 Spandan V. Shah
Spandan V. Shah Cordelia Manickam
Cordelia Manickam Daniel R. Ram
Daniel R. Ram R. Keith Reeves
R. Keith Reeves- Center for Virology and Vaccine Research, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Over the past several years, new populations of innate lymphocytes have been described in mice and primates that are critical for mucosal homeostasis, microbial regulation, and immune defense. Generally conserved from mice to humans, innate lymphoid cells (ILC) have been divided primarily into three subpopulations based on phenotypic and functional repertoires: ILC1 bear similarities to natural killer cells; ILC2 have overlapping functions with TH2 cells; and ILC3 that share many functions with TH17/TH22 cells. ILC are specifically enriched at mucosal surfaces and are possibly one of the earliest responders during viral infections besides being involved in the homeostasis of gut-associated lymphoid tissue and maintenance of gut epithelial barrier integrity. Burgeoning evidence also suggests that there is an early and sustained abrogation of ILC function and numbers during HIV and pathogenic SIV infections, most notably ILC3 in the gastrointestinal tract, which leads to disruption of the mucosal barrier and dysregulation of the local immune system. A better understanding of the direct or indirect mechanisms of loss and dysfunction will be critical to immunotherapeutics aimed at restoring these cells. Herein, we review the current literature on ILC with a particular emphasis on ILC3 and their role(s) in mucosal immunology and the significance of disrupting the ILC niche during HIV and SIV infections.
Introduction
Innate lymphoid cells (ILC) encompass a broad diversity of cell types including the nominal subtypes ILC1, ILC2, ILC3, and in some descriptions also include traditional natural killer (NK) cells and lymphoid tissue inducer cells, all of which arise from a common lymphoid progenitor. A common consensus in the field favors grouping of these cells based on the dependence on transcription factors, as well as by production of major cytokine classes (1–4). ILC1 and NK cells rely on the transcription factor T-bet and produce type I cytokines, such as IFN-γ and TNF-α, but notably ILC1 lack the complex cytotoxic functions inherent to NK cells. ILC2 are classified by their dependence on GATA3, and their production of IL-5 and IL-13 (5, 6), and finally ILC3 are generally identified by their dependence on RORγt and AHR, and secretion of IL-17 and IL-22 (7). Interestingly, through the expression of their respective cytokines and dependence on transcription factors for their development, the three ILC groups (1–3) show strong commonalities with TH1, TH2, and TH17/TH22 cells, respectively (8). It is important to note that the classification scheme remains somewhat fluid and grouping is not absolute, as NK cells and ILC1 do not always require T-bet (9), and ILC2 and ILC3 can both convert to ILC1 (10), underscoring the inherent plasticity of these cell types. To further complicate matters, innate subsets of lymphoid cells may also include mucosal-associated invariant T cells (11), which express a semi-invariant T cell receptor and defined phenotypically as CD3+Vα7.2 TCR+CD161high cells in humans (12, 13). In addition to their cellular and functional plasticity, ILC have a wide tissue distribution and thus are thought to be some of the earliest responders to infections and other inflammatory stimuli, but the full mechanisms involved are still poorly understood. Striking observations have revealed that lentiviral infection leads to the depletion of functional ILC3 in gut mucosae (14–16), and increased microbial translocation from the gut lumen and an overt disruption of epithelial tissue integrity in HIV+ individuals is linked to a massive loss of IL-17-producing gut-resident lymphocytes (17). It is now becoming increasingly clear that reduced IL-17 and IL-22 production during infection cannot be attributed solely to the loss of TH17/TH22 cells and that early depletion of ILC may also contribute to this process.
ILC Phenotypes and Distribution
Although ILC are typified by their unique plasticity and their descriptive definitions are somewhat fluid, some generally accepted phenotypic nomenclatures have been established. ILC are usually identified as negative for common lymphocyte lineage markers (Lin−) that are otherwise distinct from NK cells and can usually be distinguished as such by higher expression of the IL-7 receptor, CD127 (1–4). However, even these definitions can vary significantly as “Lin” markers differ depending on the animal species (Table 1). For instance, in mice the Lin group may include CD3, CD4, CD8, CD11b, CD11c, CD14, CD19, B220, FcεRI, TER119 antigen, and GR1, whereas in humans, the Lin group may include CD1a, CD3, CD11c, CD14, CD16, CD19, CD34, CD123, TCRαβ, TCRγδ, BDCA2, and FcεRI. Burkhard et al. (18) recommends using CD5 marker in order to exclude small levels of contaminating T cells, especially for analyzing ILC3 populations. Regardless, these exclusion criteria remove T, B, NK, and dendritic cells, as well as other myeloid/granulocyte-derived cells and stem cells. ILC in rhesus macaque models align most closely to patterns seen in humans, but partly due to variability in cross-reactive reagents, may be more simply defined by excluding CD3, FcεRI, CD14, CD20, and NK cell-related markers, such as NKG2A or NKp46 (15, 16, 19). It is also important to note that exclusion of Lin markers may vary significantly between laboratories. Several other factors are used to characterize ILC, including the presence of various cytokines mentioned above and utilization of key transcription factors and receptors (1–5, 20). The co-expression of NKp46 and NK1.1 classifies mouse ILC1 subsets including related NK cells from other ILC groups whereas the expression of transcription factors, namely T-bet and Eomes, can be used to distinguish ILC1 and NK cells from each other (21–23). Loosely, in mice, NK cells are T-bet+Eomes+ while ILC1 are T-bet+Eomes− cells, although exceptions to this classification occur (9). Based on the nomenclature proposed by different reports (1, 2), ILC1 can be more comprehensively phenotyped as Lin−CD127+RORγt−T-bet+IL-1R+ cells in mice, and Lin−CD127+ICOS+RORγt−T-bet+IL-1R+ cells in humans. ILC2 are described as Lin−CD25+CD127+ICOS+THY1+SCA1+ST2+ cells in mice and Lin−CD25loCD127+CD161+ICOS+CRTH2+ST2+ in humans. Similarly, ILC3 may be identified as Lin−CD25+CD127+CD117+THY1+NKp46+/−RORγt+IL-1R+ in mice and Lin−CD127+CD161+/−CD117+NKp46+/−NKp44+/−RORγt+IL-1R+IL-23R+ in humans. ILC may also be partially identified by receptors of cytokines to which they are responsive—IL-12Rβ2+ (ILC1), IL-17RB+ (ILC2), and IL-23R+ (ILC3), but due to issues with antibody specificity may best be shown molecularly or in functional assays. Collectively, these phenotypic descriptions of ILC populations continue to evolve, and while there is generally a good consensus about the definition of ILC2 and ILC3, what truly defines ILC1 is still somewhat unclear. Currently there are no unique markers or complete phenotypes that uniquely identify ILC1, and the field is still limited to their identification via exclusion criteria—i.e., cells that are not NK cells, ILC2, or ILC3. Functionally ILC1 are identified as IFN-γ-producing cells that are distinct from NK cells through their low cytotoxic potential. Understandably, these factors make the study of ILC1 particularly cumbersome. Indeed, a recent profiling of ILC across tissues using mass spectrometry by Simoni et al. (24) indicated lack of ILC1 as described previously by other groups (25, 26). Instead, they described a unique intra-epithelial ILC1-like cells (ieILC1) that matched the description by Fuchs et al. (27).
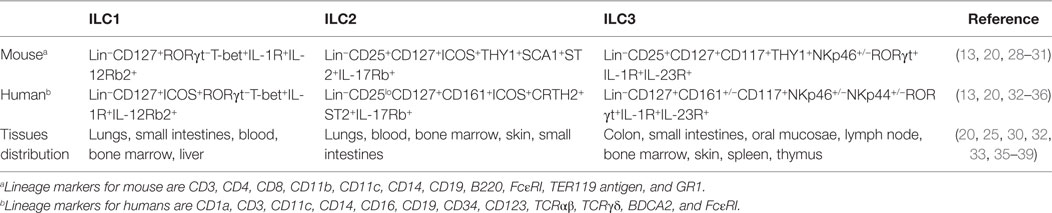
Table 1. Phenotypic markers and tissue distribution for innate lymphoid cell (ILC) groups.
Although ILC are generally found systemically, they are disparately distributed by subpopulation and are particularly enriched in mucosal sites and secondary lymphoid organs (Table 1). ILC have been identified in the lungs (ILC1, ILC2), colon (ILC3), small intestine (ILC1, ILC3), oral mucosae (ILC3), as well as in bone marrow, blood (ILC1, ILC2), lymph nodes (ILC3), liver (ILC1), and even in embryonic tissues (40), although the ILC-related NK cells tend to be much more broadly distributed (8). How ILC populations are maintained and replenished is unfortunately not well defined. Tissue-resident ILC predominantly replenish by self-renewal (40), though evidence suggests that common precursor cells from the bone marrow, or elsewhere, may also contribute to ILC homeostasis via cell recruitment (22, 41).
Role of ILC in Gastrointestinal (GI)-Related Diseases and Repair
Innate lymphoid cell populations are constitutively present in the GI tract and lymphoid tissues but differ in their compartmental distribution (42, 43). In healthy humans, ILC1 are the major population in the upper compartment of the GI tract while ILC3 are elevated in ileum and colon (44). The local distribution of ILC within the GI tract also differ—ILC1 predominate the intra-epithelial compartment of the intestine (27, 41, 42), while ILC2 are present in fat-associated lymphoid clusters in the intestinal mesentery and in significant numbers in lamina propria of small intestine where ILC3 is the dominant population (44–46). ILC3 are also enriched in the isolated lymphoid follicles, crypto- patches, and perifollicular area of Peyer’s patches at steady state (20, 47).
In the healthy gut, ILC3 are thought to be one of the major cell populations contributing to overall homeostasis. This is, in part, because ILC3 produce large quantities of IL-22 and IL-17 (48), and directly interact with intestinal epithelial cells to maintain an intact barrier and modulate inflammation (49, 50). IL-22 protects intestinal epithelium from inflammation and promotes wound healing by inducing STAT-3 dependent increases in production of antimicrobials by epithelial cells and epithelial cell proliferation, thus maintaining barrier integrity (51–53). In a mouse model of dextran sulfate sodium-induced ulcerative colitis, microinjection-based gene delivery of IL-22 ameliorated local inflammation through activation of STAT-3 in colonic epithelial cells, stimulation of mucus production, and goblet cell restitution (54). In IL-22−/− mice, increased intestinal damage, bacterial burden, and mortality was observed on infection with Citrobacter rodentium (52), and in humans, IL-22 has been shown to protect intestinal epithelium in IBD (55). Specifically, IL-23 responsive, IL-17/22-producing ILC protected intestinal stem cells against intestinal inflammation leading to epithelial regeneration in graft versus host disease patients who underwent bone marrow transplantation (56).
Another mechanism by which ILC regulates intestinal homeostasis is through their interaction with the commensal and/or pathogenic microbiota (48, 50, 57). Several protective mechanisms exist in the gut for the containment of commensal bacteria within intestinal sites including tight epithelial junctions, production of mucus and antimicrobial peptides, and immunological mechanisms that include ILC- and IgA-mediated immune exclusion pathways (58–63). ILC3 prevent commensal bacterial dysbiosis by IL-22-mediated induction of antimicrobial proteins (RegIIIβ, RegIIIγ, and β-defensins), element-sequestering proteins (S100A8, S100A9, and lipocalin-2) and mucins in epithelial cells leading to a strengthened intestinal epithelial barrier (49, 64–66). For example, depletion of ILC in mice led to selective peripheral dissemination of a commensal bacteria originating from host lymphoid tissues, namely Alcaligenes spp. and alcaligene-specific immune responses were found to be associated with Crohn’s disease and Hepatitis C virus-infected patients (63, 64). Further, ILC3 also are involved in the formation of gut-associated lymphoid tissues (GALT), including cryptopatches and isolated lymphoid tissues, which are important for protection against pathogens and act as niche areas of symbiosis for colonizing commensal microbiota (63). In turn, microbial products and signals were also found to be necessary for epigenetic modifications of ILC contributing to their diversity, plasticity, and maintenance of intestinal homeostasis (48, 57). This was evidenced by a study conducted by Manuzak et al. (67), describing the beneficial effects of probiotic therapy in healthy rhesus macaques by toll-like receptor (TLR) mediated downregulation of intestinal inflammatory markers and elevated ILC3 and T-follicular helper cells in colon.
Innate lymphoid cell can also act as a first line of defense at mucosal portals of entry due to their rapid production of cytokines following initial exposure to pathogens and recruitment of other innate and adaptive cells to sites of infection. ILC1 produce IFN-γ and TNF-α, both of which are important in the control of infections by intracellular pathogens such as Toxoplasma gondii (41) and Listeria monocytogenes (68). Furthermore, mice deficient in ILC3 were susceptible to intestinal pathogens including Helicobacter spp. and Clostridium difficile (69, 70). In helminthic infections, IL-25-mediated activation of ILC2 promotes a TH2 response which is important for an effective elimination of parasites (71). IL-17 is essential for the control of Candida albicans infection suggesting the importance of ILC3 in protection against oropharyngeal candidiasis in mice (72).
Given their critical roles in maintaining mucosal homeostasis, altered frequency or function of ILC during chronic disease could contribute to exacerbated intestinal inflammation. Indeed, intestinal ILC1 are elevated in IBD (73, 74), and production of IFN-γ by IL-15-activated ILC1 may play a major role in the pathogenesis of celiac and Crohn’s disease (25, 75). ILC2 along with NKT cells can also promote IL-13-mediated inflammation in an oxazolone-induced model of colitis (76). Interestingly, IL-23 responsive ILC3 can play a pathogenic role in intestinal inflammation through the production of IL-17A and IFN-γ and are also increased in patients with IBD (25, 28, 73, 74, 77–79). Given the significant protective roles ILC mediate in the GI tract, it may be important to take into account various interactions with intestinal epithelium and microbiota in achieving a balance of positive and negative ILC-related functions.
Loss of ILC in Pathogenic Lentivirus Infections
One of the hallmarks of HIV and pathogenic SIV infection is early loss of gut integrity followed by massive and rapid translocation of microbial products from the lumen of the intestine into the lamina propria, blood, lymph nodes, and liver (80–83). Indeed circulating lipopolysaccharide (LPS), sCD14, and other microbial products are now well-established biomarkers for microbial translocation and immune stimulants associated with inflammation and chronic immune activation. Because ILC, particularly ILC3, play major roles in maintaining gut integrity, tissue modeling, and repair (53, 84–86), these cells are likely critical players in the pathophysiology of HIV/SIV disease. Initial work in SIV-infected rhesus macaques by our group and others showed that ILC3 are generally restricted to mucosal tissue, express high levels of RORγt, and produce IL-17 and IL-22 much like their human counterparts, but they are depleted or otherwise dysfunctional in infection (15, 16, 87). Specifically, we showed that even 1 week following SIV infection there was up to a threefold reduction in ILC3 in colon and fourfold to ninefold reduction in jejunum and ileum (19) and that this loss was maintained during chronic infection. Surprisingly, SIV viral loads did not correlate with the loss of ILC3 (19), nor were ILC3 infected in vivo (15).
Functionally, ILC3 from SIV-infected animals took on a more cytotoxic phenotype and produced greater quantities of TNF-α, IFN-γ, and MIP-1β, but reduced levels of IL-17 (14). This cytokine profile suggests lentivirus infection may drive ILC3 plasticity toward ILC1, as has been previously described for mice (10). Similarly, a study by Xu et al. (16) clarified the kinetic changes in IL-17-producing ILC3 from intestinal epithelium by showing a reduction during acute pathogenic SIV infection (7–14 days postinfection) is followed by an increase in the total numbers of ILC (14–21 days postinfection) and eventually a gradual decline of ILC3 with disease progression after 28 days postinfection (16). Klatt et al., (87) also noted a significant depletion of all IL-17-producing lymphoid cells in rhesus macaques, but not in sooty mangabeys, where SIV replicates efficiently but does not cause significant mucosal barrier damage. This observation further underscores a potential role for ILC3 in maintaining gut homeostasis in HIV/SIV infections. Work in an HIV model of humanized mice by Zhang et al. (88) showed that persistent HIV-1 infection depleted ILC3 but effective antiretroviral therapy reversed this loss.
In human subjects, Kloverpris et al. (89) found that all three subgroups of ILC in blood were depleted during infection, but early administration of ART restored all ILC subsets. However, if ART was not administered within 5–14 days after infection, only ILC3 were partially restored while ILC1 and ILC2 remained depleted. Much like had been shown in SIV-infected macaque models (14), ILC3 loss did not occur in tonsil or other oral mucosal tissues (89). Surprisingly, they did not detect a reduction of ILC numbers in the gut, and a similar observation was made by Fernandes et al. (90). Although ILC levels in the gut during acute infection were not measured. The reason for this discrepancy between these studies and multiple macaque studies are not clear, but could be species specific. This could also be the reason for the contrasting observations made by Liyanage et al. (91), suggesting no restoration of NKp44+ cells in the rectum after ART. More recently, a study by Kramer et al. (92) showed that intestinal ILC distribution is significantly perturbed in patients even on effective antiretroviral therapy and that levels of colonic ILC3 were inversely correlated to markers of microbial translocation.
One of the proposed mechanisms leading to mucosal inflammation in HIV infection is the interaction of viral envelope gp120 with polarized epithelial cells directly disrupting epithelial tight junctions (93–95). A closer look at the effect of viral infection on epithelial cells showed that HIV-1 directly reduces transepithelial resistance, a measure of epithelial cell monolayer integrity by 30–60% without affecting its viability (93). Furthermore, functions of tight junction proteins, such as claudin 1, 2, 4, occludin, and ZO-1, were also disrupted and significantly increased inflammatory cytokines, such as TNF-α, IL-6, MCP-1, and IL-1β (93). The resulting increase in cytokine production following T cell infection may also cause intestinal barrier breakdown [(96), reviewed in Ref. (97)].
The effect of HIV-2 on the other hand is less obvious. A previous study correlated both HIV-1 and HIV-2 with microbial translocation. However, a more recent study by Fernandes et al. (98) suggests no disruption of the epithelial tight junction by HIV-2 despite active replication. How ILC-mediated mucosal maintenance may differ in less pathogenic infections such as HIV-2 remains unstudied. Collectively, these data indicate that in both HIV-infected humans and pathogenic SIV-infected rhesus macaque models, ILC3 loss in the gut occurs early, is at least partially irreversible, and is linked to mucosal dysregulation and translocation of microbial products.
Mechanisms of ILC Loss in Pathogenic HIV/SIV Infections
While the loss of ILC during HIV/SIV infection is well established, multiple groups have pursued molecular and cellular mechanisms leading to this depletion. We had previously observed that the expression of IDO1, an enzyme upregulated during SIV infection [also observed in Ref. (99)] (Figure 1) correlates negatively with CD4+TH17 cells as well as ILC3 (15). In HIV, IDO has been implicated in immunosuppressive activity (100) and dysbiosis during disease progression (101). Although the source(s) of IDO1 are not totally clear, the ability of HIV-1 TAT to induce production of IDO catabolites by dendritic cells has been described previously (102). Interestingly, increased levels of IDO1 in the gut showed a negative correlation of CD4+ T cells and ILC3 but not with NK or CD8+ T cells (15). This suggested IDO1 expression could be a negative regulator of ILC3 but not other effector cells. Furthermore, we were able to confirm that IDO catabolites caused numerical and functional depletion of ILC3 through a similar mechanism described for TH17 cells (99). Increased apoptosis leading to massive loss in total numbers of ILC3 was observed; however, the loss was not due to direct infection as no detectable SIV RNA was present in these cells. This is not surprising, as ILC do not express receptors for SIV/HIV.
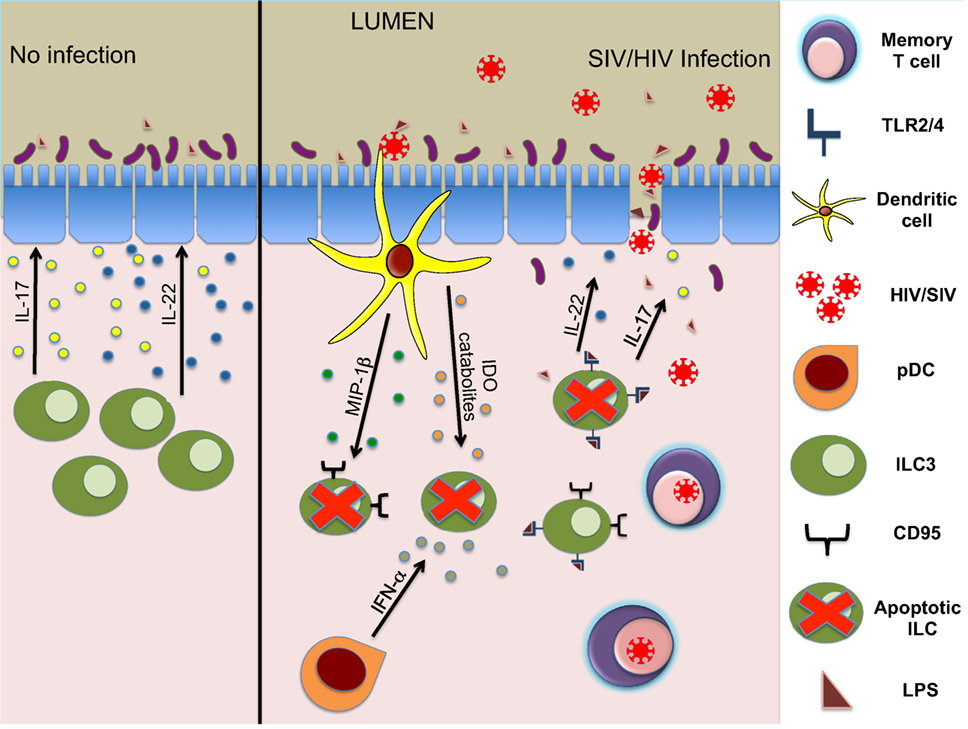
Figure 1. Mechanisms of ILC3 depletion during HIV/SIV infections. ILC3 modulate structure and homeostasis of gut epithelial cells via secretion of IL-17 and IL-22 (left panel). During the acute phase of lentivirus infection, early innate responders (DCs and other cells) secrete cytokines leading to apoptosis of ILC3. Subsequently, reduced IL-17 and IL-22 production leads to damage of gut epithelial barrier and an influx of microbial products, causing further inflammation (right panel).
Further studies indicated that the loss of ILC3 in the mucosae during acute infection was due to increased apoptosis and RORγt suppression induced by inflammatory cytokines, such as TGF-β, IL-2, IL-12, and IL-15 (19). In pathogenic SIV infection, we also showed previously that plasmacytoid DC (pDC) accumulates in the gut mucosa producing large quantities of IFN-α (103) (Figure 1). HIV-1 infection in a humanized mouse model and in vitro treatment of splenic ILC3 with IFN-α or HIV-1 significantly upregulated CD95 expression on ILC3 leading to apoptosis dependent on pDCs (88). RNA-seq analysis of ILC in human subjects with acute HIV-1 infection showed that there was a downregulation of genes associated with viability (89), and gene array analysis (87) showed that mucosal IL-17+ cells highly expressed TNF-receptor superfamily 4 (TNFRSF4, OX40), a co-stimulatory molecule involved in maintenance of mucosal lymphocytes, in comparison to IL-17− cells (104–106). Finally, ILC3 were shown to be depleted in lymphoid tissues mediated by TLRs in SIV-infected animals (107) (Figure 1). This study specifically showed that microbial translocation and resulting products like lipoteichoic acid or LPS via the TLR2/4 pathway can directly cause apoptosis in ILC3, further increasing HIV-induced disruption of GALT.
Altogether, these data indicated that a primary mechanism of ILC loss is likely apoptosis due to dysregulation of homeostatic elements on which ILC depend. One potential avenue that could be explored to restore ILC and gut integrity is IL-7-based therapies. Indeed in mice, IL-7 promoted IL-22 production during chronic LCMV infection (108); and in macaques, IL-7 therapy was shown to improve gut mucosal integrity in acute SIV-infected animals (109). Similarly, IL-7 immunotherapy in chronically infected HIV patients were associated with CD4+ T cell protective functions (108, 110, 111) and led to an overall reduced systemic inflammation (110). While these studies suggest that IL-7 plays a key role in repairing gut immunity, the precise connection to ILC is clearly understudied and needs further evaluation. Interestingly, it was also recently shown that SIV-ALVAC in combination with multiple adjuvants could induce an expansion of ILC3 (112). Whether or not this modality could be used therapeutically to restore ILC or could contribute to protective vaccine efficacy remains to be elucidated.
Conclusion and Perspectives
Innate lymphoid cell fill a unique and plastic niche of primarily tissue-resident cells that provide innate sources of typical T cell and NK cell produced cytokines, and although they clearly have a role in innate defense and homeostasis, many unknowns remain. Not the least of which being a recent report indicating that individuals lacking ILC may experience no obvious pathology as long as an intact T and B cell compartment remains (113). Specifically, regarding lentivirus infections, infection itself is not the source of depletion, but rather indirect or direct apoptosis, and while some potential mechanisms have been described herein this list is unlikely exhaustive or complete. It is also important to note that in several HIV studies no ILC depletion is observed in the gut. Regardless, whether loss is a virus-mediated subversion or an off-target effect of massive inflammation is unclear, and although ILC3 seemingly mediate gut homeostasis, their exact roles, both kinetically and functionally, in the perturbation and subsequent microbial translocation following HIV and pathogenic SIV infections are not obvious. And given the tight reciprocal relationship between gut microflora and ILC3 in mice, it will be interesting to determine if ILC3 depletion also contributes to dysbiosis. Direct evidence for these phenomena will need to be confirmed by in vivo depletion strategies in macaques, should those reagents become available. Further, HIV/SIV clearly intersects with ILC3 but whether ILC2 and ILC1 also contribute against viral pathogenesis is less clear and will require further study. Nonetheless, despite a host of unknowns, the field as a whole can appreciate the novelty of these cell populations and conclude that manipulating ILC as early responders to infection could be an attractive target for multiple infectious as well as chronic conditions.
Author Contributions
SS performed most of the writing and designed the figure. CM and DR contributed to writing of specific sections. RKR oversaw overall preparation of the manuscript, contributed to writing, and edited the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The effort of the authors on this work was support by NIH grants R01 DE026327, R01 AI120828, and R21 AI120795 (all to RKR).
References
1. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells – a proposal for uniform nomenclature. Nat Rev Immunol (2013) 13(2):145–9. doi:10.1038/nri3365
2. Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells – how did we miss them? Nat Rev Immunol (2013) 13(2):75–87. doi:10.1038/nri3349
3. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol (2016) 17(7):758–64. doi:10.1038/ni.3482
4. Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol (2016) 17(7):765–74. doi:10.1038/ni.3489
5. Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity (2012) 37(4):634–48. doi:10.1016/j.immuni.2012.06.020
6. Zhou L. Striking similarity: GATA-3 regulates ILC2 and Th2 cells. Immunity (2012) 37(4):589–91. doi:10.1016/j.immuni.2012.10.002
7. Eberl G, Colonna M, Di Santo JP, McKenzie AN. Innate lymphoid cells: a new paradigm in immunology. Science (2015) 348(6237):aaa6566. doi:10.1126/science.aaa6566
8. Fuchs A. ILC1s in tissue inflammation and infection. Front Immunol (2016) 7:104. doi:10.3389/fimmu.2016.00104
9. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity (2014) 41(3):354–65. doi:10.1016/j.immuni.2014.09.005
10. Belz GT. ILC2s masquerade as ILC1s to drive chronic disease. Nat Immunol (2016) 17(6):611–2. doi:10.1038/ni.3467
11. Gapin L. Where do MAIT cells fit in the family of unconventional T cells? PLoS Biol (2009) 7(3):e70. doi:10.1371/journal.pbio.1000070
12. Ishimori A, Harada N, Chiba A, Harada S, Matsuno K, Makino F, et al. Circulating activated innate lymphoid cells and mucosal-associated invariant T cells are associated with airflow limitation in patients with asthma. Allergol Int (2017) 66(2):302–9. doi:10.1016/j.alit.2016.07.005
13. Verykokakis M, Zook EC, Kee BL. ID’ing innate and innate-like lymphoid cells. Immunol Rev (2014) 261(1):177–97. doi:10.1111/imr.12203
14. Li H, Reeves RK. Functional perturbation of classical natural killer and innate lymphoid cells in the oral mucosa during SIV infection. Front Immunol (2013) 3:417. doi:10.3389/fimmu.2012.00417
15. Reeves RK, Rajakumar PA, Evans TI, Connole M, Gillis J, Wong FE, et al. Gut inflammation and indoleamine deoxygenase inhibit IL-17 production and promote cytotoxic potential in NKp44+ mucosal NK cells during SIV infection. Blood (2011) 118(12):3321–30. doi:10.1182/blood-2011-04-347260
16. Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol (2012) 5(6):658–69. doi:10.1038/mi.2012.39
17. Klatt NR, Funderburg NT, Brenchley JM. Microbial translocation, immune activation, and HIV disease. Trends Microbiol (2013) 21(1):6–13. doi:10.1016/j.tim.2012.09.001
18. Burkhard SH, Mair F, Nussbaum K, Hasler S, Becher B. T cell contamination in flow cytometry gating approaches for analysis of innate lymphoid cells. PLoS One (2014) 9(4):e94196. doi:10.1371/journal.pone.0094196
19. Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, et al. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog (2014) 10(12):e1004551. doi:10.1371/journal.ppat.1004551
20. Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol (2009) 10(1):75–82. doi:10.1038/ni.1681
21. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity (2012) 36(1):55–67. doi:10.1016/j.immuni.2011.11.016
22. Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. A committed precursor to innate lymphoid cells. Nature (2014) 508(7496):397–401. doi:10.1038/nature13047
23. Gronke K, Kofoed-Nielsen M, Diefenbach A. Innate lymphoid cells, precursors and plasticity. Immunol Lett (2016) 179:9–18. doi:10.1016/j.imlet.2016.07.004
24. Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, et al. Human innate lymphoid cell subsets possess tissue-type based heterogeneity in phenotype and frequency. Immunity (2017) 46(1):148–61. doi:10.1016/j.immuni.2016.11.005
25. Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol (2013) 14(3):221–9. doi:10.1038/ni.2534
26. Bernink JH, Krabbendam L, Germar K, de Jong E, Gronke K, Kofoed-Nielsen M, et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity (2015) 43(1):146–60. doi:10.1016/j.immuni.2015.06.019
27. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38(4):769–81. doi:10.1016/j.immuni.2013.02.010
28. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature (2010) 464(7293):1371–5. doi:10.1038/nature08949
29. Eberl G, Littman DR. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science (2004) 305(5681):248–51. doi:10.1126/science.1096472
30. Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity (2008) 29(6):958–70. doi:10.1016/j.immuni.2008.11.001
31. Halim TY, MacLaren A, Romanish MT, Gold MJ, McNagny KM, Takei F. Retinoic-acid-receptor-related orphan nuclear receptor alpha is required for natural helper cell development and allergic inflammation. Immunity (2012) 37(3):463–74. doi:10.1016/j.immuni.2012.06.012
32. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol (2011) 12(11):1045–54. doi:10.1031/ni.2131
33. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol (2011) 12(11):1055–62. doi:10.1038/ni.2104
34. Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity (2012) 37(4):649–59. doi:10.1016/j.immuni.2012.08.015
35. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature (2009) 457(7230):722–5. doi:10.1038/nature07537
36. Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. J Exp Med (2010) 207(2):281–90. doi:10.1084/jem.20091509
37. Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JP, Gudjonsdottir SD, Skov L, et al. Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Br J Dermatol (2014) 170(3):609–16. doi:10.1111/bjd.12658
38. Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol (2014) 134(9):2351–60. doi:10.1038/jid.2014.146
39. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A (2010) 107(24):10961–6. doi:10.1073/pnas.1005641107
40. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350(6263):981–5. doi:10.1126/science.aac9593
41. Klose CS, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell (2014) 157(2):340–56. doi:10.1016/j.cell.2014.03.030
42. Kim CH, Hashimoto-Hill S, Kim M. Migration and tissue tropism of innate lymphoid cells. Trends Immunol (2016) 37(1):68–79. doi:10.1016/j.it.2015.11.003
43. Kim M, Kim CH. Colonization and effector functions of innate lymphoid cells in mucosal tissues. Microbes Infect (2016) 18(10):604–14. doi:10.1016/j.micinf.2016.06.005
44. Spencer SP, Wilhelm C, Yang Q, Hall JA, Bouladoux N, Boyd A, et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science (2014) 343(6169):432–7. doi:10.1126/science.1247606
45. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013) 502(7470):245–8. doi:10.1038/nature12526
46. Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature (2010) 463(7280):540–4. doi:10.1038/nature08636
47. Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature (2013) 494(7436):261–5. doi:10.1038/nature11813
48. Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Berard M, Kleinschek M, et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol (2011) 12(4):320–6. doi:10.1038/ni.2002
49. Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature (2015) 528(7583):560–4. doi:10.1038/nature16460
50. Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity (2012) 37(4):601–10. doi:10.1016/j.immuni.2012.10.003
51. Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med (2009) 206(7):1465–72. doi:10.1084/jem.20082683
52. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med (2008) 14(3):282–9. doi:10.1038/nm1720
53. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol (2015) 33:747–85. doi:10.1146/annurev-immunol-032414-112123
54. Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest (2008) 118(2):534–44. doi:10.1172/JCI33194
55. Li LJ, Gong C, Zhao MH, Feng BS. Role of interleukin-22 in inflammatory bowel disease. World J Gastroenterol (2014) 20(48):18177–88. doi:10.3748/wjg.v20.i48.18177
56. Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, et al. Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity (2012) 37(2):339–50. doi:10.1016/j.immuni.2012.05.028
57. Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell (2016) 166(5):1231–46.e13. doi:10.1016/j.cell.2016.07.043
58. Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol (2010) 28:623–67. doi:10.1146/annurev-immunol-030409-101330
59. Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol (2010) 10(3):159–69. doi:10.1038/nri2710
60. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334(6053):255–8. doi:10.1126/science.1209791
61. Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson MA, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science (2009) 325(5940):617–20. doi:10.1126/science.1172747
62. Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol (2008) 6(10):776–88. doi:10.1038/nrmicro1978
63. Obata T, Goto Y, Kunisawa J, Sato S, Sakamoto M, Setoyama H, et al. Indigenous opportunistic bacteria inhabit mammalian gut-associated lymphoid tissues and share a mucosal antibody-mediated symbiosis. Proc Natl Acad Sci U S A (2010) 107(16):7419–24. doi:10.1073/pnas.1001061107
64. Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science (2012) 336(6086):1321–5. doi:10.1126/science.1222551
65. Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science (2014) 345(6202):1254009. doi:10.1126/science.1254009
66. Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol (2011) 12(5):383–90. doi:10.1038/ni.2025
67. Manuzak JA, Hensley-McBain T, Zevin AS, Miller C, Cubas R, Agricola B, et al. Enhancement of microbiota in healthy macaques results in beneficial modulation of mucosal and systemic immune function. J Immunol (2016) 196(5):2401–9. doi:10.4049/jimmunol.1502470
68. Reynders A, Yessaad N, Vu Manh TP, Dalod M, Fenis A, Aubry C, et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat- lymphoid cells. EMBO J (2011) 30(14):2934–47. doi:10.1038/emboj.2011.201
69. Powell N, Walker AW, Stolarczyk E, Canavan JB, Gokmen MR, Marks E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity (2012) 37(4):674–84. doi:10.1016/j.immuni.2012.09.008
70. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B, et al. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe (2015) 18(1):27–37. doi:10.1016/j.chom.2015.06.011
71. Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity (2014) 41(2):283–95. doi:10.1016/j.immuni.2014.06.016
72. Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol (2013) 190(2):521–5. doi:10.4049/jimmunol.1202924
73. Forkel M, Mjosberg J. Dysregulation of group 3 innate lymphoid cells in the pathogenesis of inflammatory bowel disease. Curr Allergy Asthma Rep (2016) 16(10):73. doi:10.1007/s11882-016-0652-3
74. Fuchs A, Colonna M. Innate lymphoid cells in homeostasis, infection, chronic inflammation and tumors of the gastrointestinal tract. Curr Opin Gastroenterol (2013) 29(6):581–7. doi:10.1097/MOG.0b013e328365d339
75. Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology (2011) 140(6):1756–67. doi:10.1053/j.gastro.2011.02.016
76. Camelo A, Barlow JL, Drynan LF, Neill DR, Ballantyne SJ, Wong SH, et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol (2012) 47(11):1198–211. doi:10.1007/s00535-012-0591-2
77. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med (2011) 208(6):1127–33. doi:10.1084/jem.20101712
78. Cox JH, Kljavin NM, Ota N, Leonard J, Roose-Girma M, Diehl L, et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol (2012) 5(1):99–109. doi:10.1038/mi.2011.54
79. Eken A, Singh AK, Treuting PM, Oukka M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol (2014) 7(1):143–54. doi:10.1038/mi.2013.33
80. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol (2003) 21:335–76. doi:10.1146/annurev.immunol.21.120601.141126
81. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog (2010) 6(8):e1001052. doi:10.1371/journal.ppat.1001052
82. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
83. Evans TI, Li H, Schafer JL, Klatt NR, Hao XP, Traslavina RP, et al. SIV-induced translocation of bacterial products in the liver mobilizes myeloid dendritic and natural killer cells associated with liver damage. J Infect Dis (2016) 213(3):361–9. doi:10.1093/infdis/jiv404
84. Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol (2011) 12(1):21–7. doi:10.1038/ni.1962
85. Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol (2012) 30:647–75. doi:10.1146/annurev-immunol-020711-075053
86. Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity (2011) 34(1):122–34. doi:10.1016/j.immuni.2010.12.009
87. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol (2012) 5(6):646–57. doi:10.1038/mi.2012.38
88. Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, et al. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J Clin Invest (2015) 125(9):3692–703. doi:10.1172/JCI82124
89. Kloverpris HN, Kazer SW, Mjosberg J, Mabuka JM, Wellmann A, Ndhlovu Z, et al. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity (2016) 44(2):391–405. doi:10.1016/j.immuni.2016.01.006
90. Fernandes SM, Pires AR, Ferreira C, Foxall RB, Rino J, Santos C, et al. Enteric mucosa integrity in the presence of a preserved innate interleukin 22 compartment in HIV type 1-treated individuals. J Infect Dis (2014) 210(4):630–40. doi:10.1093/infdis/jiu126
91. Liyanage NP, Gordon SN, Doster MN, Pegu P, Vaccari M, Shukur N, et al. Antiretroviral therapy partly reverses the systemic and mucosal distribution of NK cell subsets that is altered by SIVmac(2)(5)(1) infection of macaques. Virology (2014) 450–451:359–68. doi:10.1016/j.virol.2013.12.003
92. Kramer B, Goeser F, Lutz P, Glassner A, Boesecke C, Schwarze-Zander C, et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog (2017) 13(5):e1006373. doi:10.1371/journal.ppat.1006373
93. Nazli A, Chan O, Dobson-Belaire WN, Ouellet M, Tremblay MJ, Gray-Owen SD, et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog (2010) 6(4):e1000852. doi:10.1371/journal.ppat.1000852
94. Sufiawati I, Tugizov SM. HIV-associated disruption of tight and adherens junctions of oral epithelial cells facilitates HSV-1 infection and spread. PLoS One (2014) 9(2):e88803. doi:10.1371/journal.pone.0088803
95. Tugizov SM, Herrera R, Chin-Hong P, Veluppillai P, Greenspan D, Michael Berry J, et al. HIV-associated disruption of mucosal epithelium facilitates paracellular penetration by human papillomavirus. Virology (2013) 446(1–2):378–88. doi:10.1016/j.virol.2013.08.018
96. Redd AD, Dabitao D, Bream JH, Charvat B, Laeyendecker O, Kiwanuka N, et al. Microbial translocation, the innate cytokine response, and HIV-1 disease progression in Africa. Proc Natl Acad Sci U S A (2009) 106(16):6718–23. doi:10.1073/pnas.0901983106
97. Brenchley JM, Douek DC. The mucosal barrier and immune activation in HIV pathogenesis. Curr Opin HIV AIDS (2008) 3(3):356–61. doi:10.1097/COH.0b013e3282f9ae9c
98. Fernandes SM, Pires AR, Matoso P, Ferreira C, Nunes-Cabaco H, Correia L, et al. HIV-2 infection is associated with preserved GALT homeostasis and epithelial integrity despite ongoing mucosal viral replication. Mucosal Immunol (2017). doi:10.1038/mi.2017.44
99. Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med (2010) 2(32):32ra36. doi:10.1126/scitranslmed.3000632
100. Jenabian MA, El-Far M, Vyboh K, Kema I, Costiniuk CT, Thomas R, et al. Immunosuppressive tryptophan catabolism and gut mucosal dysfunction following early HIV infection. J Infect Dis (2015) 212(3):355–66. doi:10.1093/infdis/jiv037
101. Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med (2013) 5(193):193ra191. doi:10.1126/scitranslmed.3006438
102. Planes R, Bahraoui E. HIV-1 Tat protein induces the production of IDO in human monocyte derived-dendritic cells through a direct mechanism: effect on T cells proliferation. PLoS One (2013) 8(9):e74551. doi:10.1371/journal.pone.0074551
103. Reeves RK, Evans TI, Gillis J, Wong FE, Kang G, Li Q, et al. SIV infection induces accumulation of plasmacytoid dendritic cells in the gut mucosa. J Infect Dis (2012) 206(9):1462–8. doi:10.1093/infdis/jis408
104. Withers DR, Jaensson E, Gaspal F, McConnell FM, Eksteen B, Anderson G, et al. The survival of memory CD4+ T cells within the gut lamina propria requires OX40 and CD30 signals. J Immunol (2009) 183(8):5079–84. doi:10.4049/jimmunol.0901514
105. Griseri T, Asquith M, Thompson C, Powrie F. OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med (2010) 207(4):699–709. doi:10.1084/jem.20091618
106. Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, et al. Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol (2004) 172(6):3580–9. doi:10.4049/jimmunol.172.6.3580
107. Xu H, Wang X, Lackner AA, Veazey RS. Type 3 innate lymphoid cell depletion is mediated by TLRs in lymphoid tissues of simian immunodeficiency virus-infected macaques. FASEB J (2015) 29(12):5072–80. doi:10.1096/fj.15-276477
108. Pellegrini M, Calzascia T, Toe JG, Preston SP, Lin AE, Elford AR, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell (2011) 144(4):601–13. doi:10.1016/j.cell.2011.01.011
109. Ponte R, Rancez M, Figueiredo-Morgado S, Dutrieux J, Fabre-Mersseman V, Charmeteau-de-Muylder B, et al. Acute simian immunodeficiency virus infection triggers early and transient interleukin-7 production in the gut, leading to enhanced local chemokine expression and intestinal immune cell homing. Front Immunol (2017) 8:588. doi:10.3389/fimmu.2017.00588
110. Sereti I, Estes JD, Thompson WL, Morcock DR, Fischl MA, Croughs T, et al. Decreases in colonic and systemic inflammation in chronic HIV infection after IL-7 administration. PLoS Pathog (2014) 10(1):e1003890. doi:10.1371/journal.ppat.1003890
111. Vassena L, Miao H, Cimbro R, Malnati MS, Cassina G, Proschan MA, et al. Treatment with IL-7 prevents the decline of circulating CD4+ T cells during the acute phase of SIV infection in rhesus macaques. PLoS Pathog (2012) 8(4):e1002636. doi:10.1371/journal.ppat.1002636
112. Vaccari M, Gordon SN, Fourati S, Schifanella L, Liyanage NP, Cameron M, et al. Adjuvant-dependent innate and adaptive immune signatures of risk of SIVmac251 acquisition. Nat Med (2016) 22(7):762–70. doi:10.1038/nm.4105
Keywords: innate lymphoid cells, innate immunity, HIV infections, SIV, mucosal immunity
Citation: Shah SV, Manickam C, Ram DR and Reeves RK (2017) Innate Lymphoid Cells in HIV/SIV Infections. Front. Immunol. 8:1818. doi: 10.3389/fimmu.2017.01818
Received: 10 August 2017; Accepted: 04 December 2017;
Published: 13 December 2017
Edited by:
Persephone Borrow, University of Oxford, United KingdomReviewed by:
Alasdair Lelsie, Africa Health Research Institute, South AfricaJean-Pierre Routy, McGill University, Canada
Marina Cella, Washington University in St. Louis, United States
Neil Berry, National Institute for Biological Standards and Control (NIBSC), United Kingdom
Copyright: © 2017 Shah, Manickam, Ram and Reeves. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Keith Reeves, cnJlZXZlc0BiaWRtYy5oYXJ2YXJkLmVkdQ==