 Franziska M. Konrad
Franziska M. Konrad Constantin Zwergel
Constantin Zwergel Kristian-Christos Ngamsri
Kristian-Christos Ngamsri Jörg Reutershan2*
Jörg Reutershan2*
- 1Department of Anesthesiology and Intensive Care Medicine, University Hospital of Tübingen, Tübingen, Germany
- 2Department of Anesthesiology and Intensive Care Medicine, Hospital of Bayreuth, Bayreuth, Germany
Acute pulmonary inflammation is still a frightening complication in intensive care units. In our previous study, we determined that heme oxygenase (HO)-1 had anti-inflammatory effects in pulmonary inflammation. Recent literature has emphasized a link between HO-1 and the nucleotide adenosine. Since adenosine A2A- and A2B-receptors play a pivotal role in pulmonary inflammation, we investigated their link to the enzyme HO-1. In a murine model of pulmonary inflammation, the activation of HO-1 by hemin significantly decreased polymorphonuclear leukocyte (PMN) migration into the lung. This anti-inflammatory reduction of PMN migration was abolished in A2A- and A2B-knockout mice. Administration of hemin significantly reduced chemokine levels in the BAL of wild-type animals but had no effects in A2A-/- and A2B-/- mice. Microvascular permeability was significantly attenuated in HO-1-stimulated wild-type mice, but not in A2A-/- and A2B-/- mice. The activity of HO-1 rose after LPS inhalation in wild-type animals and, surprisingly, also in A2A-/- and A2B-/- mice after the additional administration of hemin. Immunofluorescence images of animals revealed alveolar macrophages to be the major source of HO-1 activity in both knockout strains—in contrast to wild-type animals, where HO-1 was also significantly augmented in the lung tissue. In vitro studies on PMN migration further confirmed our in vivo findings. In conclusion, we linked the anti-inflammatory effects of HO-1 to functional A2A/A2B-receptor signaling under conditions of pulmonary inflammation. Our findings may explain why targeting HO-1 in acute pulmonary inflammation has failed to prove effective in some patients, since septic patients have altered adenosine receptor expression.
Introduction
Acute pulmonary inflammation and its more severe form acute respiratory distress syndrome, are life-threatening complications in intensive care units (ICU) with a high mortality of 40% (1). Surviving patients have a decreased quality of life (2). Acute pulmonary inflammation can be caused either by direct lung injury, such as pneumonia, or indirect injury, such as sepsis or trauma. These different etiological factors might be the reason why, despite decades of research, no single treatment has been successful and the translation of animal models in clinical practice has failed (3). Therefore, newer studies focus on subgroups of patients because individualized treatments may be successful (3).
Polymorphonuclear leukocytes (PMNs) are part of the innate immune system and are the first cells to migrate toward inflammation. In the case of acute pulmonary inflammation, PMNs excessively migrate from the intravascular compartment through an endothelial barrier into the lung interstitium, followed by a transepithelial migration into the alveolar space. These migration steps underlay different regulation (4) and destroy the lung architecture.
Adenosine triphosphate is released in response to cellular stress and is extracellularly degraded to adenosine diphosphate by the membrane-bound nucleotidase CD39, followed by further degradation by CD73 to adenosine (5). Adenosine, a purine nucleoside, acts via the four adenosine receptors (A1, A2A, A2B, and A3). All of these adenosine receptors have been shown to be involved in inflammation (6–9); specifically, the A2A- and A2B-receptors play a crucial role in acute pulmonary inflammation (10).
The A2B-receptor, for example, exerts a protective role in ischemic lung injury (11), and an inhaled A2B-receptor agonist has been recommended for the treatment of acute lung injury (12).
Heme oxygenase (HO)-1 is a ubiquitous enzyme in the body that catalyzes the degradation of heme. HO-1 is pharmacologically inducible and known as a potent cytoprotective enzyme that is upregulated under different conditions of cellular stress (13). Although the stimulation of HO-1 in acute pulmonary inflammation has been shown to have anti-inflammatory effects in several animal studies (14, 15), clinical trials have so far failed to be convincing, showing only a trend toward a reduction in inflammatory cells in the treatment of chronic obstructive disease (COPD) or even having no effect on endotoxemia-induced inflammation (16, 17). Therefore, current research on HO-1 and its products demands further study of the function of the HO-1 enzyme in the inflammatory setting since this remains complex and still incompletely understood (18).
Recent literature suggests a link between HO-1 and adenosine receptors (19–21). Adenosine receptors play a pivotal role concerning the two hallmarks of pulmonary inflammation: PMN migration and microvascular permeability (8, 9). Therefore, we thought to investigate the link between the anti-inflammatory effects of HO-1 and the predominant pulmonary adenosine receptors A2A and A2B to potentially identify subgroups of patients where a specific stimulation of HO-1 would be a therapeutic option. Patients in ICUs sometimes have altered adenosine receptor distribution and ligand affinities (22).
Materials and Methods
Animals
We used A2A (A2A-/-) and A2B gene-deficient mice (A2B-/-) (from Dr. Katya Ravid, Boston University, School of Medicine, Department of Biochemistry, Boston, MA, USA) and obtained corresponding wild-type mice (CD1 and C57BL/6 respectively) from Charles River Laboratories (Germany). Mice were males and between 8 and 12 weeks old. All animal protocols were approved by the Animal Care and Use Committee of the University of Tübingen.
HO-1 Activator and -Inhibitor
Hemin was injected intraperitoneally (i.p.) (80 µmol/kg) 24 h before LPS inhalation to increase HO-1 activity (15). To inhibit the enzyme, SnPP (50 µmol/kg) was additionally administered i.p. 1 h before LPS inhalation (15). The specific equilibrative nucleoside transporter 1 (ENT1) antagonist NBTI was administered as described previously (9). Cobalt (III) protoporphyrin-IX-chloride (CoPP) was injected at 5 µg/g body weight i.p. 24 h before the inflammatory stimulus (23).
HO-1- Expression and Protein Level
Total RNA was isolated from murine lung samples by using pegGOLD TriFast™ (Peglab, Germany), and cDNA synthesis was performed by Bio-Rad iSkript-kit (Bio-Rad, Germany) according to the manufacturer’s direction.
We determined the expression of HO-1 in the lungs of wild-type, A2A-/-, and A2B-/- mice by RT-PCR (n = 5–8). This method was performed with the following HO-1 primers (5′-GAG ATT GAG CGC AAC AAG GA-3′ and AGC GGT AGA GCT GCT TGA ACT-3′) as described (9). 18S was used as house keeping gene (5′-GTA ACC CGT TGA ACC CCA TT-3′ and 5′-CCA TCC AAT CGG TAG TAG CG-3′).
To further verify the gene expression results on a protein level, we measured HO-1 protein expression in the lungs of mice by ELISA (ENZO Life Sciences, Lörrach, Germany) (n = 5–8). Mice inhaled LPS and HO-1 was induced, respectively, inhibited as described above.
HO-1 Activity
Lungs of mice were removed to measure the enzymatic activity of HO-1 24 h after LPS inhalation (n = 6–8). HO-1 was induced by hemin, respectively, inhibited by the additional administration of SnPP, lungs were removed and weighed and HO-1 activity buffer (twice of weight) added. The samples were frozen in liquid nitrogen. Lungs were homogenized, sonicated, and centrifugated at 18,000 g for 15 min. The supernatant was used for protein- and activity-measurement. Cytosol of the liver was obtained from 12 h fastened mice via centrifugation at 105,000 g for 27 min. The HO-1 activity assay consisted of 100 µl of the supernatant, 131 µl of HO-activity buffer, 100 µl of liver cytosol, 50 µl of glucose-6-phosphate (20 nM), and 10 µl of hemin (1 mM). After incubating for 1 h in the dark at 37°C, 500 µl chloroform was added, followed by a centrifugation of 15,000 g for 5 min. Extinction was measured at 464 and 530 nm, and HO-activity was calculated based on protein level, which was determined by a colorimetric method (bicinchoninic acid; Thermo Scientific, Rockford, IL, USA).
Immunofluorescence Images
Heme oxygenase-1 was stimulated as described above, inflammation induced and lungs of mice removed after 24 h (n = 4). The circulatory system of the lungs was flushed and lungs with 4% paraformaldehyde (PFA) for 10 min at 25 cmH2O inflated. Lungs were removed and fixed in PFA for 24 h. Rabbit polyclonal anti-HO-1 was used as primary antibody (Enzo, Life Sciences GmbH, Lörrach, Germany) to mark the enzyme and the endothelial marker von Willebrand Factor (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was employed and additionally DAPI for nuclear staining. Images were visualized by using a confocal microscope (LSM 510, Meta, Carl Zeiss). Images shown are representatives of four experiments and were analyzed using ImageJ, a public program developed at the National Institutes of Health to officially analyze scientific images.
In Vivo Migration Assay
Murine Model of Acute Lung Injury
Four to eight animals inhaled nebulized LPS as described in detail before (24). This inhalation caused acute pulmonary inflammation with a reproducible migration of PMNs into the different compartments of the lung (intravascular, interstitial, intraalveolar), an increase of chemokines, and microvascular permeability (15, 25). Control mice inhaled the solvent.
In Vivo Migration Assay
24 h after LPS inhalation, we determined PMN migration into the different compartments of the lung via a flowcytometry-based method as described in detail before (n = 5–9) (24). To mark all intravascular PMNs, a fluorescent GR-1 (clone RB6-8C5) was injected into the tail vein of mice. Lungs were flushed free of blood by injecting saline into the beating right ventricle, to remove nonadherent leukocytes from the pulmonary vasculature. PMNs from the alveolar space were determined in the BAL. Lungs were incubated with fluorescent antibodies to CD45 (clone 30-F11) and 7/4 (clone 7/4). Intravascular PMNs were now identified as CD45+, 7/4+, and GR-1+; whereas interstitial PMNs were assigned as CD45+, 7/4+, and GR-1 negative cells. Absolute cell counts were determined in the BAL and lungs. The detailed description of the method and the gating process has been described elsewhere (24).
Depletion of Ectonucleotidases CD39 and CD73
SiRNA of CD39 and CD73 (Santa Cruz Biotechnology, Heidelberg, Germany) was used to knock down gene expression of both ectonucleotidases in pulmonary epithelial cells (NCI-H441). In H441 cells, HO-1 was activated by hemin (5 µM), and cells were stimulated with LPS (100 ng/ml) for 4 h. Gene expression of interleukin-6 (IL-6) and 8 (IL8) were detected in cells by RT-PCR (n ≥ 5) by using the following primers: IL6 (5′-GAC AGC CAC TCA CCT CTT CA-3′ and 5′-CAC CAG GCA AGT CTC CTC AT-3′); IL8 (5′-TGT GGG TCT GTT GTA GGG-3′ and 5′-GTG AGG TAA GAT GGT GGC-3′); and 18S as house keeping gene (5′-GTA ACC CGT TGA ACC CCA TT-3′ and 5′-CCA TCC AAT CGG TAG TAG CG-3′).
Interleukin-6 and IL8 protein were detected in the supernatant of cells by ELISA (R&D Systems, Minneapolis, MN, USA) (n ≥ 5).
Chemokine Release
3 h after LPS inhalation, the release of CXCL1, CXCL2/3, tumor necrosis factor-α (TNFα), and IL-6 was measured in the BAL of mice (n = 5–9) by ELISA (R&D) according to the manufacture’s protocol.
Microvascular Leakage
Evans blue extravasation was determined as a marker of capillary leakage as described before (9). 6 h after LPS exposure, Evans blue (20 mg/kg, Sigma-Aldrich, Steinheim, Germany) was injected into the tail vein (n = 5–8). Intravascular Evans blue in the lungs was removed 30 min later by flushing the beating right ventricle. Lungs were homogenized and Evans blue was extracted with formamide and the final concentration determined colorimetrically (26).
In Vitro PMN Migration
We performed the in vitro transmigration assay of human PMNs through a monolayer of NCI-H441 cells (n = 6–12) or primary pulmonary endothelial cells (HMVEC-L, Lonza Walkersville, MD, USA) (n = 6–10) as described previously (9). HO-1 was induced by hemin (5 µM) and the A2A- (ZM 241385; Tocris, MO, USA) (10 ng/ml) or A2B-receptors (PSB 1115 Sigma-Aldrich, Taufkirchen, Germany) (10 ng/ml) were inhibited with the respective antagonists.
Statistical Analysis
Data are presented as the mean ± SD unless indicated otherwise. Statistical analysis was performed using GraphPad Prism version 5.3 for Windows (GraphPad Software, San Diego, CA, USA). Differences between the groups were evaluated by one-way ANOVA followed by Bonferroni post hoc test or Student t-Test to compare two groups. P < 0.05 was considered statistically significant.
Results
Gene and Protein Expression of HO-1 in the Lungs of Mice
In wild-type mice, hemin caused a significant increase in the gene expression of HO-1 in the lungs even without inflammation (all P < 0.05). Since both wild-type mice acted identically, we display only one strain in Figure 1 (CD1 in Figure 1A C57BL/6 in Figure S1 in Supplementary Material). LPS also caused a significant rise, and the additional HO-1 stimulation did not show an additive effect on the gene expression.
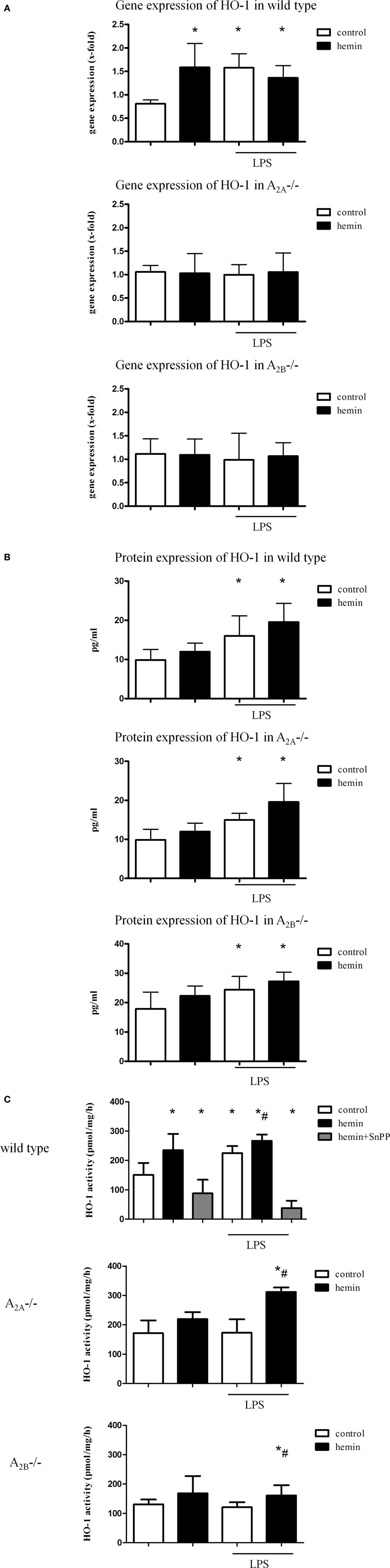
Figure 1. Heme oxygenase (HO)-1 gene and protein expression and activity in the lungs of mice. Gene (A) and protein (B) expression of HO-1 in the lungs of mice was determined. The activity of the enzyme was evaluated after the activation of HO-1 by hemin and the inhibition by additional administration of SnPP (C). Data are presented as the mean ± SD; without LPS inhalation, n = 4–8 other groups, n = 5–8; *P < 0.05 vs. control without LPS; #P < 0.05 vs. LPS-treated mice.
Neither inflammation nor HO-1 activation by hemin changed the expression of HO-1 in A2A-/- and A2B-/- mice.
Protein expression of HO-1 in the lungs of mice confirmed the significant increase in the enzyme in LPS-treated wild-type animals (all P < 0.05) (Figure 1B; Figure S1B in Supplementary Material). Surprisingly, the augmentation after inflammation was also apparent in A2A-/- and A2B-/- mice at the protein level.
Influence of A2A-/- and A2B-/- Receptor Signaling on the Activity of HO-1
To further verify our findings from the gene and protein expression experiments, we measured the activity of the HO-1 enzyme. In wild-type animals, hemin increased HO-1 activity without inflammation, whereas the inhibition caused a significant reduction (all P < 0.05) (Figure 1C; Figure S1C in Supplementary Material). Inflammation also enhanced HO-1 activity, and the additional activation of the enzyme showed an additive effect.
In A2A-/- and A2B-/- mice, hemin did not influence the activity of HO-1 without inflammation. Additionally, LPS did not change the activity, but additional hemin administration caused a significant increase in HO-1 activity in both strains (all P < 0.05).
Induction of HO-1 in Alveolar Macrophages and Lung Tissue
To evaluate the unclear source of HO-1 activity, we performed immunofluorescence staining of lung sections. Without inflammation, HO-1 was only slightly increased in wild-type animals after hemin treatment (Figure S2 in Supplementary Material). Inflammation significantly increased the expression of HO-1 in lung tissue (P < 0.05) (Figure 2). In wild-type animals, hemin further increased HO-1 in lung tissue and alveolar macrophages (P < 0.05).
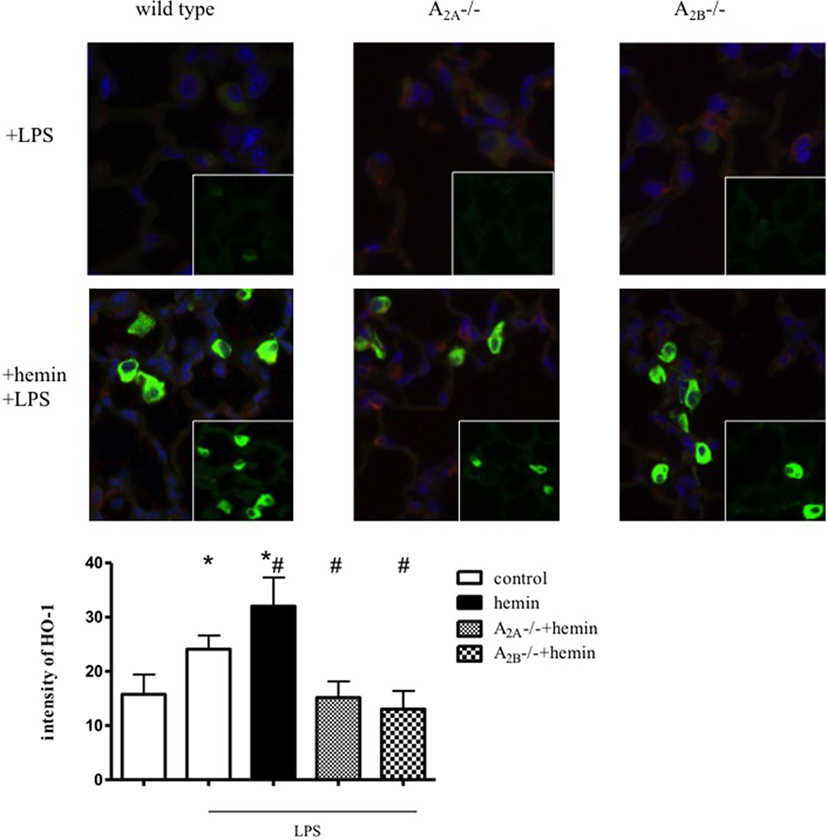
Figure 2. Determination of heme oxygenase (HO)-1 expression by immunofluorescence. Mice were treated with LPS, and HO-1 was induced by hemin in wild-type, A2A-/-, and A2B-/- mice (n = 4). Images are representatives of four experiments with similar results (original magnification, 63×). HO-1 was stained with a specific antibody and appears green, nuclei were stained with DAPI and emerge blue, cytokeratin appears red. Intensity was measured only in the lung tissue, excluding alveolar macrophages. Data are presented as the mean ± SD; n = 6; *P < 0.05 vs. control; #P < 0.05 vs. LPS-treated mice.
In A2A-/- and A2B-/- mice, inflammation and the administration of hemin did not change HO-1 expression in lung tissue. The activation of HO-1 increased HO-1 expression in alveolar macrophages in these mice, explaining our findings from the protein expression experiment and the determination of the activity of the enzyme. We had previously determined that HO-1 is expressed in pulmonary tissue, alveolar macrophages, and PMNs, but the anti-inflammatory effects of HO-1 in acute pulmonary inflammation are based on the expression of the enzyme in the tissue (15).
Anti-inflammatory Effect of HO-1 on PMN Migration Depends on Adenosine Receptor Signaling
In wild-type animals, stimulation or inhibition of HO-1 did not influence PMN counts in animals without inflammation (Figure S3 in Supplementary Material). LPS inhalation significantly increased the migration of PMNs into the intravascular, interstitial, and alveolar space, whereas the stimulation of HO-1 by hemin significantly reduced the PMN influx into all three compartments (all P < 0.05) (Figures 3A,B). Inhibition of HO-1 increased the attachment of PMNs to the endothelium and significantly enhanced PMN migration into the lung interstitium and alveolar space.
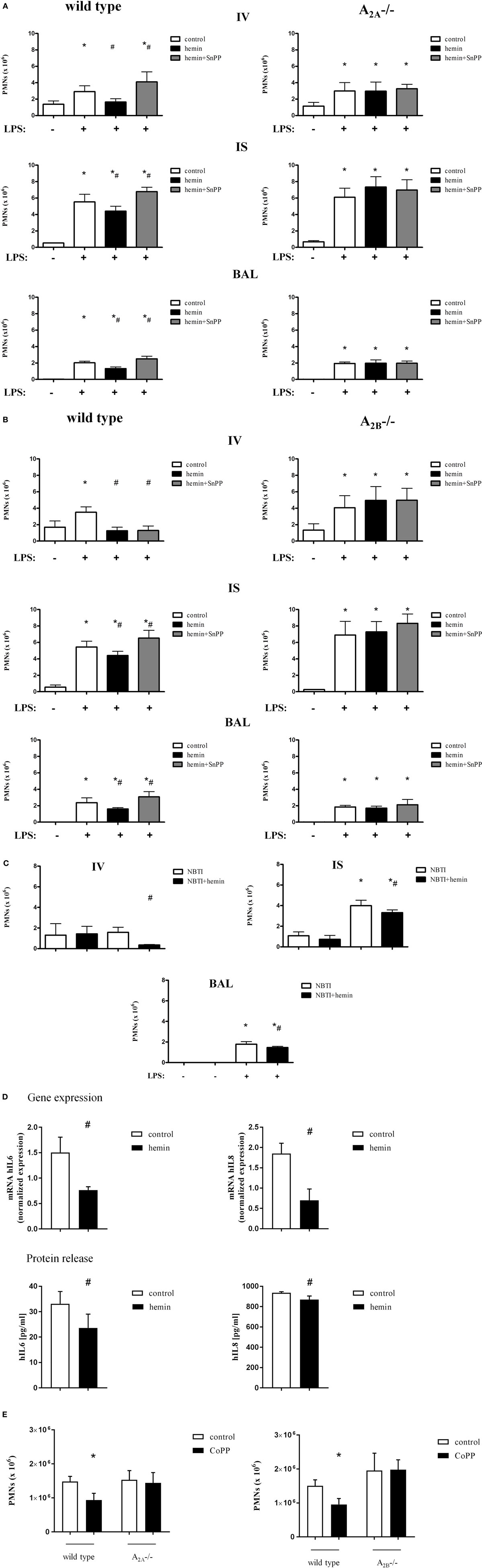
Figure 3. Migration of polymorphonuclear neutrophils (PMNs) into the different compartments of the lung after activation of heme oxygenase (HO)-1. HO-1 was induced by hemin and inhibited by the additional administration of SnPP in wild-type, A2A-/- (A), and A2B-/- mice (B). Mice were treated with LPS, and the migration of PMNs into the different compartments of the lung (IV, intravascular space; IS, interstitial space; BAL, bronchoalveolar space) was evaluated. The cellular uptake of adenosine was inhibited by the specific equilibrative nucleoside transporter 1 (ENT1) inhibitor NBTI and the effect of HO-1 induction by hemin on PMN migration into the different compartments of the lung was evaluated (C). Extracellular adenosine levels were reduced by the depletion of the ectonucleotidases CD39 and CD73 in human pulmonary epithelial cells. The impact of HO-1 stimulation and inflammation with LPS was investigated by the determination of human interleukin 6 and 8 in these cells (D) by RT-PCR. Protein concentrations of both chemokines were detected in the supernatant. PMN counts were determined after LPS inhalation in the BAL of wild-type, A2A-/-, and A2B-/- mice after HO-1 stimulation by CoPP (E) (n = 7–8). Data are presented as the mean ± SD; without LPS inhalation, n = 4 other groups, n = 5–9; *P < 0.05 vs. control without LPS; #P < 0.05 vs. LPS-treated mice.
Without inflammation, neither stimulation nor inhibition of the enzyme influenced the number of PMNs in the lungs of A2A-/- mice (Figure S3A in Supplementary Material). LPS caused a significant increase in PMNs in the intravascular, interstitial, and intra-alveolar space (all P < 0.05) (Figure 3A). In contrast to wild-type animals, stimulation and inhibition of HO-1 did not affect PMN counts in any compartment of the lung.
Next, we investigated the impact of the adenosine receptor A2B on the enzyme HO-1 in this setting (Figure S3B in Supplementary Material). Similar to the wild-type animals of the A2A-/- mice, the wild-type animals of these knockout mice showed a reduction in the number of PMNs in all three compartments of the lung in response to HO-1 stimulation (Figure 3B). Inhibition of the enzyme significantly reversed this effect in the lung interstitial and alveolar space (all P < 0.05). In A2B-/- mice, neither activation nor inhibition of the enzyme affected PMN counts in any of the lung compartments, highlighting the importance of A2A- and A2B-receptor signaling in the anti-inflammatory effects of HO-1.
As previously shown by our group, A2A-/- and A2B-/- mice have an augmented inflammatory reaction to LPS inhalation compared to wild-type animals (8, 27). In the present study, we did not directly compare wild type, A2A-/- and A2B-/- mice. The experimental setting was rather based on the identification of the effects of stimulating HO-1 in these mice. Therefore, the composition of the study groups has been shown to affect the results (9, 28).
Anti-inflammatory Effects of HO-1 Do Not Depend on Extracellular Adenosine Levels
Elevation of extracellular adenosine has anti-inflammatory effects in acute inflammation (29). To investigate if the anti-inflammatory effects of hemin are mainly based on changes in extracellular adenosine levels or are specifically mediated by A2A-/A2B-receptor signaling, we performed additional experiments. The ENT1 is a transmembrane protein that mediates the cellular uptake of adenosine (30). We increased the extracellular levels of adenosine by the specific ENT1 antagonist NBTI and investigated the therapeutic effect of hemin. There were no differences in PMN migration into the lung between the groups without LPS (Figure 3C). After inflammation, the PMN counts were significantly increased in the lung interstitium and the alveolar space and were significantly reduced by the administration of hemin (all P < 0.05), suggesting that the anti-inflammatory effects of hemin are not based on increased adenosine uptake in our model.
To further prove the A2A- and A2B- dependency of the anti-inflammatory effects of HO-1, we investigated the impact of lower extracellular adenosine levels on the anti-inflammatory effects of the enzyme. CD39 and CD73 are the enzymes that convert precursor nucleotides to adenosine; therefore, the knockdown of both enzymes decreases extracellular adenosine detrimental. In our experiments, hemin still exhibited anti-inflammatory effects concerning the expression and the release of IL6 and IL8 after the knockdown of both ectonucleotidases in human pulmonary epithelial cells (Figure 3D). These results confirm that hemin has still anti-inflammatory effects at reduced adenosine levels.
Anti-inflammatory Effects of the Enzyme HO-1 Depend on A2A- and A2B-Receptor Signaling Independent from the HO-1 Stimulus
CoPP, as an alternative to hemin, was used to stimulate HO-1. These separate experiments were performed to exclude that the observed effects of mice lacking the A2A-, respectively, the A2B-receptor are specific for HO-1 induction by hemin. All mice inhaled LPS and PMN migration into the alveolar space was evaluated (Figure 3E). In both wild-type mice, the administration of CoPP decreased migrated PMNs into the alveolar space as described before (23). In A2A-/- and A2B-/- mice, the administration of CoPP did not affect PMN counts, concordantly to our results using hemin for HO-1 stimulation.
HO-1-Reduced Chemokine Release Is Linked to A2A- and A2B-Receptor Signaling
Inhalation of LPS significantly increased TNFα, IL6 and the most potent chemoattractive chemokines for PMNs, CXCL1, and CXL2/3 (all P < 0.05). Hemin reduced all four chemokine levels in both wild-type strains (Figures 4A,B), confirming our results from the in vivo PMN migration assay.
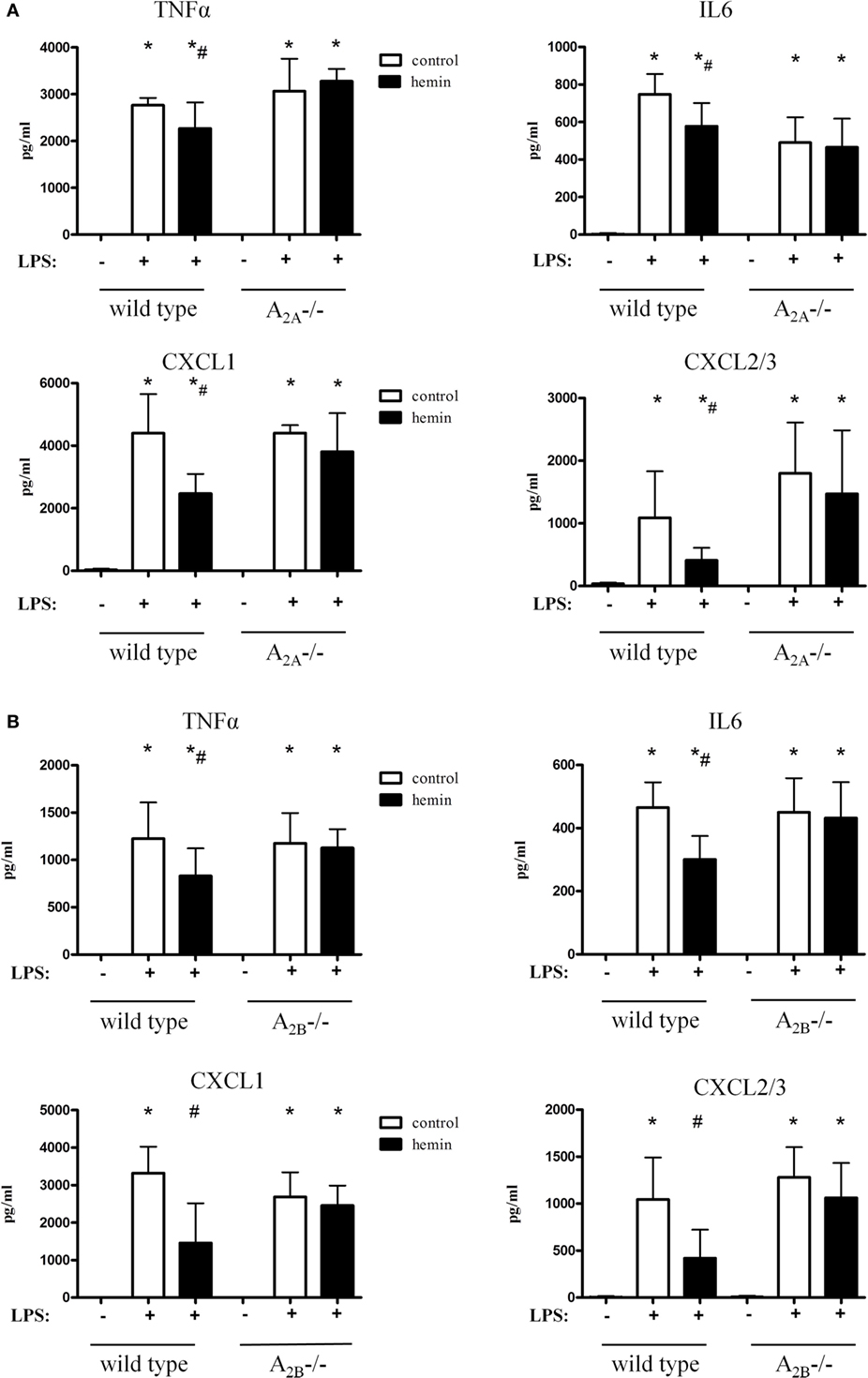
Figure 4. Chemokine release in the bronchoalveolar lavage fluid of mice after activation of heme oxygenase (HO)-1. HO-1 was induced by hemin in wild-type, A2A-/- (A), and A2B-/- mice (B). Levels of TNFα, IL6, and the most PMN chemoattractive chemokines CXCL1 and CXCL2/3 were determined in the BAL of mice. Data are presented as the mean ± SD; without LPS inhalation, n = 4 other groups, n = 5–9; *P < 0.05 vs. control without LPS; #P < 0.05 vs. LPS-treated mice.
In A2A-/- animals, the inflammation caused a significant rise of all chemokines (Figure 4A). Activation of HO-1 did not affect the release of chemokines in these mice, thereby further verifying our findings from the in vivo transmigration assay.
Hemin did not affect the chemokine levels in A2B-/- mice, highlighting and further emphasizing the A2A- and A2B-dependent anti-inflammatory effect of HO-1 (Figure 4B).
HO-1-Reduced Microvascular Permeability Depends on A2A- and A2B-Receptor Signaling
Extravasation of Evans blue as an indicator for the permeability of albumin and, therefore, a marker for capillary leakage was assessed (Figures 5A,B). Stimulation and inhibition of the enzyme HO-1 without inflammation did not affect microvascular permeability (Figure S4 in Supplementary Material). LPS inhalation increased Evans blue significantly, whereas the stimulation of HO-1 preserved capillary leakage almost to baseline (all P < 0.05) (Figure 5A). As a control, inhibition of the enzyme was shown to further increase the microvascular permeability in wild-type mice.
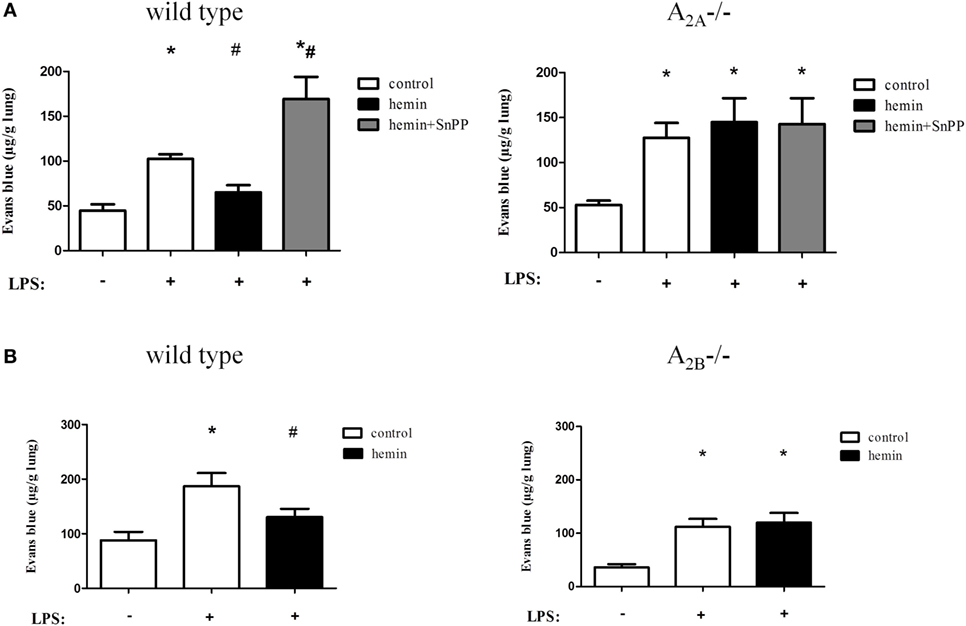
Figure 5. Anti-inflammatory effects of heme oxygenase (HO)-1 on microvascular permeability. Capillary leakage was assessed by Evans blue extravasation, and the influence of HO-1 in wild-type, A2A-/- (A), and A2B-/- (B) mice was investigated. HO-1 was induced by hemin and inhibited by the additional administration of SnPP. Data are presented as the mean ± SD; without LPS inhalation, n ≥ 4 other groups, n = 5–8; *P < 0.05 vs. control without LPS; #P < 0.05 vs. LPS-treated mice.
Inflammation increased the extravasation of Evans blue in A2A-/- (Figure 5A) and A2B-/- mice (Figure 5B). Activation and inhibition of HO-1 did not affect capillary leakage in A2A-/- mice. We also demonstrated that HO-1 activation in A2B-/- mice had no effect on microvascular permeability, highlighting the pivotal role of A2A-/- and A2B-/- receptor signaling in conjunction with the enzyme HO-1 in the appearance of the second hallmark of acute pulmonary inflammation.
In Vitro PMN Migration Assay
In this assay, isolated human PMNs migrate through a monolayer of pulmonary epithelial/endothelial cells along a chemotactic gradient. Since the anti-inflammatory effect of HO-1 in the setting of acute pulmonary inflammation depends on the expression of the enzyme in lung tissue (15), the pulmonary epithelium/endothelium was treated with hemin and specific A2A/A2B-receptor antagonists. Hemin treatment of epithelial (Figure 6A) and endothelial cells (Figure 6B) significantly reduced the number of migrated PMNs (all P < 0.05). After treatment with the specific A2A/A2B-receptor antagonists, the stimulation of HO-1 was ineffective in reducing the PMN influx in both cell lines, confirming our in vivo experiments.
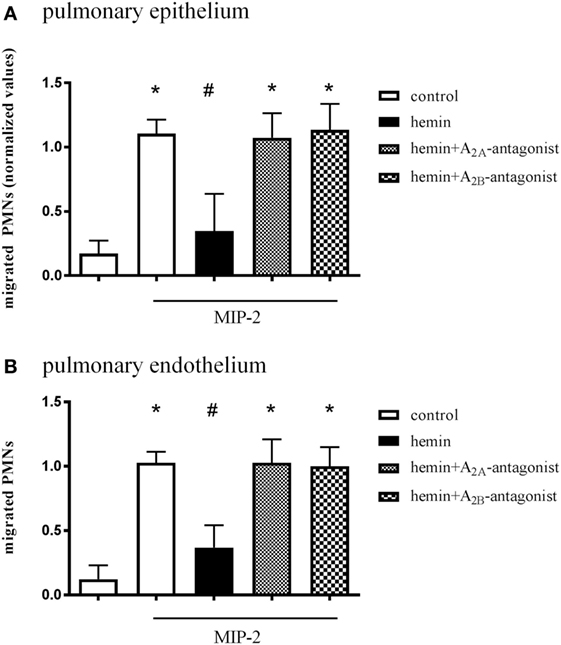
Figure 6. Effect of A2A/A2B-receptor signaling on pulmonary epithelium and endothelium. In vitro transmigration assay of human PMNs through a pulmonary epithelial (A) and endothelial (B) monolayer. Epithelium and endothelium were treated with hemin and specific A2A/A2B-receptor antagonists, and the migration of PMNs through a monolayer of human epithelium/endothelium was measured. The migration of PMNs was initiated via the chemokine MIP-2 (CXCL2/3) at indicated wells. Data are presented as the mean ± SD; n = 6–12; *P < 0.05 vs. control; #P < 0.05 vs. MIP-2-treated wells.
Discussion
Gene polymorphism in the HO-1 gene promoter (HMOX1) has been shown to be associated with increased HO-1 levels and a reduced risk of acute pulmonary distress syndrome (31). HO-1, the inducible form of HO, is upregulated in tissue during cellular stress and influences inflammation in different settings (14, 32–35).
However, the mechanism by which HO-1 elicits this cytoprotection remains incompletely understood but might be a combination of the removal of heme (a pro-oxidant iron chelate) and the enzymatic generation of biologically active end products with anti-inflammatory effects (36). The end products include iron, biliverdin, and carbon monoxide (CO). CO, as an inhaled gas, has been used to induce HO-1 with anti-inflammatory effects in several studies including those on endotoxemia (37), ventilator-induced lung injury (38), and ischemia/reperfusion injury (39).
In the presented study, we used hemin to induce HO-1. In a previous study from our group, we demonstrated the impact of this agent on the anti-inflammatory effects in acute pulmonary inflammation in terms of PMN migration, microvascular permeability, chemokine release, oxidative burst, and the formation of stress fibers (15). Additionally, we highlighted the impact of the topical administration of hemin with a significantly lower required dose and simultaneously more anti-inflammatory effects. These observations were in accordance with the actual literature, where aerosol therapy in critical care medicine is considered fundamental (40) and there is convincing evidence that nebulized administration of agents elicits higher concentrations and, therefore, better efficiency than systemic application (41–43). Additionally, typical side effects of systemic hemin administration such as headaches, nausea, and vomiting might be less prominent using the topical administration (44). Furthermore, hemin has been verified to induce the expression of HO-1 in humans (44) and is already successfully in use for the treatment of acute intermittent porphyria (45). To ensure a clinically relevant study, we also used hemin for the induction of HO-1 in the presented study.
Here, we confirmed the anti-inflammatory effects of HO-1 in acute pulmonary inflammation in two different mouse strains. We used the common inbred strain C57BL/6, which are almost genetically homogenous, and the outbred CD1 mouse strain, which has a higher genetic diversity. CD1 mice are outbred mice with a complex genetic history, comparable to a human founder population (46). Recent literature on acute pulmonary inflammation notes the importance of genetic variation in this disease (47, 48). We, therefore, demonstrated the pivotal role of hemin as an inductor of HO-1 in a condition of acute pulmonary inflammation independent of slight genetic variations. By the additional inhibition of HO-1 in selected experiments, which resulted in aggravation of the inflammation, we verified that the detected results were based on the enzyme HO-1.
Adenosine receptor signaling plays a crucial anti-inflammatory role in inflammation by protecting tissue from inflammation-related damage (49). These receptors modulate chemokine production and secretion (50). Our data link the anti-inflammatory effects of HO-1 on PMN migration, chemokine release, and microvascular permeability to A2A/2B-signaling. Extracellular adenosine uptake plays a pivotal role in pulmonary inflammation (51). In the presented study, HO-1 still had anti-inflammatory effects after the specific inhibition of the cellular adenosine uptake by the transmembrane protein ENT1 and, therefore, an elevation of extracellular adenosine. HO-1 stimulation also exhibited anti-inflammatory effects at reduced adenosine levels, suggesting rather a direct effect of stimulating HO-1 on A2A/2B-receptors than an adenosine-dependent mechanism.
The link between the adenosine system and HO-1 is in line with the findings of Reichelt et al. (20). They investigated the influence of a nonspecific adenosine receptor agonist on the expression of HO-1 and the enzyme activity in ischemic hearts of mice. Weis et al. showed that downregulation of HO-1 resulted in a decreased expression of the anti-inflammatory A2A-receptor in macrophages (21). Additionally, adenosine was found to perceptibly induce HO-1 and attenuate LPS-induced TNFα levels via the A2A-receptor (19).
The expression of HO-1 is induced by hemin and a multiplicity of pro-oxidant stimuli including oxidative stress, inflammatory cytokines, heat shock, and ischemia-reperfusion (34, 37, 52). Therefore, the transcription of HO-1 can be activated by many stimuli. The HO-1 promoter has a plurality of different response elements, which bind the corresponding activated transcription factors [reviewed in Ref. (53)]. Therefore, the current literature suggests that the transcription factor activity of HO-1 through its multiple stimulators is indirectly regulated through activation of signaling cascades that are dependent on protein (de)phosphorylation, reduction-oxidation (redox) reactions, or both. A variety of protein kinases A and G, tyrosine kinases and the enzyme of the PI3K/AKT/Nrf2 (phosphatidylinositide 3-kinase, AKT, nuclear factor (erythroid-derived 2)-like 2) pathways are involved in the activation of HMOX1 (HO-1 gene) (54–56). Furthermore, HO-1 expression in macrophages was enhanced after LPS stimulation when PKA was additionally inhibited (57). As one of the principle mediators in this setting, the MAPK (mitogen-activated protein kinase) cascades seem to be involved. MAPKs are a ubiquitously expressed signal transduction supersystem that regulates countless cellular processes such as growth, motility, differentiation, and apoptosis. HO-1 inducers are hypothesized to activate the MAPKs. The MAPK supersystem consists of three subdivided primary signaling pathways that can then induce the following transcription factors: HSF1 (heat shock transcription factor), Nfr2 (nuclear factor erythroid 2-related factor 2), AP (activator protein)-1, and NFκB (nuclear factor). These transcription activators can then induce the HO-1 gene HMOX1 either directly or through an association with another DNA-binding protein (54). Still, this pathway is quite complex and much remains unknown. For example, some HO-1 activators such as cadmium activate all three MAPK pathways (58), while others are more specific (59). However, the activation of a particular MAPK does not automatically lead to HMOX1 induction, highlighting the complexity of the system.
Al-Huseini et al. described the signaling cascade for HO-1 in dendritic cells (60). They found that HO-1-regulated dendritic cell maturation and function in conditions of inflammation are mediated via p38-MAPK. As described above, this MAPK can again in turn induce the transcription of HO-1. Sepsis or LPS-induced autophagy prevented hepatocellular death in part via an HO-1 p38-MAPK signaling pathway (61).
The signaling cascades of adenosine are also quite complex and actually include several possibilities in different settings but adenosine has also been shown to act via multiple signaling pathways in the same setting. For example, Che et al. demonstrated that the A2A-receptor mediated increased collagen production in hepatic fibrosis via PKA and p38-MAPK (62). In Alzheimer’s disease, adenosine controls the neurotoxicity of the β-amyloid peptide via the p38-MAPK but not the PKA-pathway (63).
Therefore, the signaling pathways of HO-1 and adenosine intersect at PKA and MAPKs but are both still incompletely understood.
Our studies suggest that, in our model, HO-1 stimulation is not efficient if the adenosine receptor A2A or A2B is not expressed and that this might be regulated on a transcriptional level.
Furthermore, the presented link between HO-1 and the adenosine receptors results in new insights on the interaction and connection of different systems involved in terms of inflammation (Figure 7).
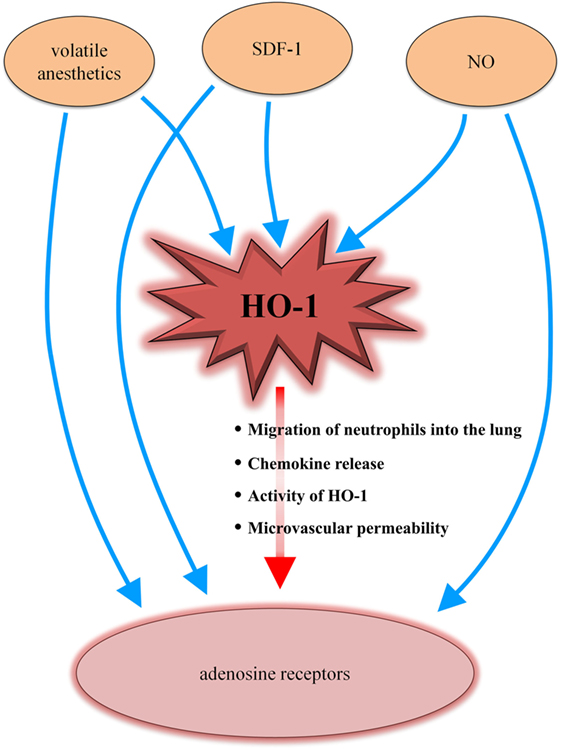
Figure 7. Heme oxygenase (HO)-1/adenosine receptor axis in the context of different inflammatory-mediated systems. Volatile anesthetics influence the HO-1 enzyme as well as adenosine receptors. The chemokine stromal cell-derived factor (SDF)-1 and nitric oxide (NO) have been shown to interact with HO-1 and adenosine receptors. Our data now complete the puzzle by adding a connection between HO-1 and adenosine receptors (red arrow), explaining the findings of several other studies.
Volatile anesthetics, such as sevoflurane or its precursor isoflurane, induce HO-1 (64, 65). Additionally, the adenosine receptors play a pivotal mechanistic role in terms of the volatile anesthetic-associated effects (66, 67). The chemokine stromal cell-derived factor (SDF)-1 induces HO-1 (68) and the anti-inflammatory effects of SDF-1 have been linked to adenosine receptor signaling (69). HO-1 expression has been shown to be modulated by nitric oxide (NO) (70). NO is connected to the adenosine pathway, which is further described in the ALANO pathway (adenosine/l-arginine/NO). Adenosine activates adenosine receptors, resulting in increased l-arginine transport and NO synthesis (71, 72). Our results now link these different studies together by presenting a new connection between HO-1 and the adenosine receptors.
The importance of the A2A/2B-dependent signaling for the anti-inflammatory effects of HO-1 further highlights the clinical importance of the presented study. Recent literature has demanded the identification of subgroups of patients to determine specific treatment strategies for acute pulmonary inflammation (1, 3). Since septic patients have altered adenosine receptor expression and ligand affinity (22), the adenosine receptor expression and ligand affinity of these patients should be explored before a therapy with hemin is considered.
Conclusion
Our data link the anti-inflammatory effects of HO-1 to adenosine A2A/2B-receptor signaling and, therefore, may explain why targeting HO-1 in acute pulmonary inflammation has failed to prove effective in some patients. Furthermore, the correlation between HO-1 and adenosine receptor signaling provides a deeper insight into inflammatory signaling and represents a new correlation that can explain the link between other enzymatic/chemotactic settings under conditions of inflammation.
Ethics Statement
All animal protocols were approved by the Animal Care and Use Committee of the University of Tübingen.
Author Contributions
All authors made substantial contributions to this article. CZ, K-CN, and FK mainly contributed by participation in the data acquisition, analysis, and interpretation. FK and JR contributed to the conception and design of the study, as well as the analysis and interpretation of the data. FK and JR wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
A2B gene-deficient mice were kindly provided by Professor Katya Ravid (Boston University, School of Medicine, Department of Biochemistry, Boston, MA, USA).
Funding
This work was supported by the Else Kröner-Fresenius-Stiftung 2014_A148 (to FK) and the fortune program F1211340 (to K-CN).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01874/full#supplementary-material.
References
1. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest (2012) 122(8):2731–40. doi:10.1172/JCI60331
2. Herridge MS, Tansey CM, Matte A, Tomlinson G, Diaz-Granados N, Cooper A, et al. Functional disability 5 years after acute respiratory distress syndrome. N Engl J Med (2011) 364(14):1293–304. doi:10.1056/NEJMoa1011802
3. Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J (2011) 87(1031):612–22. doi:10.1136/pgmj.2011.118398
4. Razavi HM, Wang le F, Weicker S, Rohan M, Law C, McCormack DG, et al. Pulmonary neutrophil infiltration in murine sepsis: role of inducible nitric oxide synthase. Am J Respir Crit Care Med (2004) 170(3):227–33. doi:10.1164/rccm.200306-846OC
5. Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol (2007) 178(12):8127–37. doi:10.4049/jimmunol.178.12.8127
6. Ngamsri KC, Wagner R, Vollmer I, Stark S, Reutershan J. Adenosine receptor A1 regulates polymorphonuclear cell trafficking and microvascular permeability in lipopolysaccharide-induced lung injury. J Immunol (2010) 185(7):4374–84. doi:10.4049/jimmunol.1000433
7. Wagner R, Ngamsri KC, Stark S, Vollmer I, Reutershan J. Adenosine receptor A3 is a critical mediator in LPS-induced pulmonary inflammation. Am J Physiol Lung Cell Mol Physiol (2010) 299(4):L502–12. doi:10.1152/ajplung.00083.2010
8. Konrad FM, Witte E, Vollmer I, Stark S, Reutershan J. Adenosine receptor A2b on hematopoietic cells mediates LPS-induced migration of PMNs into the lung interstitium. Am J Physiol Lung Cell Mol Physiol (2012) 303(5):L425–38. doi:10.1152/ajplung.00387.2011
9. Konrad FM, Neudeck G, Vollmer I, Ngamsri KC, Thiel M, Reutershan J. Protective effects of pentoxifylline in pulmonary inflammation are adenosine receptor A2A dependent. FASEB J (2013) 27(9):3524–35. doi:10.1096/fj.13-228122
10. Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) (2009) 24:298–306. doi:10.1152/physiol.00022.2009
11. Densmore JC, Schaid TR, Jeziorczak PM, Medhora M, Audi S, Nayak S, et al. Lung injury pathways: adenosine receptor 2B signaling limits development of ischemic bronchiolitis obliterans organizing pneumonia. Exp Lung Res (2017) 43(1):38–48. doi:10.1080/01902148.2017.1286697
12. Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J Immunol (2015) 195(4):1815–24. doi:10.4049/jimmunol.1401957
13. Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A (1997) 94(20):10919–24. doi:10.1073/pnas.94.20.10919
14. Wu SY, Tang SE, Ko FC, Wu GC, Huang KL, Chu SJ. Valproic acid attenuates acute lung injury induced by ischemia-reperfusion in rats. Anesthesiology (2015) 122(6):1327–37. doi:10.1097/ALN.0000000000000618
15. Konrad FM, Knausberg U, Hone R, Ngamsri KC, Reutershan J. Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol (2016) 9(1):98–111. doi:10.1038/mi.2015.39
16. Mayr FB, Spiel A, Leitner J, Marsik C, Germann P, Ullrich R, et al. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. Am J Respir Crit Care Med (2005) 171(4):354–60. doi:10.1164/rccm.200404-446OC
17. Bathoorn E, Slebos DJ, Postma DS, Koeter GH, van Oosterhout AJ, van der Toorn M, et al. Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J (2007) 30(6):1131–7. doi:10.1183/09031936.00163206
18. Ryter SW, Choi AM. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res (2016) 167(1):7–34. doi:10.1016/j.trsl.2015.06.011
19. Haschemi A, Wagner O, Marculescu R, Wegiel B, Robson SC, Gagliani N, et al. Cross-regulation of carbon monoxide and the adenosine A2a receptor in macrophages. J Immunol (2007) 178(9):5921–9. doi:10.4049/jimmunol.178.9.5921
20. Reichelt ME, Shanu A, Willems L, Witting PK, Ellis NA, Blackburn MR, et al. Endogenous adenosine selectively modulates oxidant stress via the A(1) receptor in ischemic hearts. Antioxid Redox Signal (2009) 11(11):2641–50. doi:10.1089/ars.2009.2644
21. Weis N, Weigert A, von Knethen A, Brune B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol Biol Cell (2009) 20(5):1280–8. doi:10.1091/mbc.E08-10-1005
22. Kreth S, Kaufmann I, Ledderose C, Luchting B, Thiel M. Reduced ligand affinity leads to an impaired function of the adenosine A2A receptor of human granulocytes in sepsis. J Cell Mol Med (2009) 13(5):985–94. doi:10.1111/j.1582-4934.2008.00530.x
23. Konrad FM, Braun S, Ngamsri KC, Vollmer I, Reutershan J. Heme oxygenase-1 attenuates acute pulmonary inflammation by decreasing the release of segmented neutrophils from the bone marrow. Am J Physiol Lung Cell Mol Physiol (2014) 307(9):L707–17. doi:10.1152/ajplung.00145.2014
24. Reutershan J, Basit A, Galkina EV, Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2005) 289(5):L807–15. doi:10.1152/ajplung.00477.2004
25. Reutershan J, Stockton R, Zarbock A, Sullivan GW, Chang D, Scott D, et al. Blocking p21-activated kinase reduces lipopolysaccharide-induced acute lung injury by preventing polymorphonuclear leukocyte infiltration. Am J Respir Crit Care Med (2007) 175(10):1027–35. doi:10.1164/rccm.200612-1822OC
26. Wang LF, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, et al. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med (2002) 165(12):1634–9. doi:10.1164/rccm.2110017
27. Reutershan J, Cagnina RE, Chang D, Linden J, Ley K. Therapeutic anti-inflammatory effects of myeloid cell adenosine receptor A2a stimulation in lipopolysaccharide-induced lung injury. J Immunol (2007) 179(2):1254–63. doi:10.4049/jimmunol.179.2.1254
28. Csoka B, Koscso B, Toro G, Kokai E, Virag L, Nemeth ZH, et al. A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes (2014) 63(3):850–66. doi:10.2337/db13-0573
29. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med (2005) 202(11):1493–505. doi:10.1084/jem.20050177
30. Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch (2004) 447(5):735–43. doi:10.1007/s00424-003-1103-2
31. Sheu CC, Zhai RH, Wang ZX, Gong MN, Tejera P, Chen F, et al. Heme oxygenase-1 microsatellite polymorphism and haplotypes are associated with the development of acute respiratory distress syndrome. Intensive Care Med (2009) 35(8):1343–51. doi:10.1007/s00134-009-1504-6
32. Chen X, Wei SY, Li JS, Zhang QF, Wang YX, Zhao SL, et al. Overexpression of heme oxygenase-1 prevents renal interstitial inflammation and fibrosis induced by unilateral ureter obstruction. PLoS One (2016) 11(1):e0147084. doi:10.1371/journal.pone.0147084
33. Ren H, Hao JL, Liu TT, Zhang DY, Lv HM, Song E, et al. Hesperetin suppresses inflammatory responses in lipopolysaccharide-induced RAW 264.7 cells via the inhibition of NF-kappa B and activation of Nrf2/HO-1 pathways. Inflammation (2016) 39(3):964–73. doi:10.1007/s10753-016-0311-9
34. Wu SH, Chen XQ, Lu J, Wang MJ. BML-111 attenuates renal ischemia/reperfusion injury via peroxisome proliferator-activated receptor-alpha-regulated heme oxygenase-1. Inflammation (2016) 39(2):611–24. doi:10.1007/s10753-015-0286-y
35. Zhang B, Xie SJ, Su ZL, Song SY, Xu H, Chen G, et al. Heme oxygenase-1 induction attenuates imiquimod-induced psoriasiform inflammation by negative regulation of Stat3 signaling. Sci Rep (2016) 6:21132. doi:10.1038/srep21132
36. Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity – heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med (2000) 28(2):289–309. doi:10.1016/S0891-5849(99)00223-3
37. Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med (2000) 6(4):422–8. doi:10.1038/74680
38. Hoetzel A, Schmidt R, Vallbracht S, Goebel U, Dolinay T, Kim HP, et al. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med (2009) 37(5):1708–15. doi:10.1097/CCM.0b013e31819efa31
39. Amersi F, Shen XD, Anselmo D, Melinek J, Iyer S, Southard DJ, et al. Ex vivo exposure to carbon monoxide prevents hepatic ischemia/reperfusion injury through p38 MAP kinase pathway. Hepatology (2002) 35(4):815–23. doi:10.1053/jhep.2002.32467
40. Dhanani J, Fraser JF, Chan HK, Rello J, Cohen J, Roberts JA. Fundamentals of aerosol therapy in critical care. Crit Care (2016) 20:269. doi:10.1186/s13054-016-1448-5
41. Goldstein I, Wallet F, Nicolas-Robin A, Ferrari F, Marquette CH, Rouby JJ. Lung deposition and efficiency of nebulized amikacin during Escherichia coli pneumonia in ventilated piglets. Am J Respir Crit Care Med (2002) 166(10):1375–81. doi:10.1164/rccm.200204-363OC
42. Lu Q, Girardi C, Zhang M, Bouhemad B, Louchahi K, Petitjean O, et al. Nebulized and intravenous colistin in experimental pneumonia caused by Pseudomonas aeruginosa. Intensive Care Med (2010) 36(7):1147–55. doi:10.1007/s00134-010-1879-4
43. Lu Q, Luo RB, Bodin L, Yang JX, Zahr N, Aubry A, et al. Efficacy of high-dose nebulized colistin in ventilator-associated pneumonia caused by multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Anesthesiology (2012) 117(6):1335–47. doi:10.1097/ALN.0b013e31827515de
44. Bharucha AE, Kulkarni A, Choi KM, Camilleri M, Lempke M, Brunn GJ, et al. First-in-human study demonstrating pharmacological activation of heme oxygenase-1 in humans. Clin Pharmacol Ther (2010) 87(2):187–90. doi:10.1038/clpt.2009.221
45. Siegert SWK, Holt RJ. Physicochemical properties, pharmacokinetics, and pharmacodynamics of intravenous hematin: a literature review. Adv Ther (2008) 25(9):842–57. doi:10.1007/s12325-008-0094-y
46. Aldinger KA, Sokoloff G, Rosenberg DM, Palmer AA, Millen KJ. Genetic variation and population substructure in outbred CD-1 mice: implications for genome-wide association studies. PLoS One (2009) 4(3):e4729. doi:10.1371/journal.pone.0004729
47. Sapru A, Wiemels JL, Witte JS, Ware LB, Matthay MA. Acute lung injury and the coagulation pathway: potential role of gene polymorphisms in the protein C and fibrinolytic pathways. Intensive Care Med (2006) 32(9):1293–303. doi:10.1007/s00134-006-0223-5
48. Sapru A, Liu KD, Wiemels J, Hansen H, Pawlikowska L, Poon A, et al. Association of common genetic variation in the protein C pathway genes with clinical outcomes in acute respiratory distress syndrome. Crit Care (2016) 20:151. doi:10.1186/s13054-016-1330-5
49. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414(6866):916–20. doi:10.1038/414916a
50. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature (2014) 509(7500):310–7. doi:10.1038/nature13085
51. Eltzschig HK. Extracellular adenosine signaling in molecular medicine. J Mol Med (Berl) (2013) 91(2):141–6. doi:10.1007/s00109-013-0999-z
52. Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol (2003) 24(8):449–55. doi:10.1016/S1471-4906(03)00181-9
53. Maines MD. The heme oxygenase system: update 2005. Antioxid Redox Signal (2005) 7(11–12):1761–6. doi:10.1089/ars.2005.7.1761
54. Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol (2007) 36(2):166–74. doi:10.1165/rcmb.2006-0340TR
55. Wu J, Pan X, Fu H, Zheng Y, Dai Y, Yin Y, et al. Effect of curcumin on glycerol-induced acute kidney injury in rats. Sci Rep (2017) 7(1):10114. doi:10.1038/s41598-017-10693-4
56. Ye J, Piao H, Jiang J, Jin G, Zheng M, Yang J, et al. Polydatin inhibits mast cell-mediated allergic inflammation by targeting PI3K/Akt, MAPK, NF-kappaB and Nrf2/HO-1 pathways. Sci Rep (2017) 7(1):11895. doi:10.1038/s41598-017-12252-3
57. Lee JH, Jung NH, Lee BH, Kim SH, Jun JH. Suppression of heme oxygenase-1 by prostaglandin E2-protein kinase A-A-kinase anchoring protein signaling is central for augmented cyclooxygenase-2 expression in lipopolysaccharide-stimulated RAW 264.7 macrophages. Allergy Asthma Immunol Res (2013) 5(5):329–36. doi:10.4168/aair.2013.5.5.329
58. Alam J, Wicks C, Stewart D, Gong P, Touchard C, Otterbein S, et al. Mechanism of heme oxygenase-1 gene activation by cadmium in MCF-7 mammary epithelial cells. Role of p38 kinase and Nrf2 transcription factor. J Biol Chem (2000) 275(36):27694–702. doi:10.1074/jbc.M004729200
59. Ning W, Song R, Li C, Park E, Mohsenin A, Choi AM, et al. TGF-beta1 stimulates HO-1 via the p38 mitogen-activated protein kinase in A549 pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol (2002) 283(5):L1094–102. doi:10.1152/ajplung.00151.2002
60. Al-Huseini LM, Aw Yeang HX, Hamdam JM, Sethu S, Alhumeed N, Wong W, et al. Heme oxygenase-1 regulates dendritic cell function through modulation of p38 MAPK-CREB/ATF1 signaling. J Biol Chem (2014) 289(23):16442–51. doi:10.1074/jbc.M113.532069
61. Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology (2011) 53(6):2053–62. doi:10.1002/hep.24324
62. Che J, Chan ES, Cronstein BN. Adenosine A2A receptor occupancy stimulates collagen expression by hepatic stellate cells via pathways involving protein kinase A, Src, and extracellular signal-regulated kinases 1/2 signaling cascade or p38 mitogen-activated protein kinase signaling pathway. Mol Pharmacol (2007) 72(6):1626–36. doi:10.1124/mol.107.038760
63. Canas PM, Porciuncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci (2009) 29(47):14741–51. doi:10.1523/JNEUROSCI.3728-09.2009
64. Hoetzel A, Leitz D, Schmidt R, Tritschler E, Bauer I, Loop T, et al. Mechanism of hepatic heme oxygenase-1 induction by isoflurane. Anesthesiology (2006) 104(1):101–9. doi:10.1097/00000542-200601000-00016
65. Schmidt R, Tritschler E, Hoetzel A, Loop T, Humar M, Halverscheid L, et al. Heme oxygenase-1 induction by the clinically used anesthetic isoflurane protects rat livers from ischemia/reperfusion injury. Ann Surg (2007) 245(6):931–42. doi:10.1097/01.sla.0000256891.45790.4d
66. Tas PW, Eisemann C, Roewer N. Indirect activation of adenosine A1 receptors in cultured rat hippocampal neurons by volatile anaesthetics. Eur J Anaesthesiol (2005) 22(9):694–702. doi:10.1017/S0265021505001158
67. Granja TF, Kohler D, Schad J, de Oliveira CB, Konrad F, Hoch-Gutbrod M, et al. Adenosine receptor Adora2b plays a mechanistic role in the protective effect of the volatile anesthetic sevoflurane during liver ischemia/reperfusion. Anesthesiology (2016) 125(3):547–60. doi:10.1097/ALN.0000000000001234
68. Deshane J, Chen S, Caballero S, Grochot-Przeczek A, Was H, Li Calzi S, et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med (2007) 204(3):605–18. doi:10.1084/jem.20061609
69. Ferrari D, Gulinelli S, Salvestrini V, Lucchetti G, Zini R, Manfredini R, et al. Purinergic stimulation of human mesenchymal stem cells potentiates their chemotactic response to CXCL12 and increases the homing capacity and production of proinflammatory cytokines. Exp Hematol (2011) 39(3):360–74, 374.e1–5. doi:10.1016/j.exphem.2010.12.001
70. Hoetzel A, Welle A, Schmidt R, Loop T, Humar M, Ryter SW, et al. Nitric oxide-deficiency regulates hepatic heme oxygenase-1. Nitric Oxide (2008) 18(1):61–9. doi:10.1016/j.niox.2007.10.003
71. Farias M, Puebla C, Westermeier F, Jo MJ, Pastor-Anglada M, Casanello P, et al. Nitric oxide reduces SLC29A1 promoter activity and adenosine transport involving transcription factor complex hCHOP-C/EBP alpha in human umbilical vein endothelial cells from gestational diabetes. Cardiovasc Res (2010) 86(1):45–54. doi:10.1093/cvr/cvp410
Keywords: heme oxygenase-1, pulmonary inflammation, neutrophil, PMN, hemin
Citation: Konrad FM, Zwergel C, Ngamsri K-C and Reutershan J (2017) Anti-inflammatory Effects of Heme Oxygenase-1 Depend on Adenosine A2A- and A2B-Receptor Signaling in Acute Pulmonary Inflammation. Front. Immunol. 8:1874. doi: 10.3389/fimmu.2017.01874
Received: 05 September 2017; Accepted: 08 December 2017;
Published: 20 December 2017
Edited by:
Fabrice Cognasse, Etablissement Français du Sang Rhône-Alpes-Auvergne and GIMAP EA3064 Université de Lyon, FranceReviewed by:
Saurabh Aggarwal, University of Alabama at Birmingham, United StatesSusan M. Bueno, Pontificia Universidad Católica de Chile, Chile
Copyright: © 2017 Konrad, Zwergel, Ngamsri and Reutershan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jörg Reutershan, am9lcmcucmV1dGVyc2hhbkBrbGluaWt1bS1iYXlyZXV0aC5kZQ==