 Peter Vaupel
Peter Vaupel Gabriele Multhoff
Gabriele Multhoff- Department of Radiation Oncology, Klinikum rechts der Isar der Technischen Universität München, Munich, Germany
In this minireview, we aim to highlight key factors of the tumor microenvironment, including adenosine, lactate, acidosis, vascular endothelial growth factor, phosphatidylserine, high extracellular K+ levels, and tumor hypoxia with respect to antitumor immune functions. Most solid tumors have an immature chaotic microvasculature that results in tumor hypoxia. Hypoxia is a key determinant of tumor aggressiveness and therapy resistance and hypoxia-related gene products can thwart antitumor immune responses.
Introduction
Tumor angiogenesis, a key “hallmark of cancer” (1), is necessary for a sufficient supply of solid tumors with nutrients and oxygen and removal of metabolic waste products during tumor progression (2). The tumor as well as its “immunological” microenvironment including specialized lymphocytes and myeloid cells which are attracted by tumor cells contribute to vessel growth primarily via vascular endothelial growth factor (VEGF), but also exert immunosuppressive activities. Due to rapid tumor growth and an overload of proangiogenic factors, tumor vessels are often immature with poorly interconnected endothelial cells, leaky membranes, dead ends, and loosely attached pericytes (3). This chaotic tumor microvasculature can result in an inefficient blood-borne delivery, uneven distribution, and compromised penetration and distribution of macromolecules (e.g., antibodies, cytokines) and immune cells from tumor microvessels through the interstitial space to cancer cells. To reach all viable tumor cells in an effective dose, macromolecules and antitumor immune cells are negatively affected by several barriers to vascular, transvascular, and interstitial transport. A detailed description of these barriers has been previously presented (4–8).
Vascular transport includes the convective transport within abnormal vascular networks, significant arteriovenous shunt perfusion, and pronounced spatio-temporal heterogeneities. Transvascular transport of macromolecules is hindered by an impaired transluminal convective transport (extravasation) caused by an elevated interstitial fluid pressure (IFP 5 to 40 mmHg in tumors vs. −3 to +1 mmHg in most normal tissues) and intravasation back to the vascular compartment due to critically high IFPs, i.e., “back convection” from the interstitial space into the circulation. Interstitial hypertension additionally hinders interstitial transport of antibodies, delivery of cytokines and immune cells with antitumor activity through stopping, and even reverting of the microvascular blood flow, diversion of blood flow from the center to the periphery of tumors. Enlarged interstitial volumes, increased interstitial transport distances, and a reduction of the hydrostatic pressure gradient between intravascular space and interstitial compartment further impair an adequate delivery [for a review see Ref. (8)].
Newly formed microvessels in most solid tumors do not conform to the morphology of the vasculature of normal tissues. Tumor microvessels show many structural and functional abnormalities (5). These abnormalities not only directly or indirectly cause the abovementioned “biophysical” barriers for delivery of antitumor immune therapies but also have a negative impact on oxygen delivery to solid tumors (with substantial spatial and temporal heterogeneities). As a consequence, the metabolic tumor microenvironment (TME) is characterized by a critical oxygen (O2) depletion (hypoxia, anoxia), extracellular acidosis, substantially elevated adenosine (ADO) and lactate concentrations, and nutrient deprivation (4–8).
Hypoxia crucially contributes to genetic instability, intratumoral heterogeneity, malignant progression, tumor stem cell maintenance, sustained angiogenesis, development of treatment resistance, and metabolic reprogramming upon triggering the switch to HIF-1α-dependent phenotypes (9–11). In addition, tumor hypoxia/hypoxic stress and downstream effects of HIF-1α-activation can serve as major drivers for recruitment, activation, polarization, and expansion of immune-suppressive stromal cell populations causing an impediment to antitumor (innate and adaptive) immunity and cancer immunotherapy. In this minireview, the role of major “HIF-downstream factors” of the TME and egress of intracellular K+ upon tumor cell death inhibiting local functions and survival of immune cells, thus leading to tumor immune escape, will be discussed.
Hypoxic Stress Factors Counteracting Local Antitumor Immune Responses
Earlier in vivo investigations of the effects of components of the TME on gene expression have elicited a substantial downregulation of a large number of microRNAs that are associated with the regulation and function of the immune system in hypoxic tumor areas (e.g., accumulation of cytotoxic CD8+ T cells in highly vascularized, normoxic areas vs. exclusion of cytotoxic CD8+ T cells from viable hypoxic tumor areas in vivo) raising the possibility that hypoxic tumor regions represent an immune-privileged tumor niche (12).
Major factors involved in local immune-suppressive actions exerted by hypoxia-/HIF-driven downstream factors include (a) ADO generation and accumulation in the extracellular space, (b) lactate accumulation, (c) extracellular acidosis, (d) overexpression of VEGF and activation of VEGF receptor (VEGF-R), and (e) externalization of phosphatidylserine (PS) on the outer membrane leaflet (13).
ADO Inhibits Antitumor Immune Responses
Adenosinergic effects on cancer, stromal, and immune cells have been summarized recently (14, 15). Briefly, upon hypoxic stress, cancer cells release ATP4− through PANX-1 channels and/or exocytosis followed by ADO generation with subsequent ADO accumulation in the (positively charged) extracellular space of hypoxic tumor cells (10–100 µM, extracellular ADO). For comparison, normal tissues exhibit extracellular ADO levels in the range of 10–100 nM. ADO generation is mainly controlled by the activity of HIF-sensitive, membrane associated “tandem ecto-enzymes” CD39/CD73. Accumulation is further supported by an inhibition of the “downhill” ADO-re-uptake transporter ENT-1, and by an inactivation of the ADO-to-inosine conversion (through inhibition of ADO-deaminase). In severely hypoxic tumors regions, ADO concentrations up to 100 µM have been measured (Figure 1). Inhibitory effects of free ADO on innate and adaptive immune responses are multifactorial with a major emphasis on the proliferative and cytolytic antitumor activity of CD4+ helper, CD8+ cytotoxic T, and natural killer (NK) cells (Figure 2). Furthermore, high extracellular ADO levels can impact on the antigen-presenting activity of dendritic cells (DCs) and can activate immunosuppressive cells such as Tregs, myeloid-derived suppressor cells (MDSCs), and M2 macrophages. Actions of extracellular ADO are mediated upon binding to surface receptors, mainly the A2A receptor on immune and cancer cells. HIF-1α-sensitive A2A receptors are coupled to Gs-proteins, activate adenylylcyclase, and increase intracellular cyclic adenosine monophosphate (cAMP) levels. A more detailed description of the relevant suppressive effects of ADO on the antitumor immune responses has been presented in Ref. (15).
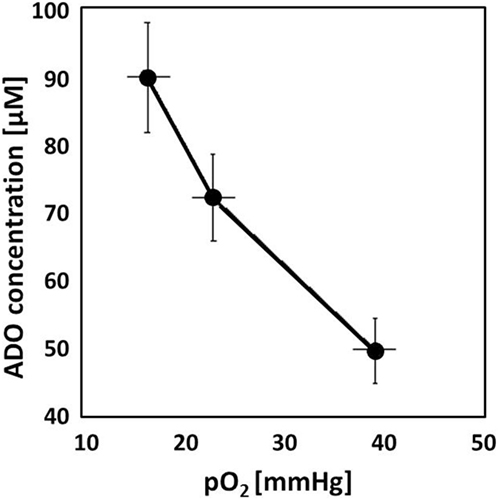
Figure 1. Adenosine (ADO) concentration measured in experimental tumors (DS-sarcomas, n = 26) as a function of the tissue oxygenation status. With decreasing mean tumor pO2 values, ADO accumulates in the tumor reaching ~100 µM in severely hypoxic tumors. For comparison, ADO levels in normal tissues are in the range of 10–100 nM (16, 17).
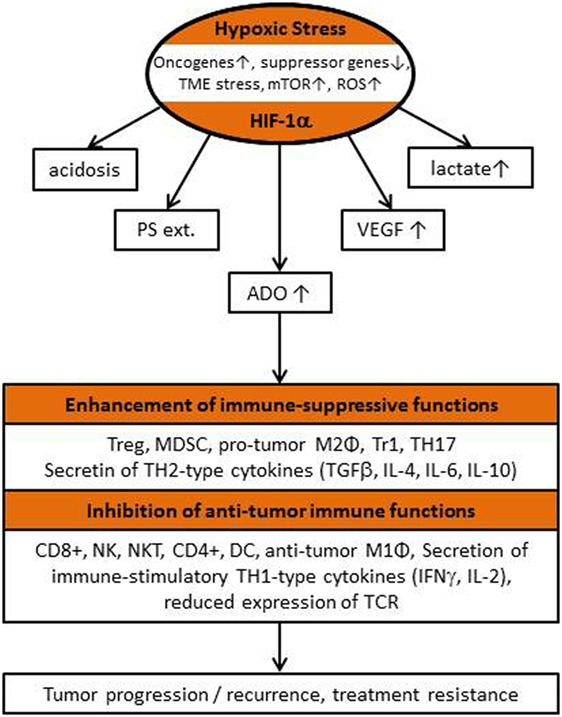
Figure 2. Flow chart describing “classical” hypoxia-/HIF-1α-driven features of the tumor microenvironment (TME) responsible for the local inhibition of antitumor immunity, for tumor progression/recurrence and poor patient outcome (see also list of abbreviations).
Lactate Accumulation Impedes Antitumor Immunity
Lactate (lac−) accumulation (up to 40 mM in heterogeneously distributed subvolumes of human cancers) (5) is a secondary consequence of hypoxia upon HIF-1α-induced reprogramming of metabolic pathways [e.g., upregulation of the glycolytic enzyme lactate dehydrogenase A (LDH-A) and accelerated glycolysis, “Warburg effect”]. Lactate per se, plentiful in hypoxic TMEs, mainly blocks proliferation, tumor infiltration, and cytokine production of T cells, inhibits the cytotoxic activity of NK, NKT, and CD8+ T cells, and increases the number of MDSCs that inhibit NK cell mediated cytotoxicity. Lactate, thus, can strongly repress antitumor immunity (18–21). In general, lactate actions are initiated after binding to the cell surface lactate receptor, GPR81 (syn. HCA-1), Gi-mediated signaling and subsequent decrease of cAMP levels.
Acidosis Opposes Antitumor Immune Responses
Extracellular acidosis in tumors mainly is a consequence of (a) an upregulation/acceleration of glycolysis [“Warburg effect,” associated with intensive conversion of glucose to lactic acid and inefficient adenosine triphosphate (ATP) production, but importantly very fast energy supply], (b) intensified glutaminolysis, (c) ketogenesis, (d) increased ATP-hydrolysis, (e) hydration of CO2 derived from oxidative metabolism and highly active pentose phosphate pathway, and (f) bicarbonate depletion in the extracellular space. Major mechanisms involved in the immune-suppressive action of acidosis (extracellular pH ≤ 6.8) include an inhibition of the proliferative and cytotoxic activity of NK cells and CD8+ T cells, secretion of TH1-type cytokines (e.g., IFN-γ) and a reduction in the expression density of T cell receptors (TCRs) (21–27).
VEGF Overexpression and VEGF-R Activation Counteract Antitumor Immunity
Vascular endothelial growth factor is one of the major players that drives tumor angiogenesis and, therefore, supports tumor progression and metastasis (28). Hypoxia-/HIF-1α directly or indirectly (in the latter case via ADO, lactate or acidosis) can drive the expression of VEGF and activate VEGF-R, thus promoting tumor evasion from immune surveillance (24–27). VEGF also can negatively affect growth and maturation of immature granulocyte–macrophage progenitors and DC precursors and, thus, prevent T cell stimulation. In addition VEGF can recruit immunosuppressive cells such as M2 macrophages into the tumor stroma which give raise to tumor-associated macrophages with immunosuppressive capacity (29). Other VEGF-triggered immunosuppressive mechanisms are mediated through Treg cells, pro-tumor M2 macrophages, MDSCs, and/or the presentation of immune checkpoint inhibitors, such as PDL1 or CTLA-4 on tumor and effector cells. Antiangiogenic therapy, using inhibitors targeting the VEGF/VEGF-R pathway may also exert beneficial effects on the reactivation of immune responses (in addition to the debatable “normalization of the tumor vasculature” theory), as discussed recently (15).
Externalization of PS Stimulates Immune-Suppressive Mechanisms
Under non-stress conditions, PS is selectively found on the inner leaflet of plasma membranes. Following environmental stress or in aging cells PS gets externalized to the outer membrane leaflet by scramblases. Externalized PS on dying normal cells acts as a dominant, anti-inflammatory eat-me signal for phagocytosis (efferocytosis) that allows controlled apoptotic cell death (30). Pathologically, the immunosuppressive activity of externalized PS has been converted into pro-inflammatory signals that can also support tumor progression. Upon hypoxic stress, the localization of PS is severely dysregulated in tumor and stromal cells (infiltrating MDSCs within the tumor stroma included) (30). If the controlled apoptotic clearance of tumor cells fails, secondary necrosis can be initiated which in turn can induce chronic inflammation and autoimmunity (31). A rapid removal of PS exposing apoptotic by phagocytosis is pivotal to prevent inflammatory responses and the maintenance of tolerogenic signals during homeostasis which in turn can downregulate antitumor immune responses. Furthermore, binding of PS exposing tumor-derived exosomes to PS-receptors (e.g., TIM-receptors on immune cells) (32), can also induce evolutionary conserved immune-suppressive signals (enhanced TGF-β and interleukin-10 secretion) that can further inactivate antitumor immune responses.
Immune Suppression by Elevated Extracellular K+
Tumor cell death (apoptosis or necrosis as a consequence of sustained hypoxia) leads to an egress of intracellular ions into the extracellular compartment impacting on antitumor T cells, on many other immune cells, and on the efficacy of immune therapies for cancer patients (33, 34). High K+ concentrations in the interstitial space [approx. 40 mM (physiological range: 3.5–5 mM)] can impair Akt–mTOR phosphorylation signaling of the TCR, thus inhibiting production of effector cytokines by T cells (e.g., IFN-γ). These recently published data have identified a novel “ionic checkpoint blockade” acting on T cell effector function upon release of cellular contents during tumor cell death (33, 34).
Therapeutic Strategies Counteracting the Immunosuppressive Activities of Hypoxia-Associated Factors in the TME
Therapeutic strategies alleviating immunosuppressive (and pro-tumor) activities of ADO have been described earlier [(14, 15) with Supplementary Material http://journal.frontiersin.org/article/10.3389/fimmu.2016.00332]. In a most recent communication, Vijayan et al. have extensively reviewed the current approaches (35).
Targeting immunosuppressive actions of VEGF have been summarized in depth earlier (15, 28, 36, 37).
Therapeutic targets inhibiting immunosuppressive activities of lactate have recently been discussed (20, 38, 39). Promising strategies include inhibition and knockdown of monocarboxylate transporters, small molecule LDH-A inhibitors, and inhibition of the cell surface lactate receptor GPR81 (40, 41).
Treatments targeting tumor acidosis include small molecules and antibodies interfering with pH regulating systems. Major regulators involved are several H+ transporters, proton pump ATPases, and carbonic anhydrases CAIX and CAXII, which can be blocked using corresponding pharmacological inhibitors (42). Preventing acidosis based on systemic buffer therapy using bicarbonate, imidazoles, or lysine has improved responses to immune therapies (22, 43). The use of inhibitors of CAIX and other new promising approaches offering new possibilities have recently been discussed in detail (44, 45).
Reversal of the PS-induced antitumor immunosuppression can be stimulated by PS-targeting therapeutics [e.g., Anx A5, bavituximab, see Table S1B in Supplementary Material (15)].
Impaired T cell functions following egress of intracellular K+ into the interstitial space upon tumor cell death can be reversed via the overexpression of voltage-gated K+ channels type Kv1.3 (34).
Conclusion
From the data presented, it is evident that hypoxia-/HIF-1α-driven features of the TME, such as ADO and lactate accumulation, extracellular acidosis, VEGF overexpression, and VEGF-R activation, and PS-externalization from the inner to the outer leaflet of tumor cells or tumor-derived exosomes are accomplices (“fatal sextet” of TME) sabotaging spontaneous and therapeutically induced antitumor immune responses. Therapeutic strategies counteracting the immune-suppressive activities of these adverse factors have been reviewed recently (14, 15).
Author Contributions
PV and GM equally contributed to the writing of this minireview.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Anett Lange for her help in preparation of the manuscript and the design of Figure 2.
Funding
The study was funded in part by DFG (SFB824/3; STA1520/1-1), MAP, BMBF (01GU0823; 02NUK038A), and BMWi (ZF4320102CS7). This work was supported by the German Research Foundation (DFG) and the Technische Universität München within the funding Programme Open Access Publishing.
Abbreviations
ADO, adenosine; A2AR, A2BR adenosine receptors; Anx A5, annexin A5; ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; CD39, ectonucleoside triphosphate diphosphohydrolase 1; CD73, ecto 5′-nucleotidase; CD4+, helper T cell; CD8+, cytotoxic T cell; DC, dendritic cell; ENT-1, equilibrative nucleoside transporter 1; Gs, Gi stimulatory/inhibitory G-protein; GPR81, G-protein receptor 81 (cell surface lactate receptor); HCA-1, hydroxycarboxylic acid receptor 1 (syn. GPR81); HIF, hypoxia-inducible (transcription) factor; IL, interleukin; IFN-γ, interferon γ; LDH-A, lactate dehydrogenase A; MCT, monocarboxylate transporter; Mϕ, macrophage; MDSC, myeloid-derived suppressor cell; mTOR, mechanistic (“mammalian”) target of rapamycin; NK, natural killer cell; NKT, natural killer-like T cell; PANX, pannexin (ATP channel); PDL1, programmed cell death 1 protein ligand; pO2, oxygen partial pressure; PS, phosphatidylserine; ROS, reactive oxygen species; TCR, T cell receptor; TGF-β, transforming growth factor β; TAM, tumor-associated macrophage; TIM, T cell immunoglobulin and mucin domain (PS surface receptor); TME, tumor microenvironment; TNF-α, tumor necrosis factor-α; Tr1, Type 1 regulatory T cell; VEGF, vascular endothelial growth factor; VEGF-R, VEGF receptor.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Rivera LB, Bergers G. CANCER. Tumor angiogenesis, from foe to friend. Science (2015) 349:694–5. doi:10.1126/science.aad0862
3. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell (2011) 146:873–87. doi:10.1016/j.cell.2011.08.039
4. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res (1989) 49:6449–65.
5. Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol (2004) 14:198–206. doi:10.1016/j.semradonc.2004.04.008
6. Vaupel P. Physiological mechanisms of treatment resistance. In: Molls M, Vaupel P, Nieder C, Anscher MS, editors. The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies. Berlin, Heidelberg, New York: Springer (2009). p. 273–90.
7. Vaupel P. Pathophysiological and vascular characteristics of solid tumors in relation to drug delivery. In: Kratz F, Senter P, Steinhagen H, editors. Drug Delivery in Oncology. (Vol. 1), Weinheim: Wiley-VCH (2012). p. 33–64.
8. Multhoff G, Vaupel P. Radiation-induced changes in microcirculation and interstitial fluid pressure affecting the delivery of macromolecules and nanotherapeutics to tumors. Front Oncol (2012) 2:165. doi:10.3389/fonc.2012.00165
9. Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Methods Enzymol (2004) 381:335–54. doi:10.1016/S0076-6879(04)81023-1
10. Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist (2008) 13(Suppl 3):21–6. doi:10.1634/theoncologist.13-S3-21
11. Mayer A, Vaupel P. Hypoxia, lactate accumulation, and acidosis: siblings or accomplices driving tumor progression and resistance to therapy? Adv Exp Med Biol (2013) 789:203–9. doi:10.1007/978-1-4614-7411-1_28
12. Marotta D, Karar J, Jenkins WT, Kumanova M, Jenkins KW, Tobias JW, et al. In vivo profiling of hypoxic gene expression in gliomas using the hypoxia marker EF5 and laser-capture microdissection. Cancer Res (2011) 71:779–89. doi:10.1158/0008-5472.CAN-10-3061
13. Vaupel P, Multhoff G. Hypoxia-/HIF-1a-driven factors of the tumor microenvironment impeding antitumor immune responses and promoting malignant progression. Adv Exp Med Biol (Forthcoming 2018).
14. Vaupel P, Multhoff G. Adenosine can thwart antitumor immune responses elicited by radiotherapy: therapeutic strategies alleviating protumor ADO activities. Strahlenther Onkol (2016) 192:279–87. doi:10.1007/s00066-016-0948-1
15. Vaupel P, Multhoff G. Commentary: a metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol (2016) 7:332. doi:10.3389/fimmu.2016.00332
16. Vaupel P, Mayer A. Can respiratory hyperoxia mitigate adenosine-driven suppression of antitumor immunity? Ann Transl Med (2015) 3:292. doi:10.3978/j.issn.2305.5839.2015.09.23
17. Vaupel P, Mayer A. Hypoxia-driven adenosine accumulation: a crucial microenvironmental factor promoting tumor progression. Adv Exp Med Biol (2016) 876:177–83. doi:10.1007/978-1-4939-3023-4_22
18. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol (2013) 191:1486–95. doi:10.4049/jimmunol.1202702
19. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab (2016) 24:657–71. doi:10.1016/j.cmet.2016.08.011
20. Romero-Garcia S, Moreno-Altamirano MM, Prado-Garcia H, Sanchez-Garcia FJ. Lactate contribution to the tumor microenvironment: mecha-nisms, effects on immune cells and therapeutic relevance. Front Immunol (2016) 7:52. doi:10.3389/fimmu.2016.00052
21. Pötzl J, Roser D, Bankel L, Homberg N, Geishauser A, Brenner CD, et al. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-gamma and induces NK cell-dependent lymphoma control without other immunotherapies. Int J Cancer (2017) 140:2125–33. doi:10.1002/ijc.30646
22. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi:10.1158/0008-5472.CAN-11-1272
23. Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene (2017) 36:439–45. doi:10.1038/onc.2016.225
24. Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends Immunol (2015) 36:240–9. doi:10.1016/j.it.2015.02.005
25. Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E, Bergers G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep (2015) 11:577–91. doi:10.1016/j.celrep.2015.03.055
26. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res (2017) 23:587–99. doi:10.1158/1078-0432.CCR-16-0387
27. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev (2017) 276:121–44. doi:10.1111/imr.12528
28. Cardones AR, Banez LL. VEGF inhibitors in cancer therapy. Curr Pharm Des (2006) 12:387–94. doi:10.2174/138161206775201910
29. Johnson B, Osada T, Clay T, Lyerly H, Morse M. Physiology and therapeutics of vascular endothelial growth factor in tumor immunosuppression. Curr Mol Med (2009) 9:702–7. doi:10.2174/156652409788970634
30. Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ (2016) 23:962–78. doi:10.1038/cdd.2016.11
31. Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:191–201. doi:10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K
32. Baghdadi M, Jinushi M. The impact of the TIM gene family on tumor immunity and immunosuppression. Cell Mol Immunol (2014) 11:41–8. doi:10.1038/cmi.2013.57
33. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature (2016) 537:539–43. doi:10.1038/nature19364
34. Gurusamy D, Clever D, Eil R, Restifo NP. Novel “elements” of immune suppression within the tumor microenvironment. Cancer Immunol Res (2017) 5:426–33. doi:10.1158/2326-6066.CIR-17-0117
35. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17(12):709–24. doi:10.1038/nrc.2017.86
36. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol (2015) 5:202. doi:10.3389/fonc.2015.00202
37. McIntyre A, Harris AL. Metabolic and hypoxic adaptation to anti-angiogenic therapy: a target for induced essentiality. EMBO Mol Med (2015) 7:368–79. doi:10.15252/emmm.201404271
38. Miranda-Goncalves V, Granja S, Martinho O, Honavar M, Pojo M, Costa BM, et al. Hypoxia-mediated upregulation of MCT1 expression supports the glycolytic phenotype of glioblastomas. Oncotarget (2016) 7:46335–53. doi:10.18632/oncotarget.10114
39. Baltazar F, Pinheiro C, Morais-Santos F, Azevedo-Silva J, Queiros O, Preto A, et al. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol Histopathol (2014) 29:1511–24. doi:10.14670/HH-29.1511
40. Lee YJ, Shin KJ, Park SA, Park KS, Park S, Heo K, et al. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget (2016) 7:70898–911. doi:10.18632/oncotarget.12286
41. Roland CL, Arumugam T, Deng D, Liu SH, Philip B, Gomez S, et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res (2014) 74:5301–10. doi:10.1158/0008-5472.CAN-14-0319
42. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer (2017) 17:577–93. doi:10.1038/nrc.2017.77
43. Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res (2016) 76:1381–90. doi:10.1158/0008-5472.CAN-15-1743
44. Böhme I, Bosserhoff AK. Acidic tumor microenvironment in human melanoma. Pigment Cell Melanoma Res (2016) 29:508–23. doi:10.1111/pcmr.12495
Keywords: antitumor immunity, adenosine, acidosis, lactate, vascular endothelial growth factor, phosphatidylserine, tumor hypoxia, potassium ions
Citation: Vaupel P and Multhoff G (2017) Accomplices of the Hypoxic Tumor Microenvironment Compromising Antitumor Immunity: Adenosine, Lactate, Acidosis, Vascular Endothelial Growth Factor, Potassium Ions, and Phosphatidylserine. Front. Immunol. 8:1887. doi: 10.3389/fimmu.2017.01887
Received: 13 September 2017; Accepted: 11 December 2017;
Published: 21 December 2017
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Francois Ghiringhelli, INSERM, FranceHaidong Dong, Mayo Clinic Minnesota, United States
Copyright: © 2017 Vaupel and Multhoff. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Vaupel, cGV0ZXIudmF1cGVsQHR1bS5kZQ==