 Mizuha Kosugi-Kanaya1,2,3
Mizuha Kosugi-Kanaya1,2,3 Satoshi Ueha1,2*
Satoshi Ueha1,2* Jun Abe1,2
Jun Abe1,2 Shigeyuki Shichino1,2
Shigeyuki Shichino1,2 Francis H. W. Shand1,2Teppei Morikawa4Makoto Kurachi5
Francis H. W. Shand1,2Teppei Morikawa4Makoto Kurachi5 Yusuke Shono6Naoto Sudo1,2Ai Yamashita1,2
Yusuke Shono6Naoto Sudo1,2Ai Yamashita1,2 Fumiko Suenaga1,2Akihiro Yokoyama1,2Wang Yong1,2
Fumiko Suenaga1,2Akihiro Yokoyama1,2Wang Yong1,2 Masahiro Imamura7
Masahiro Imamura7 Takanori Teshima3
Takanori Teshima3 Kouji Matsushima1,2
Kouji Matsushima1,2
- 1Department of Molecular Preventive Medicine, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 2CREST, Japan Science and Technology Agency, Tokyo, Japan
- 3Department of Hematology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
- 4Department of Pathology, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 5Department of Microbiology, University of Pennsylvania School of Medicine, Philadelphia, PA, United States
- 6Department of Immunology, Memorial Sloan-Kettering Cancer Center, New York, NY, United States
- 7Department of Hematology, Sapporo Hokuyu Hospital, Sapporo, Japan
Chronic graft-versus-host disease (cGVHD) is a major complication in long-term survivors of allogeneic hematopoietic stem cell transplantation (allo-HSCT). Graft-derived T cells (TG) have been implicated in the induction of cGVHD; however, the extent of their contribution to the pathogenesis of cGVHD remains unclear. Using a mouse model of cGVHD, we demonstrate that TG predominate over hematopoietic stem cell-derived T cells generated de novo (THSC) in cGVHD-affected organs such as the liver and lung even at day 63 after allo-HSCT. Persisting TG, in particular CD8+ TG, not only displayed an exhausted or senescent phenotype but also contained a substantial proportion of cells that had the potential to proliferate and produce inflammatory cytokines. Host antigens indirectly presented by donor HSC-derived hematopoietic cells were involved in the maintenance of TG in the reconstituted host. Selective depletion of TG in the chronic phase of disease resulted in the expansion of THSC and thus neither the survival nor histopathology of cGVHD was ameliorated. On the other hand, THSC depletion caused activation of TG and resulted in a lethal TG-mediated exacerbation of GVHD. The findings presented here clarify the pathological role of long-lasting TG in cGVHD.
Introduction
Chronic graft-versus-host disease (cGVHD) remains a major cause of long-term morbidity, mortality, and poor quality of life following allogeneic hematopoietic stem cell transplantation (allo-HSCT) (1, 2). One hurdle in the development of preventive and therapeutic strategies against cGVHD is the complex origins and maintenance mechanisms of the pathogenic immune cells that are involved in this protracted disease. Despite clinical and experimental evidence that mature T cells in the graft (TG) are responsible for tissue injury during acute GVHD (aGVHD), the contribution of TG to the pathogenesis of cGVHD remains poorly understood.
After allo-HSCT, host-reactive TG are primed by host-type professional antigen-presenting cells (APCs) via the direct allo-recognition pathway in the secondary lymphoid organs, where they expand and differentiate into host-reactive effector TG (3, 4). Effector TG then redistribute to the target organs of aGVHD, such as the skin, liver, intestine, and hematopoietic and lymphoid tissues, where they impair host tissue function. Of note, an indirect allo-recognition pathway in which donor bone marrow (BM)-derived APCs repopulate peripheral tissues, uptake host-type antigens, and present them to donor T cells maximizes GVHD in a CD8-dependent GVHD model (5). Interestingly, in vivo early T cell depletion by antithymocyte globulin (6, 7) or post-transplant cyclophosphamide (8–10) reduces cGVHD rather than aGVHD. These clinical observations suggest a role for TG in the development of cGVHD, but this is difficult to examine in a human setting, and experimental evidence demonstrating the contribution of long-lasting TG to the development of cGVHD is lacking.
Since non-hematopoietic cells in the target organs remain as host-type even after allo-HSCT, TG are continuously exposed to cognate antigens, which theoretically induce deletion, anergy, or replicative senescence in TG (11, 12). In contrast to TG, which lack de novo replenishment, donor HSC-derived T cells (THSC) that have undergone thymic selection are continuously replenished from the thymus, and thus THSC rather than TG have been implicated in the pathogenesis of cGVHD (13–15). On the other hand, Hossain et al. (16) have shown that functional TG persisted up to 100 days after allo-HSCT in a cGVHD model and that the persisting TG confer protection against murine cytomegalovirus infection. This finding suggests that persisting TG could be a functional population with a role in the pathogenesis of cGVHD.
In the present study, using a minor-mismatched allo-HSCT model in which the GVHD recipients display histopathology characteristic of cGVHD, we characterized the kinetics, function, and antigen reactivity of TG and THSC. Selective depletion of TG or THSC in the chronic phase of disease revealed that persisting TG suppress the accumulation of THSC in cGVHD-affected organs, whereas THSC suppress the lethal activation of TG in affected organs.
Materials and Methods
Mice
Female C57BL/6J (B6; H-2b, CD45.2, Thy1.2) mice were purchased from CLEA Japan. C57BL/6J.SJL (B6.SJL; Ptprca Pepcb, CD45.1, Thy1.2), C3H.SW-H2b (B6; H-2b, CD45.2, Thy1.2), and C57BL/6J-Igha-Thy1a-Gpia (B6.Thy1a; CD45.2, Thy1.1) mice were purchased from the Jackson Laboratory. B6 background congenic strains were crossed in-house to obtain CD45.1+ CD45.2+ Thy1.2+ and CD45.1+ CD45.2+ Thy1.1+ congenic strains. All animal experiments were conducted in accordance with institutional guidelines with the approval of the Animal Care and Use Committee of the University of Tokyo.
Transplantation and Assessment of GVHD
Cell preparation and allo-HSCT were performed as described previously (17, 18) with some modifications. In brief, T-cell-depleted BM (TCD BM) was prepared by depleting Thy1+ mature T cells from BM using an autoMACS system (Miltenyi Biotec). Splenic T cells were negatively enriched from splenocytes by autoMACS, using antibodies against CD11b, CD11c, B220, Ter-119, NK1.1, and c-kit. Recipients were lethally irradiated (9 Gy, split into two doses given 3 h apart) on day −1, then injected intravenously with 5 × 106 TCD BM cells with or without 3–4 × 106 splenic T cells on day 0. The development of systemic GVHD was quantified by measuring weight loss and using a clinical GVHD scoring system, as described previously (19).
Histological Analyses
For the assessment of pathological changes in tissues, 4–6-µm formalin-fixed paraffin sections were stained with H&E and assessed by a pathologist (Teppei Morikawa; blinded to experimental group) using a scoring system described previously (20–22). For immunohistological analyses, the left lobes of the lung were inflated by infusion of 500 µl OCT compound intratracheally before lung tissue was harvested. Acetone-fixed 6- to 8-µm cryosections were incubated sequentially with primary antibodies and the appropriate fluorochrome-labeled secondary antibodies after blocking. Sections were mounted with Prolong Gold Antifade Reagent (Life Technologies) and visualized using an SP-5 confocal microscope (Leica Microsystems).
Cell Preparation
Single cell suspensions were prepared from the liver, lung, spleen, BM and thymus after systemic transcardial perfusion with PBS. Liver cells were prepared by pressing liver tissue through a 200-µm stainless steel mesh (23). The right lobe of lung tissue was cut into small fragments and digested for 1 h at 37°C with 0.2% collagenase D (Roche, Penzberg, Germany) and 2,000 U/ml DNase I (Calbiochem, La Jolla, CA, USA). BM cells were flushed from femurs using a needle and syringe. Spleen and thymus tissue were pressed through a 70-µm cell strainer. Non-hematopoietic cells and cell debris were removed by 40% Percoll (GE healthcare) phase separation, and erythrocytes were removed using ACK lysing buffer.
Flow Cytometry
Labeled and purified antibodies were purchased from BD Biosciences, BioLegend, or eBioscience (Table 1). Single cell suspensions were incubated sequentially with anti-CD16/32 (to block Fc receptors) then primary antibodies. Data were collected on a Gallios flow cytometer (Beckman Coulter) and analyzed using FlowJo software (Tree Star). For intracellular cytokine analysis, 5–10 × 105 leukocytes were restimulated with ionomycin (1 µg/ml) and PMA (25 ng/ml) in the presence of Brefeldin A (10 µg/ml) for 4.5–5 h at 37°C. Following staining for surface antigens, cells were stained for intracellular cytokines using a Cytofix/Cytoperm kit (BD Bioscience), according to the manufacturer’s instructions. For short-term pulse BrdU labeling, mice were injected intraperitoneally with 1 mg/mouse BrdU (Sigma-Aldrich) in 100 µl PBS 1 h before sacrifice. BrdU incorporation was examined using a BrdU flow kit (BD Biosciences), according to the manufacturer’s instructions. For the intracellular staining of Foxp3 and Ki-67, 1 × 106 leukocytes were stained for surface antigens and then were fixed and permeabilized with Foxp3 Fix/Perm Buffer (eBioscience) according to the manufacturer’s instructions.

Table 1. List of antibodies used for flow cytometry.
Ex Vivo and In Vivo Proliferation Assays
Chronic graft-versus-host disease was induced by transferring CD45.1+ Thy1.1+ TCD BM and CD45.1+ Thy1.2+ splenic T cells. Single cell suspensions of the liver and lung were prepared on day 49, and total T cells were enriched by negative selection with antibodies against CD11b, CD31, CD140a, CD326, B220, NK1.1, and Ter-119. TG and THSC were enriched from the T cell fraction using positive selection with antibodies against Thy1.2 and Thy1.1, respectively, and used as secondary donors in in vivo proliferation assays, or as responders in the ex vivo proliferation assay. Non-hematopoietic cells were prepared from the lung of untreated C3H.SW-H2b mice or day 49 BMT recipients by negative selection with antibodies against CD45 and Ter-119. CD3− NK1.1− hematopoietic cells were prepared from the spleen of untreated C3H.SW-H2b mice or day 49 BMT recipients by negative selection with antibodies against CD3 and NK1.1. In an ex vivo proliferation assay, TG and THSC were labeled with 5 µM CFSE and were cocultured with the non-hematopoietic cells or CD3− NK1.1− hematopoietic cells at a ratio of 1:10 for 3 days. In an in vivo proliferation assays, CFSE-labeled TG and THSC were equally mixed and transferred intravenously into day 49 BMT recipients reconstituted with CD45.2 TCD BM.
In Vivo T Cell Depletion
To deplete TG or THSC selectively, recipient mice expressing various combinations of the congenic markers Thy1.1 and Thy1.2 were injected intraperitoneally with purified rat IgG2b anti-mouse Thy1.2 mAb (clone 30H12, BioXcell) on days 21 (200 µg) and 24 (100 µg) after allo-HSCT, followed by weekly administration (200 µg) from days 31–63.
Statistical Analyses
All values are expressed as mean ± SEM. All data are representative of results obtained in at least two independent internally controlled experiments, where intergroup effects observed within each experiment were consistent across all experiments. Survival curves for GVHD were plotted using Kaplan–Meier estimates and compared by log-rank analysis (Prism version 5.0; GraphPad Prism Software). Comparisons between two groups were performed by unpaired two-tail Student’s t-tests. Multiple comparisons were performed by one-way ANOVA with Dunnett’s post-test. P-Values less than 0.05 were considered to be statistically significant.
Results
Establishment of a Clinically Relevant Model of cGVHD
We first established a B6→C3H.SW minor-mismatched cGVHD model to facilitate identification and manipulation of TG and THSC populations using CD45 and Thy1 congenic markers. Lethally irradiated C3H.SW recipients were transplanted with TCD BM alone (“BMT group”) or TCD BM with splenic T cells (“cGVHD group”) from B6 donors. In the cGVHD group, body weight loss and GVHD score became progressively worse from day 14 onward (Figures 1A,B), and cGVHD mice had a significantly lower survival rate (Figure 1C). At day 63, the histology of the salivary glands (SG), skin, liver, and lung revealed inflammatory cell infiltration and fibrotic changes, which are the most relevant histological features in the diagnosis of human cGVHD (Figures 1D,E) (24, 25). The pathological scores for these organs were higher in cGVHD mice compared to BMT control mice, reaching statistical significance in the SG and liver (Figure 1F). These results demonstrate that cGVHD mice in the B6→C3H.SW minor-mismatched allo-HSCT model develop pathology that recapitulates human cGVHD.
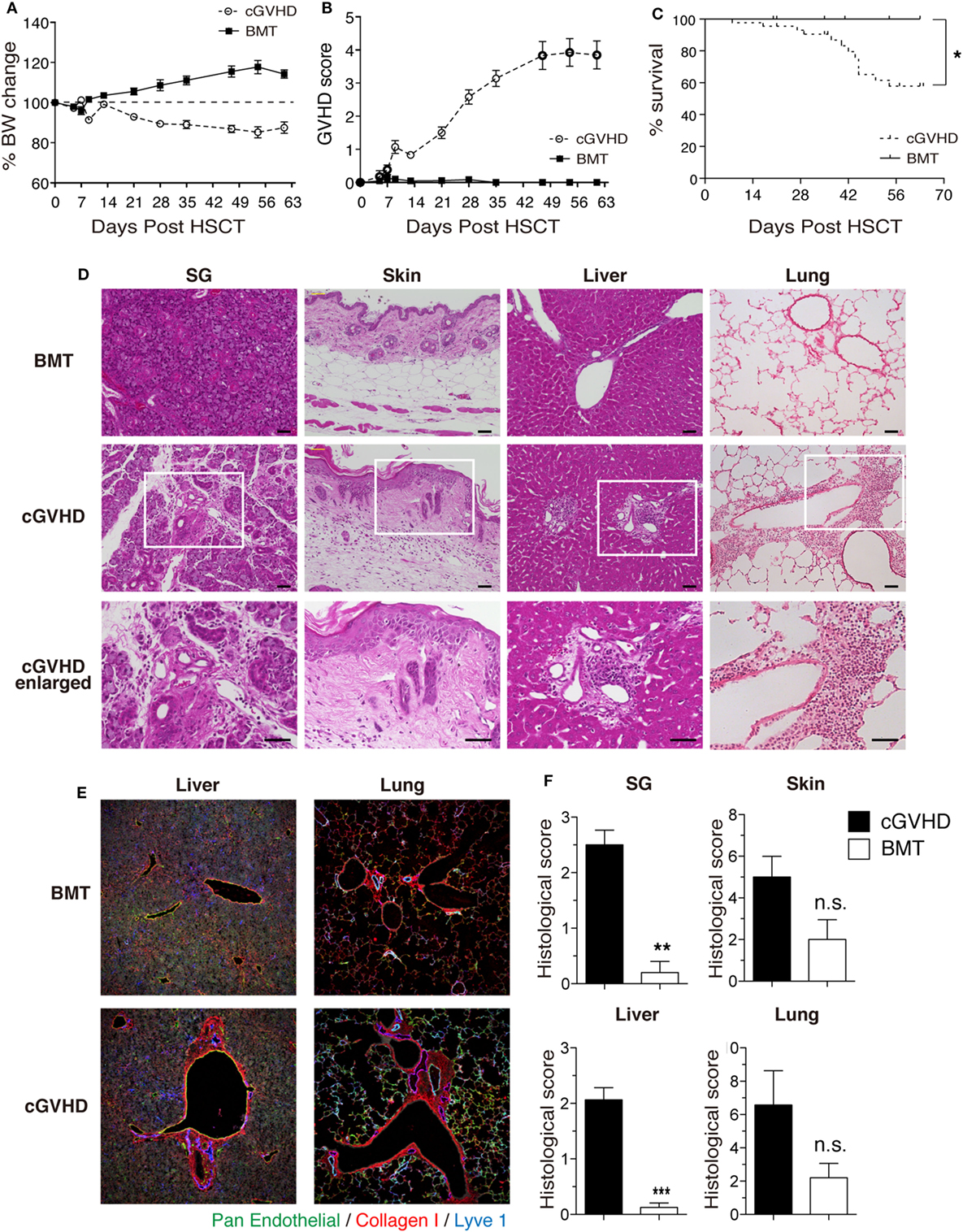
Figure 1. Establishment of a clinically relevant model of chronic graft-versus-host disease (cGVHD). Lethally irradiated C3H.SW mice were transplanted with T-cell-depleted bone marrow (TCD BM) alone or TCD BM plus 3–4 × 106 T cells from the spleens of B6 mice. (A) Body weight, (B) GVHD score, and (C) survival were monitored for 63 days after transplantation. Data are combined from three independent experiments (n = 21 for BMT control group; n = 29 (A,B); or n = 44 (C) for cGVHD group). (D) Salivary glands (SG), skin, liver, and lung were collected 63 days after transplantation for examination of pathology by HE staining. Scale bars, 50 µm. (E) Immunofluorescent staining of liver and lung sections. Green, pan-endothelial; red, collagen; and blue, Lyve1. (F) Histological scores from SG, skin, liver, and lung. Data are combined from two independent experiments (n = 5 for BMT control group; n = 8 for cGVHD groups). *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.001 (cGVHD vs. BMT).
TG Predominate over THSC in cGVHD-Affected Organs
We next examined the kinetics of TG and THSC in our cGVHD model using CD45 congenic markers (Figure 2A). The number of CD4+ CD8+ thymocytes of CD45.1+ CD45.2+ TCD BM origin (THSC) in cGVHD mice increased markedly between days 21 and 35 (Figure 2B). In peripheral tissues such as the liver, lung, and spleen, detectable numbers of THSC appeared from day 21 after HSCT, but within the CD8+ T cell populations of these tissues, TG outnumbered THSC approximately 10-fold for the duration of our experiments (Figure 2C). A similar trend was observed in CD4+ T cell populations. Interestingly, TG were more abundant than THSC within CD4+ Foxp3+ regulatory T cells in the liver by day 35; however, there was a trend toward THSC dominance by day 63 (Figure 2D). Immunofluorescent staining of the liver showed that both Foxp3+ CD4+ TG and Foxp3+ CD4+ THSC were uniformly distributed throughout the T cell pool at day 63 (Figure 2E). These results suggest that TG rather than THSC persist as the dominant CD8+ T cell population, while THSC gradually become the dominant CD4+ T cell population, particularly within the Foxp3+ CD4+ regulatory T cell populations in cGVHD-affected organs.
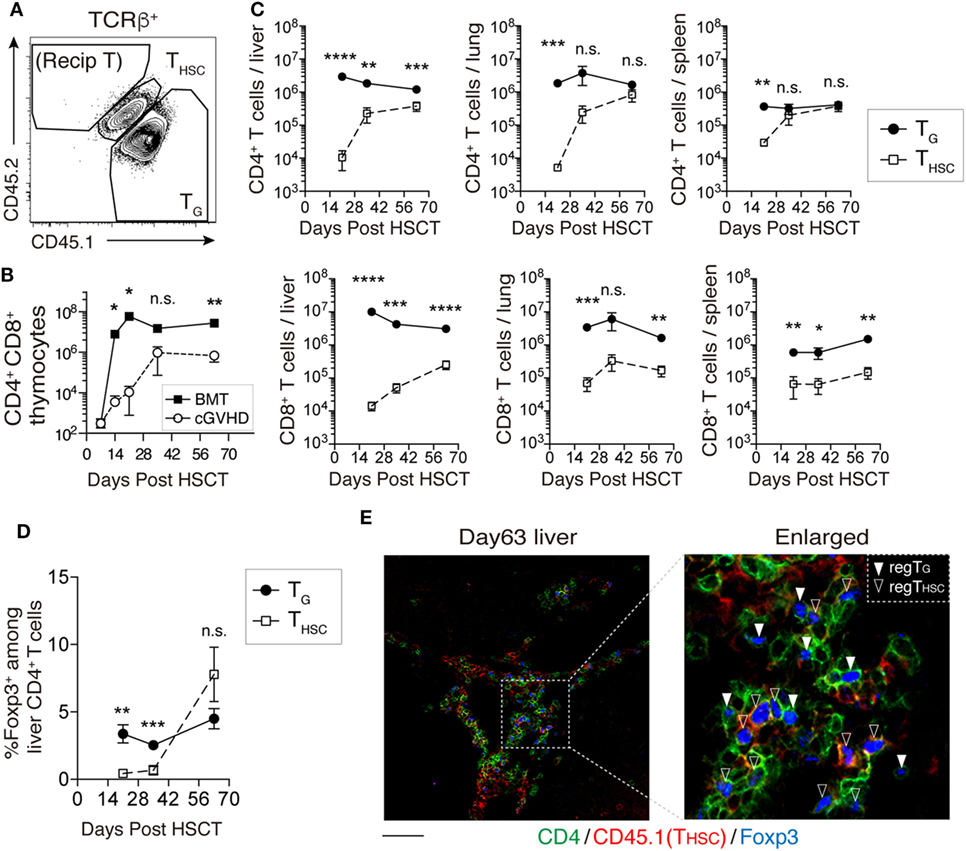
Figure 2. TG predominate over THSC in chronic graft-versus-host disease (cGVHD)-affected organs. CD45.1+ CD45.2+ T-cell-depleted bone marrow (TCD BM) and CD45.1+ CD45.2− spleen T cells from B6 donor mice were transferred into CD45.1− CD45.2+ C3H.SW-recipient mice. (A) Representative flow cytometry plot of TCRβ+ T cells showing how congenic markers were used to detect T cells from distinct origins. CD45.1+ CD45.2+ cells represent THSC cells derived from BM HSC. CD45.1+ CD45.2− T cells represent TG derived from mature T cells in the graft. (B) The number of CD4+ CD8+ thymocytes of CD45.1+ CD45.2+ TCD BM origin (THSC) was used as an indicator of immune reconstitution in the thymus. Representative data are shown from one of two independent experiments (n = 4). (C) Numbers of TG and THSC collected from the liver (per gram), lung, and spleen following allogeneic hematopoietic stem cell transplantation (allo-HSCT). (D) The proportion of Foxp3+ cells within the liver CD4+ cell population following allo-HSCT. Data represent mean ± SEM (n = 5–7 per time point) and are combined from two independent experiments. (E) CD45.1+ TCD BM and CD45.2+ spleen T cells from B6 donor mice were transferred into C3H.SW-recipient mice, and the localization of CD4+ Foxp3+ T cells of CD45.1+ (THSC) or CD45.1− (TG) origin in the liver was analyzed on day 63. Green, CD4; red, CD45.1; and blue, Foxp3. Scale bar, 50 µm. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; and ****P ≤ 0.0001 [cGVHD vs. BMT (B) or TG vs. THSC (C)].
TG Remain Functional during cGVHD
We next compared the phenotype and function of TG and THSC in the liver at day 63. Compared to their THSC counterparts, CD8+ TG expressed high levels of the exhaustion markers PD-1 and LAG-3, the senescence/terminal differentiation marker KLRG-1 (11, 26), and low levels of IL-7Rα, which is inversely correlated with alloreactivity (27) (Figure 3A). Expression levels of the early activation marker CD69 were comparable between TG and THSC (Figure 3A). However, we did not observe significant differences in surface marker expression between CD4+ TG and CD4+ THSC (Figure 3B). Upon ex vivo stimulation of liver-infiltrating T cells with PMA and ionomycin, a substantial proportion of CD8+ TG produced IFNγ and TNFα at levels that were equal to those produced by their THSC counterparts (Figure 3C). Because TG far outnumbered THSC in cGVHD-affected organs, the majority of cells with potential to produce inflammatory cytokines in cGVHD-affected organs were of TG origin (Figure 3D). A similar trend was observed for CD4+ TG and THSC (Figures 3C,D). These results suggest that a substantial proportion of CD4+ and CD8+ TG persist as a functional population in the target organs of cGVHD.
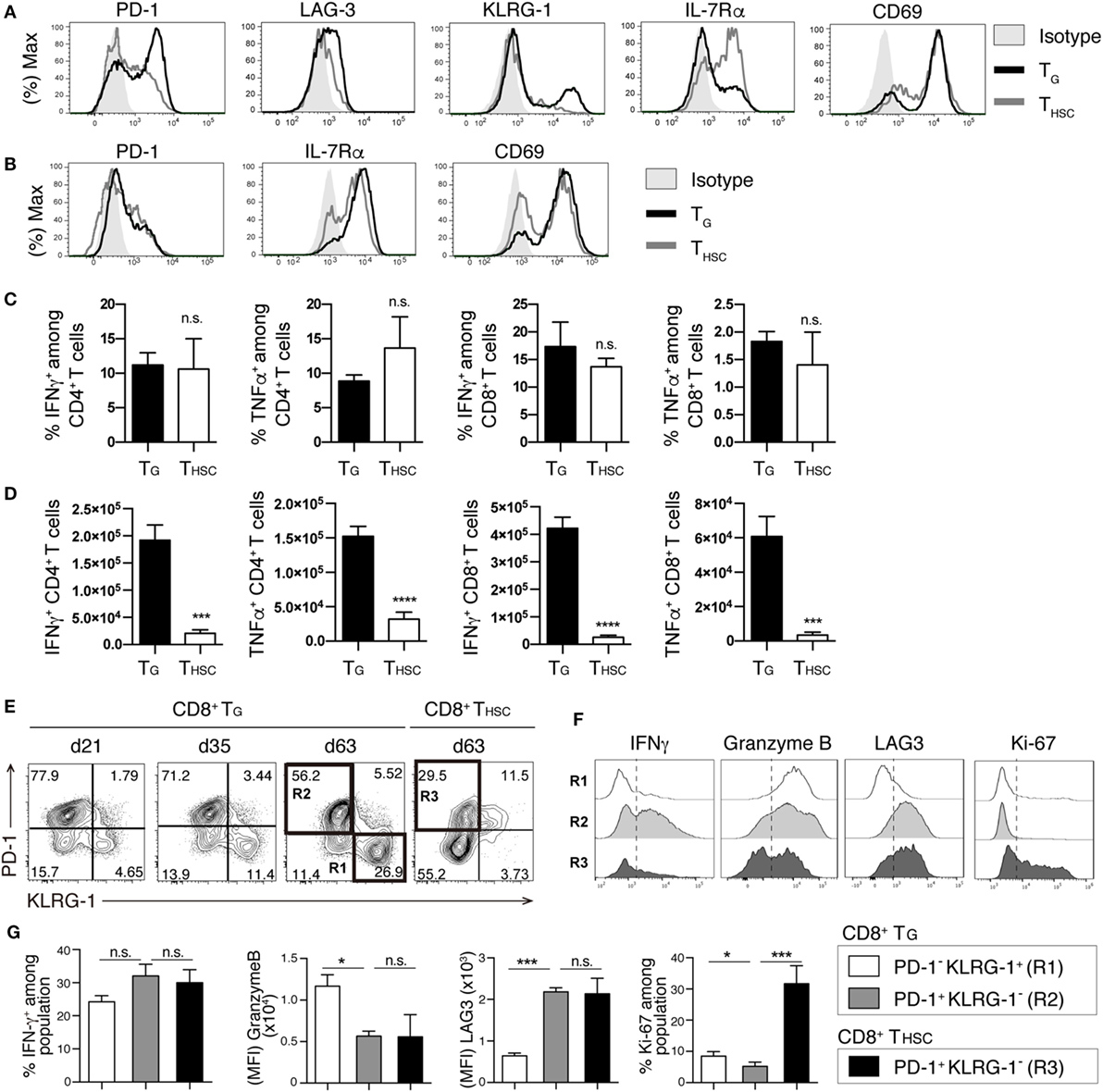
Figure 3. TG remain functional during chronic graft-versus-host disease (cGVHD). T cells were collected from the liver 63 days after allogeneic hematopoietic stem cell transplantation (allo-HSCT). Immunophenotype of CD8+ (A) or CD4+ (B) TG and THSC from cGVHD recipients. Representative data are shown from one of two independent experiments (n = 6). (C,D) Liver T cells were restimulated with PMA and ionomycin ex vivo and were analyzed for the intracellular IFNγ and TNFα 63 days after allo-HSCT. (C) The percentage of IFNγ+ or TNFα+ cells within the TG or THSC populations and (D) the absolute numbers of these cells. (E) Time-course analyses of PD-1 and KLRG-1 expression on liver-infiltrating TG and THSC. (F,G) Populations R1, R2, and R3 from (E) were analyzed for the expression of IFNγ, Granzyme B, LAG-3, and Ki-67. Representative plots are shown from one of two independent experiments (n = 6). Data represent mean ± SEM (n = 6) from one of two independent experiments. *P ≤ 0.05; ***P ≤ 0.001; and ****P ≤ 0.0001.
Interestingly, the proportion of PD-1− KLRG-1+ cells within the CD8+ TG population increased between day 21 and day 63 (Figure 3E). To examine whether these PD-1− KLRG-1+ cells are functionally distinct from the PD-1+ KLRG-1− cells that make up the majority of CD8+ TG in early phase, we analyzed the expression of functional molecules and the proliferation marker Ki-67 by these two populations at day 60. The PD-1+ KLRG-1− population displayed higher IFNγ production and LAG3 expression than the PD-1− KLRG-1+ population, whereas the PD-1− KLRG-1+ population displayed higher granzyme B and Ki-67 expression than the PD-1+ KLRG-1− population (Figures 3F,G). We also compared the functional profiles of the PD-1+ KLRG-1− populations from CD8+ TG and THSC. Although expression levels of LAG3 and granzyme B were equivalent between TG and THSC, PD-1+ KLRG-1− THSC had lower IFNγ production and significantly higher expression of Ki-67 than their TG counterparts (Figure 3G). These results suggest that among CD8+ TG the PD-1+ KLRG-1− and PD-1− KLRG-1+ populations are functionally distinct and that CD8+ THSC have a higher proliferating potential than CD8+ TG even when PD-1 expression is equivalent.
Active Proliferation Contributes to the Maintenance of TG during the Chronic Phase
We next investigated the proliferative responses of TG and THSC by 1 h in vivo BrdU labeling on day 63 after allo-HSCT. The proportion of proliferating (BrdU+) cells among liver-infiltrating CD4+ T cells was significantly lower within the TG population than in the THSC population, whereas there was no significant difference in the proportion of BrdU+ CD8+ T cells (Figure 4A). However, in terms of cell number, CD4+ BrdU+ TG were present in equivalent numbers to CD4+ BrdU+ THSC, while BrdU+ CD8+ TG far outnumbered BrdU+ CD8+ THSC (Figure 4B). To further investigate the proliferative potential of TG and THSC, we prepared TG and THSC populations from the liver and lung of cGVHD mice and adoptively transferred them into phase-matched BMT recipients on day 49 after allo-HSCT (Figure 4C). A CFSE-dilution assay demonstrated that a significant proportion of CD4+ and CD8+ TG, and an even greater proportion of THSC proliferated 3 days after transfer into secondary BMT recipients (Figures 4D,E). These results demonstrate that TG maintain proliferative potential during the chronic phase.
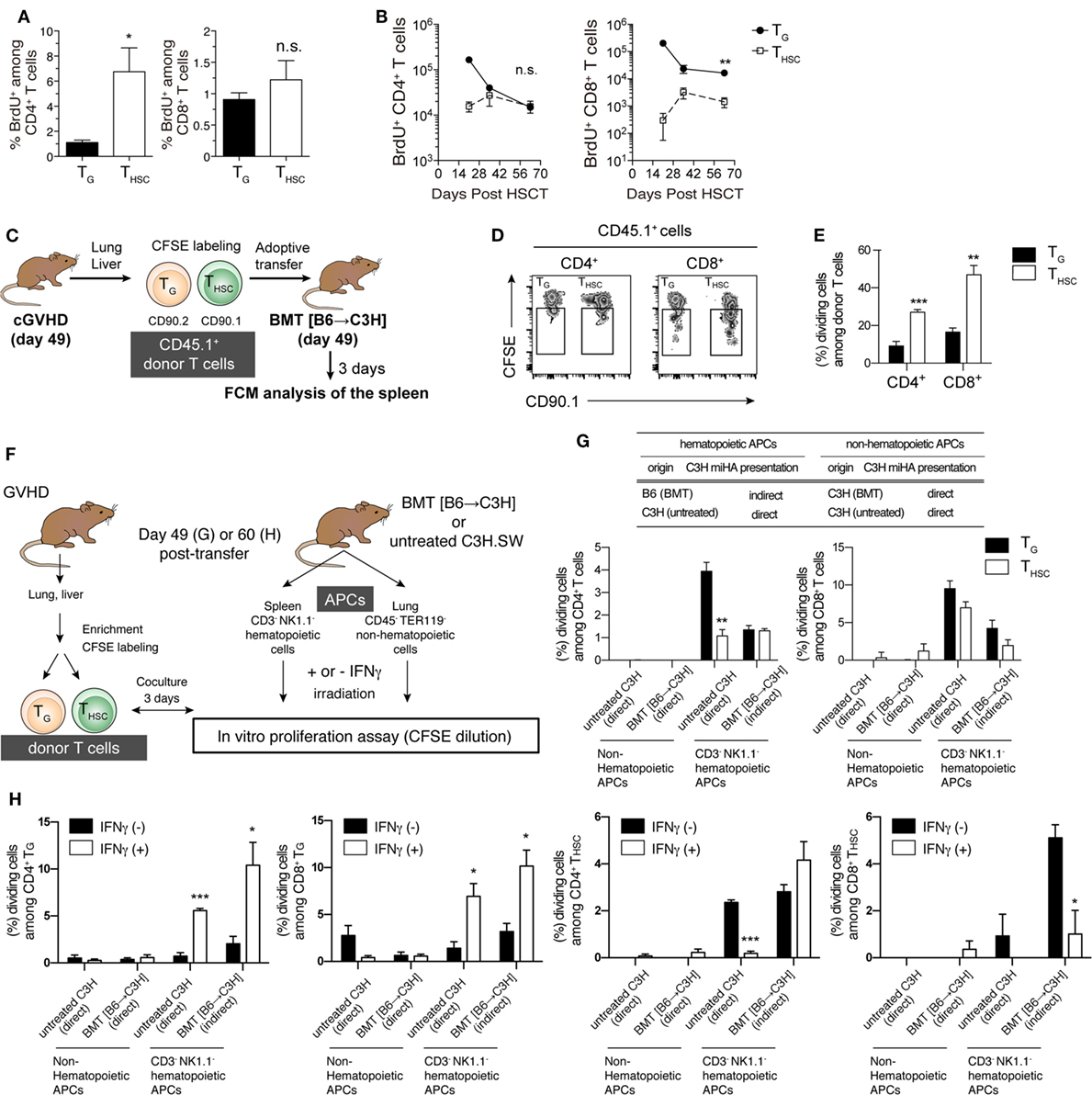
Figure 4. Proliferative potential and allo-recognition pathway of TG and THSC. (A,B) Recipients were injected with BrdU 1 h before sacrifice to enable measurement of cell proliferation in liver T cells. (A) Percentage of BrdU+ CD4+ or CD8+ T cells within the TG or THSC populations on day 63. (B) Time-course analysis of the BrdU+ TG and THSC populations in the liver. (C) Schematic of in vivo proliferation assay. CD45.1+ Thy1.1− TG or CD45.1+ Thy1.1+ THSC were prepared from the liver and lung of chronic graft-versus-host disease (cGVHD) recipients on day 49. These cells were equally mixed, CFSE-labeled, and transferred to phase-matched BMT recipients that had been reconstituted with CD45.2+ T-cell-depleted bone marrow (TCD BM). (D) Representative flow cytometry plots showing CFSE dilution in CD45.1+ donor cells, which consist of Thy1.1− Thy1.2+ TG and Thy1.1+ Thy1.2− THSC, in the spleen 3 days after secondary transfer. (E) Percentages of TG or THSC that underwent one or more cell division. (F) Schematic of in vitro proliferation assay. TG and THSC were cocultured with lung non-hematopoietic cells or spleen CD3− NK1.1− hematopoietic cells, which were prepared from either untreated C3H mice or BMT recipient C3H mice reconstituted with TCD BM from B6 mice, to determine allo-recognition pathway. (G) Percentage of TG or THSC that underwent one or more cell division after 3 days of culture. (H) T cells and antigen-presenting cells (APCs) were prepared from cGVHD or BMT recipients on day 60 as described in (F), then APCs were pretreated with 10 U/mL IFNγ for 16 h. Data show the percentage of TG or THSC that underwent one or more cell division after 3 days of culture. Data represent mean ± SEM [n = 6 (A,B), n = 4 (E), n = 3 (G,H)] from one of two independent experiments. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001 [(B,E,G); TG vs. THSC, (H); and IFNγ (−) vs. IFNγ (+)].
TG Recognize Host Antigens through both Direct and Indirect Pathways
In the chronic phase of GVHD models, APCs are classified into host radioresistant non-hematopoietic cells or donor BM-derived hematopoietic cells. The former directly present complexes of host-type minor histocompatibility antigen (miHA)-derived peptide and MHC molecule to donor T cells, whereas the latter uptake host-type miHA from host radioresistant non-hematopoietic cells and present miHA-peptide/MHC complexes to donor-T cells through an indirect pathway. To investigate the allo-recognition pathway that contributes to the maintenance of TG and THSC populations after allo-HSCT, we cocultured TG or THSC with BMT recipient-derived APCs 49 days after allo-HSCT. To examine the direct pathway, we used radio-resistant host-derived non-hematopoietic cells that present endogenous host-type antigens (Figures 4F,G). To examine the indirect pathway, we used donor-derived T cell/NK cell-depleted hematopoietic cells containing professional APCs, which uptake and then present host antigens. We also used untreated C3H.SW mouse-derived hematopoietic cells and non-hematopoietic cells to examine the direct pathway that is involved in the early induction of effector TG. CFSE-dilution assays demonstrated that neither CD4+ nor CD8+ TG proliferated in response to coculture with non-hematopoietic cells prepared from BMT recipient or untreated C3H.SW mice. In contrast, both CD4+ and CD8+ TG proliferated when cocultured with hematopoietic cells from C3H.SW mice (direct pathway) and to a lesser extent when cocultured with hematopoietic cells from BMT recipients (indirect pathway) (Figure 4G). THSC displayed a similar proliferative response except that a small proportion of CD8+ THSC proliferated in response to coculture with non-hematopoietic cells. Considering that host-type hematopoietic APCs are completely absent in mice suffering cGVHD (data not shown), these results suggest that indirect host antigen presentation by donor-derived hematopoietic cells may be involved in the maintenance of TG in cGVHD-affected organs.
Since inflammatory factors such as IFNγ modify antigen processing by inducing expression of immunoproteasomes and MHC molecules (28), we performed similar donor T cell and APC ex vivo coculture experiments with APCs that had been pretreated with 10 U/mL IFNγ for 16 h, which induced the expression of MHC class I and MHC class II on both hematopoietic and non-hematopoietic APCs. IFNγ pretreatment did not affect antigen presentation by non-hematopoietic APCs (Figure 4H). However, IFNγ pretreatment of hematopoietic APCs significantly increased the proliferation of CD4+ and CD8+ TG in both direct and indirect pathway-dependent settings. In contrast, IFNγ pretreatment of hematopoietic APCs significantly suppressed the direct pathway-dependent proliferation of CD4+ THSC and indirect pathway-dependent proliferation of CD8+ THSC. These results suggest that IFNγ expression in cGVHD-affected tissues may cause prolonged dominance of TG among CD8+ T cells.
Selective Depletion of TG or THSC Failed to Ameliorate the Pathology of cGVHD
To determine the involvement of TG and THSC in the pathogenesis of cGVHD, we selectively depleted TG, THSC, or both TG and THSC from cGVHD mice using Thy1.1/1.2 congenic markers and an anti-Thy1.2 depleting mAb (Figure 5A). This protocol resulted in >95% depletion of Thy1.2+ T cells from cGVHD-affected organs from day 35 to the end of our experiments (Figure 5B). Unexpectedly, survival rate, fibrotic changes, and inflammatory cell infiltration in cGVHD-affected organs were not ameliorated by the depletion of TG (Figures 5C–E), despite the predominance of functionally competent TG in the cGVHD-affected organs of untreated cGVHD mice. These results suggest that TG alone are not responsible for the development and/or maintenance of the histopathology of cGVHD. On the other hand, survival was markedly reduced in the THSC-depleted group, in which all mice died within approximately 3 weeks of initial antibody treatment (Figure 5F). This lethal exacerbation of GVHD was not due to the depletion of Thy1+ HSCs by the anti-Thy1.2+ antibody, because the number of CD48− CD150+ lineage− c-Kit+ Sca1+ long-term HSCs was equivalent between the cGVHD and THSC-depleted groups (Figure 5G). In addition, there was no significant difference in the frequency of Foxp3+ liver-infiltrating CD4+ T cells between the cGVHD and THSC-depleted groups, suggesting that the lethal exacerbation of GVHD in the THSC-depleted group could not be attributed to the loss of Foxp3+ regulatory T cells (Figure 5H). Importantly, simultaneous depletion of TG and THSC rescued cGVHD mice from the lethal exacerbation of disease induced by the depletion of THSC alone (Figure 5F). These results suggest that neither TG nor THSC are solely responsible for cGVHD and that the lethal exacerbation of GVHD induced by THSC depletion is mediated by TG.
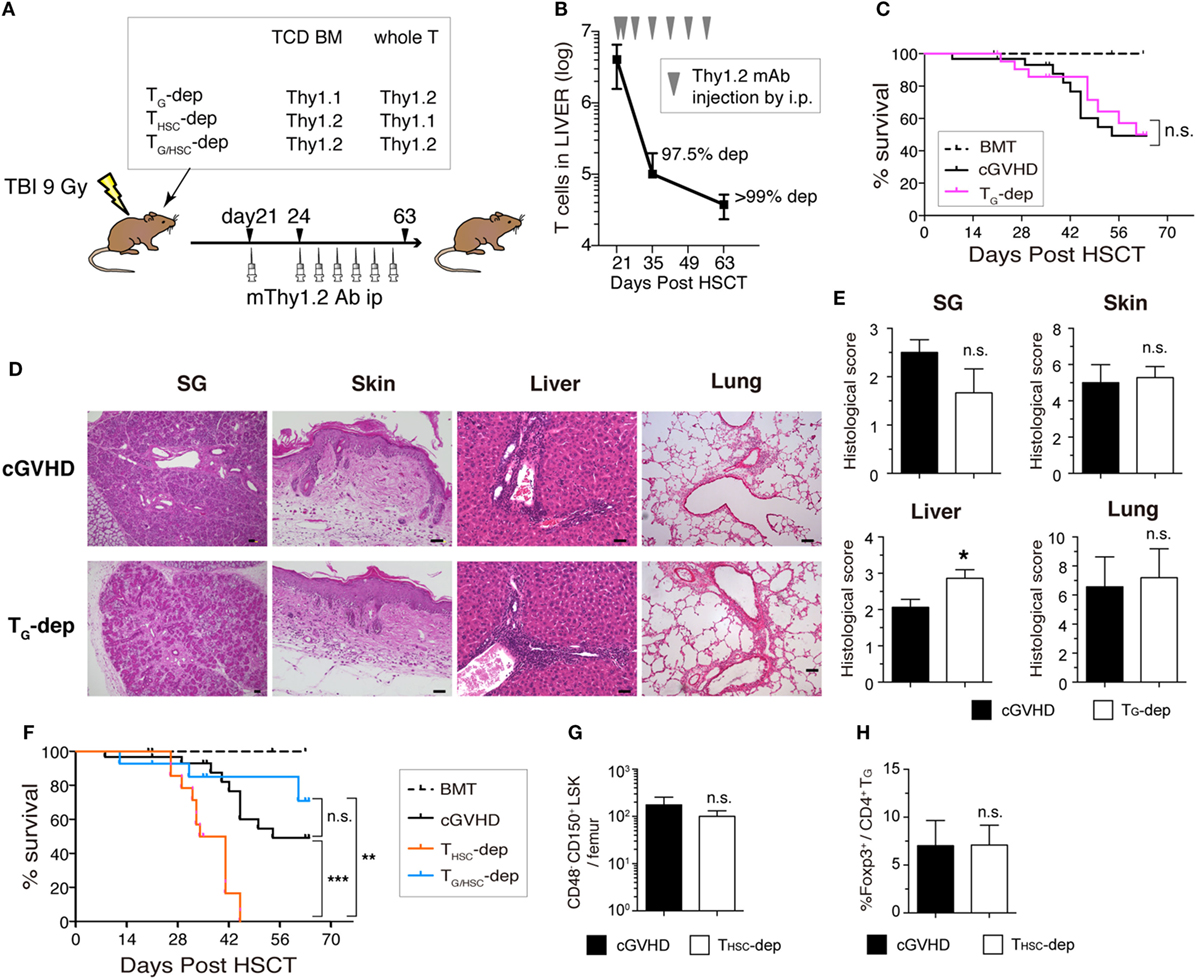
Figure 5. Both TG and THSC are involved in the pathogenesis of chronic graft-versus-host disease (cGVHD). (A) Schematic of the in vivo depletion study. TG (TG-dep), THSC (THSC-dep), or both TG and THSC (TG/HSC-dep) were selectively depleted from cGVHD mice using the Thy1.1/1.2 congenic markers and an anti-Thy1.2 depleting mAb. Recipient mice received Thy1.1+ T-cell-depleted bone marrow (TCD BM) and Thy1.2+ splenic T cells, Thy1.2+ TCD BM and Thy1.1+ splenic T cells, or Thy1.2+ TCD BM and Thy1.2+ splenic T cells from B6 donors and were then treated with an mThy1.2 antibody or a control Ab on days 21 and 24 after transplantation, and weekly thereafter until day 63. (B) Depletion efficiency of anti-Thy1.2 mAb on Thy1.2+ T cells in the liver. (C) Survival data are combined from three independent experiments (n = 12–15). (D) Representative HE images and (E) histological scoring of the salivary glands, skin, liver, and lung from TG-dep and cGVHD (n = 5) mice on day 63. Scale bars, 50 µm. (F) Survival of cGVHD, THSC-dep, and TG/HSC-dep group mice. Data are combined from three independent experiments (n = 12–15). (G) Number of HSCs (CD48− CD150+ LSK) in the BM on day 28 after transplantation (7 days after THSC depletion). (H) Proportion of Foxp3+ cells within the liver CD4+ TG population on day 28 after transplantation. Data in (B,E,G,H) represent mean ± SEM (n = 3 for the cGVHD group; n = 4 for the THSC-dep group) from one of three independent experiments. n.s., not significant; *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.001 (comparisons as indicated).
THSC Suppress Activation of TG
The observation that simultaneous depletion of TG and THSC ameliorated cGVHD while selective depletion of TG or THSC failed to ameliorate cGVHD suggests that an interaction exists between these two distinct T cell populations. To investigate this interaction further, we examined the effects of THSC depletion on the number and function of TG. Since THSC-depleted mice died within 3 weeks of depletion (day 21), analyses were performed on days 28 and 35. There were no significant differences in liver and lung TG numbers between the THSC-depleted and cGVHD groups (Figure 6A). In addition, the frequency of BrdU+ (proliferating) cells within the TG population and the expression levels of CD44, CD25, and CD69 on TG cells were equivalent between the THSC-depleted and cGVHD groups (Figures 6B,C). However, the proportion of IFNγ+ and TNFα+ cells among liver-infiltrating CD4+ and CD8+ TG cells was higher in the THSC-depleted group compared to the cGVHD group on both days 28 and 35 (Figure 6D and data not shown). These results suggest that THSC, despite being 10–100 times lesser in number than TG at the time of depletion, inhibit inflammatory cytokine production by TG and play a critical role in preventing lethal exacerbations of GVHD.
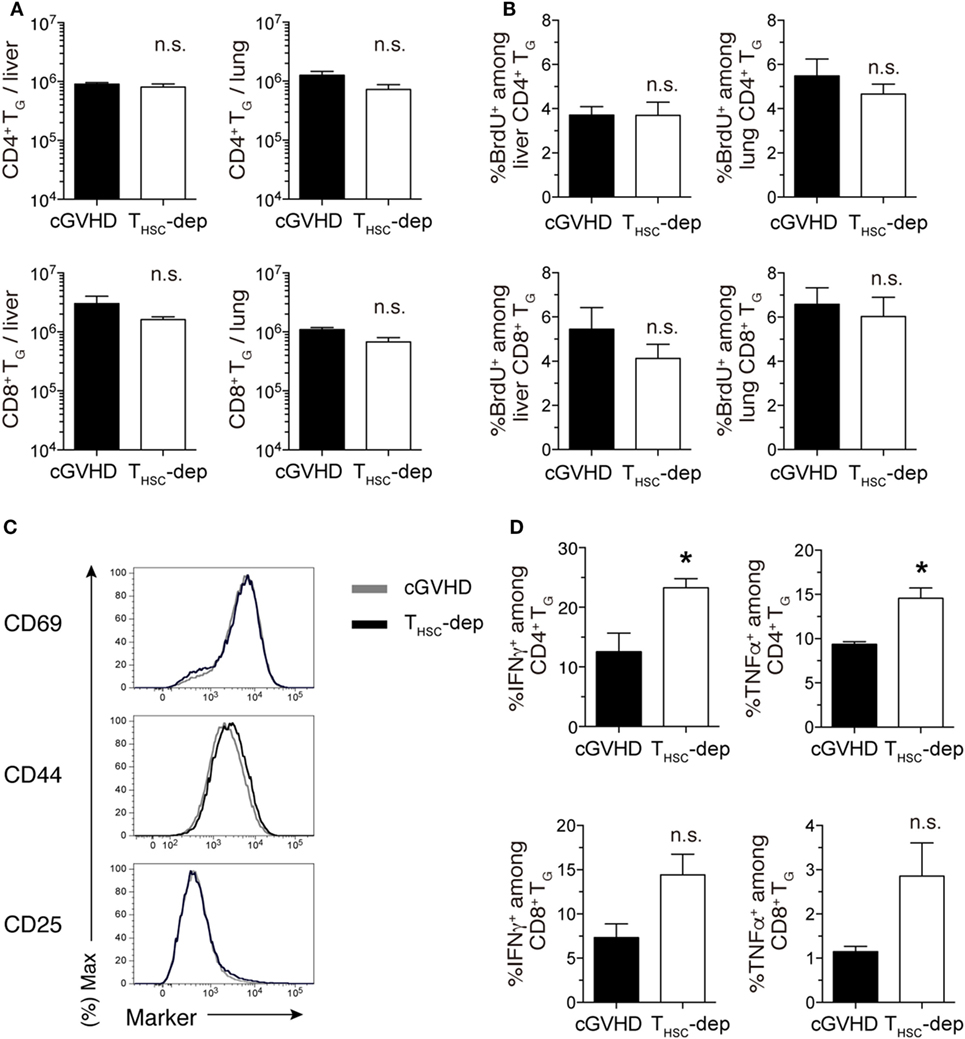
Figure 6. THSC suppress activation of TG. THSC were selectively depleted from chronic graft-versus-host disease (cGVHD) mice as indicated in Figure 5A. (A) Numbers of CD4+ and CD8+ TG from the liver and lung of THSC-dep and cGVHD on day 28, 7 days after commencing THSC depletion by mThy1.2 antibody administration. (B) Proportion of BrdU+ cells within the CD4+ or CD8+ T cell populations in the liver and lung of THSC-dep and cGVHD mice injected 1 h earlier with 1 mg of BrdU, 28 days after transplantation. Data in (A,B) represent mean ± SEM (n = 3 for the cGVHD group; n = 4 for the THSC-dep group) from one of three independent experiments. (C) Representative flow cytometry plots showing the expression of activation markers on CD8+ TG from the liver of THSC-dep or cGVHD recipients. (D) Liver T cells were collected from cGVHD or THSC-dep group mice and were restimulated with PMA and ionomycin ex vivo. Proportions of intracellular IFNγ- or TNFα-positive cells among CD4+ and CD8+ TG. Data represent mean ± SEM (n = 3 for the cGVHD group; n = 4 for the THSC-dep group) from one of three independent experiments. n.s., not significant; *P ≤ 0.05.
TG Suppress the Accumulation of THSC in cGVHD-Affected Organs
Finally, we examined the effects of TG depletion on the number and function of THSC. In kinetic studies of CD4+ and CD8+ T cell number in the liver, TG rapidly decreased after anti-Thy1.2 mAb treatment in the TG-depleted group, whereas THSC increased to numbers approximately 10-fold higher than the cGVHD group between days 35 and 63 (Figure 7A). By day 63 after allo-HSCT, the number of THSC (particularly CD4+ THSC) in the TG-depleted group had increased to levels comparable to the sum of TG and THSC numbers in the cGVHD group (Figure 7B). To determine whether this increase in THSC number was due to an increase in THSC proliferation, we performed BrdU labeling on day 63 (Figure 7C). The proportion of BrdU+ cells among liver-infiltrating CD4+ and CD8+ THSC was significantly higher in the TG-depleted group, outnumbering the sum of BrdU+ TG and THSC in the cGVHD group. There was no significant difference in the frequency of Foxp3+ cells among CD4+ T cells between the cGVHD and TG-depleted groups (Figure 7D). The majority of CD8+ THSC in the liver and lung, but not in the spleen, of TG-depleted group mice were CD44hi effector T cells and had increased expression of the activation markers CD25 and CD69 (Figure 7E). The total proportion of IFNγ+ or TNFα+ cells among liver-infiltrating CD4+ and CD8+ T cells was equivalent between TG-depleted and cGVHD group mice (Figure 7F). However, the cellular composition of these populations switched from predominately TG in the cGVHD group to predominately THSC in the TG-depleted group. Collectively, these results demonstrate that the proliferation and activation of THSC are augmented by TG depletion in cGVHD-affected organs. This change appears to compensate in part for the decrease in TG numbers and is likely to contribute to the histopathology observed in the cGVHD affected organs of the TG-depleted group.
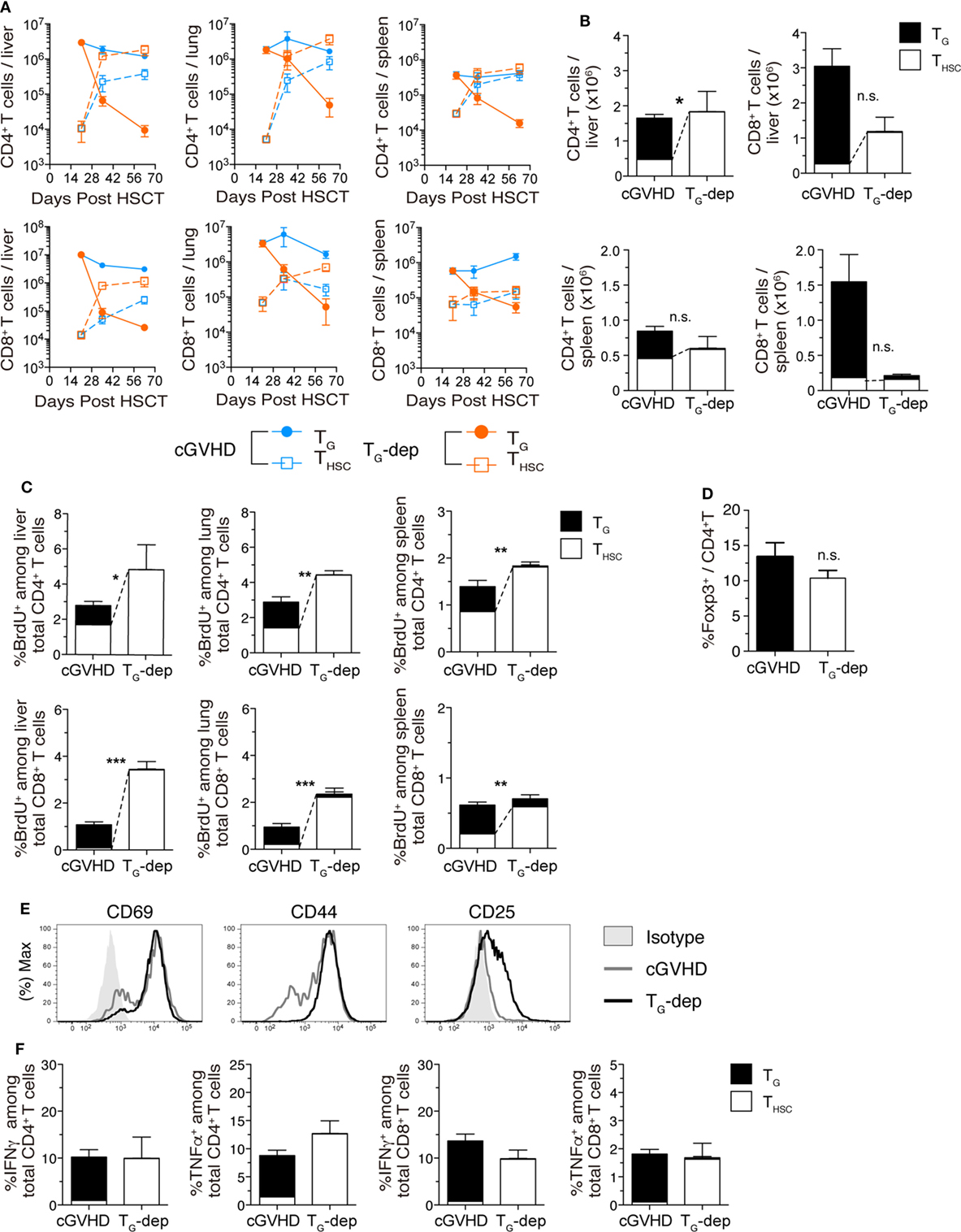
Figure 7. TG suppress accumulation of THSC. TG were selectively depleted from chronic graft-versus-host disease (cGVHD) mice as indicated in Figure 5A. (A) Numbers of CD4+ and CD8+ TG and THSC in the liver, lung, and spleen of TG-dep and cGVHD mice. (B) Numbers of CD4+ and CD8+ TG and THSC in the liver, lung, and spleen 63 days after transplantation. n.s., not significant; *P ≤ 0.05 (THSC from cGVHD vs. THSC from TG-dep). (C) Proportion of BrdU+ cells within the CD4+ or CD8+ T cell populations in the liver, lung, and spleen of TG-dep and cGVHD mice injected 1 h earlier with 1 mg of BrdU, 63 days after transplantation. Data represent mean ± SEM (n = 6 for the cGVHD group; n = 3 for the TG-dep group) from one of three independent experiments. n.s., not significant; *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.001 (THSC from cGVHD vs. THSC from TG-dep). (D) Proportion of Foxp3+ cells within the liver CD4+ T cell population on day 63 after transplantation. (E) Representative flow cytometry plots showing the expression of activation markers on CD8+ THSC from the liver and spleen of TG-dep or cGVHD recipients. (F) Liver T cells were collected from cGVHD or TG-dep group mice and were restimulated with PMA and ionomycin ex vivo. Proportions of CD4+ and CD8+ TG and THSC positive for intracellular IFNγ and TNFα were analyzed by flow cytometry. Data represent mean ± SEM (n = 6 for the cGVHD group; n = 3 for the TG-dep group) from one of three independent experiments.
Discussion
Until now, the contribution of TG to the development and maintenance of cGVHD has remained unclear. In this study, we established a model of cGVHD that recapitulates the pathology of clinical cGVHD. We demonstrated that functional TG persist and predominate over THSC in the organs affected by cGVHD. Furthermore, our depletion studies revealed the existence of an unexpected bidirectional regulatory interaction between TG and THSC: TG suppress the proliferation and accumulation of THSC in cGVHD-affected organs, whereas THSC suppress the activation of TG in affected organs, thereby preventing exacerbations of GVHD mediated by TG.
There are few mouse models of cGVHD that accurately recapitulate the histopathology and disease course of clinical cGVHD. Recently, a B6 (H2b) Recent (H2k) allo-HSCT model, which utilized cyclophosphamide and total body irradiation as a conditioning regimen, was demonstrated to induce fibrotic changes in the lungs, liver, and SG (29, 30). The developmental course of this model recapitulated cGVHD that develops without preceding aGVHD. Mice in our B6→C3H.SW minor mismatched allo-HSCT model also developed several histological aspects of clinical cGVHD, such as fibrotic changes in the skin, liver, lungs, and SG following aGVHD, which may recapitulate the acute to chronic transition in clinical GVHD.
In our cGVHD model, a large number of TG persisted in cGVHD-affected organs up to day 63 after allo-HSCT. On the other hand, peripheral reconstitution of THSC became obvious from about 4–5 weeks after allo-HSCT and gradually increased thereafter. As a result, both TG and THSC infiltrated the affected organs in late phase cGVHD with an unexpected predominance of TG. In an aGVHD model, Zhang et al. (31) have described a CD44lo CD62Lhi IL-7Rαhi CD8+ memory stem cell population that maintain CD8+ TG in the recipient for a long period of time. In contrast, in our cGVHD model, the majority of TG in affected organs had a CD44hi CD62Llo IL-7Rαlo effector phenotype. Despite the higher proportion of PD-1+ or KLRG-1+ phenotypically exhausted or senescent populations within the CD8+ TG population, the potential for inflammatory cytokine production and in situ proliferation was almost equivalent between TG and THSC. Masopust et al. (32) have reported that KLRG-1+ PD-1− senescent CD8+ T cells induced by a heterologous prime-boost vaccination show strong cytotoxic activity. Thus, KLRG-1+ PD-1− CD8+ TG may be involved in the cellular immunopathogenesis of cGVHD.
In the cGVHD affected liver and lung, a significant proportion of CD8+ TG were actively proliferating even at day 63, suggesting that TG maintenance is a dynamic process controlled by cell proliferation and cell death. Considering that TG do not proliferate in response to direct presentation of host antigens by non-hematopoietic cells, it appears that donor HSC-derived APCs capture host alloantigens and indirectly present them to TG in cGVHD-affected organs. An intriguing question that remains is whether TG clones that expanded in response to direct presentation in the acute phase cross-react to indirect presentation in the chronic phase, or whether indirect presentation-reactive TG clones are independently expanded by donor-derived APCs after repopulation of these cells in the peripheral tissues. A better understanding of the clonal response of TG after allo-HSCT and the pathological significance of this process may facilitate the early diagnosis and treatment of cGVHD.
The finding that depletion of the small THSC population potentiated the inflammatory cytokine production of TG and caused a TG-mediated acute exacerbation of GVHD suggests that reconstitution of THSC is important for limiting excessive activation of TG and promoting acute to chronic disease transition following allo-HSCT. Impaired inhibition of TG by THSC may account for the GVHD that is refractory to immunosuppressive therapy with a calcineurin inhibitor, which inhibits both TG activation and THSC development (33). However, the molecular and cellular mechanisms underlying the inhibition of TG activation by THSC remain to be elucidated.
Reconstitution of THSC is markedly delayed and suppressed in the presence of GVHD (16). In the early phase of aGVHD, CD4+ TG impair the production of common lymphoid progenitors through the destruction of the hematopoietic niche in the BM, resulting in a severe reduction in T- and B-lymphocyte genesis (17, 34). TG also impair thymopoiesis by disrupting the thymic epithelium (35). Thus, TG may suppress THSC reconstitution by impairing primary lymphoid tissues. In the present study, depletion of TG resulted in a rapid increase in the number of THSC in the liver and lung. Interestingly, increased numbers of THSC in the TG-depleted group partially compensated for the decreased numbers of TG in cGVHD-affected organs. This finding points to the possible existence of a niche that provides antigenic signals and survival factors to pathogenic T cells of TG or THSC origin. Such a “pathogenic T cell niche” might have a fixed pool capacity, meaning that TG and THSC compete with each other for space in the niche during cGVHD.
In summary, we have characterized the cellular mechanisms underlying the maintenance of pathogenic T cells in a clinically relevant cGVHD model. Both TG and THSC with the potential to proliferate and produce inflammatory cytokines infiltrated cGVHD-affected organs, with the number and/or activity of TG and THSC being reciprocally regulated by each other. In addition, our depletion studies highlight the importance of reciprocal tuning of the balance between TG and THSC, which requires the successful reconstitution of THSC, in the control of GVHD. By elucidating the interactions between TG and THSC, our findings will help guide the development of novel therapeutic strategies for the prevention and treatment of cGVHD.
Ethics Statement
All animal experiments were conducted in accordance with institutional guidelines with the approval of the Animal Care and Use Committee of the University of Tokyo.
Author Contributions
MK-K, SU, and KM participated in research design; MK-K, SU, JA, SS, FHWS, TM, NS, AY, FS, and WY conducted experiments; MK-K and SU performed data analysis; MK-K, SU, JA, SS, FHWS, MK, YS, MI, TT, and KM wrote or contributed to the writing of the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very thankful to M. Kanaya, T. Shimaoka, and S. Hashimoto for helpful discussions and suggestions. We would also like to thank S. Aoki, S. Fujita, and C. Kasahara for expert technical assistance.
Funding
This work was supported by the Japan Science and Technology Agency CREST program, Grants-in-Aid for Scientific Research (C) 13385295 (to SU), and (B) 13373346 (to KM), and Grant-in-Aid for Scientific Research on Innovative Areas 17929397 (to SU and KM) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
References
1. Wingard JR, Majhail NS, Brazauskas R, Wang Z, Sobocinski KA, Jacobsohn D, et al. Long-term survival and late deaths after allogeneic hematopoietic cell transplantation. J Clin Oncol (2011) 29:2230–9. doi:10.1200/JCO.2010.33.7212
2. Martin PJ, Counts GW, Appelbaum FR, Lee SJ, Sanders JE, Deeg HJ, et al. Life expectancy in patients surviving more than 5 years after hematopoietic cell transplantation. J Clin Oncol (2010) 28:1011–6. doi:10.1200/JCO.2009.25.6693
3. Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science (1999) 285:412–5. doi:10.1126/science.285.5426.412
4. Panoskaltsis-Mortari A, Hermanson JR, Taras E, Wangensteen OD, Serody JS, Blazar BR. Acceleration of idiopathic pneumonia syndrome (IPS) in the absence of donor MIP-1 alpha (CCL3) after allogeneic BMT in mice. Blood (2003) 101:3714–21. doi:10.1182/blood-2002-08-2465
5. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med (2004) 10:987–92. doi:10.1038/nm1089
6. Socie G, Schmoor C, Bethge WA, Ottinger HD, Stelljes M, Zander AR, et al. Chronic graft-versus-host disease: long-term results from a randomized trial on graft-versus-host disease prophylaxis with or without anti-T-cell globulin ATG-Fresenius. Blood (2011) 117:6375–82. doi:10.1182/blood-2011-01-329821
7. Kröger N, Solano C, Wolschke C, Bandini G, Patriarca F, Pini M, et al. Antilymphocyte globulin for prevention of chronic graft-versus-host disease. N Engl J Med (2016) 374:43–53. doi:10.1056/NEJMoa1506002
8. Sugita J, Kawashima N, Fujisaki T, Kakihana K, Ota S, Matsuo K, et al. HLA-haploidentical peripheral blood stem cell transplantation with post-transplant cyclophosphamide after busulfan-containing reduced-intensity conditioning. Biol Blood Marrow Transplant (2015) 21:1646–52. doi:10.1016/j.bbmt.2015.06.008
9. Mielcarek M, Furlong T, O’Donnell PV, Storer BE, McCune JS, Storb R, et al. Posttransplantation cyclophosphamide for prevention of graft-versus-host disease after HLA-matched mobilized blood cell transplantation. Blood (2016) 127:1502–8. doi:10.1182/blood-2015-10-672071
10. Kasamon YL, Bolaños-Meade J, Prince GT, Tsai H-L, McCurdy SR, Kanakry JA, et al. Outcomes of nonmyeloablative HLA-haploidentical blood or marrow transplantation with high-dose post-transplantation cyclophosphamide in older adults. J Clin Oncol (2015) 33:3152–61. doi:10.1200/JCO.2014.60.4777
11. Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity (2007) 27:670–84. doi:10.1016/j.immuni.2007.09.006
12. Asakura S, Hashimoto D, Takashima S, Sugiyama H, Maeda Y, Akashi K, et al. Alloantigen expression on non-hematopoietic cells reduces graft-versus-leukemia effects in mice. J Clin Invest (2010) 120:2370–8. doi:10.1172/JCI39165
13. Zhang Y, Hexner E, Frank D, Emerson SG. CD4+ T cells generated de novo from donor hemopoietic stem cells mediate the evolution from acute to chronic graft-versus-host disease. J Immunol (2007) 179:3305–14. doi:10.4049/jimmunol.179.5.3305
14. Sakoda Y, Hashimoto D, Asakura S, Takeuchi K, Harada M, Tanimoto M, et al. Donor-derived thymic-dependent T cells cause chronic graft-versus-host disease. Blood (2007) 109:1756–64. doi:10.1182/blood-2006-08-042853
15. Krenger W, Blazar BR, Hollander GA. Thymic T-cell development in allogeneic stem cell transplantation. Blood (2011) 117:6768–76. doi:10.1182/blood-2011-02-334623
16. Hossain MS, Roback JD, Pollack BP, Jaye DL, Langston A, Waller EK. Chronic GvHD decreases antiviral immune responses in allogeneic BMT. Blood (2007) 109:4548–56. doi:10.1182/blood-2006-04-017442
17. Shono Y, Ueha S, Wang Y, Abe J, Kurachi M, Matsuno Y, et al. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood (2010) 115:5401–11. doi:10.1182/blood-2009-11-253559
18. Suenaga F, Ueha S, Abe J, Kosugi-Kanaya M, Wang Y, Yokoyama A, et al. Loss of lymph node fibroblastic reticular cells and high endothelial cells is associated with humoral immunodeficiency in mouse graft-versus-host disease. J Immunol (2015) 194:398–406. doi:10.4049/jimmunol.1401022
19. Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood (1996) 88:3230–9.
20. Blazar BR, Taylor PA, McElmurry R, Tian L, Panoskaltsis-Mortari A, Lam S, et al. Engraftment of severe combined immune deficient mice receiving allogeneic bone marrow via in utero or postnatal transfer. Blood (1998) 92:3949–59.
21. Zhao D, Young JS, Chen Y-H, Shen E, Yi T, Todorov I, et al. Alloimmune response results in expansion of autoreactive donor CD4+ T cells in transplants that can mediate chronic graft-versus-host disease. J Immunol (2011) 186:856–68. doi:10.4049/jimmunol.1002195
22. Kaplan DH, Anderson BE, McNiff JM, Jain D, Shlomchik MJ, Shlomchik WD. Target antigens determine graft-versus-host disease phenotype. J Immunol (2004) 173:5467–75. doi:10.4049/jimmunol.173.9.5467
23. Kurachi M, Kakimi K, Ueha S, Matsushima K. Maintenance of memory CD8+ T cell diversity and proliferative potential by a primary response upon re-challenge. Int Immunol (2007) 19:105–15. doi:10.1093/intimm/dxl127
24. Imanguli MM, Atkinson JC, Mitchell SA, Avila DN, Bishop RJ, Cowen EW, et al. Salivary gland involvement in chronic graft-versus-host disease: prevalence, clinical significance, and recommendations for evaluation. Biol Blood Marrow Transplant (2010) 16:1362–9. doi:10.1016/j.bbmt.2010.03.023
25. Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health Consensus Development Project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant (2005) 11:945–56. doi:10.1016/j.bbmt.2005.09.004
27. Alpdogan O, Muriglan SJ, Eng JM, Willis LM, Greenberg AS, Kappel BJ, et al. IL-7 enhances peripheral T cell reconstitution after allogeneic hematopoietic stem cell transplantation. J Clin Invest (2003) 112:1095–107. doi:10.1172/JCI17865
28. Basler M, Kirk CJ, Groettrup M. The immuneproteasome in antigen processing and other immunological functions. Curr Opin Immunol (2013) 25:74–80. doi:10.1016/j.col.2012.11.004
29. Srinivasan M, Flynn R, Price A, Ranger A, Browning JL, Taylor PA, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood (2012) 119:1570–80. doi:10.1182/blood-2011-07-364414
30. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood (2014) 123:3988–98. doi:10.1182/blood-2014-03-562231
31. Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med (2005) 11:1299–305. doi:10.1038/nm1326
32. Masopust D, Ha S-J, Vezys V, Ahmed R. Stimulation history dictates memory CD8 T cell phenotype: implications for prime-boost vaccination. J Immunol (2006) 177:831–9. doi:10.4049/jimmunol.177.2.831
33. Jenkins MK, Schwartz RH, Pardoll DM. Effects of cyclosporine A on T cell development and clonal deletion. Science (1988) 241:1655–8. doi:10.1126/science.3262237
34. Shono Y, Shiratori S, Kosugi-Kanaya M, Ueha S, Sugita J, Shigematsu A, et al. Bone marrow graft-versus-host disease: evaluation of its clinical impact on disrupted hematopoiesis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant (2014) 20:495–500. doi:10.1016/j.bbmt.2013.12.568
Keywords: chronic graft-versus-host disease, stem cell transplantation, T cell, T cell subset, immune reconstitution
Citation: Kosugi-Kanaya M, Ueha S, Abe J, Shichino S, Shand FHW, Morikawa T, Kurachi M, Shono Y, Sudo N, Yamashita A, Suenaga F, Yokoyama A, Yong W, Imamura M, Teshima T and Matsushima K (2017) Long-Lasting Graft-Derived Donor T Cells Contribute to the Pathogenesis of Chronic Graft-versus-Host Disease in Mice. Front. Immunol. 8:1842. doi: 10.3389/fimmu.2017.01842
Received: 09 August 2017; Accepted: 05 December 2017;
Published: 18 December 2017
Edited by:
Gilles Benichou, Harvard Medical School, United StatesReviewed by:
Dennis O. Adeegbe, New York University, United StatesEmmanuel Zorn, Columbia University Medical Center, United States
Copyright: © 2017 Kosugi-Kanaya, Ueha, Abe, Shichino, Shand, Morikawa, Kurachi, Shono, Sudo, Yamashita, Suenaga, Yokoyama, Yong, Imamura, Teshima and Matsushima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Ueha, dWVoYUBtLnUtdG9reW8uYWMuanA=