 Anca Dorhoi1,2,3*
Anca Dorhoi1,2,3* Nelita Du Plessis4*
Nelita Du Plessis4*
- 1Institute of Immunology, Bundesforschungsinstitut für Tiergesundheit, Friedrich-Loeffler-Institut (FLI), Insel Riems, Germany
- 2Faculty of Mathematics and Natural Sciences, University of Greifswald, Greifswald, Germany
- 3Department of Immunology, Max Planck Institute for Infection Biology, Berlin, Germany
- 4Division of Molecular Biology and Human Genetics, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, SAMRC Centre for Tuberculosis Research, DST and NRF Centre of Excellence for Biomedical TB Research, Stellenbosch University, Tygerberg, South Africa
Heterogeneous populations of myeloid regulatory cells (MRC), including monocytes, macrophages, dendritic cells, and neutrophils, are found in cancer and infectious diseases. The inflammatory environment in solid tumors as well as infectious foci with persistent pathogens promotes the development and recruitment of MRC. These cells help to resolve inflammation and establish host immune homeostasis by restricting T lymphocyte function, inducing regulatory T cells and releasing immune suppressive cytokines and enzyme products. Monocytic MRC, also termed monocytic myeloid-derived suppressor cells (M-MDSC), are bona fide phagocytes, capable of pathogen internalization and persistence, while exerting localized suppressive activity. Here, we summarize molecular pathways controlling M-MDSC genesis and functions in microbial-induced non-resolved inflammation and immunopathology. We focus on the roles of M-MDSC in infections, including opportunistic extracellular bacteria and fungi as well as persistent intracellular pathogens, such as mycobacteria and certain viruses. Better understanding of M-MDSC biology in chronic infections and their role in antimicrobial immunity, will advance development of novel, more effective and broad-range anti-infective therapies.
Introduction
Mononuclear myeloid cells encompass various phagocyte populations exerting distinct functions during infection. From progenitors and immature myeloid cells (IMC) to mature and polarized phagocytes, subsets of myeloid regulatory cells (MRC) have been described. These populations include regulatory dendritic cells (DCs), regulatory and alternatively activated macrophages (M2-like macrophages), tumor-associated macrophages (TAM), and a unique mixture of heterogeneous cells coined myeloid-derived suppressor cells (MDSC) (1). This nomenclature indicates their origin and ability to suppress T-cell immunity (2). MDSC comprise morphologically distinct subsets, monocyte-like [monocytic MDSC (M-MDSC)] and neutrophil-like (PMN-MDSC) cells. Phenotypically, M-MDSC are HLA-DR−/lowCD11b+CD33+/highCD14+CD15− in humans and Gr-1dim/+CD11b+Ly6C+Ly6G− in mice (2). Several studies report on CD11b+Ly6C+/dimLy6Gint murine M-MDSC, a phenotype that requires further validation in additional disease models and in-depth characterization (3, 4). These cells have biochemical features characteristic of the myeloid lineage, notably abundance of products downstream of arginase 1 (ARG1), inducible nitric oxide synthase (iNOS), indoleamine dioxygenase (IDO), and cyclooxygenase (COX1) (2, 5). Unequivocal phenotypic markers for MDSC have not been identified so far, implying that cells can only be classified as MDSC upon demonstration of their lymphocyte suppressive function. This suggests that MDSC are likely underreported, particularly in conditions characterized by expansion of myeloid cells such as in infectious diseases.
Most of the information on MDSC emerges from cancer research where MDSC are associated with poor disease outcome. However, reports on myeloid suppressor cells in infection date back four decades. “Natural suppressor” cells were identified in spleens of experimentally infected animals following systemic delivery of mycobacteria, notably the vaccine strain Mycobacterium bovis Bacille Calmette–Guérin (BCG) (6). Although research on suppressor cells in cancers has flourished since then, studies in infectious diseases lagged behind. Cancer and infection share several pathophysiological features, including the non-resolving inflammation (7), which often triggers emergency hematopoiesis and expansion of MDSC (8). Given such similarities and encouraged by progress made in cancer biology, recent investigations found MDSC in communicable diseases (9–12), uncovered their interactions with microbes and emphasized critical roles in disease pathogenesis. This review focuses on M-MDSC and discusses their genesis during infection as well as interactions with immune cells, elaborating on targets and mechanisms of suppression. We will mostly describe M-MDSC biology in infections caused by M. tuberculosis, Staphylococcus aureus, hepatitis viruses [hepatitis B virus (HBV), hepatitis C virus (HCV)], and human immunodeficiency viruses (HIV) and to a lesser extent fungi and parasites (Box 1). We will use the term MDSC to refer to the total MDSC population, without further subset phenotype characterization. For studies using monocytic subsets, within the MDSC pool, we will use the acronym M-MDSC.
Box 1. Chronic infections associated with monocytic myeloid-derived suppressor cells (M-MDSC).
Monocytic myeloid-derived suppressor cells have been reported in various infections caused by bacterial and viral agents, many of them causing diseases highly relevant for the public health. Key points about the pathogen and the respective disease are presented in the following. M. tuberculosis is a Gram-positive bacterium and represents the etiologic agent of human tuberculosis (TB). TB primarily affects the lungs of millions of people, and is among the top 10 causes of death worldwide (13). Infection with M. tuberculosis frequently leads to latent TB, bacteria being contained within tissue lesions, but not eliminated. Such individuals, estimated at one-third of global population, are at risk of developing active TB upon immune suppression. S. aureus is a Gram-positive bacterium that often colonizes the human skin and nose (14). It is the leading cause of skin and soft tissue infections, pneumonia, osteomyelitis, endocarditis, and septicemia. Such conditions can manifest as acute and often long-lasting, frequently nosocomial-associated diseases, which are often resistant to antibiotics. Increased antimicrobial resistance characterizes current clinical isolates of M. tuberculosis and S. aureus. This results in significant therapy failures and economic burdens because of refractoriness to canonical chemotherapy (15). HCV and HBV are single-stranded RNA (Flaviviridae) and double-stranded DNA (Hepatdnaviridae) viruses, respectively, which cause chronic infection of the liver leading to end-stage liver disease in the absence of therapy. Prevalence of HCV and HBV in human population is high, reaching 70 million and 250 million chronic cases, respectively (16). HIV, encompassing HIV-1 and HIV-2, are lentiviruses belonging to the Retroviridae family that cause the acquired-immune deficiency syndrome (AIDS). AIDS affects more than 35 million people worldwide and the virus causes lytic infection of immune cells, primarily CD4+ lymphocytes (17). Often AIDS leads to reactivation of latent TB and such a comorbidity results in high death tolls (13).
Genesis of M-MDSC in Infectious Diseases
Expansion of M-MDSC occurs in various infectious diseases. Accumulating evidence indicate that oncogenic viruses, including HBV (18) and HCV (19–22), retroviruses, notably HIV (23, 24), simian immunodeficiency virus (SIV) (25, 26), and mouse immunodeficiency virus LP-BM (27), as well as Gram-positive bacteria, such as mycobacteria (28–30), staphylococci (31–33), enterotoxigenic bacilli (34), and Gram-negative pathogens, such as klebsiellae (35), trigger generation of M-MDSC. Fluctuation of this MDSC subset during anti-infective therapy was demonstrated in patients undergoing canonical TB chemotherapy (29), further strengthening the notion that disease progression in chronic infections is associated with expansion of M-MDSC. For some microbes, precise microbial cues and corresponding host pathways triggering M-MDSC generation or reprogramming of monocytes into M-MDSC have been elucidated (Figure 1). However, to date, for most infections, expansion of M-MDSC is explained solely by generation of inflammatory mediators during the course of the disease. Cytokines (IL-1 family members, IL-6, TNF, IL-10), lipid mediators (prostaglandin E2, PGE2), and growth factors (GM-CSF) foster generation of M-MDSC by promoting emergency myelopoiesis, skewing differentiation of progenitors into monocytes and DCs (STAT3/STAT5 activation) and promoting survival of M-MDSC (TGF-β, MCL-1-related anti-apoptotic A1) (36–40) (Figure 1). Just like in cancer, M-MDSC and populations containing M-MDSC are detectable at the site of pathology; e.g., in infected lungs in TB (29, 30, 41), pneumonia caused by Francisella tularensis (42), and influenza A virus (43, 44), in liver during HBV infection (45, 46), in skin and prosthetic bone implants during S. aureus colonization (32, 47, 48), and systemically in AIDS and sepsis (23, 24, 49). M-MDSC have also been detected in bone marrow and spleen, e.g., in TB (50), indicating their origin.
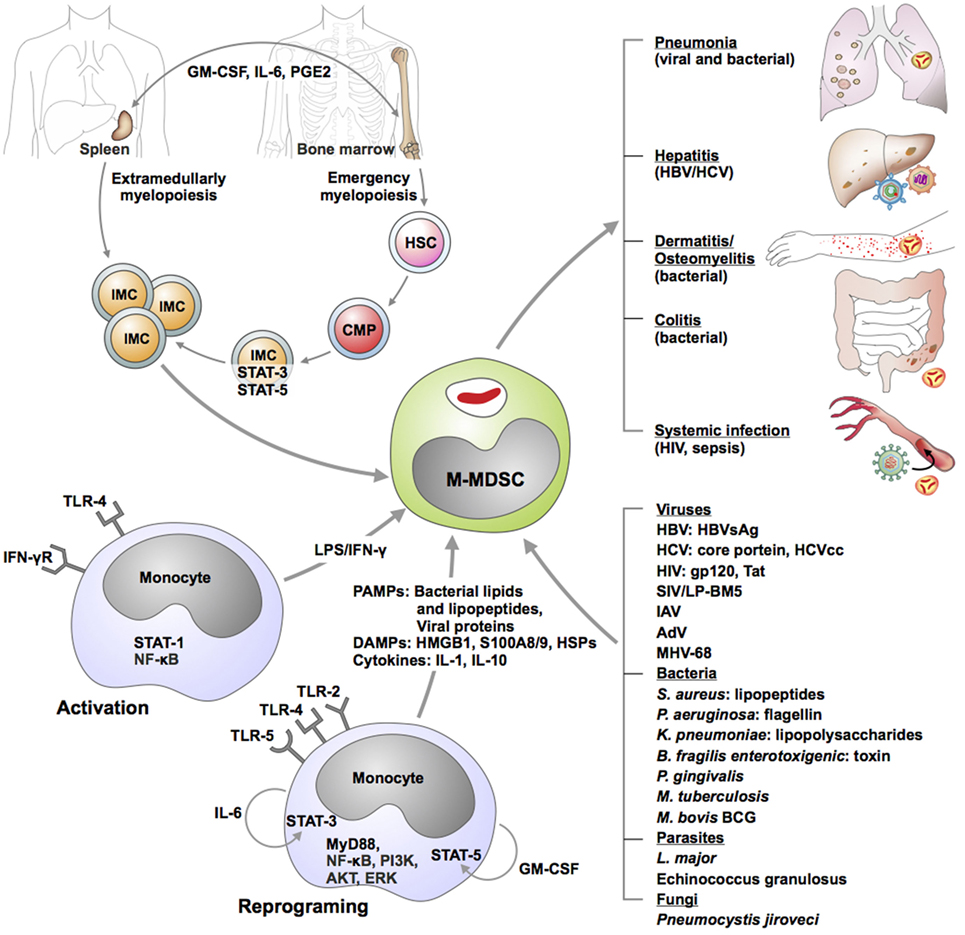
Figure 1. Genesis of monocytic myeloid-derived suppressor cells (M-MDSC) during infectious diseases. Hypothetical models were derived from ex vivo results, correlative studies in animal models as well as clinical observations. Immature myeloid cells (IMC) are generated either in bone marrow or in spleen as a consequence of emergency myelopoiesis. Growth factors, cytokines, and lipids promote progression of hematopoietic stem cells (HSC) toward common myeloid progenitor (CMP) development and subsequent IMC genesis. Combination of cytokines as well as direct stimulation of selected microbial receptors by various microorganisms may activate or reprogram circulating monocytes toward M-MDSC. M-MDSC are recruited in various organs where they exert suppressive function and modulate manifestations and outcome of the disease. Abbreviations: AdV, adenovirus; AKT, protein kinase B; ERK, extracellular signal-regulated kinase; GM-CSF, granulocyte-macrophage colony stimulating factor; gp120, glycoprotein 120; HBV, hepatitis B virus; HBVsAg, HBV soluble antigen; HCV, hepatitis C virus; HIV, human immunodeficiency virus; IAV, influenza A virus; IFN-γ, interferon gamma; IL-6, interleukin 6; LPS, lipopolysaccharide; LP-BM5, virus murine acquired-immune deficiency syndrome (AIDS); MHV-68, murine herpesvirus 68; MyD88, myeloid differentiation primary response gene 88; NF-κB, nuclear factor “kappa-light-chain-enhancer” of activated B-cells; PI3K, phosphatidylinositide 3-kinase; PGE2, prostaglandin E2; STAT, signal transducer and activator of transcription; SIV, simian immunodeficiency virus; tat, trans-activator of transcription; TLR, toll-like receptor.
Microbial Signatures and Microbial Sensors Trigger M-MDSC Genesis
Pathogen Sensors Involved in Generation of M-MDSC
Microbial signatures are detected by non-clonally distributed innate receptors termed pattern recognition receptors (PRR). PRR are grouped in families and the founder toll-like receptors (TLR) have been best characterized so far. TLR are present on the plasma membrane and within endosomes and are activated by diverse microbial structures, including lipids [e.g., TLR-4 senses lipopolysaccharide (LPS)], lipoproteins (e.g., TLR-2 senses acylated peptides) and proteins (e.g., TLR-5 senses flagellin). Generally, microbial-derived cognates of TLR-2 and -4 induce M-MDSC (20, 32, 51–53). LPS, which is the major cell wall component of Gram-negative bacteria, triggers proliferation of HSC (40) and induces M-MDSC upon pulmonary instillation or subsequent infection with Salmonella spp. or Klebsiella pneumonia (35, 51). Stimulation of human monocytes with TLR-4 agonists reprograms the cells into M-MDSC in a process dependent on STAT-3 activation (54). Crosstalk between TLR/MyD88 and JAK2/STAT5 pathways following receptor activation by LPS and GM-CSF is critical for M-MDSC generation (35, 51). The adaptor MyD88, which converges signals from multiple TLR, has also been implicated in generation of MDSC during polymicrobial sepsis (55). TLR-4 appears dispensable for sepsis-induced suppression of T cells (55) thereby indicating that IL-1, which binds IL-1R upstream of MyD88, conditions MDSC differentiation.
Several bacterial and viral agonists of TLR-2 promote M-MDSC differentiation from monocytes and in certain instances precise signaling pathways have been identified. S. aureus lipopeptides activate TLR2/6 dimers in skin cells for IL-6 production which in turn promote local MDSC accumulation (32). HCV reprograms monocytes into M-MDSC by stimulating TLR-2. More precisely, HCV core proteins or HCV cell culture-derived virions trigger TLR-2/PI3K/AKT/STAT3 pathway and this leads to cytokine production, notably IL-10 and TNF-α, and monocyte differentiation into MDSC (19–21). By contrast, TLR-3 ligation restricts HCV and LPS-induced M-MDSC differentiation (19, 52). Nonetheless, vesicular stomatitis virus activation of TLR-3 induces MDSC expansion (56). Alike TLR-3, TLR-7 activation by influenza virus blocks MDSC, including M-MDSC, accumulation in infected lungs (44). Both TLR-3 and -7 are located in endosomes. Whether signal compartmentalization, notably at the cell membrane or within endosomes, is critical for MDSC genesis remains to be established. Very little information exists on the roles of cytosolic PRR, such as nod-like receptors and AIM-like receptors, in monocyte reprogramming or M-MDSC generation. Moreover, many pathogens, notably mycobacteria, simultaneously stimulate multiple PRR (57) and the net outcome of such innate recognition on M-MDSC in TB awaits clarification.
Host alarmins that activate PRR have also been implicated in MDSC generation. S100A proteins, high-mobility-group-protein B1 and heat-shock proteins bind the receptor for advanced glycation products (RAGE), TLR-2, and TLR-4. In cancer and autoimmune diseases, these ligands have been associated with increased dynamics of MDSC, including M-MDSC (58–60). Just like microbial-derived PRR agonists, alarmins may induce cytokine release, such as IL-6 and subsequent autocrine or paracrine differentiation of immature mononuclear cells toward MDSC (61). In chronic infections, for instance, in TB patients, S100A8/9 proteins are abundant in the lung (62). These alarmins besides driving recruitment of MDSC (63) bind RAGE and subsequently upregulate ARG1, a key suppressive enzyme in M-MDSC (2). Since tissue damage often occurs during microbial insult, PRR stimulation by host-derived danger molecules along with microbial-derived agonists could contribute to the regulation of MRC. Similarly, synergy between microbial products, such as LPS, and inflammatory cytokines, notably IFN-γ, restricts differentiation of DCs and fosters genesis of M-MDSC in the bone marrow (64).
Microbial Factors Required for M-MDSC Genesis
For many microbes, the precise pathways required for M-MDSC genesis are not known. Mycobacteria induce accumulation of such cells irrespective of key virulence features, notably the type VII secretion system. M-MDSC have been reported for both M. tuberculosis and the vaccine BCG (29, 30, 50, 65). Mycobacterial glycolipids appear sufficient to induce these regulatory monocytes, as indicated by the presence of MDSC in animals inoculated with complete Freund’s adjuvant (66). In contrast to mycobacteria, non-colitogenic bacteria and oncogenic gut species (Fusobacterium nucleatum, pks+ Escherichia coli) do not trigger M-MDSC, whereas enterotoxigenic Bacillus fragilis employs the toxin to prime epithelial cells for IL-17 and M-MDSC expansion (34). HIV and SIV infection triggers accumulation of M-MDSC in the blood and their reduction in the bone marrow, which correlates with plasma viral loads and disease progression (25, 49). Several HIV viral factors promote expansion of the M-MDSC or reprogramming of monocytes. Human monocytes stimulated with HIV gp120 (23, 24) and/or with Tat proteins (54) acquire T-cell suppressive activity. This differentiation requires autocrine release of IL-6 and activation of STAT-3 (23, 54). HBV surface antigen similarly triggers differentiation of human monocytes toward M-MDSC in an autocrine manner depending on activation of the kinase ERK and the transcription factor STAT-3 (18). The necessity of specific kinases, such as ERK (18) and AKT (19, 20) for microbial-induced M-MDSC generation resembles kinase signatures of MDSC in cancer (67). Similarly, STAT-3 is required for M-MDSC in cancer (68) as well as during infection with HIV (23, 54), HCV (20, 22), and stimulation with bacterial LPS (54). For many bacterial (Mycobacterium spp., F. tularensis, Porphyromonas gingivalis) (29, 30, 42, 50, 69) and viral pathogens [vaccinia virus, lymphocoriomeningitis virus (LCMV), MCMV, murine gamma virus, LP-BM5] (70–72), and protozoa (Leishmania spp.)(73, 74), the host pathways or microbial signatures required for M-MDSC genesis are still undefined.
Inflammation Drives M-MDSC Generation during Infection
A common denominator in infection and cancer biology is the inflammation. Whereas physiological inflammation protects the host and restores homeostasis, in exuberant acute infections and chronic processes, inflammation often becomes pathologic and leads to disease manifestation. In such a scenario, inflammation-induced pathology becomes life-threatening. M-MDSC are primarily associated with chronic infections; however, they have been also reported in acute infectious diseases. Genesis of this myeloid regulatory subset is uncoupled from a specific phase of an infectious process. For instance, F. tularensis triggers IMC with M-MDSC features during acute, but not sub-acute, non-lethal infection (42). In polymicrobial sepsis M-MDSC are present early, as well as at late stages of sepsis, during the suppressive phase (55, 75). In infection with the LCMV, acute strains (Armstrong) do not induce M-MDSC, whereas chronic strains (Clone 13) induce suppressive myeloid cells (71).
Certain transcription factors and inflammatory mediators are critical for generation of MRC in infections. These requirements resemble those observed for MDSC in cancer (63). In sepsis, myeloid specific deletion of the myeloid differentiation-related transcription factor nuclear factor I-A, or deletion of the transcription factor C/EBPβ, result in reduction of MDSC, including M-MDSC (76, 77). Pro-inflammatory cytokines, notably IL-6, TNF-α, and IL-1, drive generation of MDSC in various infection models. In viral infections, including HIV (23) and HBV (18), IL-6 reprograms monocytes into suppressor cells. The same cytokine drives accumulation of M-MDSC in S. aureus skin infection and into the lungs subsequent to LPS instillations (32, 35). TNF promotes differentiation of MDSC in chronic inflammation (37, 78), likely through membrane expression of TNFR2, as shown in sterile inflammation (79). TNF signaling contributes to M-MDSC generation in HCV infection (19) and regulates M-MDSC dynamics and activity also in murine mycobacterial infection (80). Besides cytokines, pro-inflammatory lipids such as the eicosanoid PGE2 are highly abundant in the TB-susceptible mouse strain C3HeB/FeJ (81) and these animals also accumulate M-MDSC (41). Interestingly, application of a COX2 inhibitor which lowers PGE2 levels rescues C3HeB/FeJ from TB lethality (81), thereby suggesting that this lipid may be critical for genesis of host-detrimental MDSC in TB. In addition, PGE2 positively regulates enzymatic pathways critical for the suppressive function of the MDSC, including iNOS, IDO1, and IL-10. COX2 crosstalks with the IL-1/IL-1R pathway, as well as with IFN I pathway, which has been revealed in TB and flu (82, 83). The positive cross-regulation between COX2 and IL-1 may affect M-MDSC genesis. IL-1/IL-1R pathway drives accumulation of M-MDSC in BCG-vaccinated mice (65). IL-1β also regulates PMN-MDSC generation by itself and during fungal disease (84). Activation of specific inflammasomes for release of bioactive IL-1β has not yet been related to MDSC induction during infectious diseases. However, the NLRP3 inflammasome drives MDSC accumulation in cancer (85). To what extent key inflammatory molecules, including IL-1β and the downstream inflammasome platforms, may affect generation and accumulation of M-MDSC in other chronic infections than TB remains to be established.
As a corollary, various stimuli trigger M-MDSC generation and expansion during microbial insult. Additional pathways will likely be uncovered as the research into M-MDSC in infection expands. Recent studies indicate that GM-CSF licenses monocytes for suppressive activity upon further stimulation with PRR agonists or cytokines (86). Such a two-step process likely occurs during infection. Furthermore, fate-mapping studies are imperative to elucidate whether bone marrow or extramedullary myelopoiesis are unique sites for M-MDSC expansion or whether this myeloid subset can self-maintain in situ, at the site of the infection. Furthermore, the signals triggering recruitment of M-MDSC at the site of the pathology require further elucidation. Panoply of chemokines and alarmins are generated during infection. These, along with factors known to drive MDSC accumulation in cancer may be essential for MDSC dynamics in infected tissue. For instance, both PGE2 and TGF-β upregulate CXCR2 and CXCR4 expression in M-MDSC in cancers and they may be critical for the accumulation of such cells toward CXCL12 or CCL2 gradients at the site of infection, as it has been demonstrated in tumors (63, 87–89).
M-MDSC in Pathophysiology of Chronic Infections
M-MDSC Immunosuppressive Mechanisms and Cellular Interactions
Myeloid regulatory cells regulate host immunity through interaction with immune and non-immune cells (90) (Figure 2). This link is typically bi-directional: e.g., T-cells also regulate MRC expansion and activity, to induce tissue healing and remodeling (91, 92). Here, we describe current information on monocytic MDSC immunosuppressive machinery and interaction with archetypal immune cells (Table 1).
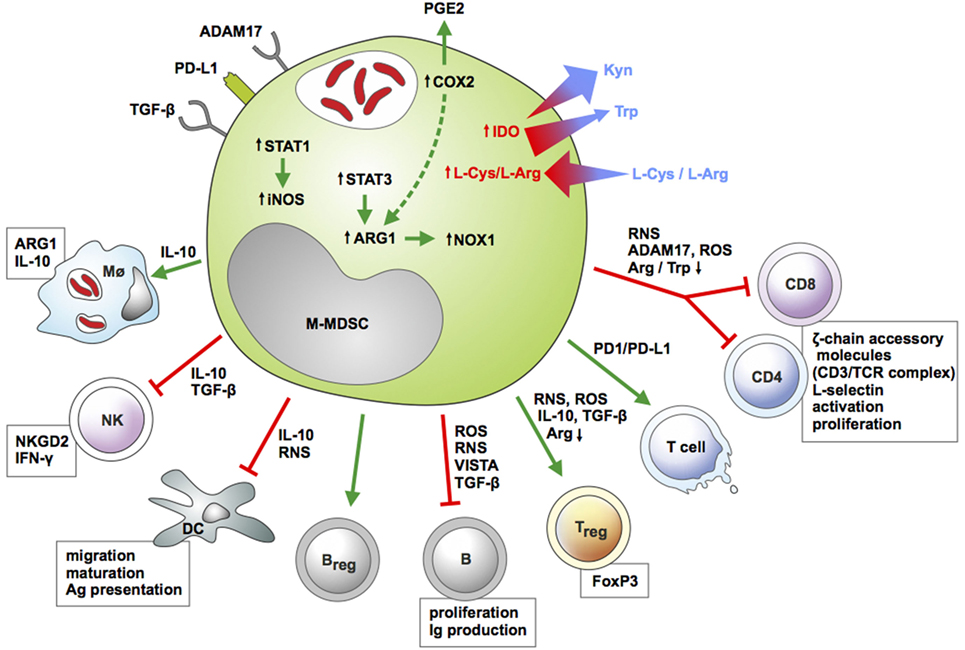
Figure 2. Features of monocytic myeloid-derived suppressor cells (M-MDSC) and their interactions with immune cells during infection. M-MDSC express membrane-bound inhibitory receptors and upregulate enzymatic pathways [inducible nitric oxide synthase (iNOS), ARG1, COX2, IDO] conferring suppressive activity toward multiple myeloid and lymphoid cell subsets. The key function of M-MDSC is suppression of T-cell immunity. M-MDSC restrict proliferation and release of cytokines by effector CD4 and CD8 lymphocytes and induce apoptotic cell death in these cells. In addition, these myeloid regulatory cells induce regulatory T and B cells, while limiting antibody release and proliferation of conventional B cells. M-MDSC alter activity of NK cells and antigen-presenting cells (APCs) and induce polarization of macrophages toward a regulatory phenotype. Color-coded arrows indicate induction/activation (green) or suppression (red), and molecules employed by M-MDSC for such effects are highlighted. Size- and color-coded arrows indicate gradient fluxes for selected essential amino acids. Boxes indicate cellular functions or pathways modulated by M-MDSC. Abbreviations: ADAM17, ADAM metallopeptidase domain 17; ARG1, arginase 1; CD, cluster of differentiation; COX2, cyclooxygenase 2; DC, dendritic cell; IDO1, indoleamine dioxygenase 1; IFN-γ, interferon gamma; IL-10, interleukin 10; iNOS, inducible nitric oxide synthase; Kyn, kynurenine; l-Arg, l-arginine; l-Cys, l-cysteine; MΦ, macrophage; NK, natural killer cell; NKGD2, killer cell lectin like receptor K1; NOX1, NADPH oxidase 1; PGE2, prostaglandin E2, PD-L1, programmed-death ligand 1; RNS, reactive nitrogen species; ROS, reactive oxygen species; STAT, signal transducer and activator of transcription; TGF-β, transforming growth factor beta; Trp, tryptophan; VISTA, V-domain Ig suppressor of T-cell Activation.
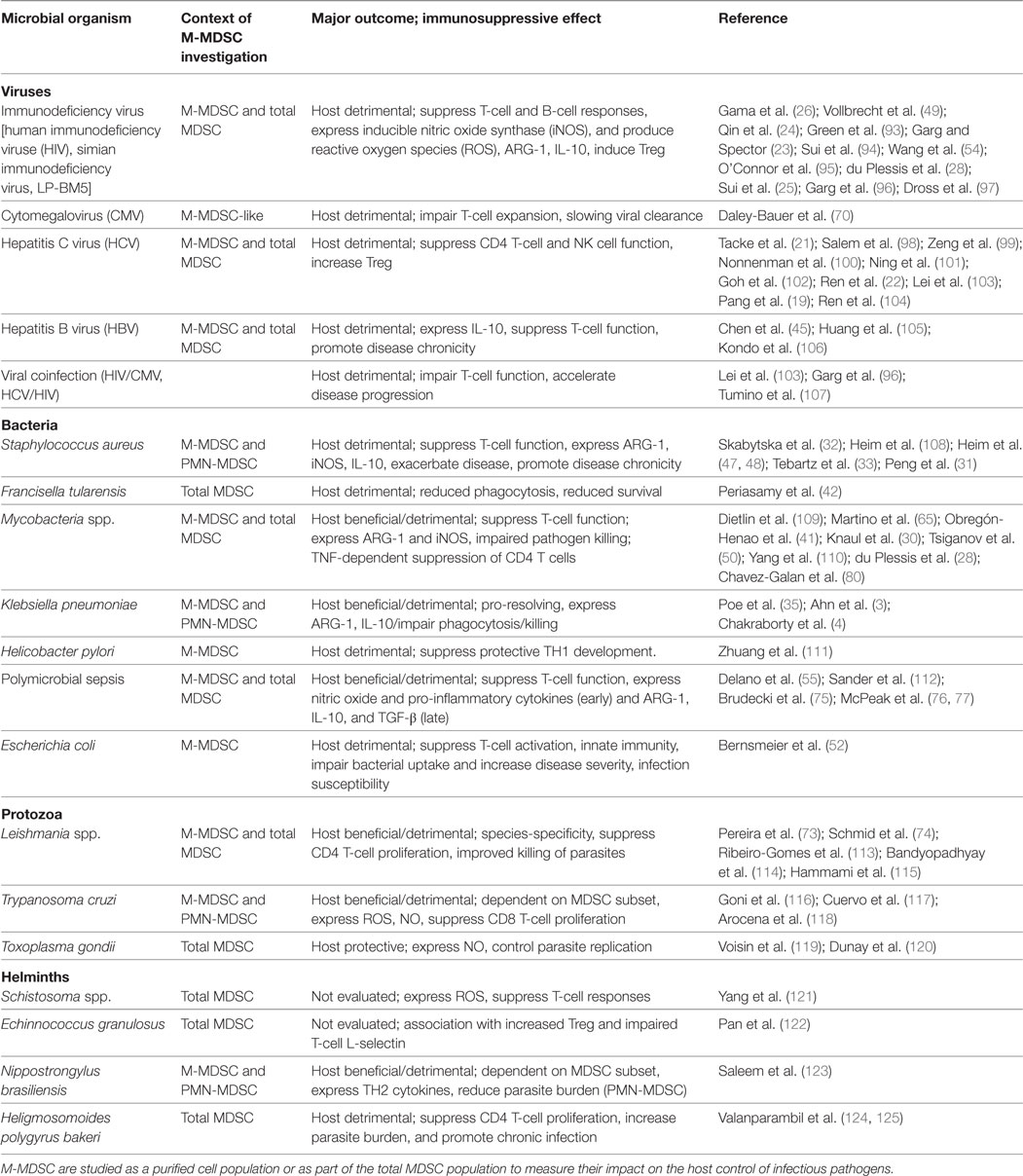
Table 1. Impact of monocytic myeloid-derived suppressor cells (M-MDSC) on infectious disease outcome and their immunosuppressive effects.
T Cells
Immunosuppression by MDSC has the potential to inhibit innate and adaptive immune cell activation, proliferation, viability, trafficking, and cytokine production. M-MDSC utilize a variety of suppressive mechanisms and likely differ in their ability to initiate antigen-specific versus non-specific suppression (126, 127). Each immune suppressive function is determined by the type of MRC, the microenvironmental components and the state of T-cell activation, favoring the probability that non-specific and antigen-specific suppressive mechanisms may coincide. Although not the focus of this review, as an example, PMN-MDSC can present peptides to T cells, but their low expression of major histocompatibility complex (MHC) II and costimulatory molecules, suggest they might only affect CD8 T-cell responses in an antigen-specific manner, as reported during retrovirus infection (128). This idea is supported by reports on MDSC-mediated inhibition of antigen-specific CD8 T-cell responses in tumors, likely due to the MHC I-restricted nature of cancer MDSC (2, 127, 129, 130). In infection, antigen-specific immunosuppression of CD8 T cells by M-MDSC is restricted to polymicrobial sepsis (131), HCV (21), HBV (46), murine encephalomyelitis virus (132), SIV and HIV infections (26), and LCMV infection (71). Data on the effect of MDSC on CD4 T helper cell (TH) subsets during infectious diseases are limited, but do exist as a result of the MHC-independent suppressive effects of MDSC in the context of HCV (21), HIV (24), and murine encephalomyelitis virus infection (132). During BCG-induced pleurisy, transmembrane TNF on M-MDSC restricts proliferation of CD4 T cells via interaction with lymphocyte-expressed TNFR2 (80). Results on MDSC interaction with TH17 and TH2 polarized CD4 T cells are contradictory and reports exist of mainly PMN-MDSC-mediated induction and suppression of TH17 responses in cancer, autoimmunity and infection (133–138), likely indicating that the combination of mediators present in the microenvironment determines the final outcome. In turn, TH1 and TH2 are involved in the expansion and activation of MDSC in cancer and also hepatitis (137, 139). Interestingly, recent findings suggest that CD1d-restricted natural killer T cells can convert immunosuppressive murine-MDSC into immune stimulating APCs following influenza virus infection, via their interaction with CD40 (140).
Regulatory T cells (Treg) are equally important components of the host immunoregulatory network. Data suggest reciprocal regulation of MDSC and Treg through mechanisms involving presence of IL-10, TGF-β, IL-4Rα, p47phox, PD-L1, TGF-β, and CD40–CD40L interactions, ARG1 induction and CCR-5-mediated recruitment (91, 126, 141–144). Interactions between total MDSC and Treg in cancer are well described (145, 146) with Treg depletion reducing MDSC immunosuppression by lowering their expression of PD-L1 and IL-10 production (147). Evidence of interaction in non-cancerous models, including type-1 diabetes, cardiac allograft and airway hyper-responsiveness, also exist (148–150). More specifically, the induction of Treg by M-MDSC, has also been described during HIV infection and shown to contribute to host immunosuppression (23, 49, 54). Data by O’Connor suggest reciprocal crosstalk between M-MDSC and Treg during LP-BM5-induced murine AIDS. Here, M-MDSC subsets display differential suppression of T- and B-cells, thereby indicating functionally overlapping, but distinguishable, immunosuppressive effects (27, 95). Incubation of M-MDSC from peripheral blood of HIV-1-infected individuals, even those on antiretroviral therapy with undetectable viremia, with CD4 T cells from healthy individuals, significantly increased differentiation of Foxp3 Treg, whereas depletion of MDSC significantly increased IFN-γ production by CD4 T cells (54).
B Cells
Information on MDSC interaction with B-cells only recently started to accumulate. In autoimmune disease, M-MDSC inhibit B-cell proliferation and antibody production via an iNOS and a PGE2-induced pathway (151). However, opposing data demonstrated that the total MDSC population promotes proliferation and differentiation of immunoglobulin-A-producing immunosuppressive plasma B-cells via cell contact in mouse tumor models (152). In infectious diseases, M-MDSC suppressed B-cell responsiveness to retroviral infection in mice via iNOS and the negative immune checkpoint regulator V-domain Ig Suppressor of T-cell Activation (VISTA) (72, 93).
Myeloid Cells
Data on MDSC interaction with myeloid cells, such as DC, neutrophils, and macrophages in infectious diseases, are equally restricted, with reports mainly revealing that their inhibitory effects are exacerbated by cross-regulation with macrophages at tumor sites. In lung infections, such as Pneumocystis pneumonia (PcP), M-MDSC expressing PD-L1 are induced and impair alveolar macrophage (AM) phagocytic activity while increasing AM expression of PD-1 (153). MDSC interaction with neutrophils has been described in mice infected with K. pneumoniae or challenged with LPS, demonstrating that MDSC efferocytose infected, apoptotic neutrophils (35). Furthermore, M-MDSC suppress DC maturation, antigen uptake, migration, and TH1 cytokine production following administration of a DC vaccine for malignant melanoma (154). Similar findings were reported following LPS stimulation and in hepatocellular carcinoma, where both MDSC subsets reduced expression ofMHC II, stimulatory molecules on DC, and cytokine production (64, 155). It stands to reason that these MDSC-induced modifications, affecting DC-mediated activation of T cells and antigen uptake, could also be effective in infectious diseases and warrant further investigation.
Natural Killer (NK) Cells
Reports on MDSC-mediated impairment of NK cell function emanate mainly from the cancer field. NK cells are critical to the innate immune system, exhibit cytotoxic and cytolytic functions, and target pathogens and malignant cells. In tumors, M-MDSC and also a population containing M-MDSC, inhibit cytotoxic activity and cytokine production by NK cells through cell contact-dependent mechanisms involving membrane-bound TGF-β and NKp30 ligand (156–158). NK cell-mediated suppression by total HLA-DRloCD33+CD11blo MDSC has also been reported in chronic HCV infection and it is mediated via an ARG1-dependent inhibition of mammalian target of rapamycin (102).
Kinetics, Interference with Immunity, and Impact on Disease Outcome
The immune inhibitory functions of M-MDSC have extensive consequences on disease outcome (Table 1). According to current understanding, the class of pathogen and the immune mediators present, collectively determine pathogen persistence versus clearance. M-MDSC have versatile roles in infection, with either beneficial or detrimental outcomes for the host depending on the pathogen and the course of infection. During long-lasting infections, MDSC may even exhibit dual roles depending on the disease stage. E.g., M-MDSC are host-protective in certain fulminant acute infections by restricting immunopathology (35, 112, 159). During late sepsis, the immature total MDSC population aggravates disease (76, 77, 160). M-MDSC may, however, be harmful in acute infection with intracellular microbes, notably francisellae (42). Alternatively, M-MDSC may be detrimental to the host, irrespective of the phase of the disease, as reported in AIDS (25). By limiting anti-viral immunity early, these regulatory monocytes foster disease progression, while provoking disease exacerbation during the chronic HIV infection.
Viruses
Viral infections are known for their induction of pro-inflammatory mediators associated with the generation of MDSC. E.g., M-MDSC are increased in both clinical and experimental viral infections, such as HIV, SIV, and LP-BM5 (25–27, 49, 93, 94). During these retroviral infections, increased levels of M-MDSC are likely detrimental to disease outcome and facilitate pathogen survival, when considering the TH1 immunosuppressive effect and correlation to viral load and CD4 T-cell count (24, 49, 54, 95). Interestingly, HIV infection-mediated expansion of M-MDSC in peripheral blood mononuclear cells may also negatively affect containment of other concurrent infections, as reported for cytomegalovirus (CMV) infection (96). Recruitment of M-MDSC-like cells were also reported for murine CMV mono-infection and shown to impair viral clearance (70). Information on MDSC in HCV infections has been variable, but largely provides evidence of unfavorable effects on host protective immunity (19, 22, 104). Increased MDSC frequencies positively correlate with HCV viral load and decreased CD8 T-cell function (21, 99). Reports show that elevated levels of immature Lin−HLA-DR−CD33+CD11b+ MDSC, consisting of M-MDSC and PMN-MDSC, in chronic HCV-infected patients, decline following successful IFN-α treatment (98), while treatment-naive HCV-infected individuals show significantly increased liver- and circulating MDSC frequencies compared to treated and uninfected individuals (99, 161). Nonetheless, other in vivo investigations failed to show significant MDSC elevations or an association with viral load (100). Ning et al. also provided evidence of increased M-MDSC in HCV-infected patients; however, this was correlated with age and not viral load, suggesting that the immune response caused by viral replication, rather than the virus itself, is responsible for increased M-MDSC (101). HBV infections are also associated with induction of MDSC. HLA-DR−/lowCD14+ M-MDSC occur at higher frequency in peripheral blood of chronic HBV-infected patients and suppress HBV-specific CD8 T-cell cytotoxicity (105). Suppressive MDSC are also increased in murine HBV infection (45) and drive CD8 T-cell exhaustion via their crosstalk with γδT-cells (46). M-MDSC accumulate during viral coinfections, but frequencies appear to be similar with those observed in mono-infections (103). E.g., elevated number of MDSC were reported for HCV/HIV (103) and shown to regulate excessive IFN-γ production in HIV/CMV coinfected individuals (96).
Bacteria
Bacterial infections are often associated with excessive inflammation or low-grade chronic production of pro-inflammatory cytokines and chemokines known to induce the expansion and activation of MDSC. E.g., chronic S. aureus infection in mice is sustained by M-MDSC and PMN-MDSC expressing ARG1, iNOS, and IL-10 which foster an immunosuppressive environment and impair monocyte/macrophage responsiveness (33, 47, 48, 108). Similarly, during infections with intracellular bacteria, such as F. tularensis, MDSC frequencies correlate with the extent of tissue pathology, loss of pulmonary function, and host mortality (42). Several reports demonstrate that inoculation of mice with BCG or infection with M. tuberculosis induce M-MDSC that diminish pathogen control and promote disease lethality (50, 65, 109). Obregón-Henao provided new evidence, demonstrating accumulation of ARG1-producing MDSC in M. tuberculosis-infected mice (41). Similar findings were reported in human TB, with increased immunosuppressive M-MDSC in TB patients and individuals with recent exposure to TB patients (28, 110). More recently, a protective role of M-MDSC in early stages of BCG-induced pleurisy was reported (80). This effect has been linked to TNF-dependent suppression of CD4+ T-cell inflammation. MDSC were also highly induced following infection with a clinical isolate of multidrug-resistant K. pneumoniae. These M-MDSC express anti-inflammatory surface markers and displayed compromised phagocytic abilities (3). Impairment of IL-10 production from total MDSC inhibited resolution of K. pneumoniae-induced inflammation (4). H. pylori-mediated inflammation of the gastric mucosa also promoted an influx of M-MDSC that countered host protective TH1 immune responses (111). In addition, MDSC gradually increase after polymicrobial sepsis (75–77), with M-MDSC mainly promoting sepsis-induced mortality early during infection (75).
Fungi
TH17-polarized immunity is generally required for protection against fungal infections; however, fungi modulate host immunity by inducing immunosuppressive MDSC which could also benefit the host by reducing hyperinflammatory responses (84). The majority of studies only report the induction of PMN-MDSC following infection with pathogenic fungi, such as Candida albicans and Aspergillus fumigatus (84, 162). In line with this, treatment of mice with yeast-derived antigens, such as β-glucan specific to dectin-1, reduced accumulation of PMN-MDSC but not M-MDSC and significantly decreased tumor burden (163).
Protozoa
Induction of potent TH1 immunity is generally sufficient to protect the host against debilitating protozoal expansion and pathology. While MDSC are typically detrimental to diseases requiring a robust host protective TH1 response, MDSC induction could in fact be beneficial during infections triggering inflammation-mediated tissue damage. For example, chronic and acute protozoan infections with L. major or Trypanosoma cruzi, mediate induction of M-MDSC which protect against pathology and parasite load, despite suppression of T-cell proliferation (73, 116, 118), although contradictory evidence have been reported (117). Similar results were shown in a mouse model of Toxoplasma gondii infection, where the total MDSC population induced hyporesponsiveness and were required for resistance against the pathogen (119). Corroborating work demonstrated that the absence of cells resembling total MDSC during acute T. gondii infection resulted in extensive intestinal necrosis due to the host TH1 inflammatory response (119, 120). More recent data on L. donovani provided evidence of the expansion of myeloid cells, likely a combination of M-MDSC and PMN-MDSC, in the spleens of infected BALB/c and C57BL/6 mice. These cells exhibit TH1 immunosuppressive features and their immunosuppressive capacity is reduced following soluble leishmanial antigen vaccination (114, 115).
Helminths
Helminths characteristically cause stable, long-term infections with severe host immunomodulatory consequences, such as triggering TH2 host immune polarization. Several helminth species and their excretory/secretory products induce accumulation of M-MDSC, including Schistosoma spp. (121), Echinnococcus granulosus (122), and Nippostrongylus brasiliensis (123). Important work in a mouse model of Heligmosomoides polygyrus bakeri infection revealed the induction of a MDSC subset, likely comprising M-MDSC and PMN-MDSC, with TH2 immunosuppressive capabilities that exacerbate infection and worm burden (124, 125). Another important consideration during helminth infections is the host protective effect of MDSC-mediated suppression of TH1 immunity and induction of TH2 immunity. E.g., MDSC mediate enhanced pathogen clearance in a model of N. brasiliensis infection, although this appears to be specific to the granulocytic subset and might increase host susceptibility to diseases requiring TH1 for protection (123).
Monocytic myeloid-derived suppressor cells have been investigated only in a number of infections. In some circumstances, this MRC subset emerges as a regulator of disease pathogenesis. Based on depletion studies in animal models and correlative studies in humans undergoing anti-infective therapy, M-MDSC have both host-destructive and -protective roles. They promote establishment and progression of HIV/SIV (24, 25, 49), LCMV (71), staphylococcal prosthetic complications (33, 48, 108), and TB (29, 30) (Table 1). On the contrary, several studies indicate that this MRC subset protects from immunopathology, particularly in certain acute bacterial infections (35) and in protozoal infection (73), but also at distinct stages of viral infection with vaccinia virus (164). In such circumstances, M-MDSC contribute to resolution of inflammation or prevent disease flares. Such dual roles may correlate with biology of M-MDSC, notably their interaction with pathogens.
Phagocytic M-MDSC Harboring Pathogens
Subcellular compartmentalization of microbes within M-MDSC, as well as how pathogens modulate cell death patterns or metabolic features of these monocytic cells have not been fully elucidated. Since MDSC are phagocytes, an alternative function of M-MDSC is as a reservoir for invading pathogens. Initial evidence of impaired pathogen elimination came from a mouse model showing that mycobacteria, notably BCG, are phagocytosed by CD11b+Ly6CintLy6G− MDSC (65). Despite NO production, they were unable to kill M. bovis or the nonpathogenic M. smegmatis and suppressed T-cell activation. More recent data demonstrate that murine MDSC, induced following M. tuberculosis infection, display dose-dependent phagocytic and endocytic capabilities (30). Considering that M. tuberculosis survival in phagocytes is attributed to host-derived lipids, and since these serve as their primary carbon source via the glyoxylate shunt, it is tempting to speculate that MDSC provide niche for pathogen persistence. This assumption is supported by the finding that MDSC highly express complement receptor-3 CD11b and receptors for oxidized lipid (oxLDL)-uptake (CD36 and LOX-1) (165), which assist M. tuberculosis engulfment (166, 167). MDSC-resembling cells were shown to contain microbes, such as Escherichia coli and L. major (52, 55, 73, 113).
Other investigators report on defects in MDSC phagocytic potential under conditions of persistent stimulation or chronic inflammation (168). M-MDSC displayed reduced uptake of F. tularensis in comparison to naïve bone marrow-derived macrophages or AM (42) and poor phagocytic/killing potential of K. pneumoniae (3). MDSC may also impair the phagocytic potential of other innate cells. For example, the phagocytic ability of AM is significantly reduced in the presence of MDSC from PcP-infected mice. These adverse effects on AM are dependent on MDSC expressing PD-L1 and induction of PD-1 expression in AM during PcP infection (153, 169). Nonetheless, others failed to show any significant impact of MDSC on macrophage phagocytic potential (170).
Besides harboring bacterial pathogens, M-MDSC may support replication of viruses. Retroviruses, including SIV (25), LP-BM5 (93), and HIV (24) have been detected within this monocytic subset in macaques, mice, and humans, respectively. M-MDSC may traffic and interact with lymphocytes and thereby contribute to viral spread, besides limiting functionality of T lymphocytes.
Conclusion and Outlook
Many open questions and challenges for MDSC research remain. In particular, evidence on human MDSC subset characterization and their place in the spectrum of the myeloid lineage are still conflicting. In mice, TAM differentiation from M-MDSC may be accomplished to some extent based on positivity of TAM for F4/80 and their low or negative expression of Ly6C along with higher transcript levels for IRF8, M-CSF, and reduced ER-stress markers (2, 36, 171, 172). A detailed comparison between activated tissue macrophages and M-MDSC has not been conclusively conducted in infection. Lineage-tagging studies and phenotype stability are currently lacking and, therefore, tracing M-MDSC development in infection is either hypothetical or based on ex vivo observations and extrapolations from cancer models. Furthermore, a detailed understanding of the pathogen- and host-derived signals modulating MDSC induction and function will assist in the development of their therapeutic application. Specifically, the factors mediating suppression of host immunity in an antigen-specific manner need to be better understood to exploit drugs inhibiting MDSC in infections where these cells favor pathogen survival or limit optimal host responses. Moreover, pathogen responses, including stress and adaptation, to M-MDSC have not been investigated yet.
Although several therapeutic approaches involving re-purposed agents, mostly all-trans retinoic acid, effectively reverse MDSC immunosuppressive features in murine infection models of TB (30) and sepsis (173) as well as in few ex vivo human studies in HBV (18), comprehensive human clinical studies are required to systematically assess the safety, efficacy, dose, and timing of such interventions. Same rationale may improve vaccination in case of live vaccine, notably BCG and viral vector-based vaccines against HIV, known to trigger M-MDSC (65, 94). Furthermore, considering the diagnostic and prognostic potential of MDSC in the cancer field, these myeloid regulatory subsets should be considered for their potential role in biomarker development for infectious diseases.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Helga Keßler and Helena Kuivaniemi for the editorial assistance and Diane Schad for assistance with the graphics work. AD acknowledges the European Cooperation in Science and Technology program “Mye-EUNITER”; NDP acknowledges the “ICIDR” Biology and Biosignatures of Anti-Tuberculosis Treatment Response (NIH U01 AI115619).
References
1. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res (2007) 67(1):425. doi:10.1158/0008-5472
2. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
3. Ahn D, Peñaloza H, Wang Z, Wickersham M, Parker D, Patel P, et al. Acquired resistance to innate immune clearance promotes Klebsiella pneumoniae ST258 pulmonary infection. JCI Insight (2016) 1(17):e89704. doi:10.1172/jci.insight.89704
4. Chakraborty K, Raundhal M, Chen BB, Morse C, Tyurina YY, Khare A, et al. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat Commun (2017) 8:13944. doi:10.1038/ncomms13944
5. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13(10):739–52. doi:10.1038/nrc3581
6. Bennett JA, Rao VS, Mitchell MS. Systemic bacillus Calmette-Guerin (BCG) activates natural suppressor cells. Proc Natl Acad Sci U S A (1978) 75(10):5142–4. doi:10.1073/pnas.75.10.5142
7. Nathan C, Ding A. Nonresolving inflammation. Cell (2010) 140(6):871–82. doi:10.1016/j.cell.2010.02.029
8. Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S, Totaro MG, et al. RORC1 regulates tumor-promoting “emergency” granulo-monocytopoiesis. Cancer Cell (2015) 28(2):253–69. doi:10.1016/j.ccell.2015.07.006
9. Ost M, Singh A, Peschel A, Mehling R, Rieber N, Hartl D. Myeloid-derived suppressor cells in bacterial infections. Front Cell Infect Microbiol (2016) 6:37. doi:10.3389/fcimb.2016.00037
10. Goh C, Narayanan S, Hahn YS. Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev (2013) 255(1):210–21. doi:10.1111/imr.12084
11. Ray A, Chakraborty K, Ray P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front Cell Infect Microbiol (2013) 3:52. doi:10.3389/fcimb.2013.00052
12. O’Connor MA, Rastad JL, Green WR. The role of myeloid-derived suppressor cells in viral infection. Viral Immunol (2017) 30(2):82–97. doi:10.1089/vim.2016.0125
13. WHO. Global Tuberculosis Report 2016. World Health Organization (2016). Available from: http://www.who.int/tb/publications/global_report/en/
14. Thammavongsa V, Kim HK, Missiakas D, Schneewind O. Staphylococcal manipulation of host immune responses. Nat Rev Microbiol (2015) 13(9):529–43. doi:10.1038/nrmicro3521
15. O’Neill J. Tackling Drug-resistance Infections Globally: Final Report and Recommendations. Review on Antimicrobial Resistance. (2016). Available from: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf
16. WHO. Global Hepatitis Report 2017. World Health Organization (2017). Available from: http://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/
17. WHO. HIV/AIDS Fact Sheet. World Health Organization (2017). Available from: http://www.who.int/mediacentre/factsheets/fs360/en/
18. Fang Z, Li J, Yu X, Zhang D, Ren G, Shi B, et al. Polarization of monocytic myeloid-derived suppressor cells by hepatitis B surface antigen is mediated via ERK/IL-6/STAT3 signaling feedback and restrains the activation of T cells in chronic hepatitis B virus infection. J Immunol (2015) 195(10):4873–83. doi:10.4049/jimmunol.1501362
19. Pang X, Song H, Zhang Q, Tu Z, Niu J. Hepatitis C virus regulates the production of monocytic myeloid-derived suppressor cells from peripheral blood mononuclear cells through PI3K pathway and autocrine signaling. Clin Immunol (2016) 164:57–64. doi:10.1016/j.clim.2016.01.014
20. Zhai N, Li H, Song H, Yang Y, Cui A, Li T, et al. Hepatitis C virus induces MDSCs-like monocytes through TLR2/PI3K/AKT/STAT3 signaling. PLoS One (2017) 12(1):e0170516. doi:10.1371/journal.pone.0170516
21. Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR, et al. Myeloid suppressor cells induced by hepatitis C virus suppress T-cell responses through the production of reactive oxygen species. Hepatology (2012) 55(2):343–53. doi:10.1002/hep.24700
22. Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology (2016) 148(4):377–86. doi:10.1111/imm.12616
23. Garg A, Spector SA. HIV type 1 gp120-induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J Infect Dis (2014) 209(3):441–51. doi:10.1093/infdis/jit469
24. Qin A, Cai W, Pan T, Wu K, Yang Q, Wang N, et al. Expansion of monocytic myeloid-derived suppressor cells dampens T cell function in HIV-1-seropositive individuals. J Virol (2013) 87(3):1477–90. doi:10.1128/jvi.01759-12
25. Sui Y, Frey B, Wang Y, Billeskov R, Kulkarni S, McKinnon K, et al. Paradoxical myeloid-derived suppressor cell reduction in the bone marrow of SIV chronically infected macaques. PLoS Pathog (2017) 13(5):e1006395. doi:10.1371/journal.ppat.1006395
26. Gama L, Shirk EN, Russell JN, Carvalho KI, Li M, Queen SE, et al. Expansion of a subset of CD14highCD16negCCR2low/neg monocytes functionally similar to myeloid-derived suppressor cells during SIV and HIV infection. J Leukoc Biol (2012) 91(5):803–16. doi:10.1189/jlb.1111579
27. O’Connor MA, Fu WW, Green KA, Green WR. Subpopulations of M-MDSCs from mice infected by an immunodeficiency-causing retrovirus and their differential suppression of T- vs B-cell responses. Virology (2015) 485:263–73. doi:10.1016/j.virol.2015.07.020
28. du Plessis N, Jacobs R, Gutschmidt A, Fang Z, van Helden PD, Lutz MB, et al. Phenotypically resembling myeloid derived suppressor cells are increased in children with HIV and exposed/infected with Mycobacterium tuberculosis. Eur J Immunol (2017) 47(1):107–18. doi:10.1002/eji.201646658
29. du Plessis N, Loebenberg L, Kriel M, von Groote-Bidlingmaier F, Ribechini E, Loxton AG, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent Mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med (2013) 188(6):724–32. doi:10.1164/rccm.201302-0249OC
30. Knaul JK, Jorg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190(9):1053–66. doi:10.1164/rccm.201405-0828OC
31. Peng KT, Hsieh CC, Huang TY, Chen PC, Shih HN, Lee MS, et al. Staphylococcus aureus biofilm elicits the expansion, activation and polarization of myeloid-derived suppressor cells in vivo and in vitro. PLoS One (2017) 12(8):e0183271. doi:10.1371/journal.pone.0183271
32. Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, et al. Cutaneous innate immune sensing of toll-like receptor 2–6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity (2014) 41(5):762–75. doi:10.1016/j.immuni.2014.10.009
33. Tebartz C, Horst SA, Sparwasser T, Huehn J, Beineke A, Peters G, et al. A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol (2015) 194(3):1100–11. doi:10.4049/jimmunol.1400196
34. Thiele Orberg E, Fan H, Tam AJ, Dejea CM, Destefano Shields CE, Wu S, et al. The myeloid immune signature of enterotoxigenic Bacteroides fragilis-induced murine colon tumorigenesis. Mucosal Immunol (2017) 10(2):421–33. doi:10.1038/mi.2016.53
35. Poe SL, Arora M, Oriss TB, Yarlagadda M, Isse K, Khare A, et al. STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol (2013) 6(1):189–99. doi:10.1038/mi.2012.62
36. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi:10.1016/j.it.2016.01.004
37. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest (2012) 122(11):4094–104. doi:10.1172/JCI64115
38. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res (2007) 67(9):4507–13. doi:10.1158/0008-5472.CAN-06-4174
39. Haverkamp JM, Smith AM, Weinlich R, Dillon CP, Qualls JE, Neale G, et al. Myeloid-derived suppressor activity is mediated by monocytic lineages maintained by continuous inhibition of extrinsic and intrinsic death pathways. Immunity (2014) 41(6):947–59. doi:10.1016/j.immuni.2014.10.020
40. Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, et al. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell (2007) 131(5):994–1008. doi:10.1016/j.cell.2007.09.047
41. Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. Gr1intCD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS One (2013) 8(11):e80669. doi:10.1371/journal.pone.0080669
42. Periasamy S, Avram D, McCabe A, MacNamara KC, Sellati TJ, Harton JA. An immature myeloid/myeloid-suppressor cell response associated with necrotizing inflammation mediates lethal pulmonary tularemia. PLoS Pathog (2016) 12(3):e1005517. doi:10.1371/journal.ppat.1005517
43. De Santo C, Salio M, Masri SH, Lee LY, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest (2008) 118(12):4036–48. doi:10.1172/jci36264
44. Jeisy-Scott V, Davis WG, Patel JR, Bowzard JB, Shieh WJ, Zaki SR, et al. Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS One (2011) 6(9):e25242. doi:10.1371/journal.pone.0025242
45. Chen S, Akbar SM, Abe M, Hiasa Y, Onji M. Immunosuppressive functions of hepatic myeloid-derived suppressor cells of normal mice and in a murine model of chronic hepatitis B virus. Clin Exp Immunol (2011) 166(1):134–42. doi:10.1111/j.1365-2249.2011.04445.x
46. Kong X, Sun R, Chen Y, Wei H, Tian Z. γδT cells drive myeloid-derived suppressor cell-mediated CD8+ T cell exhaustion in hepatitis B virus-induced immunotolerance. J Immunol (2014) 193(4):1645–53. doi:10.4049/jimmunol.1303432
47. Heim CE, Vidlak D, Kielian T. Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol (2015) 98(6):1003–13. doi:10.1189/jlb.4VMA0315-125RR
48. Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, Kielian T. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol (2015) 194(8):3861–72. doi:10.4049/jimmunol.1402689
49. Vollbrecht T, Stirner R, Tufman A, Roider J, Huber RM, Bogner JR, et al. Chronic progressive HIV-1 infection is associated with elevated levels of myeloid-derived suppressor cells. AIDS (2012) 26(12):F31–7. doi:10.1097/QAD.0b013e328354b43f
50. Tsiganov EN, Verbina EM, Radaeva TV, Sosunov VV, Kosmiadi GA, Nikitina IY, et al. Gr-1dimCD11b+ immature myeloid-derived suppressor cells but not neutrophils are markers of lethal tuberculosis infection in mice. J Immunol (2014) 192(10):4718–27. doi:10.4049/jimmunol.1301365
51. Arora M, Poe SL, Oriss TB, Krishnamoorthy N, Yarlagadda M, Wenzel SE, et al. TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol (2010) 3(6):578–93. doi:10.1038/mi.2010.41
52. Bernsmeier C, Triantafyllou E, Brenig R, Lebosse FJ, Singanayagam A, Patel VC, et al. CD14+CD15-HLA-DR- myeloid-derived suppressor cells impair antimicrobial responses in patients with acute-on-chronic liver failure. Gut (2017). doi:10.1136/gutjnl-2017-314184
53. Van Rompaey N, Le Moine A. Myeloid-derived suppressor cells: characterization and expansion in models of endotoxemia and transplantation. Methods Mol Biol (2011) 677:169–80. doi:10.1007/978-1-60761-869-0_12
54. Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, Elgazzar MA, et al. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS (2016) 30(10):1521–31. doi:10.1097/qad.0000000000001083
55. Delano MJ, Scumpia PO, Weinstein JS, Coco D, Nagaraj S, Kelly-Scumpia KM, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med (2007) 204(6):1463–74. doi:10.1084/jem.20062602
56. Liu C, Zhang C, Lu H, Cai J, Wang Z, Chen J, et al. Poly(I:C) induce bone marrow precursor cells into myeloid-derived suppressor cells. Mol Cell Biochem (2011) 358(1–2):317–23. doi:10.1007/s11010-011-0982-3
57. Dorhoi A, Kaufmann SH. Perspectives on host adaptation in response to Mycobacterium tuberculosis: modulation of inflammation. Semin Immunol (2014) 26(6):533–42. doi:10.1016/j.smim.2014.10.002
58. Bunt SK, Clements VK, Hanson EM, Sinha P, Ostrand-Rosenberg S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through toll-like receptor 4. J Leukoc Biol (2009) 85(6):996–1004. doi:10.1189/jlb.0708446
59. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205(10):2235–49. doi:10.1084/jem.20080132
60. Thaxton JE, Liu B, Zheng P, Liu Y, Li Z. Deletion of CD24 impairs development of heat shock protein gp96-driven autoimmune disease through expansion of myeloid-derived suppressor cells. J Immunol (2014) 192(12):5679–86. doi:10.4049/jimmunol.1302755
61. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120(2):457–71. doi:10.1172/jci40483
62. Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med (2013) 188(9):1137–46. doi:10.1164/rccm.201304-0803OC
63. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
64. Greifenberg V, Ribechini E, Rossner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol (2009) 39(10):2865–76. doi:10.1002/eji.200939486
65. Martino A, Badell E, Abadie V, Balloy V, Chignard M, Mistou MY, et al. Mycobacterium bovis bacillus Calmette-Guerin vaccination mobilizes innate myeloid-derived suppressor cells restraining in vivo T cell priming via IL-1R-dependent nitric oxide production. J Immunol (2010) 184(4):2038–47. doi:10.4049/jimmunol.0903348
66. Wang Z, Jiang J, Li Z, Zhang J, Wang H, Qin Z. A myeloid cell population induced by Freund adjuvant suppresses T-cell-mediated antitumor immunity. J Immunother (2010) 33(2):167–77. doi:10.1097/CJI.0b013e3181bed2ba
67. Gato-Canas M, Martinez de Morentin X, Blanco-Luquin I, Fernandez-Irigoyen J, Zudaire I, Liechtenstein T, et al. A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget (2015) 6(29):27160–75. doi:10.18632/oncotarget.4746
68. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
69. Su L, Xu Q, Zhang P, Michalek SM, Katz J. Phenotype and function of myeloid-derived suppressor cells induced by Porphyromonas gingivalis infection. Infect Immun (2017) 85(8):e00213-17. doi:10.1128/IAI.00213-17
70. Daley-Bauer LP, Wynn GM, Mocarski ES. Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity (2012) 37(1):122–33. doi:10.1016/j.immuni.2012.04.014
71. Norris BA, Uebelhoer LS, Nakaya HI, Price AA, Grakoui A, Pulendran B. Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity. Immunity (2013) 38(2):309–21. doi:10.1016/j.immuni.2012.10.022
72. Rastad JL, Green WR. Myeloid-derived suppressor cells in murine AIDS inhibit B-cell responses in part via soluble mediators including reactive oxygen and nitrogen species, and TGF-beta. Virology (2016) 499:9–22. doi:10.1016/j.virol.2016.08.031
73. Pereira WF, Ribeiro-Gomes FL, Guillermo LV, Vellozo NS, Montalvao F, Dosreis GA, et al. Myeloid-derived suppressor cells help protective immunity to Leishmania major infection despite suppressed T cell responses. J Leukoc Biol (2011) 90(6):1191–7. doi:10.1189/jlb.1110608
74. Schmid M, Zimara N, Wege AK, Ritter U. Myeloid-derived suppressor cell functionality and interaction with Leishmania major parasites differ in C57BL/6 and BALB/c mice. Eur J Immunol (2014) 44(11):3295–306. doi:10.1002/eji.201344335
75. Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun (2012) 80(6):2026–34. doi:10.1128/IAI.00239-12
76. McPeak MB, Youssef D, Williams DA, Pritchett C, Yao ZQ, McCall CE, et al. Myeloid cell-specific knockout of NFI-A improves sepsis survival. Infect Immun (2017) 85(4):e00066-17. doi:10.1128/IAI.00066-17
77. McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, et al. Frontline science: myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. J Leukoc Biol (2017) 102(2):191–200. doi:10.1189/jlb.4HI1216-537R
78. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity (2013) 38(3):541–54. doi:10.1016/j.immuni.2013.02.007
79. Polz J, Remke A, Weber S, Schmidt D, Weber-Steffens D, Pietryga-Krieger A, et al. Myeloid suppressor cells require membrane TNFR2 expression for suppressive activity. Immun Inflamm Dis (2014) 2(2):121–30. doi:10.1002/iid3.19
80. Chavez-Galan L, Vesin D, Uysal H, Blaser G, Benkhoucha M, Ryffel B, et al. Transmembrane tumor necrosis factor controls myeloid-derived suppressor cell activity via TNF receptor 2 and protects from excessive inflammation during BCG-induced pleurisy. Front Immunol (2017) 8:999. doi:10.3389/fimmu.2017.00999
81. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis (2013) 208(2):199–202. doi:10.1093/infdis/jit152
82. Coulombe F, Divangahi M. Targeting eicosanoid pathways in the development of novel anti-influenza drugs. Expert Rev Anti Infect Ther (2014) 12(11):1337–43. doi:10.1586/14787210.2014.966082
83. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511(7507):99–103. doi:10.1038/nature13489
84. Rieber N, Singh A, Oz H, Carevic M, Bouzani M, Amich J, et al. Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe (2015) 17(4):507–14. doi:10.1016/j.chom.2015.02.007
85. van Deventer HW, Burgents JE, Wu QP, Woodford RM, Brickey WJ, Allen IC, et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res (2010) 70(24):10161–9. doi:10.1158/0008-5472.CAN-10-1921
86. Ribechini E, Hutchinson JA, Hergovits S, Heuer M, Lucas J, Schleicher U, et al. Novel GM-CSF signals via IFN-γR/IRF-1 and AKT/mTOR license monocytes for suppressor function. Blood (2017) 1:947–60. doi:10.1182/bloodadvances.2017006858
87. Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med (2015) 66:97–110. doi:10.1146/annurev-med-051013-052304
88. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13(1):23–35. doi:10.1016/j.ccr.2007.12.004
89. Zhang H, Li Z, Wang L, Tian G, Tian J, Yang Z, et al. Critical role of myeloid-derived suppressor cells in tumor-induced liver immune suppression through inhibition of NKT cell function. Front Immunol (2017) 8:129. doi:10.3389/fimmu.2017.00129
90. Zhang S, Ma X, Zhu C, Liu L, Wang G, Yuan X. The role of myeloid-derived suppressor cells in patients with solid tumors: a meta-analysis. PLoS One (2016) 11(10):e0164514. doi:10.1371/journal.pone.0164514
91. Lee C-R, Kwak Y, Yang T, Han JH, Park S-H, Ye MB, et al. Myeloid-derived suppressor cells are controlled by regulatory T cells via TGF-β during murine colitis. Cell Rep (2016) 17(12):3219–32. doi:10.1016/j.celrep.2016.11.062
92. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget (2016) 7(2):1168–84. doi:10.18632/oncotarget.6662
93. Green KA, Cook WJ, Green WR. Myeloid-derived suppressor cells in murine retrovirus-induced AIDS inhibit T- and B-cell responses in vitro that are used to define the immunodeficiency. J Virol (2013) 87(4):2058–71. doi:10.1128/jvi.01547-12
94. Sui Y, Hogg A, Wang Y, Frey B, Yu H, Xia Z, et al. Vaccine-induced myeloid cell population dampens protective immunity to SIV. J Clin Invest (2014) 124(6):2538–49. doi:10.1172/jci73518
95. O’Connor MA, Vella JL, Green WR. Reciprocal relationship of T regulatory cells and monocytic myeloid-derived suppressor cells in LP-BM5 murine retrovirus-induced immunodeficiency. J Gen Virol (2016) 97(2):509–22. doi:10.1099/jgv.0.000260
96. Garg A, Trout R, Spector SA. Human immunodeficiency virus type-1 myeloid derived suppressor cells inhibit cytomegalovirus inflammation through interleukin-27 and B7-H4. Sci Rep (2017) 7:44485. doi:10.1038/srep44485
97. Dross SE, Munson PV, Kim SE, Bratt DL, Tunggal HC, Gervassi AL, et al. Kinetics of myeloid-derived suppressor cell frequency and function during Simian Immunodeficiency Virus infection, combination antiretroviral therapy, and treatment interruption. J Immunol (2017) 198(2):757–66. doi:10.4049/jimmunol.1600759
98. Salem ML, Zidan A-AA, Abou Senna M, Abou Al-Azm AR, Albatei H, Aldemelaawy M, et al. Frequencies of circulating myeloid derived suppressor cells and dendritic cells in Egyptian patients with chronic Hepatitis C Virus infection undergoing treatment with IFN-α-based therapy. J Immunother Cancer (2013) 1(1):248. doi:10.1186/2051-1426-1-s1-p248
99. Zeng Q-L, Yang B, Sun H-Q, Feng G-H, Jin L, Zou Z-S, et al. Myeloid-derived suppressor cells are associated with viral persistence and downregulation of TCR ζ chain expression on CD8(+) T cells in chronic hepatitis C patients. Mol Cells (2014) 37(1):66–73. doi:10.14348/molcells.2014.2282
100. Nonnenmann J, Stirner R, Roider J, Jung MC, Schrödl K, Bogner JR, et al. Lack of significant elevation of myeloid-derived suppressor cells in peripheral blood of chronically hepatitis C virus-infected individuals. J Virol (2014) 88(13):7678–82. doi:10.1128/JVI.00113-14
101. Ning G, She L, Lu L, Liu Y, Zeng Y, Yan Y, et al. Analysis of monocytic and granulocytic myeloid-derived suppressor cells subsets in patients with hepatitis C virus infection and their clinical significance. Biomed Res Int (2015) 2015:8. doi:10.1155/2015/385378
102. Goh CC, Roggerson KM, Lee HC, Golden-Mason L, Rosen HR, Hahn YS. Hepatitis C virus-induced myeloid-derived suppressor cells suppress NK cell IFN-gamma production by altering cellular metabolism via arginase-1. J Immunol (2016) 196(5):2283–92. doi:10.4049/jimmunol.1501881
103. Lei AH, Yang Q, Cai WP, Liu YF, Lan Y, Qin AP, et al. Clinical significance of myeloid-derived suppressor cells in human immunodeficiency virus-1/hepatitis C virus-coinfected patients. Scand J Immunol (2016) 83(6):438–44. doi:10.1111/sji.12429
104. Ren JP, Wang L, Zhao J, Wang L, Ning SB, El Gazzar M, et al. Decline of miR-124 in myeloid cells promotes regulatory T-cell development in hepatitis C virus infection. Immunology (2017) 150(2):213–20. doi:10.1111/imm.12680
105. Huang A, Zhang B, Yan W, Wang B, Wei H, Zhang F, et al. Myeloid-derived suppressor cells regulate immune response in patients with chronic hepatitis B virus infection through PD-1-induced IL-10. J Immunol (2014) 193(11):5461–9. doi:10.4049/jimmunol.1400849
106. Kondo Y, Shimosegawa T. Significant roles of regulatory T cells and myeloid derived suppressor cells in Hepatitis B Virus persistent infection and Hepatitis B Virus-related HCCs. Int J Mol Sci (2015) 16(2):3307–22. doi:10.3390/ijms16023307
107. Tumino N, Casetti R, Fabbri G, Cimini E, Romanelli A, Turchi F, et al. In HIV/HCV co-infected patients T regulatory and myeloid-derived suppressor cells persist after successful treatment with directly acting antivirals. J Hepatol (2017) 67(2):422–4. doi:10.1016/j.jhep.2017.03.036
108. Heim CE, Vidlak D, Scherr TD, Kozel JA, Holzapfel M, Muirhead DE, et al. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol (2014) 192(8):3778–92. doi:10.4049/jimmunol.1303408
109. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, van der Veen RC. Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leukoc Biol (2007) 81(5):1205–12. doi:10.1189/jlb.1006640
110. Yang B, Wang X, Jiang J, Zhai F, Cheng X. Identification of CD244-expressing myeloid-derived suppressor cells in patients with active tuberculosis. Immunol Lett (2014) 158(1–2):66–72. doi:10.1016/j.imlet.2013.12.003
111. Zhuang Y, Cheng P, Liu XF, Peng LS, Li BS, Wang TT, et al. A pro-inflammatory role for Th22 cells in Helicobacter pylori-associated gastritis. Gut (2015) 64(9):1368–78. doi:10.1136/gutjnl-2014-307020
112. Sander LE, Sackett SD, Dierssen U, Beraza N, Linke RP, Muller M, et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J Exp Med (2010) 207(7):1453–64. doi:10.1084/jem.20091474
113. Ribeiro-Gomes FL, Roma EH, Carneiro MB, Doria NA, Sacks DL, Peters NC. Site-dependent recruitment of inflammatory cells determines the effective dose of Leishmania major. Infect Immun (2014) 82(7):2713–27. doi:10.1128/IAI.01600-13
114. Bandyopadhyay S, Bhattacharjee A, Banerjee S, Halder K, Das S, Paul Chowdhury B, et al. Glycyrrhizic acid-mediated subdual of myeloid-derived suppressor cells induces antileishmanial immune responses in a susceptible host. Infect Immun (2015) 83(12):4476–86. doi:10.1128/IAI.00729-15
115. Hammami A, Abidin BM, Charpentier T, Fabie A, Duguay AP, Heinonen KM, et al. HIF-1alpha is a key regulator in potentiating suppressor activity and limiting the microbicidal capacity of MDSC-like cells during visceral leishmaniasis. PLoS Pathog (2017) 13(9):e1006616. doi:10.1371/journal.ppat.1006616
116. Goni O, Alcaide P, Fresno M. Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1(+))CD11b(+)immature myeloid suppressor cells. Int Immunol (2002) 14(10):1125–34. doi:10.1093/intimm/dxf076
117. Cuervo H, Guerrero NA, Carbajosa S, Beschin A, De Baetselier P, Girones N, et al. Myeloid-derived suppressor cells infiltrate the heart in acute Trypanosoma cruzi infection. J Immunol (2011) 187(5):2656–65. doi:10.4049/jimmunol.1002928
118. Arocena AR, Onofrio LI, Pellegrini AV, Carrera Silva AE, Paroli A, Cano RC, et al. Myeloid-derived suppressor cells are key players in the resolution of inflammation during a model of acute infection. Eur J Immunol (2014) 44(1):184–94. doi:10.1002/eji.201343606
119. Voisin M-B, Buzoni-Gatel D, Bout D, Velge-Roussel F. Both expansion of regulatory GR1(+) CD11b(+) myeloid cells and anergy of T lymphocytes participate in hyporesponsiveness of the lung-associated immune system during acute toxoplasmosis. Infect Immun (2004) 72(9):5487–92. doi:10.1128/IAI.72.9.5487-5492.2004
120. Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity (2008) 29(2):306–17. doi:10.1016/j.immuni.2008.05.019
121. Yang Q, Qiu H, Xie H, Qi Y, Cha H, Qu J, et al. A Schistosoma japonicum infection promotes the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. J Immunol (2017) 198(12):4716–27. doi:10.4049/jimmunol.1601860
122. Pan W, Zhou HJ, Shen YJ, Wang Y, Xu YX, Hu Y, et al. Surveillance on the status of immune cells after Echinnococcus granulosus protoscoleces infection in Balb/c mice. PLoS One (2013) 8(3):e59746. doi:10.1371/journal.pone.0059746
123. Saleem SJ, Martin RK, Morales JK, Sturgill JL, Gibb DR, Graham L, et al. Cutting edge: mast cells critically augment myeloid-derived suppressor cell activity. J Immunol (2012) 189(2):511–5. doi:10.4049/jimmunol.1200647
124. Valanparambil RM, Tam M, Gros PP, Auger JP, Segura M, Gros P, et al. IRF-8 regulates expansion of myeloid-derived suppressor cells and Foxp3+ regulatory T cells and modulates Th2 immune responses to gastrointestinal nematode infection. PLoS Pathog (2017) 13(10):e1006647. doi:10.1371/journal.ppat.1006647
125. Valanparambil RM, Tam M, Jardim A, Geary TG, Stevenson MM. Primary Heligmosomoides polygyrus bakeri infection induces myeloid-derived suppressor cells that suppress CD4+ Th2 responses and promote chronic infection. Mucosal Immunol (2017) 10(1):238–49. doi:10.1038/mi.2016.36
126. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi:10.1158/0008-5472.can-05-1299
127. Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol (2001) 166(9):5398–406. doi:10.4049/jimmunol.166.9.5398
128. Drabczyk-Pluta M, Werner T, Hoffmann D, Leng Q, Chen L, Dittmer U, et al. Granulocytic myeloid-derived suppressor cells suppress virus-specific CD8+ T cell responses during acute Friend retrovirus infection. Retrovirology (2017) 14(1):42. doi:10.1186/s12977-017-0364-3
129. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172(2):989–99. doi:10.4049/jimmunol.172.2.989
130. Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells. Adv Exp Med Biol (2007) 601:213–23. doi:10.1007/978-0-387-72005-0_22
131. Lai D, Qin C, Shu Q. Myeloid-derived suppressor cells in sepsis. Biomed Res Int (2014) 2014:8. doi:10.1155/2014/598654
132. Bowen JL, Olson JK. Innate immune CD11b+Gr-1+ cells, suppressor cells, affect the immune response during Theiler’s virus-induced demyelinating disease. J Immunol (2009) 183(11):6971–80. doi:10.4049/jimmunol.0902193
133. Yi H, Guo C, Yu X, Zuo D, Wang XY. Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol (2012) 189(9):4295–304. doi:10.4049/jimmunol.1200086
134. Hammerich L, Warzecha KT, Stefkova M, Bartneck M, Ohl K, Gassler N, et al. Cyclic adenosine monophosphate-responsive element modulator alpha overexpression impairs function of hepatic myeloid-derived suppressor cells and aggravates immune-mediated hepatitis in mice. Hepatology (2015) 61(3):990–1002. doi:10.1002/hep.27571
135. Obermajer N, Wong JL, Edwards RP, Chen K, Scott M, Khader S, et al. Induction and stability of human Th17 cells require endogenous NOS2 and cGMP-dependent NO signaling. J Exp Med (2013) 210(7):1433–45. doi:10.1084/jem.20121277
136. Yazawa T, Shibata M, Gonda K, Machida T, Suzuki S, Kenjo A, et al. Increased IL-17 production correlates with immunosuppression involving myeloid-derived suppressor cells and nutritional impairment in patients with various gastrointestinal cancers. Mol Clin Oncol (2013) 1(4):675–9. doi:10.3892/mco.2013.134
137. Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene (2017) 36(5):639–51. doi:10.1038/onc.2016.229
138. Park MJ, Lee SH, Kim EK, Lee EJ, Park SH, Kwok SK, et al. Myeloid-derived suppressor cells induce the expansion of regulatory B cells and ameliorate autoimmunity in the sanroque mouse model of systemic lupus erythematosus. Arthritis Rheumatol (2016) 68(11):2717–27. doi:10.1002/art.39767
139. Cripps JG, Wang J, Maria A, Blumenthal I, Gorham JD. Type 1 T helper cells induce the accumulation of myeloid-derived suppressor cells in the inflamed Tgfb1 knockout mouse liver. Hepatology (2010) 52(4):1350–9. doi:10.1002/hep.23841
140. Mussai F, De Santo C, Cerundolo V. Interaction between invariant NKT cells and myeloid-derived suppressor cells in cancer patients: evidence and therapeutic opportunities. J Immunother (2012) 35(6):449–59. doi:10.1097/CJI.0b013e31825be926
141. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi:10.1158/0008-5472.can-09-1882
142. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi:10.4049/jimmunol.1201018
143. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68(13):5439–49. doi:10.1158/0008-5472.can-07-6621
144. Yang R, Cai Z, Zhang Y, Yutzy WH, Roby KF, Roden RB. CD80 in immune suppression by mouse ovarian carcinoma-associated Gr-1+CD11b+ myeloid cells. Cancer Res (2006) 66(13):6807–15. doi:10.1158/0008-5472.can-05-3755
145. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology (2008) 135(1):234–43. doi:10.1053/j.gastro.2008.03.020
146. Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol (2012) 132(4):1239–46. doi:10.1038/jid.2011.416
147. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, et al. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One (2010) 5(1):e8922. doi:10.1371/journal.pone.0008922
148. Deshane J, Zmijewski JW, Luther R, Gaggar A, Deshane R, Lai JF, et al. Free radical-producing myeloid-derived regulatory cells: potent activators and suppressors of lung inflammation and airway hyperresponsiveness. Mucosal Immunol (2011) 4(5):503–18. doi:10.1038/mi.2011.16
149. Nakamura T, Nakao T, Ashihara E, Yoshimura N. Myeloid-derived suppressor cells recruit CD4(+)/foxp3(+) regulatory T cells in a murine cardiac allograft. Transplant Proc (2016) 48(4):1275–8. doi:10.1016/j.transproceed.2015.10.060
150. Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol (2010) 185(10):5828–34. doi:10.4049/jimmunol.0903636
151. Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, et al. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol (2015) 97(3):573–82. doi:10.1189/jlb.4A0314-139R
152. Xu X, Meng Q, Erben U, Wang P, Glauben R, Kuhl AA, et al. Myeloid-derived suppressor cells promote B-cell production of IgA in a TNFR2-dependent manner. Cell Mol Immunol (2017) 14(7):597–606. doi:10.1038/cmi.2015.103
153. Lei GS, Zhang C, Lee CH. Myeloid-derived suppressor cells impair alveolar macrophages through PD-1 receptor ligation during Pneumocystis pneumonia. Infect Immun (2015) 83(2):572–82. doi:10.1128/iai.02686-14
154. Poschke I, Mao Y, Adamson L, Salazar-Onfray F, Masucci G, Kiessling R. Myeloid-derived suppressor cells impair the quality of dendritic cell vaccines. Cancer Immunol Immunother (2012) 61(6):827–38. doi:10.1007/s00262-011-1143-y
155. Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang G-J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol (2011) 46(2):156–64. doi:10.3109/00365521.2010.516450
156. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (2009) 50(3):799–807. doi:10.1002/hep.23054
157. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol (2009) 182(1):240–9. doi:10.4049/jimmunol.182.1.240
158. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res (2014) 20(15):4096–106. doi:10.1158/1078-0432.ccr-14-0635
159. Derive M, Bouazza Y, Alauzet C, Gibot S. Myeloid-derived suppressor cells control microbial sepsis. Intensive Care Med (2012) 38(6):1040–9. doi:10.1007/s00134-012-2574-4
160. Martire-Greco D, Rodriguez-Rodrigues N, Castillo LA, Vecchione MB, de Campos-Nebel M, Cordoba Moreno M, et al. Novel use of all-trans-retinoic acid in A model of lipopolysaccharide-immunosuppression to decrease the generation of myeloid-derived suppressor cells by reducing the proliferation of CD34+ precursor cells. Shock (2017) 48(1):94–103. doi:10.1097/shk.0000000000000812
161. Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res (2014) 74(3):727–37. doi:10.1158/0008-5472.can-13-2584
162. Singh A, Lelis F, Braig S, Schäfer I, Hartl D, Rieber N. Differential regulation of myeloid-derived suppressor cells by Candida species. Front Microbiol (2016) 7:1624. doi:10.3389/fmicb.2016.01624
163. Albeituni SH, Ding C, Liu M, Hu X, Luo F, Kloecker G, et al. Yeast-derived particulate β-glucan treatment subverts the suppression of myeloid-derived suppressor cells by inducing PMN-MDSC apoptosis and M-MDSC differentiation to APC in cancer. J Immunol (2016) 196(5):2167–80. doi:10.4049/jimmunol.1501853
164. Fortin C, Huang X, Yang Y. NK cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J Immunol (2012) 189(4):1843–9. doi:10.4049/jimmunol.1200584
165. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3(11):1236–47. doi:10.1158/2326-6066.cir-15-0036
166. Dodd CE, Pyle CJ, Glowinski R, Rajaram MV, Schlesinger LS. CD36-mediated uptake of surfactant lipids by human macrophages promotes intracellular growth of Mycobacterium tuberculosis. J Immunol (2016) 197(12):4727–35. doi:10.4049/jimmunol.1600856
167. Philips JA, Rubin EJ, Perrimon N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science (2005) 309(5738):1251–3. doi:10.1126/science.1116006
168. Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 91(1):167–81. doi:10.1189/jlb.0311177
169. Zhang C, Lei GS, Shao S, Jung HW, Durant PJ, Lee CH. Accumulation of myeloid-derived suppressor cells in the lungs during Pneumocystis pneumonia. Infect Immun (2012) 80(10):3634–41. doi:10.1128/iai.00668-12
170. Lacotte S, Slits F, Orci LA, Meyer J, Oldani G, Delaune V, et al. Impact of myeloid-derived suppressor cell on Kupffer cells from mouse livers with hepatocellular carcinoma. Oncoimmunology (2016) 5(11):e1234565. doi:10.1080/2162402X.2016.1234565
171. Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest (2014) 124(6):2626–39. doi:10.1172/JCI74056
172. Thevenot PT, Sierra RA, Raber PL, Al-Khami AA, Trillo-Tinoco J, Zarreii P, et al. The stress-response sensor chop regulates the function and accumulation of myeloid-derived suppressor cells in tumors. Immunity (2014) 41(3):389–401. doi:10.1016/j.immuni.2014.08.015
Keywords: myeloid-derived suppressor cells, infection, inflammation, tuberculosis, human immunodeficiency virus, Staphylococcus, viral hepatitis
Citation: Dorhoi A and Du Plessis N (2018) Monocytic Myeloid-Derived Suppressor Cells in Chronic Infections. Front. Immunol. 8:1895. doi: 10.3389/fimmu.2017.01895
Received: 13 September 2017; Accepted: 11 December 2017;
Published: 04 January 2018
Edited by:
Geanncarlo Lugo-Villarino, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Leslie Chavez-Galan, National Institute of Respiratory Diseases, MexicoSimona Stäger, Institut national de la recherche scientifique (INRS), Canada
Prabir Ray, University of Pittsburgh School of Medicine, United States
Copyright: © 2018 Dorhoi and Du Plessis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anca Dorhoi, YW5jYS5kb3Job2lAZmxpLmRl;
Nelita Du Plessis, bmVsaXRhQHN1bi5hYy56YQ==