 Misako Matsumoto
Misako Matsumoto Yohei Takeda
Yohei Takeda Megumi Tatematsu
Megumi Tatematsu Tsukasa Seya
Tsukasa Seya- Department of Vaccine Immunology, Hokkaido University Graduate School of Medicine, Sapporo, Japan
Pattern recognition receptors (PRRs) play a crucial role in the innate immune system and contribute to host defense against microbial infection. PRR-mediated antimicrobial signals provide robust type-I IFN/cytokine production and trigger inflammation, thereby affecting tumor progression and autoimmune diseases. Accumulating evidence demonstrates that among the PRRs, only the signaling pathway of endosomal toll-like receptor 3 (TLR3) induces no systemic inflammation and mediates cross-priming of antigen-specific CD8+ T cells by dendritic cells. Treatment with a newly developed TLR3-specific ligand, ARNAX, along with tumor-associated antigens (TAAs), induces tumor-specific cytotoxic T lymphocytes, modulates the tumor microenvironment to establish Th1-type antitumor immunity, and leads to tumor regression without inflammation in mouse tumor models. Combination therapy using ARNAX/TAA and PD-1/PD-L1 blockade potently enhances antitumor response and overcomes anti-PD-1/PD-L1 resistance. In this review, we will discuss the TLR3-mediated signaling in antitumor immunity and its application to cancer immunotherapy.
Introduction
The innate immune system senses pathogen- and host-derived nucleic acids to maintain host homeostasis (1). Nucleic-acid-sensing innate immune receptors can be classified into two groups: (1) direct antiviral receptors that induce robust type-I IFN/cytokine production (2–4) and (2) dendritic cell (DC)-priming receptors that induce adaptive immunity (5, 6). The former consists of endosomal toll-like receptor (TLR) 7, 8, and 9, cytoplasmic RNA sensors retinoic acid-inducible gene-I (RIG-I), and melanoma differentiation-associated protein 5 (MDA5) and DNA sensors cGAS and STING, whose activation is closely associated with systemic IFN/cytokinemia and autoimmune diseases resulting from the recognition of endogenous RNA/DNA (7–9). The resultant IFN/cytokines induce inflammation and trans-activate antigen (Ag)-presenting DCs. On the other hand, TLR3 belongs to the latter group due to its restricted expression in myeloid DCs and usage of the signaling adaptor protein toll-IL-1 receptor-containing adaptor molecule-1 (TICAM-1) (also named TRIF) (10). The TLR3–TICAM-1 pathway predominantly works in professional Ag-presenting DCs to cross-prime CD8+ T cells as well as to induce production of Th1-type cytokines/chemokines (11, 12). However, polyinosinic:polycytidylic acid [poly(I:C)] that activates both TLR3 and MDA5 has been used as a TLR3 ligand. Studies with a recently developed TLR3-specific agonist, ARNAX, demonstrate that TLR3–TICAM-1 signaling primarily induces DC-priming without systemic cytokine production (13–15). The results suggest that TLR3-specific signal is non-inflammatory and RNA-driven inflammation is rooted in the systemic cytoplasmic pathway (Table 1). Hence, in the context of DC-priming, targeting endosomal TLR3 is a promising strategy for induction of antitumor immunity.
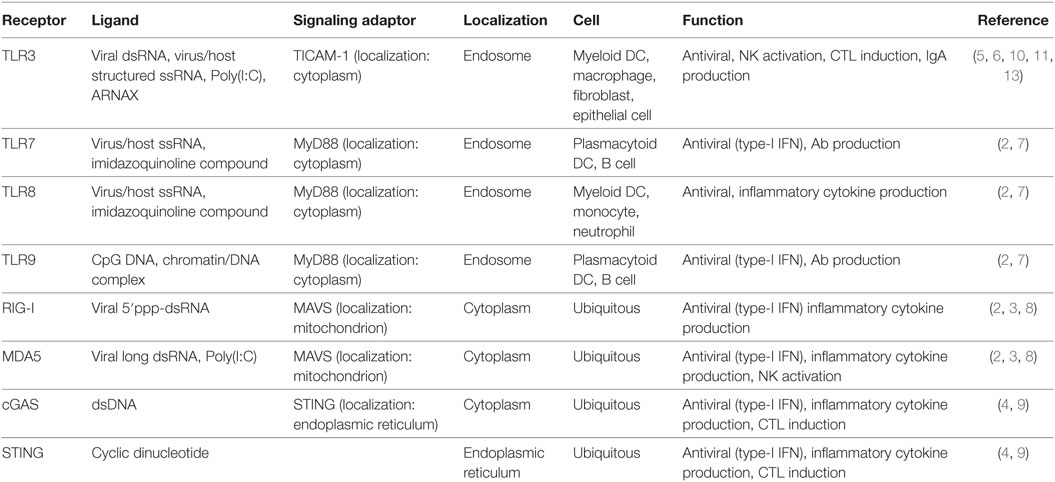
Table 1. Nucleic-acid-sensing innate immune receptors.
Toll-like receptor 3 is expressed on endosomal membranes in myeloid DCs, as well as on both cell and endosomal membranes in macrophages, fibroblasts, and some kinds of epithelial cells (16). Professional Ag-presenting DCs, including mouse CD8α+ and CD103+ DCs as well as human CD141+ DCs, highly express TLR3 (17, 18). TLR3 recognizes virus-derived double-stranded RNA (dsRNA) and virus- or host-derived single-stranded RNA having incomplete stem structures (19). Upon ligand recognition within endosomes, TLR3 oligomerizes and recruits the adaptor molecule TICAM-1, which activates the transcription factors NF-κB, IRF3, and AP-1, leading to the production of cytokines (IL-6, TNF-α, IL-12) and type-I IFN (especially IFN-β) (20, 21). TBK-1 is critical for IRF3 activation in the TICAM-1 signalosome, which resides in perinuclear regions as speckles following the dissociation from endosomal TLR3 (22). TICAM-1 is expressed in various cells and tissues at low levels, and TICAM-1 activation is tightly regulated by the conformational context of protein–protein associations (23). Spatiotemporal regulation of the TLR3–TICAM-1 pathway might be important for triggering non-inflammation and Th1-type adaptive immune responses.
ARNAX Signaling Pathway
ARNAX is a synthetic DNA–dsRNA hybrid molecule consisting of 140 bp of measles virus vaccine strain-derived dsRNA with a 5′ GpC-type phosphorothioated oligodeoxynucleotides (ODNs) cap (Figure 1A) (13, 24). DNA–RNA conjunction sites and dsRNA regions are relatively resistant to nucleases (25) and measles virus-derived dsRNA fails to induce RNA interference against host cell-derived RNAs, suggesting a stable and safer structure. The GpC ODN cap guides dsRNA to TLR3-positive cells for endocytosis, where the dsRNA activates TLR3 (26). dsRNA with a length of >90 bp is required for sufficient activation and signal transduction of the TLR3–TICAM-1 pathway (18). These ARNAX structural motifs do not stimulate cytoplasmic RNA sensors, RIG-I and MDA5, or DNA sensors: they are ubiquitously expressed all over the body.
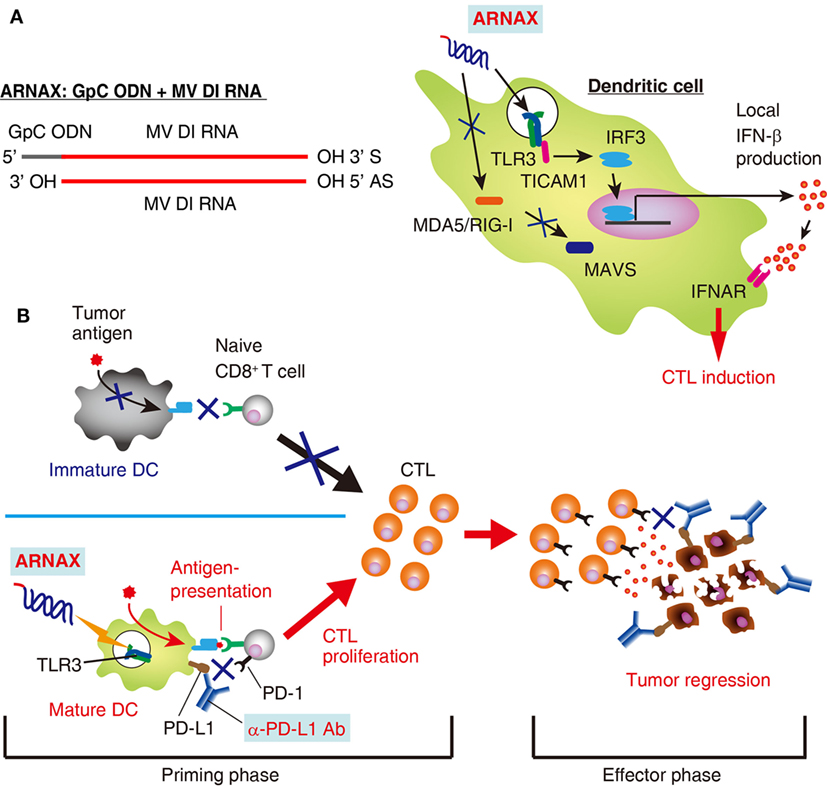
Figure 1. Structure and function of ARNAX. (A) Structure and signaling pathway of ARNAX. ARNAX activates endosomal toll-like receptor 3 (TLR3), but not cytoplasmic MDA5/RIG-I. The TLR3–TICAM-1–IRF3–IFN-β signaling axis is indispensable in dendritic cells (DCs) for ARNAX-mediated cytotoxic T lymphocyte (CTL) induction. (B) ARNAX therapy enhances antitumor responses in conjunction with PD-1/PD-L1 blockade. Tumors are self-originating and essentially lack adjuvant. In the absence of adjuvant, DCs remain immature state (immature DC) and fail to induce tumor-associated antigen (TAA)-specific CTLs (upper left panel). ARNAX activates TLR3 in DCs to induce maturation and cross-priming of TAA-specific CTLs in lymphoid tissues (priming phase) (lower left panel). PD-1/PD-L1 blockade potentiates ARNAX-mediated CTL induction in the priming phase and reinvigorates tumor infiltrating CTLs in the effector phase (right panel).
The viral dsRNA analogue poly(I:C)—a well-known ligand for TLR3—additionally activates the cytoplasmic dsRNA sensor MDA5 (27, 28), resulting in systemic and robust production of type-I IFNs/cytokines that causes undesirable inflammation. In contrast to poly(I:C), ARNAX induces marginal inflammatory cytokine/IFN-β production in a TLR3–TICAM-1-dependent fashion, demonstrating that the TLR3–TICAM-1 pathway contributes to minimal and local cytokine release to effectively prime DCs. Furthermore, the Th1-type cytokine IL-12 is substantially induced by ARNAX, as per poly(I:C), in a TLR3-dependent manner upon subcutaneous injection in mice (29). Non-inflammatory features of ARNAX and its Th1-skewing profile can be attributed to the restricted expression profile and early endosome localization of TLR3 as well as the TICAM-1 signalosome composition.
ARNAX activates professional Ag-presenting DCs to cross-prime CD8+ T cells. The TLR3–TICAM-1–IRF3–IFN-β signaling axis in DCs is indispensable for ARNAX-induced Ag-specific CD8+ T-cell priming (Figure 1A) (15). MAVS (signaling adaptor of RIG-I-like receptors), MyD88 (adaptor of all TLRs except for TLR3), and STING (adaptor of DNA sensors) do not contribute to ARNAX-induced cross-priming of CD8+ T cells (30). Importantly, DC-mediated local, but not systemic, IFN-β production is sufficient for CD8+ T-cell cross-priming (15, 31, 32), although the molecular determinants that regulate cross-priming downstream of the IFN-α/β receptor remains undefined. Thus, ARNAX is a non-inflammatory DC-priming adjuvant that specifically targets the TLR3–TICAM-1 pathway.
Induction of Antitumor Immunity
The presence of functional tumor-specific cytotoxic T lymphocytes (CTLs) in the tumor microenvironment is mandatory for tumor regression (33). ARNAX can induce tumor-specific CTLs by activating Ag-presenting DCs. In mouse implant tumor models EG7 and MO5 (OVA expressing T lymphoma and melanoma, respectively), injection of ARNAX along with OVA regresses tumor growth (15). OVA-specific CD8+ T cells proliferate in both lymphoid tissues and within tumors. Furthermore, CD8α+ DCs and CD8+ T cells increasingly infiltrate into tumor. Numerous genes associated with antitumor immunity are significantly upregulated in whole EG7 tumors during the ARNAX + OVA therapy (15). Chemokine genes responsible for recruiting DCs and T cells (Ccl4, Ccl5, Ccl27) (34), NK/T-cell function-related genes (Gzmb, Prf1, Fasl), cell adhesion-related genes, and cytokine receptor genes such as IL2rb and IL12rb1 are also upregulated. ARNAX and TAA therapy thus fosters Th1-type antitumor immunity in these tumor models. Hence, vaccine immunotherapy with TLR3 adjuvant enables to establish antitumor immunity against certain tumor types.
Notably, ARNAX monotherapy induces tumor growth retardation (15). It is likely that DCs internalize tumor cell debris, which contains TAAs, and cross-prime CD8+ T cells via TLR3-mediated activation of DCs (35). Alternatively, TLR3 signaling may facilitate the infiltration of preexisting tumor-reactive CTLs into tumor sites by inducing chemokine production. Mouse CD8α+ DCs and human CD141+ DCs express the C-type lectin-like receptor CLEC9A on their cell surface, which is involved in phagocytosis of dead cell debris that contains TAAs (36, 37). The quality of TAAs and their efficient delivery to DCs are important factors influencing the validation of vaccine immunotherapy with TLR3 adjuvant.
Modulation of the Tumor Microenvironment
The tumor microenvironment strongly affects tumor progression and antitumor immunity (38–40). Tumor-associated macrophages (TAMs), granulocytic or monocytic myeloid-derived suppressor cells (G- or M-MDSCs), and regulatory T cells are major constituents of the immunosuppressive tumor microenvironment (41, 42). The extent and composition of immune cell infiltration within tumors considerably differ among tumor types (42, 43). Accumulating evidence suggests that modulation of the tumor microenvironment from immunosuppressive to immunosupportive is a crucial factor for the success of cancer immunotherapy (44–46). It has emerged from several studies that TLR activation changes the properties of the tumor microenvironment (47–49). Among the TLR ligands, TLR2 ligand enhances the survival of M-MDSCs and their differentiation into macrophages, which augments the immunosuppressive activity of M-MDSCs toward CD8+ T cells through iNOS expression from macrophages (49). In contrast, TLR3 ligand converts TAMs from an M2- to an M1-like phenotype (47). When activated with poly(I:C), TAMs robustly produced TNF-α in 3LL (mouse lung carcinoma cell line) tumor in mice, resulting in tumor cell death and growth suppression. The TLR3–TICAM-1 pathway is critical for poly(I:C)-induced tumor regression via stromal macrophages in the 3LL tumor mouse model. In addition, G-MDSCs act as effector but not suppressor cells upon activation with TLR3 ligand in the EL4 tumor model (48). G-MDSCs produce reactive oxygen species through the TLR3–TICAM-1 pathway, leading to tumor growth inhibition (48). Furthermore, the proportion of G-MDSCs in EG7 tumors is greatly decreased by ARNAX + TAA therapy (15), which makes CTL unexhausted. Thus, TLR3 signaling functionally ameliorates the tumor microenvironment to potentiate antitumor immunity.
Combination Therapy of ARNAX with Anti-PD-1/PD-L1 Antibodies
Recent advances in cancer immunotherapy with checkpoint inhibitors have shown durable antitumor responses and good prognoses in patients with melanoma and non-small cell lung cancer, but only ~20% of patients with solid tumors respond to checkpoint blockade (50–54). The presence of preexisting tumor-specific CD8+ T cells and their infiltration into tumor sites are required for responsiveness to PD-1/PD-L1 blockade therapy (51). Many cancer patients, however, have limited numbers of tumor-specific CTLs, if at all, as well as/or the presence of therapy-resistant tumor microenvironments (55). The appearance and amount of mutation-associated neo-antigens in tumors correlates with sensitivity to PD-1 blockade (56–58), which is likely associated with preexisting tumor-specific CTLs (59). However, molecular determinants of tumor cells that define CTL induction by the immune system are still unclear. In this setting, vaccine immunotherapy that potently induces tumor-specific CD8+ T cells through DC-priming is a feasible approach to overcome primary resistance to PD-1/PD-L1 blockade.
In anti-PD-L1 antibody unresponsive mouse tumors, vaccine immunotherapy using ARNAX and tumor Ag decreases tumor progression irrespective of PD-L1 levels on tumor cells (15). Combination therapy with anti-PD-L1 antibody and ARNAX + TAA induces an antitumor response more effectively than anti-PD-L1 antibody monotherapy, especially in tumors expressing high levels of PD-L1. Priming of tumor-specific CD8+ T cells in lymphoid tissues and the infiltration of CD8+ T cells into the tumor site are greatly enhanced by combination therapies. DCs and macrophages in lymphoid tissues express PD-L1 at low or intermediate levels (60, 61). It is conceivable that blockade of the PD-1/PD-L1 pathway augments TAA-specific CD8+ T-cell induction from ARNAX + TAA therapy at the priming phase, and infiltrated CTLs are reinvigorated at the effector phase (62, 63) (Figure 1B). Tumor-associated DCs and CD11b+ immunosuppressive myeloid cells express high levels of PD-L1 within tumors (60), which is independent of efficacy of ARNAX + TAA therapy in several mouse tumor models. Although PD-L1 levels on tumor cells and infiltrated myeloid cells are one of the predictive biomarkers for responsiveness to PD-1/PD-L1 blockade (64, 65), tumor PD-L1 level is not always applicable for a prognostic biomarker to the ARNAX therapy. Potent induction of tumor-specific CD8+ T cells with DC-priming adjuvant and PD-1/PD-L1 blockade is important for infiltration and reinvigoration of CD8+ T cells within tumors.
Conclusion
To overcome the unresponsiveness of tumors to anti-PD-1/PD-L1 therapy, many cancer immunotherapy approaches have been conducted. Vaccine immunotherapy with TAA and the DC-priming adjuvant ARNAX generates tumor-specific CTLs with minimal essential cytokine production, which appears to avoid exacerbating adverse effects observed in certain population of patients treated with checkpoint inhibitors, such as the onset of autoimmune diseases. Up until now, numerous trials of peptide vaccine therapies have been performed unsuccessfully (66). The main factor contributing to the ineffectiveness is thought to be the usage of inflammatory, as opposed to DC-priming, adjuvants in peptide vaccine therapies. On the other hand, several preclinical studies with vaccine immunotherapies using poly(I:C) as a DC-priming adjuvant have been approved (67–70). However, poly(I:C) triggers undesirable inflammation caused by cytokine toxicity (67). The introduction of the non-inflammatory adjuvant ARNAX to peptide vaccine immunotherapy and/or combination therapy with PD-1 blockade appears to be a promising strategy to overcome anti-PD-1 resistance. Notably, induction of tumor cell death by radiation, chemotherapy, and oncolytic viruses appear to liberate TAAs from tumors (71–74), which may cooperate with ARNAX to induce polyclonal tumor-reactive CTLs and facilitate suppression of tumor growth.
In cancer immunotherapy, acquired resistance to newly developed therapies is a subject of intense discussion. In the case of TLR3 adjuvant therapy, the acquisition of resistance to CTL activity by tumor cells should be investigated. Evaluation of the quality and quantity of TAAs in individual tumors could be a therapeutic prerequisite for TLR3 adjuvant therapy, but this prediction has not been confirmed. Further studies elucidating the mechanism of tumor evasion from CTL cytotoxicity and the development of appropriate protocols for TLR3 adjuvant therapy would prove useful in this field to allow for complete tumor regression in cancer patients.
Author Contributions
MM, YT, MT, and TS conceived and designed the experiments. YT and MT performed the experiments. MM and TS wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Drs. H. Shime, M. Azuma, H. Takaki, K. Funami, K. Takashima and F. Nishikawa for invaluable discussions. We had a non-profit support from Nobelpharma Co., Ltd. through the university-company contract, which we acknowledge gratefully.
Funding
This work was supported in part by Grants-in Aid from the Ministry of Education, Science, and Culture and the Ministry of Health, Labour, and Welfare of Japan, as well as by Grants-in Aid from the Drug Discovery Support Promotion Project from Japan Agency for Medical Research and Development (AMED) (grant number 16nk0101327h0002), the Akiyama Life Science Foundation, and the Uehara Memorial Foundation.
References
1. Desmet CJ, Ishii KJ. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat Rev Immunol (2012) 12:479–91. doi:10.1038/nri3247
2. Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol (2006) 7:131–7. doi:10.1038/ni1303
3. Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Viral RNA detection by RIG-I-like receptors. Curr Opin Immunol (2015) 32:48–53. doi:10.1016/j.coi.2014.12.012
4. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol (2016) 17:1142–9. doi:10.1038/ni.3558
5. Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature (2005) 433:887–92. doi:10.1038/nature03326
6. Takaki H, Kure S, Oshiumi H, Sakoda Y, Suzuki T, Ainai A, et al. Toll-like receptor 3 in nasal CD103+ dendritic cells is involved in immunoglobulin A production. Mucosal Immunol (2017) 61:107–13. doi:10.1038/mi.2017.48
7. Krieg AM, Vollmer J. Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev (2007) 220:251–69. doi:10.1111/j.1600-065X.2007.00572.x
8. Kato H, Fujita T. RIG-I-like receptors and autoimmune diseases. Curr Opin Immunol (2015) 37:40–5. doi:10.1016/j.coi.2015
9. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med (2014) 371:507–18. doi:10.1056/NEJMoa1312625
10. Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Del Rev (2008) 60:805–12. doi:10.1016/j.addr.2007.11.005
11. Azuma M, Ebihara T, Oshiumi H, Matsumoto M, Seya T. Cross-priming for antitumor CTL induced by soluble Ag + polyI:C depends on the TICAM-1 pathway in mouse CD11c(+)/CD8α(+) dendritic cells. Oncoimmunology (2012) 1:581–92. doi:10.4161/onci.19893
12. Azuma M, Takeda Y, Nakajima H, Sugiyama T, Ebihara T, Oshiumi H, et al. Biphasic function of TLR3 adjuvant on tumor and spleen dendritic cells promotes tumor T cell infiltration and regression in a vaccine model. Oncoimmunology (2016) 5:e1188244. doi:10.1080/2162402X.2016.1188244
13. Matsumoto M, Tatematsu M, Nishikawa F, Azuma M, Ishii N, Morii-Sakai A, et al. Defined TLR3-specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nat Commun (2015) 6:6280. doi:10.1038/ncomms7280
14. Seya T, Takeda Y, Matsumoto M. Tumor vaccines with dsRNA adjuvant ARNAX induces antigen-specific tumor shrinkage without cytokinemia. Oncoimmunology (2015) 5:e1043506. doi:10.1080/2162402X.2015.1043506
15. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-specific adjuvant relieves innate resistance to PD-L1 blockade without cytokine toxicity in tumor vaccine immunotherapy. Cell Rep (2017) 19:1874–87. doi:10.1016/j.celrep.2017.05.015
16. Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, et al. Subcellular localization of toll-like receptor 3 in human dendritic cells. J Immunol (2003) 171:3154–62. doi:10.4049/jimmunol.171.9.4934-b
17. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med (2010) 207:1247–60. doi:10.1084/jem.20092140
18. Jelinek I, Leonard JN, Price GE, Brown KN, Meyer-Manlapat A, Goldsmith PK, et al. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J Immunol (2011) 186:2422–9. doi:10.4049/jimmunol.1002845
19. Tatematsu M, Nishikawa F, Seya T, Matsumoto M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat Commun (2013) 4:1833. doi:10.1038/ncomms2857
20. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in toll-like receptor 3-mediated interferon β-induction. Nat Immunol (2003) 4:161–7. doi:10.1038/ni886
21. Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301:640–3. doi:10.1126/science.1087262
22. Funami K, Sasai M, Ohba Y, Oshiumi H, Seya T, Matsumoto M. Spatiotemporal mobilization of TICAM-1 in response to dsRNA. J Immunol (2007) 179:6867–72. doi:10.4049/jimmunol.179.10.6867
23. Tatematsu M, Ishii A, Oshiumi H, Horiuchi M, Inagaki F, Seya T, et al. A molecular mechanism for Toll/IL-1 receptor domain-containing adaptor molecule-1-mediated IRF-3 activation. J Biol Chem (2010) 285:20128–36. doi:10.1074/jbc.M109.099101
24. Shingai M, Ebihara T, Begum NA, Okabe M, Akazawa T, Miyamoto Y, et al. Differential type I interferon (IFN) inducing abilities of wild-type vs. vaccine strains of measles virus. J Immunol (2007) 179:6123–33. doi:10.4049/jimmunol.179.9.6123
25. Mochizuki S, Higuchi S, Sakurai K. ssDNA–dsRNAs are cleaved at the next to its chimera-junction point by an unknown RNase activity. Biochem Biophys Res Commun (2012) 428:433–7. doi:10.1016/j.bbrc.2012.10.100
26. Itoh K, Watanabe A, Funami K, Seya T, Matsumoto M. The clathrin-mediated endocytic pathway participates in dsRNA-induced IFN-beta production. J Immunol (2008) 181:5522–9. doi:10.4049/jimmunol.181.8.5522
27. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature (2006) 441:101–5. doi:10.1038/nature04734
28. Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A (2006) 103:8459–64. doi:10.1073/pnas.0603082103
29. Gerner MY, Heltemes-Harris LM, Fife BT, Mescher MF. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol (2013) 191:1011–5. doi:10.4049/jimmunol.1300652
30. Zevini A, Olagnier D, Hiscott J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol (2017) 38:194–205. doi:10.1016/j.it.2016.12.004
31. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med (2011) 208:2005–16. doi:10.1084/jem.20101159
32. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol (2014) 12:e1001759. doi:10.1371/journal.pbio.1001759
33. Seya T, Matsumoto M. The extrinsic RNA-sensing pathway for adjuvant immunotherapy of cancer. Cancer Immunol Immunother (2009) 58:1175–84. doi:10.1007/s00262-008-0652-9
34. Bachmann MF, Kopf M, Marsland BJ. Chemokines: more than just road signs. Nat Rev Immunol (2006) 6:159–64. doi:10.1038/nri1776
35. Kroemer G, Galluzzi L, Keppo O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
36. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8α+ dendritic cells. J Exp Med (2010) 207:1261–71. doi:10.1084/jem.20092618
37. Zhang JG, Czabotar PE, Policheni AN, Caminschi I, Wan SS, Kitsoulis S, et al. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity (2012) 36:646–57. doi:10.1042/BJ20121819
38. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
39. Coussens LM, Zitvogel L, Palucks AK. Neutralizing tumor-promoting chronic inflammation: a major bullet? Science (2013) 339:286–91. doi:10.1126/science.1232227
40. Joyce JA, Fearon DT. T cell exhaustion, immune privilege, and tumor microenvironment. Science (2015) 348:74–9. doi:10.1126/science.aaa6204
41. Peranzoni E, Zilio S, Marigo I, Dolcetti L, Zanovello P, Mandruzzato S, et al. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr Opin Immunol (2010) 22:238–44. doi:10.1016/j.coi.2010.01.021
42. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi:10.1038/ni.2703
43. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi:10.4049/jimmunol.181.8.5791
44. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol (2013) 25:268–76. doi:10.1016/j.coi.2013.02.009
45. Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162:1257–70. doi:10.1016/j.cell.2015.08.015
46. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature (2016) 539:443–7. doi:10.1038/nature20554
47. Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A (2012) 109:2066–71. doi:10.1073/pnas.1113099109
48. Shime H, Matsumoto M, Seya T. Double-stranded RNA promotes CTL-independent tumor cytolysis mediated by CD11b+Ly6G+ intratumor myeloid cells through the TICAM-1 signaling pathway. Cell Death Differ (2016) 24:385–96. doi:10.1038/cdd.2016.131
49. Shime H, Maruyama A, Yoshida S, Takeda Y, Matsumoto M, Seya T. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. Oncoimmunology (2017):e1373231. doi:10.1080/2162402X.2017.1373231
50. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. N Engl J Med (2013) 369:134–44. doi:10.1056/NEJMoa1305133
51. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
52. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi:10.1038/nature14011
53. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161:205–14. doi:10.1016/j.cell.2015.03.030
54. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348:56–61. doi:10.1126/science.aaa8172
55. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell (2016) 165:35–44. doi:10.1016/j.cell.2016.02.065
56. Rizvi N, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi:10.1126/science.aaa1348
57. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi:10.1126/science.aaa4971
58. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi:10.1126/science.aan6733
59. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 5:135–46. doi:10.1038/nrc3670
60. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9:562–7. doi:10.1038/nm863
61. Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med (2016) 8:328rv4. doi:10.1126/scitranslmed.aad7118
62. Keir ME, Butte MJ, Freeman GJ, Sharp AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
63. Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol (2014) 35:51–60. doi:10.1016/j.it.2013.10.001
64. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression via 3’-UTR disruption in multiple cancers. Nature (2016) 534:402–6. doi:10.1038/nature18294
65. Kleinovink JW, van Hall T, Ossendorp F, Fransen MF. PD-L1 immune suppression in cancer: tumor cells or host cells? Oncoimmunology (2017) 6:e1325982. doi:10.1080/2162402X.2017.1325982
66. Rosenberg SA, Yang JC. Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10:909–15. doi:10.1038/nm1100
67. Lampkin BC, Levine AS, Levy H, Krivit W, Hammond D. Phase II trial of a complex polyriboinosinic-polyribocytidylic acid with poly-L-lysine and carboxymethyl cellulose in the treatment of children with acute leukemia and neuroblastoma: a report from the children’s cancer study group. Cancer Res (1985) 45:5904–9.
68. Mbow ML, De Gregorio E, Valiante NM, Rappuoli R. New adjuvants for human vaccines. Curr Opin Immunol (2010) 22:411–6. doi:10.1016/j.coi.2010.04.004
69. Galluzzi L, Vacchelli E, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: experimental toll-like receptor agonists for cancer therapy. Oncoimmunology (2012) 1:699–716. doi:10.4161/onci.20696
70. Sabbatini P, Tsuji T, Ferran L, Ritter E, Sedrak C, Tuballes K, et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res (2012) 18:6497–508. doi:10.1158/1078-0432.CCR-12-2189
71. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity (2014) 41:843–52. doi:10.1016/j.immuni.2014.10.019
72. van der Sluis TC, van Duikeren S, Huppelschoten S, Jordanova ES, Beyranvand Nejad E, Sloots A, et al. Vaccine-induced tumor necrosis factor-producing T cells synergize with cisplatin to promote tumor cell death. Clin Cancer Res (2015) 21:781–94. doi:10.1158/1078-0432.CCR-14-2142
73. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer (2016) 16:219–33. doi:10.1038/nrc.2016.16
74. Dovedi SJ, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res (2017) 23:5514–26. doi:10.1158/1078-0432.CCR-16-1673
Keywords: adjuvant, cancer immunotherapy, checkpoint inhibitors, cross-priming, dendritic cells, double-stranded RNA, innate immunity, toll-like receptor 3
Citation: Matsumoto M, Takeda Y, Tatematsu M and Seya T (2017) Toll-Like Receptor 3 Signal in Dendritic Cells Benefits Cancer Immunotherapy. Front. Immunol. 8:1897. doi: 10.3389/fimmu.2017.01897
Received: 17 October 2017; Accepted: 12 December 2017;
Published: 21 December 2017
Edited by:
Matteo Bellone, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Mercedes Beatriz Fuertes, Instituto de Biología y Medicina Experimental (IBYME), ArgentinaEnrico Proietti, Istituto Superiore di Sanità, Italy
Copyright: © 2017 Matsumoto, Takeda, Tatematsu and Seya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Misako Matsumoto, bWF0dW1vdG9AcG9wLm1lZC5ob2t1ZGFpLmFjLmpw;
Tsukasa Seya, c2V5YS10dUBwb3AubWVkLmhva3VkYWkuYWMuanA=
†Present address: Megumi Tatematsu, Dr. von Hauner Children’s Hospital, Ludwig-Maximilians University, Kubus Research Center, Munchen, Germany