 Mariangela Lecciso1*†
Mariangela Lecciso1*† Darina Ocadlikova1*†
Darina Ocadlikova1*† Sabina Sangaletti2
Sabina Sangaletti2 Sara Trabanelli3
Sara Trabanelli3 Elena De Marchi4
Elena De Marchi4 Elisa Orioli4
Elisa Orioli4 Anna Pegoraro4Paola Portararo2
Anna Pegoraro4Paola Portararo2 Camilla Jandus3Andrea Bontadini5Annarita Redavid1Valentina Salvestrini1
Camilla Jandus3Andrea Bontadini5Annarita Redavid1Valentina Salvestrini1 Pedro Romero3Mario P. Colombo2
Pedro Romero3Mario P. Colombo2 Francesco Di Virgilio4Michele Cavo1
Francesco Di Virgilio4Michele Cavo1 Elena Adinolfi4†
Elena Adinolfi4† Antonio Curti1†
Antonio Curti1†
- 1Department of Experimental, Diagnostic and Specialty Medicine, Institute of Hematology L. and A. Seràgnoli, S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy
- 2Istituto Nazionale dei Tumori (IRCCS), Milan, Italy
- 3Ludwig Cancer Research Center, Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland
- 4Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, Ferrara, Italy
- 5Immunohematology Service and Blood Bank, Policlinico S.Orsola Malpighi, Bologna, Italy
Chemotherapy-induced immunogenic cell death can favor dendritic cell (DC) cross-priming of tumor-associated antigens for T cell activation thanks to the release of damage-associated molecular patterns, including ATP. Here, we tested the hypothesis that in acute myeloid leukemia (AML), ATP release, along with its well-known immune stimulatory effect, may also contribute to the generation of an immune suppressive microenvironment. In a cohort of AML patients, undergoing combined daunorubicin and cytarabine chemotherapy, a population of T regulatory cells (Tregs) with suppressive phenotype, expressing the immune checkpoint programmed cell death protein 1 (PD-1), was significantly increased. Moving from these results, initial in vitro data showed that daunorubicin was more effective than cytarabine in modulating DC function toward Tregs induction and such difference was correlated with the higher capacity of daunorubicin to induce ATP release from treated AML cells. DCs cultured with daunorubicin-treated AML cells upregulated indoleamine 2,3-dioxygenase 1 (IDO1), which induced anti-leukemia Tregs. These data were confirmed in vivo as daunorubicin-treated mice show an increase in extracellular ATP levels with increased number of Tregs, expressing PD-1 and IDO1+CD39+ DCs. Notably, daunorubicin failed to induce Tregs and tolerogenic DCs in mice lacking the ATP receptor P2X7. Our data indicate that ATP release from chemotherapy-treated dying cells contributes to create an immune suppressive microenvironment in AML.
Introduction
The cancer cell death induced by some chemotherapeutic agents, especially anthracyclines, such as daunorubicin (DNR), stimulates an effective antitumor T-cell immune response in solid tumors (1–3) and leukemias (1), including acute myeloid leukemia (AML). Such a death process, named immunogenic cell death, is characterized by intracellular modifications as well as alterations of tumor microenvironment, which elicit antitumor immune response and may account, at least in part, for the therapeutic effect of antineoplastic drugs (4). Among the different mechanisms underlying immunogenic cell death, autophagy-dependent extracellular release of ATP from chemotherapy-treated dying tumor cells is a key priming factor (5). Indeed, ATP, acting at the purinergic P2X7 receptor (P2X7R), drives dendritic cells (DCs) activation, thereby enabling the cross-presentation mode (5, 6).
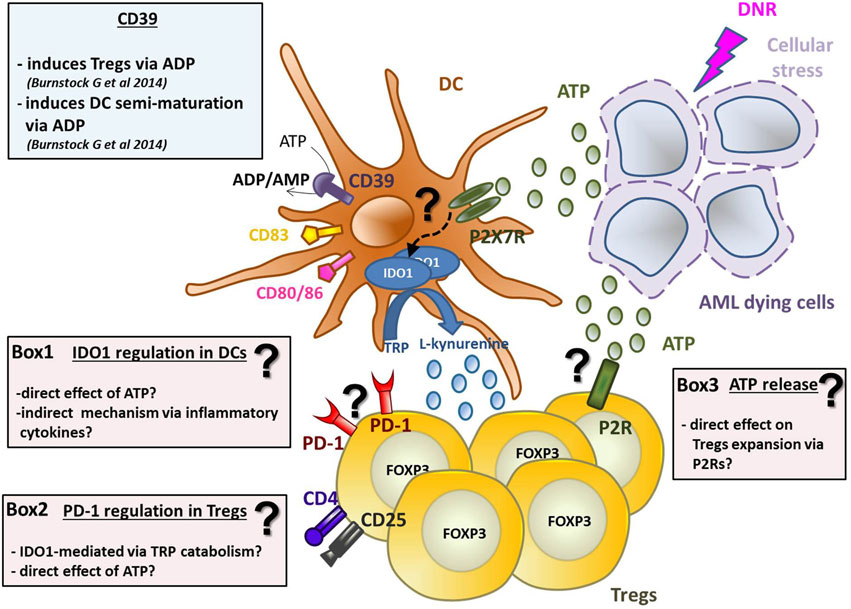
Graphical Abstract. ATP released from chemotherapy-treated dying leukemia cells has tolerogenic effects by inducing IDO1-expressing DCs and increasing Tregs. DNR-treated AML dying cells release ATP, which induces DCs activation and maturation (in particular CD80, CD83, and CD86 upregulation) and IDO1 expression via purinergic P2X7 receptor. The mechanism of IDO1 upregulation is still unknown (see Box 1 for hypotheses). IDO1 catabolizes the conversion of tryptophan (TRP) into l-kynurenine inducing Tregs. Along with DCs maturation, ATP induces the upregulation of CD39, which converts ATP into ADP/AMP, known to induce semi-maturation of DCs and partial Th1 polarization of CD4+ T cells. On the other hand, AMP is known to impair maturation of DCs, thus decreasing the capacity of human DCs to prime CD8+ T cells leading to tolerance. ATP released from dying AML cells has two distinct effects on Tregs: (1) it induces their expansion and (2) PD-1 upregulation. The exact mechanisms underlying the effect of ATP on Tregs are still unclear (see Boxes 2 and 3).
More recently, some antineoplastic agents have been also associated with the generation of an immunosuppressive, rather than immunostimulant, tumor microenvironment (7–9), but the underlying mechanisms are still unknown. In particular, to our knowledge, a tolerogenic effect of ATP release from chemotherapy-treated dying tumor cells was never investigated in AML.
Acute myeloid leukemia cells have been shown to induce a suppressive microenvironment by expanding T regulatory cells (Tregs), which in turn may hamper anti-leukemia immune response (10). Although the direct activity of ATP on Tregs is well established (11–14), the contribution of ATP release from chemotherapy-treated AML cells on Tregs induction was never investigated. ATP and, more in general, inflammatory stimuli can stimulate DCs either to promote or suppress T-cell responses (15), the latter occurring through the generation of Tregs. The most relevant mechanism by which DCs induce Tregs is through the upregulation of indoleamine 2,3-dioxygenase 1 (IDO1) (15–18), an enzyme that degrades the essential amino acid tryptophan into kynurenine and is involved in the generation of an immunosuppressive microenvironment in AML (19, 20). Whether upon chemotherapy, along with its capacity of promoting DC-mediated cross-priming to tumor antigen-specific T cells, ATP may be implicated in conferring tolerogenic features to infiltrating DCs via IDO1 upregulation has not been specifically addressed.
In the present study, by moving from ex vivo analysis of T-cell composition emerging in AML patients after induction chemotherapy, we in vitro and in vivo investigated the effect of ATP release from chemotherapy-treated dying leukemia cells on the induction of an immune suppressive microenvironment in AML. In particular, we addressed the effect of ATP release from chemotherapy-treated AML cells on Tregs and DCs.
Materials and Methods
Cells
All human samples were collected from healthy donors (HD) and from newly diagnosed AML patients after informed consent (local Ethical Committee approval code: 147/2013/O/Tess). Patients’ characteristics are reported in Table S1 in Supplementary Material. AML cells were obtained as mononuclear cells isolated by Ficoll-Hypaque centrifugation (Amersham, USA) from patients’ bone marrow or peripheral blood (PB) samples, including at least 70% leukemic cells, as evaluated by morphology and FACS analysis. CD3+, CD19+, CD14+, and CD4+CD25+CD127dim/− cells were purified by magnetic separation (Miltenyi Biotec, Germany), according to manufacturer’s instructions from mononuclear cells separated from buffy coats and patients’ PB by Ficoll-Hypaque centrifugation (Amersham). Purity of cell populations was always >90%. Human HL-60 (DMSZ; ACC 3, FAB M2) and murine WEHI-3B (DMSZ; no. ACC 26) AML cell lines were maintained at 37°C and 5% CO2. HL-60 cells were cultured in RPMI 1640 medium (Lonza, Milan, Italy), supplemented with 10% heat-inactivated fetal bovine serum (Gibco-Invitrogen, USA), 2 mM l-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin (MP Biomedicals, Italy) (complete RPMI). WEHI-3B cells were cultured in Iscove modified Dulbecco’s medium (Sigma-Aldrich, USA) supplemented with 10% fetal bovine serum (Euroclone, Italy), 100 U/ml penicillin and 100 mg/ml streptomycin (Euroclone). AML cells (1 × 106/ml) were treated with DNR 500 ng/ml (Sigma-Aldrich) or cytarabine (ARA-C) 25 µg/ml (Sigma-Aldrich) for 4 h and tested for apoptosis by Annexin-V-FLUOS Apoptosis Detection Kit (Roche, Switzerland) and ATP release (see Datasheet S1 in Supplementary Material).
Ex Vivo Characterization of Leukemia-Reactive T Cells
T cells from newly diagnosed AML patients (n = 23) undergoing standard “7 + 3” induction chemotherapy regimen, including continuous infusion of ARA-C at 200 mg/m2 for 7 days and DNR at 60 mg/m2 for 3 days, were analyzed before drug administration and at different time points (+7, +14, +21, and +28) after the end of administration regimen. Due to paucity of evaluable cells, in few cases (n = 3), day 7 T cells were not analyzed. For IFN-γ intracellular detection, purified CD3+ T cells were co-cultured with autologous AML blasts, at 10:1 ratio, in complete RPMI supplemented with 10% autologous serum (autologous RPMI). CD3+ T cells either used as such or pre-activated with ionomycin (500 ng/ml; Sigma-Aldrich) and phorbol-12-myristate-13-acetate (PMA, 10 ng/ml; Sigma-Aldrich) and co-cultured with autologous CD19+ cells were used as controls. After 4 h, brefeldin A (2 µg/ml; BD Biosciences, USA) was added, followed by overnight incubation before permeabilization and staining.
In selected cases, 5 × 106 CD3+ T cells from IFN-γ-responding AML patients were collected after chemotherapy and stimulated overnight with autologous AML blasts (ratio 1:1). Cells were incubated with IFN-γ and TNF-α Catch reagent (Miltenyi) for 5 min on ice. Finally, after incubation with medium for 45 min at 37°C, cells were stained with the following human monoclonal antibodies (mAbs): IFN-γ APC, TNF-α APC (Miltenyi), CD137 PE-Cy7 (4-1BB, 4B4-1; Biolegend, USA), CD3 APC-eFluor780 (SK7; eBioscience, USA), CD8 Pacific Blue (B9.11; BD Pharmingen, USA), CD33 FITC (HIM3-4), or CD34 FITC (561) (both from Biolegend, USA) to discard blasts population from analysis. CD3+CD8+IFN-γ+TNF-α+CD137+ were sorted at BD FACS Aria cell sorter (BD Biosciences) and in vitro expanded (2–3 weeks) on a irradiated mononuclear cell feeder layer from two HD (1 × 106 cells/ml) in RPMI supplemented with human serum (HS, 8%), phytohemagglutinin (PHA, 1 µg/ml; Sigma-Aldrich) and IL-2 (150 U/ml; Roche), for 2–3 weeks. IL-2 was added to the culture every other day.
DC Generation, Maturation, and Pulsing
Human monocyte-derived DCs were generated by a 5-day culture of CD14+ cells in complete RPMI in presence of granulocyte-macrophage colony stimulation factor (50 ng/ml; GM-CSF Endogen, USA) and IL-4 (800 U/ml; Miltenyi), as previously described (21, 22). DC maturation was induced with a cocktail of cytokine made of TNF-α (10 ng/ml; Endogen), IL-6 (10 ng/ml; Endogen), IL-1β (10 ng/ml; Endogen), and 1 µg/ml PGE2 (Endogen) (23) or with ATP (1 mM; Sigma-Aldrich). For DC pulsing, chemotherapy-treated HL-60 cells were cultured for 20 h with immature DCs (2:1 ratio) in autologous RPMI. After culture, IDO1 protein expression was evaluated by Western blotting (see Datasheet S1 in Supplementary Material).
In Vitro Induction of IFN-γ-Producing Leukemia-Specific T Cells
Leukemia-reactive IFN-γ producing CD3+ T cells were evaluated after two rounds of in vitro stimulation (7 + 7 days) with autologous DCs pulsed with chemotherapy-treated HL-60 cells (ratio 10:1) in autologous RPMI. IL-2 (20 U/ml) was added on alternate days. In selected experiments, allogeneic Tregs induced by the same DCs used for T cell priming were added to cell cultures (ratio 1:1) for 5 days to test their suppressive activity. At the end of culture, T cells were tested for IFN-γ production. DCs loaded for 24 h (1:2 ratio) with HL-60 cell lysate, obtained after three cycles of cells freeze-thawing and filtering through an insulin syringe, were used as target.
Antigen-specific IFN-γ-producing CD3+ T cells were obtained as described above, except challenging of DCs with HLA-A0201-restricted Wilms’ tumor-derived peptide WT1-A (10 µg/ml; 126–134; PRIMM, Italy) for 4 h. DCs loaded with WT1-A or WT1-B (187–195) were used as targets.
In Vitro Tregs Induction
Immature DCs, chemotherapy-treated HL-60 pulsed DCs and DCs matured in presence of ATP or a cytokine cocktail were co-cultured for 5 days in autologous RPMI with allogeneic CD3+ T cells, in presence or absence of the IDO1-inhibitor 1-methyl tryptophan-L (1 mM, 1-MT-l; Sigma-Aldrich). IDO1-silenced DCs were also used (see Datasheet S1 Supplementary Material) (10). Tregs were quantified by FACS analysis. In selected experiments, purified and irradiated CD4+CD25+CD127dim/− Tregs (104/well) were added to cultures consisting of CFSE-labeled CD3+ T cells (105/well) as responders, stimulated by allogeneic immature monocyte-derived DCs (1:10 ratio), for 5 days, to test their suppressive activity.
Flow Cytometry on Human Cells
T cells, Tregs, and DCs were characterized using the following mAbs, according to manufacturer’s instructions: anti-CD8 Pacific blue (B9.11) or PE, anti-CD15S PE-CF594 (CSLEX1), anti-CD25 BV605 (2A3), anti-CD28 APC or APC-eFluor 780 (CD28.2), anti-CD80 PE-Cy 7 (L307.4), anti-CD152 (CTLA-4; clone BNI3), and anti-Ki-67 Alexa Fluor 700 (B56) from BD Bioscience; anti-CD3 PE-Cy7 or APC (UCHT1) or APC-eFluor 780 (SK7), anti-CD4 FITC (RPA-T4), anti-CD8 APC (SK1), anti-CD25 APC (SK1) or APC-eFluor 780 (CD25-4E3), anti-CD38 Alexa 700 (HIT2), anti-CD83 PE (HB15e), anti-CD127 Pe-Cyanine5 (eBioRDR5), anti-CD197 (CCR7) PE-Cy7 or Brillant Violet 421 (G043H7), and anti-HLA-DR FITC (L243) from eBioscience; anti-CD4 FITC (SFCI12T4D11) and anti-CD45RA ECD (2H4LDH11LDB9) (Beckman Coulter, USA); anti-CD39 PE/Cy7 (A1), anti-CD45RA Brilliant Violet 510 (HI100), anti-CD127 PerCP/Cy5.5 (IL-7Rα; clone A019D5), and anti-CD279 (programmed cell death protein 1; PD-1) APC or Brilliant Violet 711 (EH12.2H7) from Biolegend. LIVE/DEAD Fixable Aqua (Thermo Fisher Scientific, USA) was used to gate out dead cells. For intracellular staining (24), after cells fixation and permeabilization with 4% paraformaldehyde (VWR, USA) and 0.1% saponin (Sigma), the following mAbs were used: anti-IFN-γ PE (4S.B3), anti-Foxp3 PE or FITC (236A/E7), and anti-Ki-67 FITC (20Raj1) from eBioscience. Circulating Tregs in PB of AML patients were assessed by using Human Regulatory T Cell Whole Blood Staining Kit (eBioscience) (see Datasheet S1 in Supplementary material).
Specifically, for ex vivo post-chemotherapy patients’ Tregs screening, anti-CD3/CD4/CD25/FOXP3 mAbs were used. In selected cases (n = 6), a wider panel including anti-CD3/CD4/CD25/CD127/CD15s/CD45RA/FOXP3/PD-1/Ki67 mAbs for identification of Tregs subpopulation (Treg1/2/3) were used.
For each sample, isotype-matched irrelevant mAbs staining was used as control. At least 10,000 events were collected from each sample at Gallios Flow Cytometer (Beckman Coulter) or BD Accuri C6 (BD Biosciences). For expanded T cells, data analyses were performed by using FlowJo Single Cell Analysis Software (FlowJo LLC).
In Vivo Studies
To obtain WEHI-3B cell clones stably expressing plasma membrane luciferase, cells were transfected with the PmeLUC probe, as previously described (25). Briefly, 6 × 106 cells were suspended in electroporation buffer (Life Technologies, USA) with 3 µg of plasmid’s DNA and electroporated in a Microporator MP-100 (Digital bio, Thermo Fisher), at 1,250 V for 40 ms. Stably transfected cell clones were obtained by selection with neomycin/G418 sulfate (0.2–0.8 mg/ml; Sigma) followed by limiting dilution as previously described (26). Male 4–6 weeks old Balbc/J wt or P2X7−/− mice were subcutaneously injected with 2 × 106 WEHI-3B PmeLUC cells. Tumors became palpable approximately 7 days post-inoculum (p.i.) and were measured with a manual caliper. Tumor volume was calculated according to the following equation: π/6 [w1 × (w2)2], where w1 is the major diameter and w2 is the minor diameter. Luminescence emission was measured daily, days 7–12 from p.i. with a total body luminometer for small animals (IVIS Lumina, Caliper; Perkin Elmer, USA), as previously described (27). Mice anesthetized with 2.5% isofluorane were intra peritoneum (i.p.) injected with 150 mg/kg d-luciferin (Promega, USA) and luminescence was captured from dorsal view. Photon emission was quantified using the Living Image® software (Perkin Elmer) and averaged as photons/second/cm2/steradian (abbreviated as p/s/cm2/s). DNR (3 mg/kg, Sigma), ARA-C (50 mg/kg, Sigma), or sterile PBS vehicle (placebo) were i.p. administered at p.i. days 7 and 9. Blood samples were collected from the submandibular vein under general anesthesia immediately before sacrificing the animal (p.i. day 12). Cytokines levels were evaluated following 1:2 plasma dilution with Ciraplex CK1 mouse multi-cytokine assay kit (AushonBiosystem, distributed by TemaRicerca, Bologna, Italy) as per manufacturer’s instructions. Tumors were excised and processed for flow cytometry (see Datasheet S1 in Supplementary material). All animal procedures were approved by the University of Ferrara (Ferrara, Italy) Ethic committee and the Italian Ministry of Health in compliance with International laws and policies (EU Directive 2010/63/EU and Italian D.Lgs 26/2014; authorization number 821/2015PR to EA).
Statistical Analysis
Data were expressed as mean ± SEM of values obtained in the experiments. Statistical analyses were performed with GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA), using ANOVA or unpaired t-test. p Values <0.05 were considered statistically significant.
Results
Increased Tregs with Suppressive Phenotype Are Detectable in AML Patients after Combined DNR and ARA-C Chemotherapy
We analyzed the induction of tumor-reactive CD8+ cytotoxic T cells (CTLs) in a cohort of AML patients (n = 23) undergoing combined DNR and ARA-C chemotherapy. In 15 out of 23 patients, we observed an increase of leukemia-reactive IFN-γ-producing CD8+ T cells (Figure S1 in Supplementary Material) mostly belonging to effector memory (EM) and EM expressing CD45RA (effector memory expressing RA) subsets (Figure S2 in Supplementary Material), which highly expressed the activation marker CD38 and downregulated CD28 as compared to naïve (p = 0.03) or central memory (CM) (p = 0.03) T cells.
Along with the detection of leukemia-reactive CD8+ T cells, we observed an increase in CD4+CD25+Foxp3+ T cells after DNR plus ARA-C chemotherapy (Figure 1A) with a peak at day 21. We, then, sought to better characterize Tregs subsets at the phenotypic level. Among CD3+CD4+ T cells collected at day 21 post-chemotherapy, CD25+CD127− cells were subdivided into three different subsets (Treg1, Treg2, Treg3), according to the expression of CD15s (Figure S3 in Supplementary Material) (28, 29). As compared to HD, no differences were observed for Treg1 and Treg3 frequencies, whereas the percentage of Treg2 cells was significantly increased in AML patients after chemotherapy (Figure 1B). Treg2 cells from AML patients expressed Foxp3, which correlated with CTLA-4 and CD39 (Figure 1C), indicating a suppressive phenotype (28, 30, 31). Moreover, as compared to Treg1, Treg2 cells showed higher expression of PD-1, which identifies a novel population of Tregs with crucial suppressive activity in the tumor setting (Figure 1D) (32, 33). Intriguingly, after 21 days post-chemotherapy an increase in Ki67 expression was observed in Treg2 over Treg1 and Treg3 cells, suggesting recent activation (Figure 1E). Indeed, a time-course analysis revealed a selective increase of proliferating Ki67+ Treg2 over Treg1 and Tregs3 cell subsets at day 14 post-chemotherapy, which progressively reduced at later time points (Figure S4 in Supplementary Material).
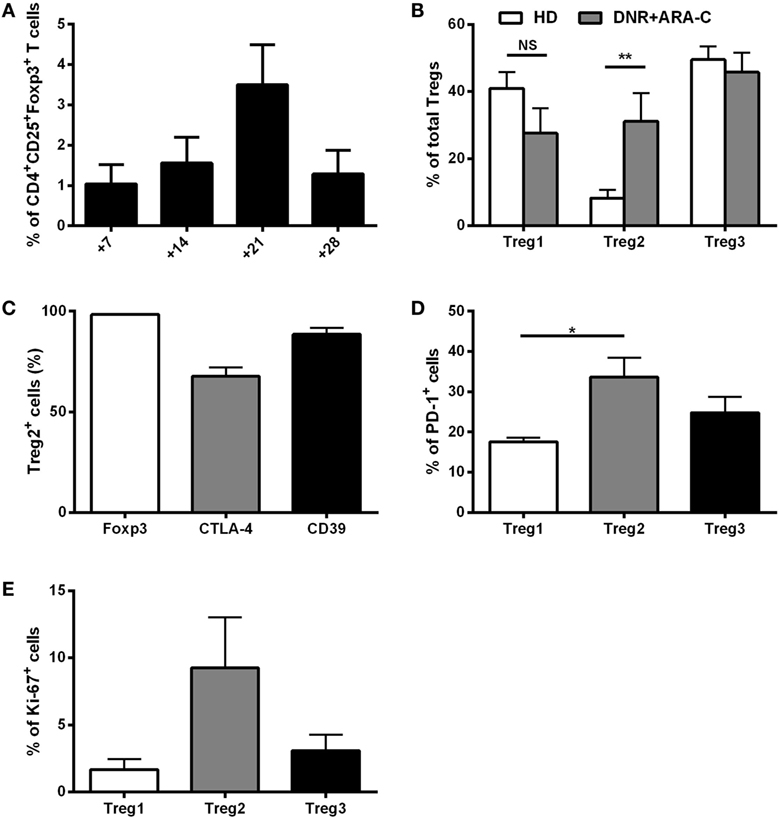
Figure 1. Ex vivo characterization of T regulatory cells (Tregs) after DNR + ARA-C chemotherapy. (A) Percentage of circulating CD4+CD25+Foxp3+ Tregs in peripheral blood of 23 acute myeloid leukemia (AML) patients at days 7, 14, 21, and 28 after chemotherapy analyzed by flow cytometry. (B) Characterization of Tregs subsets based on the expression of CD45RA and CD15s cell surface markers in AML patients at day 21 by flow cytometry (DNR + ARA-C). Total Tregs (gated on CD3+CD4+CD25+CD127dim/− T cells) are subdivided into Treg1 (CD45RA+CD25+CD15s−), Treg2 (CD45RA−CD25+CD15s+), and Treg3 (CD45RA−CD25+CD15s−) (n = 6). Healthy donors (HD) were used as negative control (n = 6). (C) Expression of Foxp3, CTLA-4, and CD39 markers in Treg2 subset of AML patients at day 21 by FACS analysis (n = 6). Expression of PD-1 (D) and Ki-67 (E) markers in the different subsets of Tregs by FACS analysis (n = 6). *p < 0.05; **p < 0.01; NS, not significant difference. Values are represented as mean ± SEM.
Taken together, the analysis of the composition of T cells, emerging in AML patients after combined DNR and ARA-C chemotherapy, reveals an increase of Tregs with a suppressive phenotype. These ex vivo data prompted us to dissect the contribution of DNR versus ARA-C in Tregs induction and to investigate a possible mechanism responsible for chemotherapy-driven induction of a suppressive microenvironment in AML.
In Vitro, DNR Is More Effective Than ARA-C at Inducing Fully Functional Tregs through DCs
Inflammatory stimuli may promote Tregs via the induction of tolerogenic DCs (16). Since chemotherapy treatment is associated with the abundant release of inflammatory signals, we tested the contribution of DNR and ARA-C in driving DCs toward Tregs induction. Chemotherapy-treated AML cells were pulsed into DCs, which were, then, used in co-culture for inducing leukemia-specific T cells and Tregs. DC-loaded with DNR-treated AML cells were more efficient than DC-loaded with ARA-C-treated cells not only, as expected, in inducing leukemia-reactive CD8+ T cells (Figure 2A), but also in increasing the frequency of CD4+CD25+Foxp3+ T cells (11.70 ± 4.15 versus 5.97 ± 3.10%, respectively; p < 0.05) (Figure 2B). DNR-induced CD4+CD25+Foxp3+ T cells were shown to act as Tregs through the complete inhibition of leukemia-specific IFN-γ production by both CD4+ and CD8+ T cells (Figure 2C). Moreover, these DC-induced Tregs also reduced T cell-mediated IFN-γ-production in response to the leukemia-associated antigen, WT1 (Figure 2D).
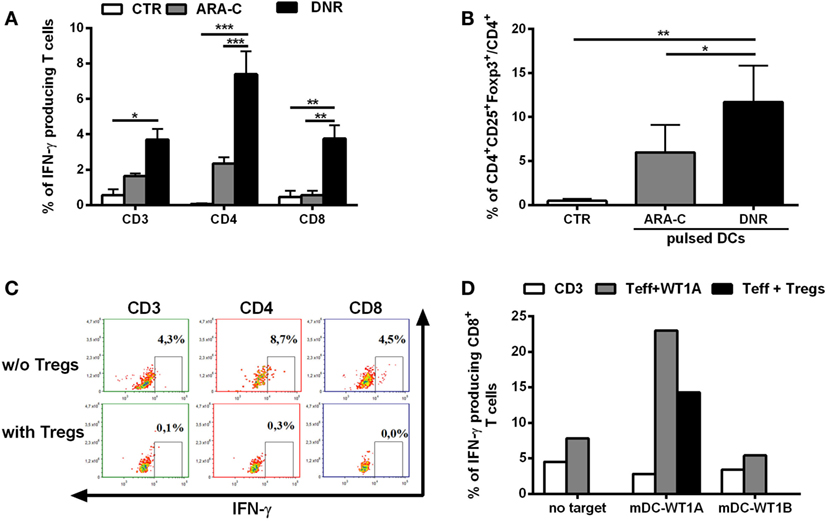
Figure 2. DNR is more efficient than ARA-C in promoting induction of functional T regulatory cells (Tregs). (A) Percentage of IFN-γ-producing CD3+, CD4+, and CD8+ leukemia-reactive T cells, previously stimulated and restimulated with DCs loaded with ARA-C- or DNR-treated HL-60 cells (ARA-C or DNR, respectively), evaluated by flow cytometry. Autologous DCs loaded with HL-60 cell lysate were used as target. Unstimulated CD3+ T cells were used as negative control of T effector cells (CTR). The values are represented as mean ± SEM of three independent experiments; *p < 0.05; **p < 0.01; ***p < 0.001. (B) Percentage of CD4+CD25+Foxp3+ T cells (gated on CD4+ T cells) induced by DCs pulsed with ARA-C- or DNR-treated HL-60 cells. Unstimulated CD3+ T cells were used as negative control (CTR); *p < 0.05; **p < 0.01. Values are represented as mean ± SEM of three independent experiments. (C) FACS analysis of inhibition of CD3+, CD4+, and CD8+ IFN-γ-producing T cells by autologous CD4+CD25+Foxp3+ Tregs. One representative experiment of three is shown. (D) Quantification of IFN-γ-producing leukemia antigen-specific CD8+ T cells, previously stimulated and restimulated with mature DCs loaded with WT1-A (Teff WT1A), in presence (Teff + Tregs) or absence of autologous CD4+CD25+Foxp3+ Tregs. Autologous DCs loaded with WT1-A or WT1-B (as antigen specificity control) were used as targets (mDC-WT1A or mDC-WT1B, respectively), T cells without target (no target) were used as negative control. Unstimulated CD3+ T cells were used as negative control of effector cells (CD3). Tregs were added to T effector cells at ratio of 1:1. One representative experiment of three is shown.
These results demonstrate that DNR is more efficient than ARA-C in promoting functional Tregs through tolerogenic DCs.
In Vitro, ATP Release from DNR-Treated Leukemia Cells Is Correlated with Tregs Generation and with IDO1 Upregulation in Mature DCs
Within tumor microenvironment, chemotherapy-treated tumor cells release a high amount of ATP, which is a major driver of DC activation and function (5, 6). To test the contribution of ATP release from chemotherapy-treated AML cells in DC-mediated Tregs induction, we preliminarily in vitro measured ATP release from dying AML cells, including cell lines and primary cells, after treatment with DNR and ARA-C. DNR and ARA-C were used at the concentrations capable to induce comparable apoptosis level, but only DNR increased ATP release (Figures 3A,B). These data suggest that the increased capacity of DNR over ARA-C to induce Tregs via DCs may be, at least in part, mediated by ATP. Indeed, when treated with ATP, DCs induced a population of CD4+CD25+CD127−/dim T cells (Figure 3C), which acted as bone fide Tregs by reducing allogeneic T-cell proliferation (Figure 3D). Interestingly, in comparison to a cocktail of pro-inflammatory cytokines, ATP treatment of DCs resulted in higher expression of PD-1 on Tregs (Figure 3E), whereas Foxp3 and CD39 expression was similar (data not shown). Moreover, Tregs obtained after culture with ATP-treated DCs expressed Ki-67 at comparable level, suggesting recent activation/proliferation (Figure 3F).
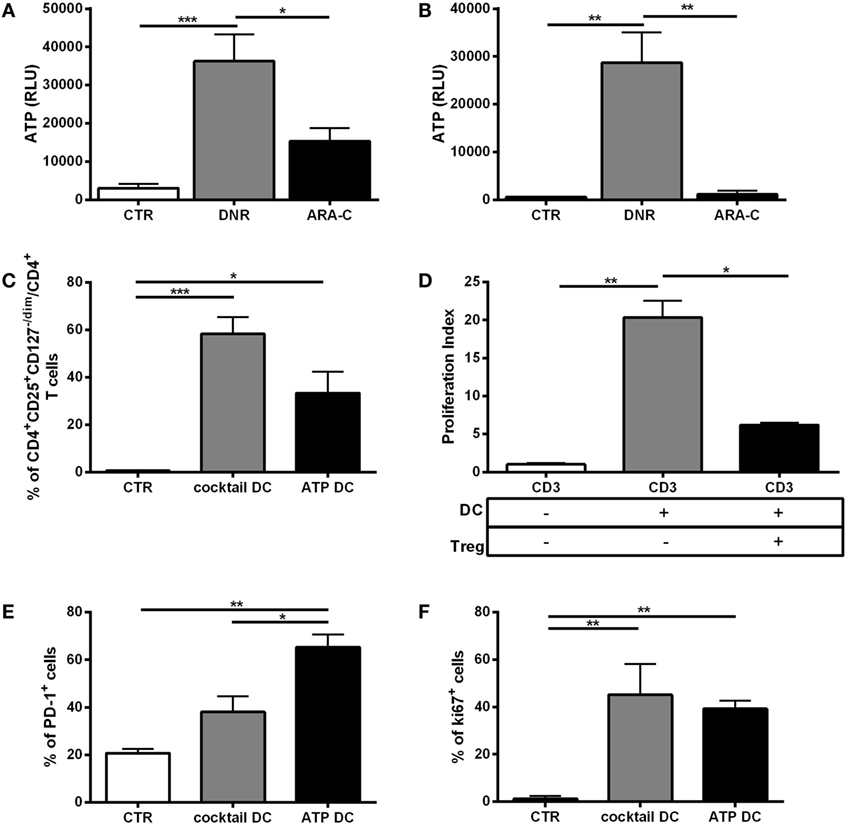
Figure 3. In vitro, ATP release from DNR-treated leukemia cells is correlated with functional T regulatory cells (Tregs) generation. Indirect measurement of ATP by quantification of emitted bioluminescence, expressed as relative light unit (RLU) in supernatants of (A) HL-60 and (B) primary acute myeloid leukemia cells after DNR (500 ng/ml) and ARA-C (25 µg/ml) treatment or untreated (CTR). The values are represented as mean ± SEM of four independent experiments; *p < 0.05; **p < 0.01; ***p < 0.001. (C) Percentage of Tregs identified as CD4+CD25+CD127−/dim cells induced after 5 days of co-culture of ATP- or cytokine cocktail-treated dendritic cells (DCs) (ATP DC or cocktail DC, respectively) with allogeneic CD3+ T cells. Unstimulated CD3+ T cells were used as negative control (CTR). The values are represented as mean ± SEM of three independent experiments; *p < 0.05; ***p < 0.001. (D) To test their immunosuppressive activity, CD4+CD25+CD127dim/− Tregs obtained at the end of the co-culture with ATP DC (Treg) were purified and added (1:10) to CD3+ T cells stimulated with mature DCs (DC). Proliferation index was evaluated by FACS analysis of CFSE staining. Unstimulated CD3+ T cells were used as negative control. Histograms represent the means ± SEM of three independent experiments; *p < 0.05; **p < 0.01. (E) Expression of PD-1 cell surface marker and (F) intracellular Ki-67 proliferation marker in CD4+CD25+CD127−/dim Tregs induced by ATP-treated DCs. DCs matured with cytokine cocktail were used as positive control, whereas CD3 alone was used as negative control (n = 3); *p < 0.05; **p < 0.01.
Among the different mechanisms used by DCs for promoting Tregs, IDO1 upregulation is crucial (15, 16). Then, we investigated the involvement of ATP in IDO1 upregulation in DCs during chemotherapy. ATP treatment upregulated IDO1 protein along with the maturation markers, CD80 and CD86 (Figures 4A,B), albeit to a lower extent than pro-inflammatory cytokines, used as positive control (21). Of note, in agreement with the different levels of ATP release (Figures 3A,B), DNR was more efficient than ARA-C in upregulating IDO1 protein (Figure 4C), along with the maturation marker CD83, which was significantly increased by DNR, and not by ARA-C treatment (p < 0.05) (Figure 4D). The inhibition of IDO1 in DCs upon DNR treatment by the IDO1-inhibitor, 1-MT-l (Figure 4E, p < 0.01) or by IDO1-specific RNA interference (data not shown) significantly reduced DC-mediated Tregs induction, thus demonstrating that DNR-induced IDO1 upregulation in DCs may drive Tregs. Taken together, these data suggest that ATP release from DNR- and not ARA-C-treated leukemia cells is correlated with Tregs generation and IDO1 upregulation in mature DCs.
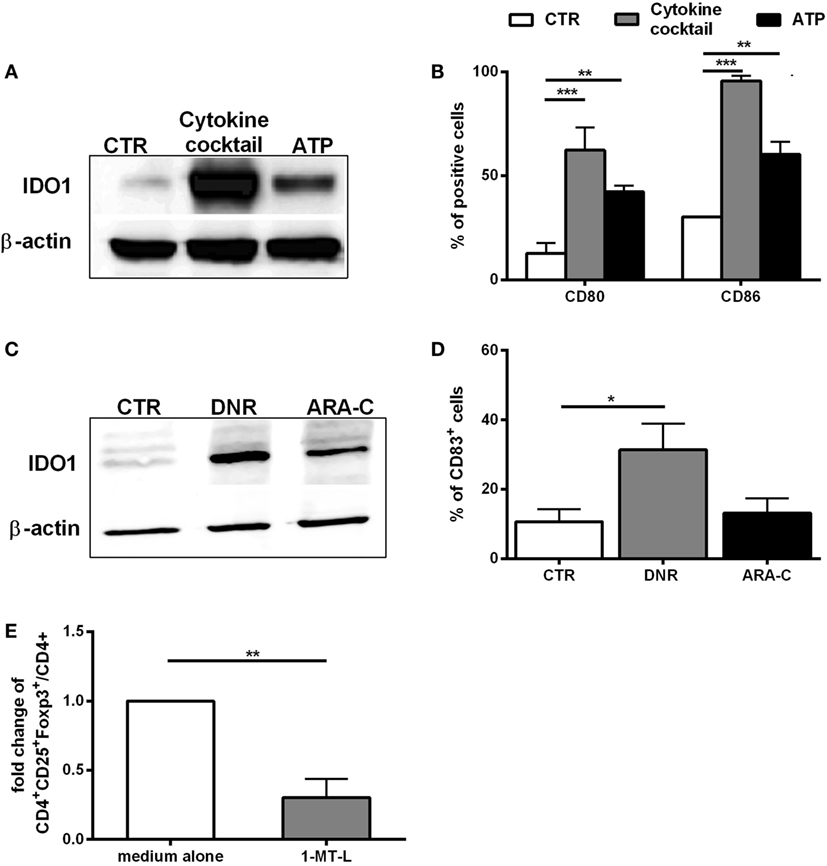
Figure 4. In vitro, ATP release from DNR-treated leukemia cells is correlated with IDO1 upregulation in mature dendritic cells (DCs). (A) Western blot analysis of IDO1 protein expression in DCs treated with ATP (1 mM; 24 h). Unloaded DCs and DCs matured with cytokine cocktail were used as negative (CTR) and positive control (cytokine cocktail). One representative experiment is shown. (B) FACS analysis of CD80 and CD86 expression in DCs treated as described in panel (A). The percentage values are represented as mean ± SEM of three independent experiments; **p < 0.01; ***p < 0.001. (C) Western blot analysis of IDO1 protein expression in DCs loaded with DNR- or ARA-C-treated HL-60 cells (DNR 500 ng/ml, ARA-C 25 µg/ml). Unloaded DCs were used as negative control (CTR). One representative experiment is shown. (D) Expression of CD83 maturation marker in DCs treated as described in panel (C). The percentage values are represented as mean ± SEM of four independent experiments; *p < 0.05. (E) Fold change of CD4+CD25+Foxp3+ T cells (gated on CD4+ T cells) induced by DCs loaded with DNR-treated HL-60 cells in presence of 1-MT-l. CD4+CD25+Foxp3+ T cells induced by DCs in absence of 1-MT-l (medium alone) were used as reference and set as 1; **p < 0.01.
DNR-Treated Mice Show Increased Level of ATP Release from Leukemia Cells
We, then, investigated in a mouse model the link between ATP release from chemotherapy-treated cells and the induction of an immune suppressive microenvironment. In line with data obtained with human AML cells (Figures 3A,B), only DNR, and not ARA-C, increased the release of ATP from murine WEHI-3B AML cells in vitro (Figure S5 in Supplementary Material).
This cell line was then transfected with PmeLUC, a luciferase expressed at the plasma membrane, which allows for ATP measure in the extracellular milieu (25). WEHI-3B PmeLUC live clones were injected in syngeneic mice, which were then treated with either DNR or ARA-C. While DNR and ARA-C treatment had comparable effect on tumor mass reduction (Figure 5A), only DNR increased ATP release (36,094.20 ± 7,420.75 p/s/cm2/sr) over control (13,950.9 ± 4,118.65 p/s/cm2/sr; p < 0.05) (Figures 5B,C). These data prompted us to correlate the composition of leukemia immune suppressive microenvironment, i.e., Tregs and tolerogenic DCs, with the different levels of ATP released upon chemotherapy.
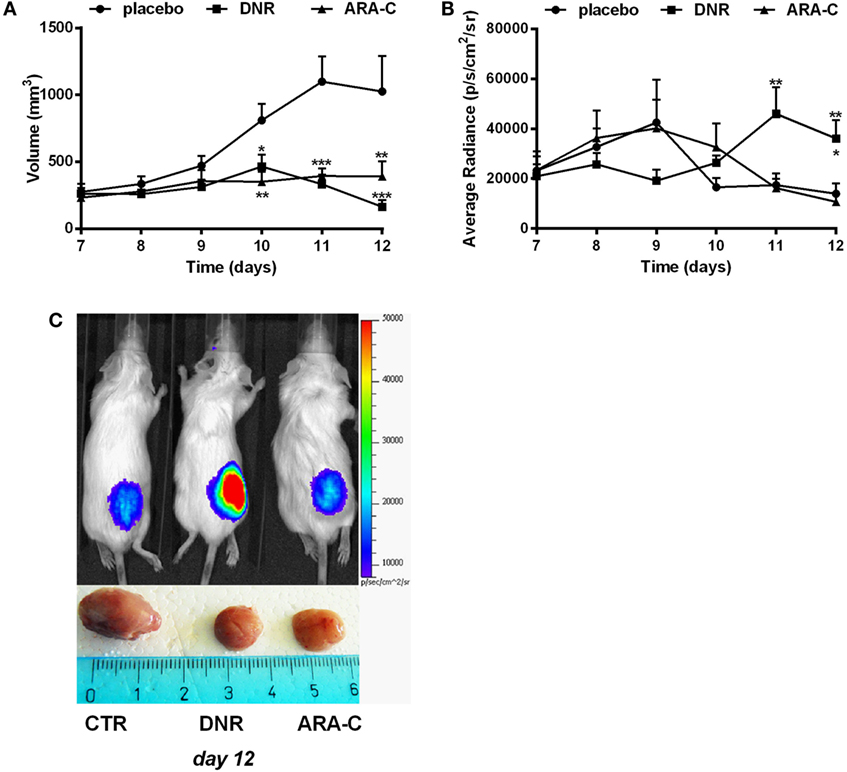
Figure 5. DNR increases ATP release in vivo. Balb/cJ mice inoculated with WEHI-3B PmeLUC cells and treated with DNR, ARA-C, or PBS vehicle (placebo) at post-inoculum days 7 and 9. (A) Tumor volume was in vivo assessed by a caliper and calculated as follows: π/6 [w1 × (w2)2] (w1 = major diameter, w2 = minor diameter). (B) Kinetic measurements of ATP levels in treated Balb/cJ mice estimated by PmeLUC luminescence. Reported data represent the average number of photons in the tumor area (p/s/cm2/s = photons/second/cm2/steradian). The values are represented as mean ± SEM of 12 mice per condition; *p < 0.05; **p < 0.01; ***p < 0.001 versus placebo and, when reported, versus ARA-C. (C) Representative pictures (n = 3) of PmeLUC luminescence emission in treated Balb/cJ mice and of ex vivo tumor volumes at post-inoculum day 12.
ATP Release from DNR-Treated Mice Increases Leukemia-Infiltrating Tregs and IDO1-Expressing DCs
Chemotherapy-induced ATP release correlates with an increase in pro-inflammatory mediators (34, 35). Accordingly, administration of DNR but not ARA-C promoted a significant increase of IFN-γ (p < 0.05), IL-1β (p < 0.01), IL-2 (p < 0.001), and IL-12 (p < 0.05) in the serum of leukemia-bearing mice (Figure S6 in Supplementary Material). To investigate the capacity of chemotherapy to elicit a suppressive, along with its activatory, effect, leukemia infiltrate was analyzed after DNR and ARA-C treatment and compared to placebo. Among CD4+ T cells, DNR treatment was associated with a significant increase of total CD25+Foxp3+ T cells as compared to placebo and ARA-C (Figure 6A). Notably, DNR, and not ARA-C, significantly increased the expression of PD-1 on CD4+CD25+Foxp3+ T cells, both as percentage of positive cells (data not shown) and as mean fluorescence intensity (Figure 6B). This finding in DNR-treated mice paralleled with the increase of PD-1-expressing Tregs in AML patients after induction chemotherapy (Figure 1D) and in Tregs obtained in vitro after culture with human ATP-treated DCs (Figure 3E). Moreover, among CD11b+MHCII+LY6C− myelo-monocytic cells (Figure S7 in Supplementary Material) only DNR, and not ARA-C treatment upregulated DC expression of CD11c (Figure 6C), which correlated with higher expression of IDO1 (Figure 6D) and MHCII (Figure 6E). Interestingly, in DNR-treated mice, CD11chigh DCs had also increased expression of CD39 (Figure 6F), which is the rate-limiting enzyme during ATP catabolism (36, 37).
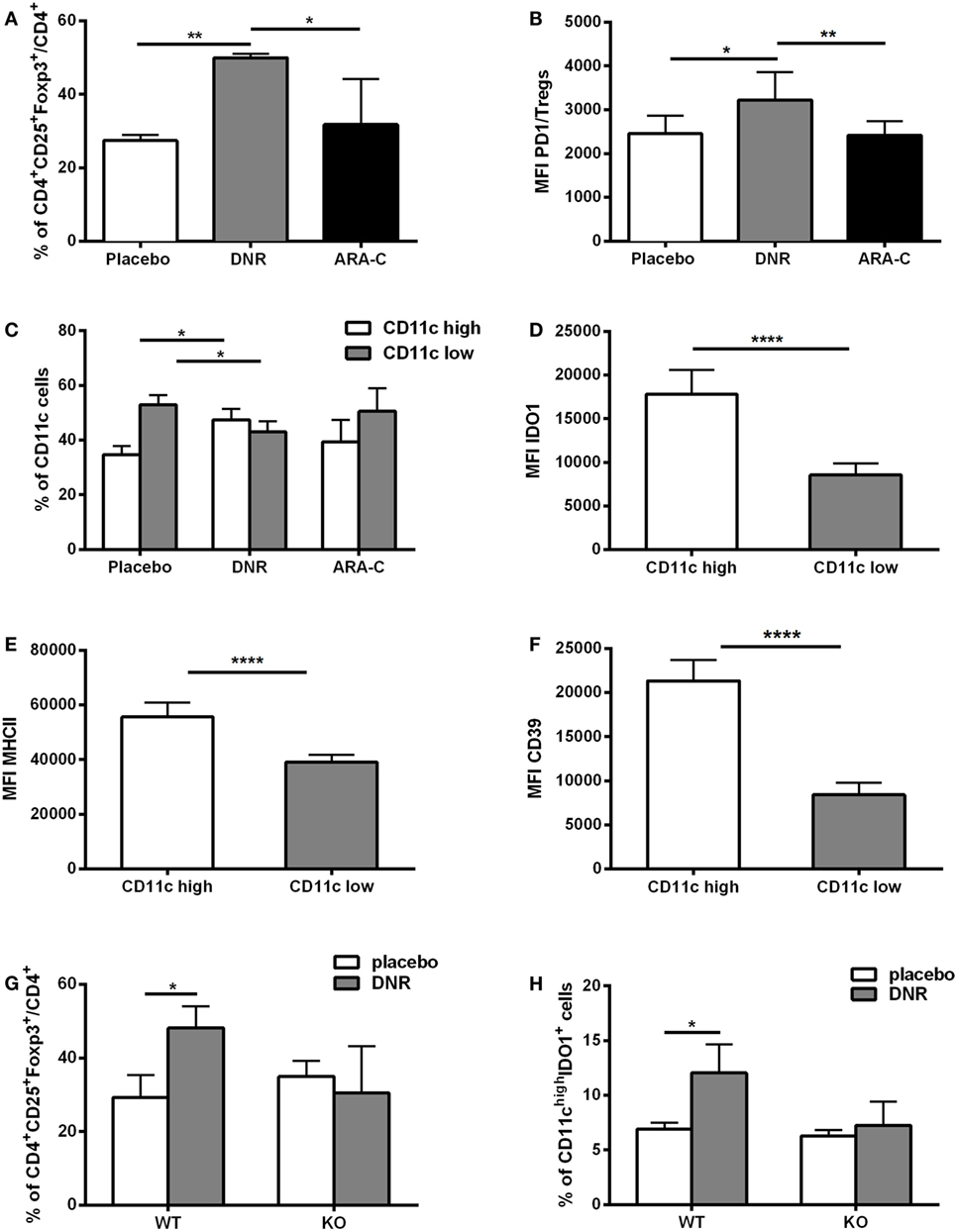
Figure 6. ATP release from DNR-treated mice increases leukemia-infiltrating T regulatory cells (Tregs) and IDO1-expressing dendritic cells (DCs). (A) Percentage of CD4+CD25+Foxp3+ infiltrating T cells (gated on CD4+ T cells) by FACS analyses. Data were obtained from six mice per condition. Mice were treated with placebo, DNR, or ARA-C as described in Figure 5; *p < 0.05; **p < 0.01. (B) The mean fluorescence intensity (MFI) of PD-1 in CD4+Foxp3+ infiltrating T cells (Tregs); *p < 0.05; **p < 0.01. (C) Percentage of infiltrating CD11clow and CD11chigh myeloid DCs by FACS analyses. Data were obtained from six mice per condition; *p < 0.05. MFI obtained by flow cytometry indicates the expression level of IDO1 (D), MHCII (E), and CD39 (F) in CD11clow and CD11chigh myeloid DCs in DNR-treated mice; ****p < 0.0001. FACS analysis of tumor-infiltrating CD4+CD25+Foxp3+ T cells (G) and CD11chighIDO1+ myeloid DCs (H) in P2X7 wild-type (WT) and knockout (KO) DNR- or placebo-treated mice. Data were obtained from six mice per condition and represented as mean ± SEM.
To demonstrate in vivo the contribution of ATP release in increasing the infiltration by Tregs and tolerogenic DCs after DNR treatment, P2X7R null mice were injected with WEHI-3B PmeLUC cells and treated with DNR. The levels of ATP after DNR treatment were similar to those observed in wild-type (WT) mice (Figure S8 in Supplementary Material). However, differently from WT mice, in P2X7 null mice DNR failed to increase CD4+CD25+Foxp3+ Tregs (Figure 6G) and IDO1-expressing CD11chigh DCs (Figure 6H). Taken together, these in vivo results demonstrate that ATP release from DNR-treated dying leukemia cells has a role in the induction of an immune suppressive microenvironment, which comprises Tregs and IDO1-expressing DCs.
Discussion
Some chemotherapeutic agents, such as DNR, induce immunogenic cell death of both solid tumors (1–3, 38) and leukemias (1, 39), allowing immune recognition and T-cell-mediated anticancer response. However, this treatment is rarely curative, especially in AML, suggesting that other mechanisms are in place. One possibility is that, besides inducing T-cell activation, such treatment could concomitantly promote the development of immune suppressive pathways, not yet exploited (7, 8). Accordingly, our data from AML patients indicate that CD8+ T cells emerging after combined DNR and ARA-C chemotherapy are not only functionally capable of leukemia recognition but are also coupled with a novel population of Tregs with suppressive phenotype, Treg2 cells, recently described according to CD15s (28, 29). Tregs are known to induce a suppressive state in tumor-infiltrating CTLs, which favors leukemia development and growth (40–42). Clinically, in AML, the persistence of increased Tregs after chemotherapy is correlated with poor clinical outcome (43), which is paralleled with the exhaustion of leukemia-specific CTLs over time in association with disease relapse (44, 45).
It has been reported that ATP regulates the function of lymphoid cells with opposite effects depending on the cellular subsets analyzed (11, 46, 47). In case of Tregs, ATP may directly elicit their suppressive function or may be metabolized by ecto-5′-nucleotidases, such as CD39 and CD73, into adenosine, which enhances Tregs activity (11–13). Our results indicate that, during chemotherapy, the levels of ATP within leukemia microenvironment are positively correlated with the frequency of Tregs. Indeed, only DNR treatment, which was capable of significantly augmenting intra-leukemia ATP level, was associated with increased Tregs. In mice lacking the ATP receptor P2X7R, DNR failed to increase Tregs, strongly supporting the crucial role of ATP in this process. Notably, in vivo ATP-stimulated Tregs showed higher expression of the immune checkpoint inhibitor PD-1, similarly to PD-1-expressing suppressive Treg2 cells expanded in chemotherapy-treated AML patients. Upon inflammation, the triggering of PD-1 on T cells negatively regulates their proliferation and cytokine production (48–50). The expression of PD-1 on Tregs correlates with their immunosuppressive activity (32, 33, 51) and the accumulation of PD1+Foxp3+ Tregs within the tumor microenvironment of solid tumors further confirms their immunosuppressive potential (32, 51). Our data confirm and extend these data to AML and provide new evidence and explanation for the correlation between the frequency of PD1+ Tregs and the level of ATP released from chemotherapy-treated leukemia cells.
ATP release from chemotherapy-treated tumor cells is a potent driver of antitumor immune response through P2X7R activation on DCs, which are induced to complete maturation and full competence in antigen presentation (6, 52, 53). Whether chemotherapy-dependent ATP release may also mediate the induction of tolerogenic features in tumor-infiltrating DCs has been poorly investigated. Our data add evidence to the hypothesis that ATP release from chemotherapy-treated tumor cells, being a potent inflammatory stimulus, may have a dual role on DC function. Along with the well-established effect on DC function toward cross-priming (53, 54), ATP may also promote the induction of tolerogenic pathways, such as IDO1. Our previous work in AML demonstrated that IDO1 mediates the conversion of naive CD4+CD25− T cells into fully competent Tregs, and that leukemic DCs inhibit the induction of leukemia-specific T cells via IDO1 (10, 19). Here, we show that ATP release from chemotherapy-treated AML cells drives IDO1 upregulation in DCs in a P2X7R-dependent manner. These DCs are in vitro fully competent at inducing through IDO1 a population of Tregs, which in turn inhibit leukemia-specific T-cell immune response. According to a previous report (51), upon chemotherapy the effect of ATP on IDO1 expression is part of a continuum, in which the DC maturation process may stand by IDO1 production to counterbalance the exceeding immune activation triggered by pro-inflammatory stimuli and associated to chemotherapy-induced immunogenic cell death (16, 18). Of note, our in vivo experiments indicate that during chemotherapy increased levels of ATP are associated with higher expression of CD39 on DCs, which regulates the rate-limiting enzymatic step of ATP catabolism (37). Such correlation is intriguing and may suggest that, within an ATP-enriched tumor microenvironment, infiltrating DCs may take part to ATP degradation by modulating on their surface the expression of ATP-specific ecto-nucleotidases.
Overall, our data indicate that chemotherapy treatment for AML, along with its well-known immunogenic effect, may also result in the induction of an immune suppressive microenvironment. This scenario, at least in part, depends on the local effects of ATP release from dying leukemia cells, which may critically influence an increase of leukemia-associated Tregs and tolerogenic DCs. A better understanding of the mechanisms leading to the induction of a leukemia immune suppressive microenvironment during chemotherapy has important clinical implications to fully exploit the immunogenic potential of anti-leukemia agents and tune their application.
Ethics Statement
For Human Samples
This study was carried out in accordance with the recommendations of Ethics Committee of the University Hospital of Bologna S. Orsola-MALPIGHI with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Ethics Committee of the University Hospital of Bologna S. Orsola-Malpighi, approval code: 147/2013/O/Tess.
For Animal Subjects
This study was carried out in accordance with the recommendations of Ethical Committee of University of Ferrara. The protocol was approved by the Ethical Committee of University of Ferrara and Italian Ministry of Health (EU Directive 2010/63/EU and Italian D.Lgs 26/2014; authorization number 821/2015PR to EA).
Author Contributions
ML and DO performed ex vivo characterization of lymphocytes, cell cultures experiments, and in vitro assays, analyzed the data, and wrote the manuscript; VS and AR analyzed IFN-γ-producing lymphocytes from patients; EM, EO, SS, AP, PP, and AB performed in vivo assays and analyzed the data; ST and CJ performed ex vivo characterization of lymphocytes and cytotoxicity test; AB, PR, and MC contributed to write the paper; MPC and FV contributed to study design and to write the paper; EA and AC designed the research, analyzed the data, wrote the manuscript, and gave the final approval for submission of the manuscript.
Conflict of Interest Statement
FV is member of the Scientific Advisory Board of Biosceptre Ltd., a Biotech involved in the development of P2X7-targeted therapies. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Conventional and Confocal Microscopy Facility of the Fondazione IRCCS Istituto Nazionale Tumori for confocal microscopy images acquisition. The authors wish to thank Dr. Metz (Lankenau Institute for Medical Research, Wynnewood, PA, USA) for kindly providing anti-human IDO1 Ab.
Funding
The research was supported by Regione Emilia-Romagna and University of Bologna (Progetto di Ricerca Regione-Università, Bando Giovani ricercatori “Alessandro Liberati”), FATRO/Fondazione Corrado e Bruno Maria Zaini-Bologna, AIL (Associazione Italiana contro le leucemie)/Section of Bologna. FV is supported by grants from AIRC, the Italian Association for Cancer Research (no. IG 13025 and IG 18581). EA is supported by grants from AIRC (MFAG11630 and IG16812).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2017.01918/full#supplementary-material.
Abbreviations
IDO1, indoleamine-2,3-dioxygenase 1; DNR, daunorubicin; AML, acute myeloid leukemia; P2X7R, P2X7 receptor; DCs, dendritic cells; Tregs, T regulatory cells; PB, peripheral blood; ARA-C, cytarabine; mAbs, monoclonal antibodies; 1-MT-l, 1-mthyltryptophano-levo; PD-1, programmed cell death protein 1; p.i., post-inoculum; p/s/cm2/s, photons/second/cm2/steradian; CTLs, cytotoxic T cells; EM, effector memory; EMRA, effector memory expressing RA; CM, central memory.
References
1. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res (2011) 71:4821–33. doi:10.1158/0008-5472.CAN-11-0950
2. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res (2011) 71:4809–20. doi:10.1158/0008-5472.CAN-11-0753
3. Suzuki Y, Mimura K, Yoshimoto Y, Watanabe M, Ohkubo Y, Izawa S, et al. Immunogenic tumor cell death induced by chemoradiotherapy in patients with esophageal squamous cell carcinoma. Cancer Res (2012) 72:3967–76. doi:10.1158/0008-5472.CAN-12-0851
4. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology (2014) 3:e955691. doi:10.4161/21624011.2014.955691
5. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334:1573–7. doi:10.1126/science.1208347
6. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res (2010) 70:855–8. doi:10.1158/0008-5472.CAN-09-3566
7. Bugaut H, Bruchard M, Berger H, Derangere V, Odoul L, Euvrard R, et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS One (2013) 8:e65181. doi:10.1371/journal.pone.0065181
8. Kanakry CG, Hess AD, Gocke CD, Thoburn C, Kos F, Meyer C, et al. Early lymphocyte recovery after intensive timed sequential chemotherapy for acute myelogenous leukemia: peripheral oligoclonal expansion of regulatory T cells. Blood (2011) 117:608–17. doi:10.1182/blood-2010-04-277939
9. Krysko O, Løve Aaes T, Bachert C, Vandenabeele P, Krysko DV. Many faces of DAMPs in cancer therapy. Cell Death Dis (2013) 4:e631. doi:10.1038/cddis.2013.156
10. Curti A, Trabanelli S, Onofri C, Aluigi M, Salvestrini V, Ocadlikova D, et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica (2010) 95:2022–30. doi:10.3324/haematol.2010.025924
11. Trabanelli S, Ocadlíková D, Gulinelli S, Curti A, Salvestrini V, Vieira RP, et al. Extracellular ATP exerts opposite effects on activated and regulatory CD4+ T cells via purinergic P2 receptor activation. J Immunol (2012) 189:1303–10. doi:10.4049/jimmunol.1103800
12. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood (2007) 110:1225–32. doi:10.1182/blood-2006-12-064527
13. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res (2015) 3:254–65. doi:10.1158/2326-6066.CIR-14-0018
14. Koo TY, Lee JG, Yan JJ, Jang JY, Ju KD, Han M, et al. The P2X7 receptor antagonist, oxidized adenosine triphosphate, ameliorates renal ischemia-reperfusion injury by expansion of regulatory T cells. Kidney Int (2017) 92:415–31. doi:10.1016/j.kint.2017.01.031
15. Steinman RM, Hawiger D, Liu K, Bonifaz L, Bonnyay D, Mahnke K, et al. Dendritic cell function in vivo during the steady state: a role in peripheral tolerance. Ann N Y Acad Sci (2003) 987:15–25. doi:10.1111/j.1749-6632.2003.tb06029.x
16. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117:1147–54. doi:10.1172/JCI31178
17. Trabanelli S, Ocadlikova D, Ciciarello M, Salvestrini V, Lecciso M, Jandus C, et al. The SOCS3-independent expression of IDO2 supports the homeostatic generation of T regulatory cells by human dendritic cells. J Immunol (2014) 192:1231–40. doi:10.4049/jimmunol.1300720
18. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother (2014) 63:721–35. doi:10.1007/s00262-014-1549-4
19. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood (2007) 109:2871–7. doi:10.1182/blood-2006-07-036863
20. Curti A, Trabanelli S, Salvestrini V, Baccarani M, Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood (2009) 113:2394–401. doi:10.1182/blood-2008-07-144485
21. Trabanelli S, Lecciso M, Salvestrini V, Cavo M, Očadlíková D, Lemoli RM, et al. PGE2-induced IDO1 inhibits the capacity of fully mature DCs to elicit an in vitro antileukemic immune response. J Immunol Res (2015) 2015:253191. doi:10.1155/2015/253191
22. Curti A, Pandolfi S, Aluigi M, Isidori A, Alessandrini I, Chiodoni C, et al. Interleukin-12 production by leukemia-derived dendritic cells counteracts the inhibitory effect of leukemic microenvironment on T cells. Exp Hematol (2005) 33:1521–30. doi:10.1016/j.exphem.2005.08.005
23. Curti A, Isidori A, Ferri E, Terragna C, Neyroz P, Cellini C, et al. Generation of dendritic cells from positively selected CD14+ monocytes for anti-tumor immunotherapy. Leuk Lymphoma (2004) 45:1419–28. doi:10.1080/10428190310001653682
24. Očadlíková D, Trabanelli S, Salvestrini V, Ciciarello M, Evangelisti C, Lecciso M, et al. CD103 marks a subset of human CD34+-derived langerin+ dendritic cells that induce T-regulatory cells via indoleamine 2,3-dioxygenase-1. Exp Hematol (2015) 43:268–76.e5. doi:10.1016/j.exphem.2014.12.007
25. Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell (2005) 16:3659–65. doi:10.1091/mbc.E05-03-0222
26. Adinolfi E, Raffaghello L, Giuliani AL, Cavazzini L, Capece M, Chiozzi P, et al. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res (2012) 72:2957–69. doi:10.1158/0008-5472.CAN-11-1947
27. Adinolfi E, Capece M, Franceschini A, Falzoni S, Giuliani AL, Rotondo A, et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res (2015) 75:635–44. doi:10.1158/0008-5472.CAN-14-1259
28. Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, et al. Sialyl Lewis x (CD15s) identifies highly differentiated and most suppressive FOXP3high regulatory T cells in humans. Proc Natl Acad Sci U S A (2015) 112:7225–30. doi:10.1073/pnas.1508224112
29. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22:679–84. doi:10.1038/nm.4086
30. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol (2004) 4:762–74. doi:10.1038/nri1457
31. Sakaguchi S, Wing K, Yamaguchi T. Dynamics of peripheral tolerance and immune regulation mediated by Treg. Eur J Immunol (2009) 39:2331–6. doi:10.1002/eji.200939688
32. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv (2015) 1:e1500845. doi:10.1126/sciadv.1500845
33. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27:1–7. doi:10.1016/j.coi.2013.12.005
34. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28:690–714. doi:10.1016/j.ccell.2015.10.012
35. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene (2017) 36:293–303. doi:10.1038/onc.2016.206
36. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol (2011) 11:201–12. doi:10.1038/nri2938
37. Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal (2014) 10:529–64. doi:10.1007/s11302-014-9427-2
38. Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene (2010) 29:482–91. doi:10.1038/onc.2009.356
39. Wemeau M, Kepp O, Tesnière A, Panaretakis T, Flament C, De Botton S, et al. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis (2010) 1:e104. doi:10.1038/cddis.2010.82
40. Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR. Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Invest (2014) 124:2425–40. doi:10.1172/JCI66375
41. Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood (2009) 114:3793–802. doi:10.1182/blood-2009-03-208181
42. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood (2010) 116:2484–93. doi:10.1182/blood-2010-03-275446
43. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of CD4? CD25? CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer (2011) 129:1373–81. doi:10.1002/ijc.25791
44. Casalegno-Garduño R, Schmitt A, Spitschak A, Greiner J, Wang L, Hilgendorf I, et al. Immune responses to WT1 in patients with AML or MDS after chemotherapy and allogeneic stem cell transplantation. Int J Cancer (2016) 138:1792–801. doi:10.1002/ijc.29909
45. Casalegno-Garduño R, Meier C, Schmitt A, Spitschak A, Hilgendorf I, Rohde S, et al. Immune responses to RHAMM in patients with acute myeloid leukemia after chemotherapy and allogeneic stem cell transplantation. Clin Dev Immunol (2012) 2012:146463. doi:10.1155/2012/146463
46. Apostolova P, Zeiser R. The role of purine metabolites as DAMPs in acute graft-versus-host disease. Front Immunol (2016) 7:439. doi:10.3389/fimmu.2016.00439
47. Salles É, Menezes MN, Siqueira R, Borges da Silva H, Amaral EP, Castillo-Méndez SI, et al. P2X7 receptor drives Th1 cell differentiation and controls the follicular helper T cell population to protect against Plasmodium chabaudi malaria. PLoS Pathog (2017) 13:e1006595. doi:10.1371/journal.ppat.1006595
48. Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol (2007) 19:309–14. doi:10.1016/j.coi.2007.04.012
49. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi:10.1084/jem.192.7.1027
51. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) (2016) 4(3):E28. doi:10.3390/vaccines4030028
52. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity (2013) 38:729–41. doi:10.1016/j.immuni.2013.03.003
53. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi:10.1038/nm.2028
Keywords: acute myeloid leukemia, chemotherapy, ATP, dendritic cell, T regulatory cells, immunosuppression
Citation: Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, Pegoraro A, Portararo P, Jandus C, Bontadini A, Redavid A, Salvestrini V, Romero P, Colombo MP, Di Virgilio F, Cavo M, Adinolfi E and Curti A (2017) ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 8:1918. doi: 10.3389/fimmu.2017.01918
Received: 13 October 2017; Accepted: 14 December 2017;
Published: 22 December 2017
Edited by:
Giovanna Schiavoni, Istituto Superiore di Sanità, ItalyReviewed by:
Carlos Alfaro, Universidad de Navarra, SpainShahram Kordasti, King’s College London, United Kingdom
Copyright: © 2017 Lecciso, Ocadlikova, Sangaletti, Trabanelli, De Marchi, Orioli, Pegoraro, Portararo, Jandus, Bontadini, Redavid, Salvestrini, Romero, Colombo, Di Virgilio, Cavo, Adinolfi and Curti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariangela Lecciso, bWFyaWFuZ2VsYS5sZWNjaXNvM0B1bmliby5pdA==;
Darina Ocadlikova, amVubnlua2FAc2V6bmFtLmN6
†These authors have contributed equally to this work.