Opsonization of C. neoformans spores by antibodies plays a critical role in the induction of cytokine and chemokine expression in microglia (48). For example, opsonizing antibodies induce microglial expression of chemokines, such as CCL2/MCP-1, CCL3/MIP-1α, and CCL4/MIP-1β, but the response is also known to be inhibited by cryptococcal capsular polysaccharides (47). In addition to antibodies, LPS and IFNγ promote the killing of opsonized and unopsonized C. neoformans by augmenting nitric oxide production without inducing phagocytosis in a microglial cell line (48). Another study showed that IFNγ is required for enhanced anticryptococcal responses when microglia are activated by intracranial injection of IL-2 and a CD40 agonistic antibody (79). Taken together, IFNγ appears to be critical for microglia to respond to C. neoformans.
Aspergillus fumigatus also causes meningitis, but little is known about responses of microglia to A. fumigatus. One study showed that CR3 expression of microglia is reduced by an A. fumigatus-derived protease, resulting in a significant decrease in phagocytosis by primary human microglia (49). The high frequency of host mortality by cerebral aspergillosis suggests that antifungal responses of microglia are not efficient, although it might be possible that IFNγ also enhances the response against Aspergillus by microglia.
Taken together, these studies suggest that microglia are not efficient in fungal clearance. Although it is not clear why microglia are not effective cells among the MPS, the specific microenvironment of the CNS, which is known as an immune-privileged site, may be involved in shaping the character of microglia. The CNS is isolated from other peripheral organs because it is separated from blood circulation by the blood–brain barrier. The physical separation of the CNS from the immune system in the rest of organs, at least in part, may contribute to the specific development and functions of microglia, distinct from the rest of TRMs.
Kidney—Renal Macrophages
In healthy kidneys, immune cells are rarely found except for resident DCs and macrophages (80). Renal macrophages are found in the tubulointerstitium (81), a compartment of the kidney bounded by the vasculature and nephrons, and comprising about 80% of kidney volume (80). Renal macrophages in adult mice are largely derived from fetal liver monocytes (11, 82) and have been extensively studied due to their involvement in immune homeostasis (83–85) and host defense against infections (29, 86).
The kidney is a main target organ in murine systemic candidiasis (87, 88), but not necessarily a primary target in human systemic candidiasis (89). Nevertheless, host resistance heavily depends on the immune system in the kidney. For example, renal macrophages, as well as possibly splenic and liver macrophage, are considered to be protective in host defense against Candida (29, 87, 90). CX3CR1-deficient mice are susceptible to Candida infection, possibly due to reduced numbers of kidney-resident and -infiltrated macrophages (91). As early as 2 h after Candida infection, renal macrophages elicit their protective responses by internalizing conidia and encasing pseudohyphal elements (91). In addition to their phagocytic ability, renal macrophages isolated from naïve mice are shown to kill Candida conidia in tissue culture (91). Besides their endogenous fungal-killing ability, kidney F4/80hi macrophages also recruit neutrophils by secreting high levels of chemokine CXCL2 in the first 24 h of systemic Candida infection in an autophagy-dependent manner (25), indeed playing a role as immune sentinels. In summary, kidney macrophages are important players in fungal clearance in murine candidiasis model.
Closing Remarks
Our knowledge on TRMs identities and functions has been greatly expanded in the last decade. Depending on the physical locations and fungal pathogens, TRMs respond in different ways. Tissue-specific factors may also have impacts on the antifungal outcome of TRMs. However, there are still many unanswered questions and technical hurdles to further advance the field. Here, we close our discussion with six questions.
(A) Do the functions of TRMs from various organs share something in common? Because TRMs are shaped by tissue-specific environments to acquire unique intracellular gene expression profile and assisted by tissue factors to enhance their antifungal response, previous studies have focused on the dissimilarity among TRMs from various organs. Yet, all TRMs are expected to play a similar role in maintaining immune surveillance and behaving as sentinels when infections occur. Thus, despite their organ-specific environments, TRMs could potentially share some functions, particularly as sentinels during infections. (B) To which extent can result from tissue culture experiments be applied to TRMs’ functions in vivo? Majority of functional studies on TRMs have been performed in tissue culture or even with cell lines. It is not clear if ex vivo behaviors of TRMs reflect those in vivo. (C) Do human TRMs behave similarly to murine TRMs? Due to the technical limits to isolate TRMs from humans, a majority of TRM studies have been carried out by using animals. Therefore, it is again not clear if and to what extent TRMs from human and murine share similar responses. (D) Are TRMs involved in allowing fungi to switch from commensal/non-pathogenic to pathogenic? TRMs’ involvement in the switching might be possible because of the localization of TRMs in tissues where commensal fungi are homed. (E) Are TRMs heterogeneous if they are within a single organ? For example, the presence of microglia subsets has been identified (92, 93). It is intriguing to explore possible cellular subsets within TRMs in a single tissue and their possibly distinct functions. To answer the question, new technologies, such as single-cell sequencing or CyTOF would be very powerful tools to answer the question. (F) How can we “specifically” deplete a certain TRM population? One of the most significant technical challenges in studying TRMs may be depleting a certain population of TRMs. Clodronate-liposome is used to deplete TRMs, but it is not specific depletion. There are genetically modified mice and antagonists of certain receptors used to particularly deplete microglia. However, what is the best method to deplete AMs or Kupffer cells, for example? These are at least several questions and challenges to overcome to better understand TRMs in fungal infections and even other pathogenic conditions.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank M. Elizabeth Deerhake and William E. Barclay for critical reading and editing of the manuscript. This study was supported by an NIH grant to M.L.S. (R01-AI088100).
References
1. Cavaillon JM. The historical milestones in the understanding of leukocyte biology initiated by Elie Metchnikoff. J Leukoc Biol (2011) 90:413–24. doi:10.1189/jlb.0211094
2. Kaufmann SH. Immunology’s foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat Immunol (2008) 9:705–12. doi:10.1038/ni0708-705
3. van Furth R, Cohn ZA, Hirsch JG, Humphrey JH, Spector WG, Langevoort HL. The mononuclear phagocyte system: a new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ (1972) 46:845–52.
4. Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol (2005) 23:901–44. doi:10.1146/annurev.immunol.23.021704.115816
5. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol (2015) 33:643–75. doi:10.1146/annurev-immunol-032414-112220
6. Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell (2014) 159:1327–40. doi:10.1016/j.cell.2014.11.023
7. Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell (2014) 159:1312–26. doi:10.1016/j.cell.2014.11.018
8. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi:10.1126/science.1194637
9. Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science (2012) 336:86–90. doi:10.1126/science.1219179
10. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38:79–91. doi:10.1016/j.immuni.2012.12.001
11. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity (2014) 40:91–104. doi:10.1016/j.immuni.2013.11.019
12. Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med (2013) 210:1977–92. doi:10.1084/jem.20131199
13. Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi:10.1016/j.immuni.2015.03.011
14. Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity (2013) 38:792–804. doi:10.1016/j.immuni.2013.04.004
15. Bain CC, Bravo-Blas A, Scott CL, Gomez Perdiguero E, Geissmann F, Henri S, et al. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol (2014) 15:929–37. doi:10.1038/ni.2967
16. Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol (2012) 13:1118–28. doi:10.1038/ni.2419
17. Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell (2014) 157:832–44. doi:10.1016/j.cell.2014.04.016
18. Haldar M, Kohyama M, So Alex Y-L, Kc W, Wu X, Briseño Carlos G, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell (2014) 156:1223–34. doi:10.1016/j.cell.2014.01.069
19. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci (2014) 17:131–43. doi:10.1038/nn.3599
20. van de Laar L, Saelens W, De Prijck S, Martens L, Scott CL, Van Isterdael G, et al. Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity (2016) 44:755–68. doi:10.1016/j.immuni.2016.02.017
21. Buchanan KL, Murphy JW. What makes Cryptococcus neoformans a pathogen? Emerg Infect Dis (1998) 4:71–83. doi:10.3201/eid0401.980109
22. Hasenberg M, Behnsen J, Krappmann S, Brakhage A, Gunzer M. Phagocyte responses towards Aspergillus fumigatus. Int J Med Microbiol (2011) 301:436–44. doi:10.1016/j.ijmm.2011.04.012
23. Schaffner A, Douglas H, Braude A. Selective protection against conidia by mononuclear and against mycelia by polymorphonuclear phagocytes in resistance to Aspergillus. Observations on these two lines of defense in vivo and in vitro with human and mouse phagocytes. J Clin Invest (1982) 69:617–31. doi:10.1172/JCI110489
24. Shoham S, Levitz SM. The immune response to fungal infections. Br J Haematol (2005) 129:569–82. doi:10.1111/j.1365-2141.2005.05397.x
25. Kanayama M, Inoue M, Danzaki K, Hammer G, He YW, Shinohara ML. Autophagy enhances NFkappaB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat Commun (2015) 6:5779. doi:10.1038/ncomms6779
26. Zhang L, Wang CC. Inflammatory response of macrophages in infection. Hepatobiliary Pancreat Dis Int (2014) 13:138–52. doi:10.1016/S1499-3872(14)60024-2
27. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity (2014) 40:642–56. doi:10.1016/j.immuni.2014.04.016
28. Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe (2009) 6:470–81. doi:10.1016/j.chom.2009.10.007
29. Lionakis MS, Swamydas M, Fischer BG, Plantinga TS, Johnson MD, Jaeger M, et al. CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J Clin Invest (2013) 123:5035–51. doi:10.1172/JCI71307
30. Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J Infect Dis (2014) 209:109–19. doi:10.1093/infdis/jit413
31. Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol (2008) 26:421–52. doi:10.1146/annurev.immunol.26.021607.090326
32. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11:762–74. doi:10.1038/nri3070
33. Griffin FM Jr. Roles of macrophage Fc and C3b receptors in phagocytosis of immunologically coated Cryptococcus neoformans. Proc Natl Acad Sci U S A (1981) 78:3853–7. doi:10.1073/pnas.78.6.3853
34. Zaragoza O, Taborda CP, Casadevall A. The efficacy of complement-mediated phagocytosis of Cryptococcus neoformans is dependent on the location of C3 in the polysaccharide capsule and involves both direct and indirect C3-mediated interactions. Eur J Immunol (2003) 33:1957–67. doi:10.1002/eji.200323848
35. McQuiston T, Luberto C, Del Poeta M. Role of sphingosine-1-phosphate (S1P) and S1P receptor 2 in the phagocytosis of Cryptococcus neoformans by alveolar macrophages. Microbiology (2011) 157:1416–27. doi:10.1099/mic.0.045989-0
36. Guillot L, Carroll SF, Homer R, Qureshi ST. Enhanced innate immune responsiveness to pulmonary Cryptococcus neoformans infection is associated with resistance to progressive infection. Infect Immun (2008) 76:4745–56. doi:10.1128/IAI.00341-08
37. Ibrahim-Granet O, Philippe B, Boleti H, Boisvieux-Ulrich E, Grenet D, Stern M, et al. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect Immun (2003) 71:891–903. doi:10.1128/IAI.71.2.891-903.2003
38. Dubourdeau M, Athman R, Balloy V, Huerre M, Chignard M, Philpott DJ, et al. Aspergillus fumigatus induces innate immune responses in alveolar macrophages through the MAPK pathway independently of TLR2 and TLR4. J Immunol (2006) 177:3994–4001. doi:10.4049/jimmunol.177.6.3994
39. Philippe B, Ibrahim-Granet O, Prevost MC, Gougerot-Pocidalo MA, Sanchez Perez M, Van der Meeren A, et al. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infect Immun (2003) 71:3034–42. doi:10.1128/IAI.71.6.3034-3042.2003
40. Madan T, Eggleton P, Kishore U, Strong P, Aggrawal SS, Sarma PU, et al. Binding of pulmonary surfactant proteins A and D to Aspergillus fumigatus conidia enhances phagocytosis and killing by human neutrophils and alveolar macrophages. Infect Immun (1997) 65:3171–9.
41. Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, et al. Aspergillus fumigatus triggers inflammatory responses by stage-specific beta-glucan display. PLoS Pathog (2005) 1:e30. doi:10.1371/journal.ppat.0010030
42. Zhang HJ, Qu JM, Shao CZ, Zhang J, He LX, Yuan ZH. Aspergillus fumigatus conidia upregulates NOD2 protein expression both in vitro and in vivo. Acta Pharmacol Sin (2008) 29:1202–8. doi:10.1111/j.1745-7254.2008.00860.x
43. Shah VB, Huang Y, Keshwara R, Ozment-Skelton T, Williams DL, Keshvara L. Beta-glucan activates microglia without inducing cytokine production in dectin-1-dependent manner. J Immunol (2008) 180:2777–85. doi:10.4049/jimmunol.180.5.2777
44. Maneu V, Yanez A, Murciano C, Molina A, Gil ML, Gozalbo D. Dectin-1 mediates in vitro phagocytosis of Candida albicans yeast cells by retinal microglia. FEMS Immunol Med Microbiol (2011) 63:148–50. doi:10.1111/j.1574-695X.2011.00829.x
45. Shah VB, Ozment-Skelton TR, Williams DL, Keshvara L. Vav1 and PI3K are required for phagocytosis of beta-glucan and subsequent superoxide generation by microglia. Mol Immunol (2009) 46:1845–53. doi:10.1016/j.molimm.2009.01.014
46. Maneu V, Noailles A, Megias J, Gomez-Vicente V, Carpena N, Gil ML, et al. Retinal microglia are activated by systemic fungal infection. Invest Ophthalmol Vis Sci (2014) 55:3578–85. doi:10.1167/iovs.14-14051
47. Goldman D, Song X, Kitai R, Casadevall A, Zhao ML, Lee SC. Cryptococcus neoformans induces macrophage inflammatory protein 1alpha (MIP-1alpha) and MIP-1beta in human microglia: role of specific antibody and soluble capsular polysaccharide. Infect Immun (2001) 69:1808–15. doi:10.1128/IAI.69.3.1808-1815.2001
48. Blasi E, Barluzzi R, Mazzolla R, Tancini B, Saleppico S, Puliti M, et al. Role of nitric oxide and melanogenesis in the accomplishment of anticryptococcal activity by the BV-2 microglial cell line. J Neuroimmunol (1995) 58:111–6. doi:10.1016/0165-5728(95)00016-U
49. Rambach G, Hagleitner M, Mohsenipour I, Lass-Florl C, Maier H, Wurzner R, et al. Antifungal activity of the local complement system in cerebral aspergillosis. Microbes Infect (2005) 7:1285–95. doi:10.1016/j.micinf.2005.04.014
50. Rambach G, Dum D, Mohsenipour I, Hagleitner M, Wurzner R, Lass-Florl C, et al. Secretion of a fungal protease represents a complement evasion mechanism in cerebral aspergillosis. Mol Immunol (2010) 47:1438–49. doi:10.1016/j.molimm.2010.02.010
51. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol (2015) 16:36–44. doi:10.1038/ni.3052
52. Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G. Pulmonary macrophages. Eur Respir J (1994) 7:1678–89.
53. Denning DW. Invasive aspergillosis. Clin Infect Dis (1998) 26:781–803; quiz 804–5. doi:10.1086/513943
54. Hohl TM, Feldmesser M. Aspergillus fumigatus: principles of pathogenesis and host defense. Eukaryot Cell (2007) 6:1953–63. doi:10.1128/EC.00274-07
56. Chelen CJ, Fang Y, Freeman GJ, Secrist H, Marshall JD, Hwang PT, et al. Human alveolar macrophages present antigen ineffectively due to defective expression of B7 costimulatory cell surface molecules. J Clin Invest (1995) 95:1415–21. doi:10.1172/JCI117796
58. Luo Y, Cook E, Fries BC, Casadevall A. Phagocytic efficacy of macrophage-like cells as a function of cell cycle and Fcgamma receptors (FcgammaR) and complement receptor (CR)3 expression. Clin Exp Immunol (2006) 145:380–7. doi:10.1111/j.1365-2249.2006.03132.x
59. Luo Y, Tucker SC, Casadevall A. Fc- and complement-receptor activation stimulates cell cycle progression of macrophage cells from G1 to S. J Immunol (2005) 174:7226–33. doi:10.4049/jimmunol.174.11.7226
60. Marr KA, Koudadoust M, Black M, Balajee SA. Early events in macrophage killing of Aspergillus fumigatus conidia: new flow cytometric viability assay. Clin Diagn Lab Immunol (2001) 8:1240–7. doi:10.1128/CDLI.8.6.1240-1247.2001
61. Braem SG, Rooijakkers SH, van Kessel KP, de Cock H, Wosten HA, van Strijp JA, et al. Effective neutrophil phagocytosis of Aspergillus fumigatus is mediated by classical pathway complement activation. J Innate Immun (2015) 7:364–74. doi:10.1159/000369493
62. Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F, et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med (2007) 204:793–804. doi:10.1084/jem.20061301
63. Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, et al. Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One (2011) 6:e15943. doi:10.1371/journal.pone.0015943
64. Dague E, Alsteens D, Latge JP, Dufrene YF. High-resolution cell surface dynamics of germinating Aspergillus fumigatus conidia. Biophys J (2008) 94:656–60. doi:10.1529/biophysj.107.116491
65. Gersuk GM, Underhill DM, Zhu L, Marr KA. Dectin-1 and TLRs permit macrophages to distinguish between different Aspergillus fumigatus cellular states. J Immunol (2006) 176:3717–24. doi:10.4049/jimmunol.176.6.3717
66. Braedel S, Radsak M, Einsele H, Latge JP, Michan A, Loeffler J, et al. Aspergillus fumigatus antigens activate innate immune cells via toll-like receptors 2 and 4. Br J Haematol (2004) 125:392–9. doi:10.1111/j.1365-2141.2004.04922.x
67. Mehrad B, Wiekowski M, Morrison BE, Chen SC, Coronel EC, Manfra DJ, et al. Transient lung-specific expression of the chemokine KC improves outcome in invasive aspergillosis. Am J Respir Crit Care Med (2002) 166:1263–8. doi:10.1164/rccm.200204-367OC
68. Jhingran A, Kasahara S, Shepardson KM, Junecko BA, Heung LJ, Kumasaka DK, et al. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog (2015) 11:e1004589. doi:10.1371/journal.ppat.1004589
69. Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J Infect Dis (2009) 200:647–56. doi:10.1086/600380
70. McQuiston TJ, Williamson PR. Paradoxical roles of alveolar macrophages in the host response to Cryptococcus neoformans. J Infect Chemother (2012) 18:1–9. doi:10.1007/s10156-011-0306-2
71. Osterholzer JJ, Milam JE, Chen GH, Toews GB, Huffnagle GB, Olszewski MA. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect Immun (2009) 77:3749–58. doi:10.1128/IAI.00454-09
72. Goodman JS, Kaufman L, Koenig MG. Diagnosis of cryptococcal meningitis. Value of immunologic detection of cryptococcal antigen. N Engl J Med (1971) 285:434–6. doi:10.1056/NEJM197108192850804
73. Gottfredsson M, Perfect JR. Fungal meningitis. Semin Neurol (2000) 20:307–22. doi:10.1055/s-2000-9394
74. Kleinschmidt-DeMasters BK. Central nervous system aspergillosis: a 20-year retrospective series. Hum Pathol (2002) 33:116–24. doi:10.1053/hupa.2002.30186
75. Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res (2006) 83:711–30. doi:10.1002/jnr.20767
76. Larsen PH, Holm TH, Owens T. Toll-like receptors in brain development and homeostasis. Sci STKE (2007) 2007:e47. doi:10.1126/stke.4022007pe47
77. Aguirre K, Miller S. MHC class II-positive perivascular microglial cells mediate resistance to Cryptococcus neoformans brain infection. Glia (2002) 39:184–8. doi:10.1002/glia.10093
78. Hill JO, Aguirre KM. CD4+ T cell-dependent acquired state of immunity that protects the brain against Cryptococcus neoformans. J Immunol (1994) 152:2344–50.
79. Zhou Q, Gault RA, Kozel TR, Murphy WJ. Protection from direct cerebral Cryptococcus infection by interferon-gamma-dependent activation of microglial cells. J Immunol (2007) 178:5753–61. doi:10.4049/jimmunol.178.9.5753
80. Kaissling B, Hegyi I, Loffing J, Le Hir M. Morphology of interstitial cells in the healthy kidney. Anat Embryol (Berl) (1996) 193:303–18. doi:10.1007/BF00186688
81. Hume DA, Gordon S. Mononuclear phagocyte system of the mouse defined by immunohistochemical localization of antigen F4/80. Identification of resident macrophages in renal medullary and cortical interstitium and the juxtaglomerular complex. J Exp Med (1983) 157:1704–9. doi:10.1084/jem.157.5.1704
82. Hoeffel G, Chen J, Lavin Y, Low D, Almeida Francisca F, See P, et al. C-Myb+ erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity (2015) 42:665–78. doi:10.1016/j.immuni.2015.03.011
83. Cao Q, Wang Y, Wang XM, Lu J, Lee VW, Ye Q, et al. Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol (2015) 26:349–63. doi:10.1681/ASN.2013121336
84. Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol (2014) 14:392–404. doi:10.1038/nri3671
85. Kim MG, Boo CS, Ko YS, Lee HY, Cho WY, Kim HK, et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant (2010) 25:2908–21. doi:10.1093/ndt/gfq183
86. Hochheiser K, Heuser C, Krause TA, Teteris S, Ilias A, Weisheit C, et al. Exclusive CX3CR1 dependence of kidney DCs impacts glomerulonephritis progression. J Clin Invest (2013) 123:4242–54. doi:10.1172/JCI70143
87. Lionakis MS, Lim JK, Lee CC, Murphy PM. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun (2011) 3:180–99. doi:10.1159/000321157
88. Spellberg B, Ibrahim AS, Edwards JE Jr, Filler SG. Mice with disseminated candidiasis die of progressive sepsis. J Infect Dis (2005) 192:336–43. doi:10.1086/430952
89. Lionakis MS. New insights into innate immune control of systemic candidiasis. Med Mycol (2014) 52:555–64. doi:10.1093/mmy/myu029
90. Hebecker B, Vlaic S, Conrad T, Bauer M, Brunke S, Kapitan M, et al. Dual-species transcriptional profiling during systemic candidiasis reveals organ-specific host-pathogen interactions. Sci Rep (2016) 6:36055. doi:10.1038/srep36055
91. Lionakis MS, Netea MG. Candida and host determinants of susceptibility to invasive candidiasis. PLoS Pathog (2013) 9:e1003079. doi:10.1371/journal.ppat.1003079
92. Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science (1988) 239:290–2. doi:10.1126/science.3276004
Keywords: tissue-resident macrophages, fungal infections, microglia, alveolar macrophages, Candida, Cryptococcus, Aspergillus
Citation: Xu S and Shinohara ML (2017) Tissue-Resident Macrophages in Fungal Infections. Front. Immunol. 8:1798. doi: 10.3389/fimmu.2017.01798
Received: 27 September 2017; Accepted: 30 November 2017;
Published: 12 December 2017
Edited by:
Amariliz Rivera, New Jersey Medical School, United StatesReviewed by:
Ilse Denise Jacobsen, Leibniz-Institut für Naturstoff-Forschung und Infektionsbiologie, Hans Knöll Institut, GermanyJoshua J. Obar, Dartmouth College, United States
George So Yap, Rutgers University, Unites States
Copyright: © 2017 Xu and Shinohara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mari L. Shinohara, bWFyaS5zaGlub2hhcmFAZHVrZS5lZHU=