 Carlos Cuesta-Mateos1,2
Carlos Cuesta-Mateos1,2 Ana Alcaraz-Serna
Ana Alcaraz-Serna Beatriz Somovilla-Crespo
Beatriz Somovilla-Crespo Cecilia Muñoz-Calleja
Cecilia Muñoz-Calleja- 1Servicio de Inmunología, Instituto de Investigación Sanitaria Hospital Universitario de La Princesa, Madrid, Spain
- 2IMMED S.L., Immunological and Medicinal Products, Madrid, Spain
- 3Department of Immunology and Oncology, Centro Nacional de Biotecnología, Consejo Superior de Investigaciones Científicas (CSIC), Madrid, Spain
Today, monoclonal antibodies (mAbs) are a widespread and necessary tool for biomedical science. In the hematological cancer field, since rituximab became the first mAb approved by the Food and Drug Administration for the treatment of B-cell malignancies, a number of effective mAbs targeting lineage-specific antigens (LSAs) have been successfully developed. Non-LSAs (NLSAs) are molecules that are not restricted to specific leukocyte subsets or tissues but play relevant pathogenic roles in blood cancers including the development, proliferation, survival, and refractoriness to therapy of tumor cells. In consequence, efforts to target NLSAs have resulted in a plethora of mAbs—marketed or in development—to achieve different goals like neutralizing oncogenic pathways, blocking tumor-related chemotactic pathways, mobilizing malignant cells from tumor microenvironment to peripheral blood, modulating immune-checkpoints, or delivering cytotoxic drugs into tumor cells. Here, we extensively review several novel mAbs directed against NLSAs undergoing clinical evaluation for treating hematological malignancies. The review focuses on the structure of these antibodies, proposed mechanisms of action, efficacy and safety profile in clinical studies, and their potential applications in the treatment of hematological malignancies.
Introduction
Cancer treatment is expanding from non-specific cytotoxic chemotherapies to targeted therapies as a consequence of increased knowledge of the pathogenesis of cancer that leads to a better design of treatments to inhibit tumor growth and spread. Most of these therapies consist in monoclonal antibodies (mAbs) that bind to specific antigens (Ags) expressed on the surface of cancer and normal cells, mediating different mechanisms of action (MOA).
IgG antibodies, which are the most commonly used in cancer immunotherapy, show two regions that determine their biologic properties: the variable fragment (Fv), responsible for interaction with Ag and the constant fragment (Fc), responsible for interaction with immune cells or molecules bringing together cells bearing the Ag (or the Ag itself) to components of innate or acquired immunity. The Fc of an antibody is responsible for half-life, antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent phagocytosis (ADCP), or complement-dependent cytotoxicity (CDC) (1, 2). Both Fv and Fc determine the different and characteristics of MOA displayed by a single mAb and its utility as immunotherapeutic agent in cancer. These MOA may work alone or combined. Briefly, a particular mAb may inhibit ligand–receptor interactions, and/or induce proapoptotic signaling, and/or activate innate immune cells or molecules triggering ADCC, ADCP, or CDC, and/or may induce tumor cell killing by targeting regulatory molecules on host immune cells (1, 2). In addition, mAbs can be used to deliver payloads such as cytotoxic agents, toxins, or radioisotopes, which are coupled to the mAb targeting tumor cells (3). One explanation to the rapid growth of mAbs as therapeutic drugs is their plasticity. Antibodies can be engineered at several levels leading to customized modulations in the Fv/Fc properties. Altering the glycosylation status is the most extended modification among all the novel mAbs under development and is used to regulate anti- and proinflammatory properties and to control the binding to Fc receptors (FcRs) to modulate ADCC (4, 5).
In the hematological malignancies field, therapeutic mAbs are especially relevant owing to accessibility to tumor cells, facilitating in vitro studies of targets and MOA. In addition, the historical knowledge of the hematopoietic differentiation Ags, usually grouped as cluster of differentiation (CD) Ags, has provided a large number of potential targets in hematological malignancies. Similar to other cancers, tumor-associated Ags recognized by therapeutic mAbs in blood cancers fall into different categories. Many of them are present at the different normal maturation steps of a given linage and this is why they are called lineage-specific antigens (LSAs). For example, B-cell differentiation is associated with the expression of CD19, CD20, CD22, and surface Ig (6). Similarly, myeloid differentiation is associated with CD33 expression (7), whereas CD3 is the hallmark of the T-cell linage (8). These LSAs show significant overlapping expression patterns between leukemia or lymphoma subtypes within the same lineage.
It could be said that most of the LSAs are clinically validated targets in antibody-based therapy. CD20 is a LSA exclusively expressed on B-cells membrane and on the majority of malignant B-cells (6, 9). The “blockbuster” antibody rituximab is the first-in-class anti-CD20 mAb approved for the treatment of B-cell non-Hodgkin lymphoma (B-NHL) and chronic lymphocytic leukemia (CLL); it is by far the most important mAb used in hematological malignancies (10–12). Since its approval in 1997, four additional mAbs targeting different CD20 epitopes and displaying several MOA have been approved by the US Food and Drug Administration (US-FDA) (13–15). These CD20-targeting therapeutic mAbs account for >30% of all current therapeutic mAbs for cancer (3) and reflect the previous tendency to develop improved antibodies against the same LSAs. The MOA of antibodies directed to CD20 are given in Table 1.
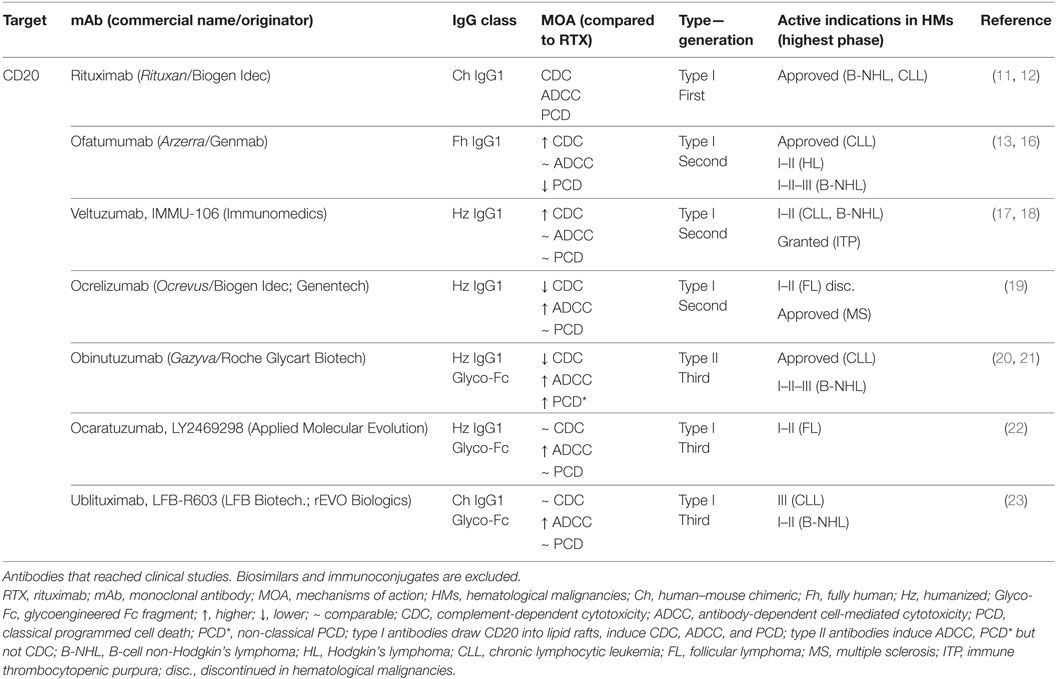
Table 1. Characteristics of antibodies directed to CD20.
The success of anti-CD20 mAbs has encouraged drug developers to propose novel LSAs, such as CD19, CD22, or CD79b (Table 2) (24–26). Despite these LSAs representing potential candidates for the treatment of B-cell cancers, antibodies directed to CD19 (MOR00208, inebilizumab, or MDX-1342) or CD22 (epratuzumab) have yielded only modest responses in clinical studies (9). This low efficacy has been attributed to high Ag internalization rates on mAb ligation (3). Consequently, CD19, CD22, and CD79 have been widely investigated for immunoconjugate therapy with promising clinical results as a single agent with no unexpected safety concerns. Finally, but beyond the scope of this review, it should be mentioned that other antibody formats, such as the bispecific T-cell engager (BiTe) blinatumomab, show promising results when targeting CD19 (27, 28). Thanks to a dual specificity for CD19 and CD3, this BiTe efficiently redirects host T-cells to CD19 expressed in tumor B-cells, although it shows neurological toxicity as treatment-related adverse event (29, 30).
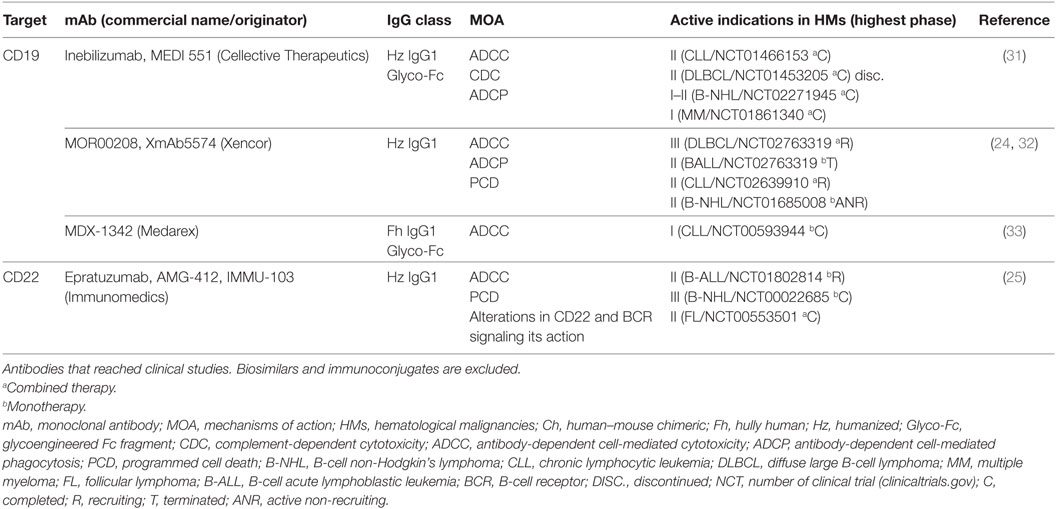
Table 2. Characteristics of antibodies directed to LSAs.
A different group of tumor Ags are the non-lineage-specific antigens (NLSAs), which comprise many molecules that are preferentially expressed by tumor cells but not restricted to specific leukocyte subsets or tissues and include, among others, oncogenic receptors, chemokine receptors (CKRs), and molecules involved in the formation and preservation of the tumor microenvironment (TME). The ubiquous expression of many NSLAs potentially enables antibodies directed to these molecules to be used in different hematological malignancies, or even universally in cancer.
Limited clinical efficacy of some mAbs targeting LSAs and the advent of patients with refractory diseases to therapies directed to LSAs boosted the research on many NLSAs with a relevant role in the pathogenesis of cancer, especially in B-cell malignancies (9, 34). Moreover, in some disorders the lack or loss of LSA expression in cell membrane may preclude the use of antibodies, thus prompting research of other potential therapeutic targets. This is the case of multiple myeloma (MM), a B-cell disorder where tumor cells do not express CD20 (35) and where novel antibodies directly targeting several NLSAs are a profound change compared with earlier treatment approaches based on anti-CD20 antibodies.
Efforts to target NLSAs have resulted in an ever-increasing number of new murine, chimeric and human antibodies with proven efficacy in preclinical models. Here, we extensively review the results of several novel mAbs directed against NLSAs undergoing clinical evaluation (Table 3). The review focuses on the structure of these antibodies, proposed MOA, efficacy, and safety profile in clinical studies, and their potential applications in the treatment of hematological cancers.
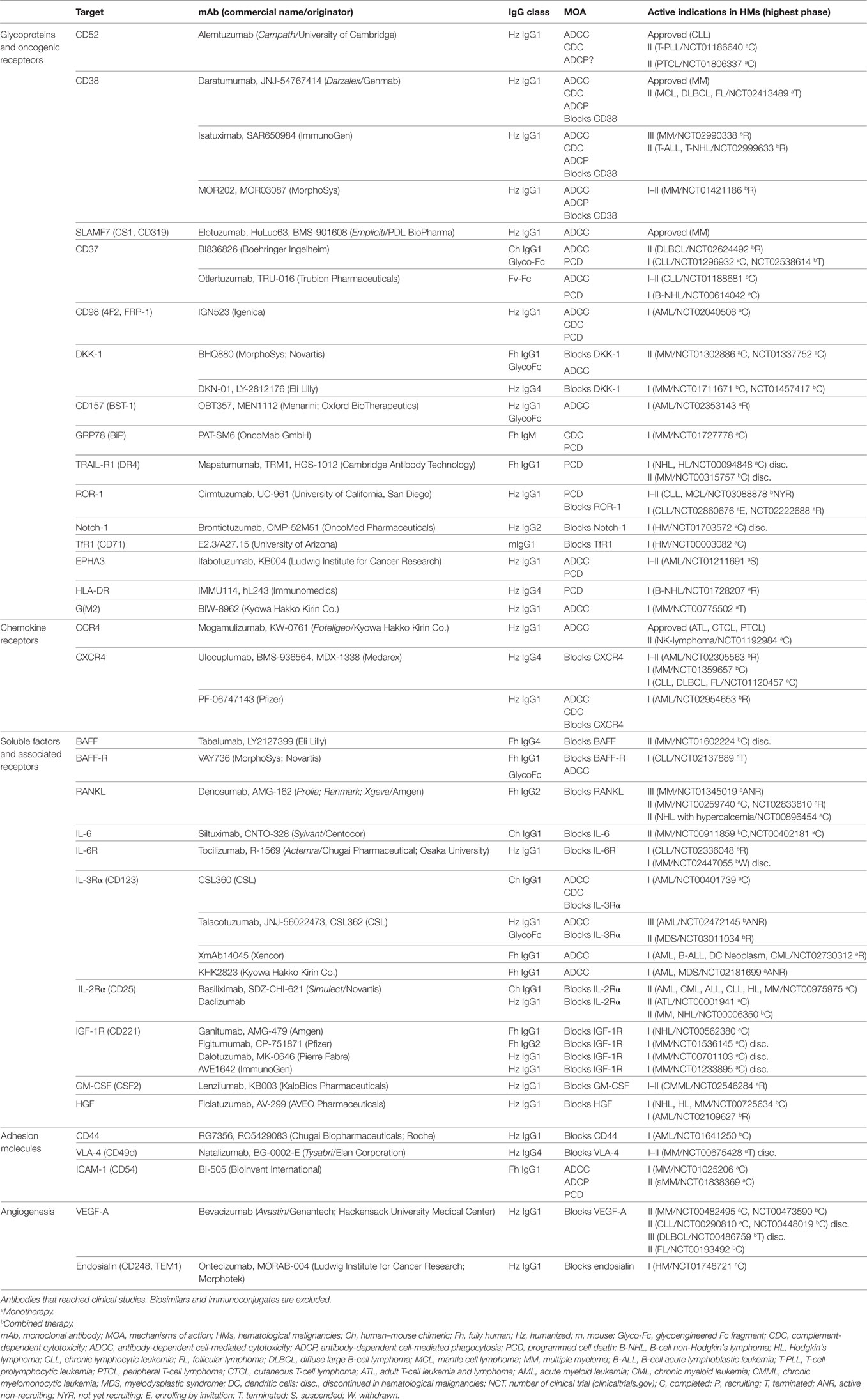
Table 3. Characteristics of antibodies directed to NLSAs.
Antibodies Targeting Glycoproteins and Oncogenic Receptors
Pathologic clonal populations express or overexpress different NLSAs which are involved in different oncogenic pathways and may confer an evolutionary advantage to the tumor. In some cases, high expression of the NLSAs is the rationale behind targeting a single receptor. In other cases, this targeting represents an optimal strategy to avoid cancer cell proliferation and survival.
CAMPATH-1 (CD52)
CD52 is a glycoprotein anchored to glycosylphosphatidylinoitol (GPI) present on the surface of mature lymphocytes, monocytes and dendritic cells (DCs) (36). CD52 expression is particularly high on T-cell prolymphocytic leukemia (T-PLL), Sézary syndrome (SS), acute lymphoblastic leukemia (ALL), CLL, and acute myeloid leukemia (AML) (36–39), which is the reason why it was selected as therapeutic target despite not having a clear role in the pathogenesis of these conditions. Nonetheless, efficacy as single agent in patients with high-risk CLL (40–42) prompted the approval of the anti-CD52 mAb alemtuzumab as front-line therapy in CLL. The main MOA of alemtuzumab are CDC and ADCC (36) which are likely to be involved in its efficacy in SS and T-PLL (39). Curiously, side effects associated with immune-suppression and infections were more frequent in B-cell than in T-cell malignancies, probably due to off-target activities (43). Despite being one of the few working weapons in T-cell malignancies, alemtuzumab was withdrawn in 2012, due to a strategic decision, and now is only available through an international compassionate use program.
CD38
In some leukocytes, this type II transmembrane glycoprotein behaves both as an ectoenzyme (NADase/ADPR cyclase) and as a receptor involved in cell adhesion, calcium flux and signal transduction (44, 45). While its expression was low to moderate on lymphoid and myeloid cells, both normal and tumor plasma cells exhibited high levels of CD38, making it an attractive target for MM (44, 45). In 2015, daratumumab, a humanized anti-CD38 IgG1 mAb, became the first mAb approved for MM (46). In preclinical models, daratumumab caused cell death through ADCC, CDC, ADCP, and blocking of CD38 that inhibits its enzymatic activity and induces apoptosis in a caspase-dependent manner (47–50). In addition, it has been recently suggested that depletion of CD38+ immunosuppressive regulatory T (Tregs) and B-cells and myeloid-derived suppressor cells (MDSCs) increase antitumor effector T-cell responses (51). Altogether, these MOA are responsible for daratumumab single-agent efficacy as demonstrated by two phase I–II trials in pretreated MM patients (NCT00574288; NCT01985126) that prompted FDA approval of daratumumab (52, 53). Moreover, daratumumab shows promising results both in the relapsing/refractory setting (rrMM) and in the upfront setting when combined with other potent MM therapeutics, including lenalidomide, dexamethasone and bortezomib (54–56). As a result, the FDA granted “Breakthrough Therapy” designation to daratumumab in combination with lenalidomide–dexamethasone or bortezomib–dexamethasone for the treatment of previously treated MM.
In the light of the aforementioned results, it is not difficult to find several anti-CD38 mAbs under clinical development. Isatuximab, with similar MOA to daratumumab, has shown promising results in ongoing phase I–II studies in rrMM both in monotherapy (NCT01084252) (57) or combined with immunomodulatory drugs (IMIDs) or dexamethasone (NCT01749969) (58). Another mAb is MOR202, which lacks CDC activity, but still shows promising results in ongoing trials both in monotherapy or in combination (NCT04121186) (59, 60). Last, but not least, anti-CD38 mAbs are attracting the interest in many other B-cell malignancies expressing surface CD38 including CLL, mantle cell lymphoma (MCL), diffuse large B-cell lymphoma (DLBCL), and transformed follicular lymphoma (FL) (NCT02413489) (44, 52, 61).
Signaling Lymphocytic Activation Molecule Family Member F7 (SLAMF7; CS1)
This glycoprotein is moderately expressed by normal plasma cells and by cytolytic lymphocyte subsets such as natural killer (NK) cells, NKT cells or CD8+ T-cells (62, 63). As their normal counterpart, MM plasma cells express SLAMF7, but at higher levels (62, 63) as a consequence of an amplification of chromosome 1q23 region, where SLAMF7 gene is located, which is very frequent in aggressive MM (62, 64). SLAMF7 expression in MM does not correlate with other high-risk cytogenetic abnormalities or the degree of disease progression (62, 63), thus validating SLAMF7 as a potential target.
The humanized IgG1 mAb elotuzumab was the first-in-class anti-SLAMF7 to be approved by the FDA in 2015, and the second antibody marketed for the treatment of MM (65). Similar to daratumumab, elotuzumab has several MOA in vitro, although it seems to predominantly act through ADCC in vivo (63, 66–68) since homozygosis for the high-affinity FcγRIIIa Val significantly prolonged median period free survival in clinical settings (69). In addition, elotuzumab is an agonistic mAb, which activates NK cells, further enhancing their cytotoxicity through a unique SLAM-associated pathway. Conversely, MM cells lack the SLAM-associated adaptor EAT-2 thus preventing proliferation upon elotuzumab binding (70, 71).
In contrast to daratuzumab, elotuzumab has demostrated limited activity as a single agent in both preclinical and clinical studies (63). The deffects on NK cell activity observed in MM patients may be explained by elotuzumab activity relying on ADCC. Also, the paradox of NK cells becoming targets may also contribute to the lack of objective responses in rrMM patients treated with elotuzumab as single-agent (72). Therefore, to reach its maximum efficacy, elotuzumab needs to be combined with other antimyeloma agents such as lenalidomide-dexamethasone (NCT00742560, NCT01239797) (66, 73, 74) or bortezomib-dexamethasone (69, 75). Currently, several studies are examining different combinations either in the upfront or the relapsed/refractory settings.
CD37
This heavily glycosylated tetraspanin is highly expressed by mature B-cells and B-cell malignancies, including CLL and NHL (76–78). The exact function of CD37 has not yet been elucidated, although it seems to be important for T-cell-dependent B-cell responses, and may be involved in both pro- and antiapoptotic signaling (78). In addition, recent evidence confirms CD37 expression on the surface of CD34+/CD38− AML stem cells (LSCs), which are considered the root of tumor drug resistance and recurrence (79). For this reason, despite initially conceived as a lineage-specific therapy for B-cell malignancies, anti-CD37 mAbs are also being tested as therapeutics in AML.
CD37 has unique properties for generating therapies as low internalization rates allows the preservation of its ADCC activity (76). For this reason, different kinds of IgG formats targeting CD37 are currently in clinical development. BI836826 is an Fc-engineered, chimeric IgG1 that mediates potent ADCC and induces apoptosis on CD37-overexpressing cells (80). This mAb is undergoing phase I–II studies for the treatment of CLL and B-NHL, either as a single agent or in combination with ibrutinib, idelalisib or rituximab. A number of anti-CD37 immunoconjugates are also in advanced clinical phases (79, 81, 82) (Table 6).
Of special interest is the modular homodimer called otlertuzumab (TRU-016) formed by a single-chain Fv linked to the hinge region and Fc domain of hIgG1 (148, 149). Otlertuzumab induces apoptosis directly via binding to the CD37 protein, which results in up-regulation of the proapoptotic protein BIM (also termed BCL2L11) (150). In addition, otlertuzumab triggers Fc-dependent cytotoxicity (ADCC) but does not induce complement activation. In B-cell malignancies, otlertuzumab has shown efficacy as a single agent or combined with other therapeutic drugs in preclinical models (151, 152) as well as in phase I (NCT00614042) and phase II (NCT01188681) studies (149, 153). Other studies in B-NHL patients (NCT01317901) further confirm that combination regimens are well tolerated and lead to higher response rates (154). As a consequence, novel clinical trials are recruiting patients to evaluate combinations with standards of care in B-NHL such as rituximab, obinutuzumab, idelalisib, and ibrutinib.
CD98
The CD98 heterodimer consists of a type II single-pass transmembrane glycoprotein (also known as 4F2 Ag heavy chain or FRP-1) with two biochemical functions depending on the coupled light chain (155). Upon binding to the cytoplasmic tail of the integrin beta-chain it mediates adhesive signals thereby controlling cell proliferation, survival, migration, epithelial adhesion and polarity. In addition, CD98 contributes to the amino acid transport processes through the binding to one of the six permease-type amino acid transporters including L-type amino acid transporter 1 and 2 (LAT-1 and LAT-2) (155, 156), whose localization and proper function rely on the CD98 heavy chain (157). Both CD98-mediated activities take place on fast-cycling cells undergoing clonal expansion, such as AML cells, where CD98 supports elevated growth rates and contributes to proliferation, survival, and metastasis (158). Few approaches target metabolic cancer, and most of them are based on small molecules against CD98-associated light chains (158). In this context, targeting CD98 heavy chain with antibodies provides an alternative approach as demonstrated by IGN523, a novel humanized anti-CD98 mAb with robust preclinical activity against established lymphoma tumor xenografts (158). IGN523 elicits strong ADCC, mild CDC, and induces lysosomal membrane permeabilization that elicits capase-3- and caspase-7-mediated apoptosis in the presence of crosslinking antibody. But the most differential feature of IGN523 is the inhibition of essential amino acid (phenylalanine) uptake by rapidly proliferating tumor cells that ultimately results in caspase-3- and -7-mediated apoptosis (158). IGN523 has been evaluated in a completed Phase I study for rrAML (NCT02040506), although results are not published yet (158, 159).
Dickkopf-1 Protein (DKK1)
This NLSA is related to the canonical Wnt/beta-catenin signaling pathway. DKK1 is a soluble inhibitor that binds simultaneously the transmembrane receptors Kremen-1 or 2 and the Wnt/beta catenin coreceptor LRP5/6 (160). This extracellular binding leads to endocytosis of the DKK1-associated complex that impairs a subsequent activation of Wnt/beta-catenin signaling. The first association between DKK-1 and cancer was described in MM patients suffering osteolytic lesions MM (160). Later, on preclinical studies have demonstrated that a neutralizing DKK mAb reduces osteolytic bone resorption, increases bone formation, and controls MM growth (161–163). BHQ880 and DKN-1 are neutralizing humanized IgG1 mAbs, which are being tested in phase I–II studies in MM. Most of the studies (NCT00741377, NCT01457417) are using anti-DKK-1 mAbs in combination with antimyeloma therapy, except the phase II study that evaluated the efficacy of BHQ880 in monotherapy in previously untreated patients with high risk smoldering MM (phase II, NCT01302886). Overall, BHQ880 was well tolerated but the clinical benefits were limited (164). For this reason, other studies were designed to test the efficacy of anti-DKK-1 antibodies in the setting of MM with bone alterations combined with specific agents such zoledronic acid (NCT00741377).
CD157 (BST-1)
This GPI-linked membrane protein has a close resemblance to CD38 and a significant role in myeloid cells trafficking and pre-B-cell growth (165–167). It is, therefore, not surprising that high levels of CD157 can be found in B-ALL cells and in most primary AML patient samples, including the LSCs compartment (168). Based on this rationale, a novel defucosylated IgG1 termed OBT357/MEN1112 validated CD157 as a therapeutic target in AML in vitro and ex vivo models (168). Now, the potent ADCC observed in preclinical phases is under evaluation in a phase I study in AML patients (NCT02353143).
Glucose-Regulated Protein 78 (GRP78; BiP; HSP5a)
Members of the heat shock protein-70 family, if expressed on the cell membrane, are NLSAs of interest in mAb-based cancer therapy. GRP78 is a protein with multiple functions related to its different cellular locations. It may control the unfolded protein response, the macroautophagia or prosurvival pathways activated by PI3K/AKT. In some circumstances like glucose starvation, hypoxia or protein malfolding, GRP78 is translocated to the membrane, where it mediates, in general, cytoprotective responses (169). Many tumor cells, including MM, overexpress GRP78 on the outer plasma membrane to promote tumor survival, proliferation, and motility and this overexpression correlates with an adverse prognosis and drug resistance (170). Interestingly, normal plasma cells do not express the molecule on their membrane (171). Based on this, GRP78 is an ideal candidate for immunotherapeutic intervention of MM. Recently, the natural fully human IgM PAT-SM6 (initially isolated from a patient with gastric cancer) was evaluated as monotherapy in a phase I study in rrMM (NCT01727778). Results show that PAT-SM6 is well tolerated but has modest clinical activity (169). PAT-SM6 lacks ADCC activity thus its MOA mainly relies on apoptosis and to a lesser extent CDC (171, 172). Interestingly, patients who received prior treatment with proteasome inhibitors responded much better to PAT-SM6 than patients who had been previously treated with IMIDs or other chemotherapeutics. Hence, future clinical studies will focus on synergistic combinations with proteasome inhibitors to induce better clinical responses (press release by Patrys).
Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Receptor-1 (TRAIL-R1; DR4)
This protein, also known as death receptor 4, is a cell surface receptor that binds TRAIL (ApoL2) and activates the extrinsic apoptotic pathway (173). After binding of either ligand or agonist antibodies to TRAIL-R1, a death-inducing signaling complex (DISC) starts the recruitment, cleavage and activation of caspases-3, -6, -7, resulting in the characteristic programmmed cell death (PCD) (173). The expression of TRAIL-R1 is minimal or absent in healthy tissues. In contrast, this receptor is frequently detected in cancer, including B-cell malignancies (174–176). This rationale boosted the development of the agonist anti-TRAIL-R1 IgG1 antibody mapatumumab (HGS-ETR1). Unlike native TRAIL, mapatumumab has longer half-life and binds specifically to TRAIL-R1 and not to the other TRAIL receptors (177). Like TRAIL, mapatumumab mediates caspase-dependent apoptosis by binding TRAIL-R1. In preclinical models of hematological malignancies, mapatumumab induced apoptosis in a wide spectrum of human cancers and promoted tumor regressions in xenograft models (175–179). Interestingly, in a recent study, the combination of mapatumumab with low dose bortezomib potentiated the uptake of myeloma cell apoptotic bodies by DC and induced antimyeloma cytotoxicity by both CD8+ T-cells and NK cells (180). Based on this, it has been suggested that mapatumumab may also promote endogenous antitumor immune responses.
Results from a phase II study (NCT00094848) demonstrated that mapatumumab was capable of producing clinical responses when used as single agent in patients with B-NHL (181), particularly FL. Of interest, immunohistochemistry analysis suggested that strong TRAIL-R1 staining in tumor specimens was not a requirement for mapatumumab activity in FL (181). In another phase II study in MM (NCT00315757), no differences in efficacy were observed between patients receiving mapatumumab plus bortezomib and the control group. What remains unclear is whether immunosuppressive effects of bortezomib could affect the ability of mapatuzumab to promote immune responses (180).
Receptor Tyrosine Kinase-Like Orphan Receptor 1 (ROR-1)
This type I membrane glycoprotein lacks catalytic activity but is essential for ligand binding and signal transduction in the non-canonical Wnt pathway. It is considered an oncofetal Ag since it is highly expressed during early embryonic development, where it modulates neurite growth, but absent in most adult tissues (182). Ubiquitously found in human cancers, ROR-1 protein is highly expressed on the surface of CLL cells and several other B-cell malignancies where it favors invasion, metastasis and therapeutic resistance (183).
Cirmtuzumab (UC-961) is the first IgG1 directed against a functional epitope of the extracellular domain of ROR-1. It blocks ROR-dependent non canonical Wnt5a signaling through ROR-1 dephosphorylation, thus blocking tumor cell proliferation, migration and survival, leading to tumor cell death by apoptosis (184, 185). Preclinical and phase I studies have shown good tolerability and moderate activity of cirmtuzumab when used as a single agent but with apparent synergistic activity with other agents like ibrutinib (184, 185). Interestingly, cirmtuzumab acts like other kinase inhibitors mobilizing ROR1-expressing CLL cells, thereby preventing progression in protective niches and providing an additional MOA (185). Currently, cirmtuzumab is facing phase I–II studies as single agent in rrCLL (NCT02222688) or in combination with ibrutinib in rrCLL or rrMCL (NCT03088878). Finally, the interest in ROR-1 as target is supported by two bi-specific antibodies (ROR1-CD3-DART and APVO425) that aim to redirect cytotoxic T-cells to ROR-expressing cells (182) and by a novel anti-ROR1 single-chain (sc) antibody able to induce apoptotic death of CLL lines and primary CLL cells (186).
Notch-1
Aberrant signaling of this Notch family transmembrane receptor has been implicated in cancer, cancer stem cells, and tumor vasculature (187). Indeed, Notch1 is a well-characterized oncoprotein in T-ALL and lymphomas where activating Notch1 mutations are responsible for approximately 60% of T-ALL cases (188, 189). In preclinical models, blocking the extracellular region of Notch1 with antibodies decreased T-ALL tumor growth by inhibiting cancer cell growth and by disrupting angiogenesis (190, 191). Brontictuzumab (OMP-52M51) is the only anti-Notch-1 antibody tested in clinical settings (NCT01703572). Although completed, the results from this study have not been published and development in hematological cancers was discontinued. Severe grade adverse events could explain this, as Notch-1 inhibition causes gastrointestinal side effects (190, 191). Finally, antibodies against DLL4, a ligand of Notch-1, are alternatives to target this pathway, including OMP-21M18 or MEDI0639. Already tested in vitro and in vivo in solid tumors, they are currently under evaluation in several ongoing clinical trials. Nonetheless, no development has been reported for blood cancers (192).
EphrinA3 (EPHA3)
This member of the ephrin subfamily of receptor protein-tyrosine kinases can be considered an oncofetal Ag since it is not expressed in normal healthy adult tissues but is overexpressed by a variety of tumor types instead, including most hematological malignancies (193), where it plays an important role in tumor cell proliferation. Ifabotuzumab (KB004) is a humanized, non-fucosylated IgG1 mAb targeting EphrinA3 which induces apoptosis and stimulates ADCC (193). One phase I–II study (NCT01211691) tested for the utility of KB004 as a single agent in patients with heavily pretreated AML. KB004 was well tolerated but the efficacy was very limited with responses observed in patients with fibrotic myeloid diseases (194). In this study, it was postulated that low expression of EPHA3 in various myeloid leukemic cell subsets or the ability of KB004 to be internalized upon Ag binding are likely explanations for KB004 ineffectiveness (194). Based on this ability to be internalized, an alternative approach targeting EPHA3 with an immunoconjugate was proposed (195).
HLA-DR
Ligation of HLA-DR by antibodies is one of the oldest approaches to eliminate hematological tumors, since most of them express high levels of this MHC class II molecule (196). Anti-HLA-DR antibodies with different MOA such as apolizumab, Lym-1, and 1D09C3 eventually had no convincing clinical response in several clinical trials and were discontinued (197). In addition, anti–HLA-DR mAbs are potent inducers of complement activation, which plays a pivotal role in the pathogenesis of mAb infusion side effects (197). To our knowledge, there is only one ongoing phase I study recruiting patients to test the safety and efficacy of an anti-HLA-DR antibody called IMMU-114 in B-cell disorders (NCT01728207). This drug is a humanized IgG4 form of murine anti–human HLA-DR mAb, L243, which recognizes a conformational epitope in the alpha chain of HLA-DR. Due to safety concerns related to the expression of HLA-DR in non-tumor cells, IMMU-114 was specifically generated to kill tumor cells avoiding CDC or ADCC (198). IMMU-114 binding to tumor B-cells results in antiproliferative effects and apoptosis and has demonstrated efficacy in preclinical models (198). Although the exact mechanism has yet to be fully elucidated, it appears to induce hyperactivation of ERK- and JNK-dependent mitogen activated protein kinase signaling pathways that may lead to mitochondrial membrane depolarization and reactive oxygen species generation. This eventually leads to an induction of tumor cell apoptosis and a reduction in tumor cell proliferation (198).
Antibodies Targeting Chemokine Receptors
Chemokines are small chemotactic cytokines that bind to specific surface seven transmembrane domain G protein-coupled receptors, or CKRs. Upon binding of their ligands, CKRs promote cell survival, proliferation, and adhesion, contributing to mammalian development and organogenesis, thus playing a central role in homeostasis and the maintenance of innate and acquired immunity (199). In cancer, CKRs may associate with tumor cells facilitating their survival, proliferation, and metastasis (200, 201). Moreover, they may also promote an immunotolerant milieu by recruiting Treg, tumor-associated macrophages (TAMs) or MDSCs that opens the way to tumor growth, angiogenesis, and immune evasion (202–205). For all these reasons, tumor-associated CKRs are considered suitable targets for cancer therapy (206). Nevertheless, generating antibodies against these Ags is particularly challenging due to, among other reasons, a complex and unstable native conformation (206, 207). So far, few anti-CKRs are under study in preclinical or early clinical phases and only one has been approved for clinical use (208).
C-C-Motif-Chemokine Receptor 4 (CCR4)
Under homeostasis, this receptor and its ligands, the chemokines CCL17 and CCL22, predominantly contribute to the biology of Th2, Th17, Treg, and skin-homing T-cells positive for cutaneous lymphocyte antigen (CLA) (209–211). In addition, CCR4 has been implicated in the pathogenesis of inflammatory diseases and cancer, being overexpressed in several T-cell disorders including adult T-cell leukemia–lymphoma (ATL), peripheral T-cell lymphoma (PTCL), and cutaneous T-cell lymphoma (CTCL) (212–214).
In cancer therapy, mogamulizumab (KW-0761) is the first approved and clinically tested antibody against a CKR and, in addition, the first glycoengineered antibody to be marketed. This IgG1 antibody is directed to the N-terminal region of human CCR4. Despite this, it does not block the interaction between CCR4 and its ligands, thereby not interfering with CCR4-mediated protumor functions or migration (215, 216). Moreover, it does not bind complement molecules either. Nevertheless, its Fc was selectively defucosylated to reach a potent ADCC via high-affinity binding to the FcγRIIIa on effector cells (215, 217). As a result, phase I and II clinical trials investigating mogamulizumab in T-cell malignancies demonstrated its effectiveness and led to the approval for use in Japan for rrATL in 2012 and rrCTCL in 2014 (208, 218). Given the safety and efficacy of mogamulizumab, different clinical studies are underway for T-cell disorders (208, 219). In addition, based on preclinical evidence, studies are being conducted to establish whether other diseases could be targeted by mogamulizumab therapy, including certain NK-cell lymphoproliferative disorders (220) and Hodgkin’s lymphoma (HL) (221). Interestingly, in HL, the majority of the cells in TME are TAMs, Tregs, and CD4+ Th2 cells recruited by chemokines secreted by tumor cells such as CCL17 (222). This infiltrate probably enables tumors to escape from immune surveillance. Therefore, it is conceivable that targeting CCR4-positive cells in HL niche might revert this immunosuppresive environment enhancing the antitumor immunity. Indeed, in CTCL patients, a single dose of mogamulizumab decreased the fraction of CCR4-positive malignant T-cells, with a concomitant reduction of CCR4+ Tregs (223). Notably, similar effects on Treg subsets were observed in melanoma patients (224). All together, these results prompted phase I–II clinical studies in solid tumors not expressing CCR4 in order to evaluate the potential of mogamulizumab as immunomodulatory drug. Finally, the lack of neutralization of CCR4-ligands interaction by mogamulizumab leaves room to novel mAbs able to target this interaction. In preclinical phases, mAb1567 and its high-affinity variant (mAb2-3) were able to abolish CCR4-mediated chemotaxis of malignant cells and Tregs (225, 226). Moreover, in vitro studies confirmed that both antibodies mediated CDC and ADCC (225, 227), whereas the derivative mAb2-3 affected Treg functions and survival by means of CD25 shedding (226).
C-X-C-Motif-Chemokine Receptor 4 (CXCR4)
This CKR and its chemokine CXCL12 (or stromal cell-derived factor 1α) regulate hematopoietic development, lymphoid tissue architecture, and hematopoietic cell trafficking. Additionally, this couple controls organogenesis and development in several tissues (228, 229), hence CXCR4 is not surprisingly overexpressed in more than 23 different human cancers and has been demonstrated to be particularly relevant in B-cell malignancies like B-ALL, CLL, or MM (230, 231), T-cell malignancies such as T-ALL (232), and myeloid malignancies like AML (233). In these conditions, CXCR4 causes tumor cell trafficking and homing into lymphoid and non-lymphoid tissues where CXCL12 is produced. Here, the couple CXCR4/CXCL12 keeps leukemic cells in close contact with stromal cells and extracellular matrix that together provide growth-promoting and antiapoptotic signals which facilitate resistance to chemotherapy and disease relapse (234–239). All together, these data strongly indicate that therapeutic strategies targeting the CXCL12–CXCR4 axis represent an attractive investigative approach to disrupt the leukemia–stromal interaction.
The first anti-CXCR4 clinically tested was ulocuplumab (BMS-936564), an IgG4 that blocks CXCL12 binding to its receptor thereby inhibiting CXCL12-induced migration and calcium flux (240). In this context, ulocuplumab is comparable to AMD3100 (Plerixafor-Mozobil), a small molecule CXCR4 inhibitor. However, ulocuplumab, but not AMD3100, induces caspase-independent apoptosis on a panel of cell lines and primary samples from AML, CLL, and MM patients (240–242). Both mechanisms contribute to the efficacy of ulocuplumab as monotherapy observed in xenograft models of the aforementioned diseases (240). The first clinical report on ulocuplumab suggests safe and significant antileukemia activity in AML patients, achieving fairly respectable complete remissions (CR/CRi) of 51%, and, notably in four patients, CR/CRi was documented after a single dose of ulocuplumab monotherapy (NCT01120457) (243). Results on other conditions are not available yet. Another IgG4 targeting CXCR4 is LY2624587, a humanized antibody deeply modified to eliminate half-antibody exchange associated with human IgG4 isotypes (244). Similar to ulocuplumab, LY2624587 inhibits CXCL12 binding to CXCR4 thus abrogating CXCR4-mediated survival and migration. The first clinical trial of LY2624587 (NCT01139788) was completed on 2011; however, results have not yet been published. Besides IgG4 isotypes, novel anti-CXCR4 antibodies with IgG1 isotype are demonstrating to be effective in preclinical phases. This is the case of hz515H7 (245) or PF-06747143. The latter was the first anti-CXCR4 mAb with an IgG1 scaffold to be evaluated in humans (NCT02954653), specifically in AML patients (246, 247). Like IgG4 formats, IgG1 antibodies are antagonist that block tumor cell chemotaxis toward CXCL12 and induce tumor cell apoptosis in either presence or absence of stromal cells (245, 246). In contrast, IgG1 isotypes trigger potent ADCC and CDC, which are involved in the antitumor effect observed in AML and CLL models as monotherapy or in combination with standard therapy (245–247). Currently, there is evidence suggesting that anti-CXCR4-IgG4 antibodies are generally safe although they induce short-term toxicity affecting the process of normal hematopoiesis with the result of myelosupression, or a deleterious effect on immune cells where CXCR4 is widely expressed (243). In addition, the off-target adverse event of hyperleukocytosis was reported in a number of patients. Finally, owing to the ubiquitous expression of CXCR4, long-term effects should be carefully evaluated, even more with the upcoming IgG1 molecules as they may trigger off-target ADCC or CDC.
CCR2 and Others
CCR2 is another CKR targeted by an antibody under clinical development. Plozalizumab (MLN-1202) is a neutralizing antibody that showed a positive effect in phase II for the treatment of bone metastases (NCT01015560) (206). Interestingly, recent preclinical evidence suggests that targeting CCR2 may be effective in the setting of AML (248) and MM (249). MM cells from patients with bone lesions overexpress CCR2, while osteoclasts secrete chemokines that act as growth factors for tumor cells. In this scenario, targeting CCR2 could reduce MM cells survival and prevent drug resistance similar to CXCR4 antagonism (249). Many other CKRs with pathogenic role in hematological malignancies were preclinically validated as good targets for mAb-based therapy. This includes antibodies against CCR7 (34, 250) and CCR9 (251).
Recent evidence on a CXCL12-neutralizing RNA oligonucleotide reveals that targeting the chemokine instead of the CKR may interfere with CXCR4-mediated drug resistance in CLL and MM (252). These data support a rationale for clinical development of mAbs targeting chemokines instead of their corresponding receptors. However, to date no mAb targeting chemokines has been included in a clinical trial for cancer therapy. There are two plausible explanations. First, targeting chemokines does not activate the host immune response against tumor cells. Second, a cell surface-restricted receptor is more efficiently targeted than delocalized secreted chemokines (206, 207). Moreover, a recent study in cynomolgus monkeys demonstrated that targeting the chemokine CCL21 with a novel mAb (QBP359) requires impractical large doses and frequent administration to maintain suppression of CCL21 in the clinical setting. In other words, it is difficult to target soluble proteins with high synthesis rates, a common characteristic to many chemokines (253).
Antibodies Targeting the Tumor Niche
Malignant cells are surrounded by different types of leukocytes and stromal cells that compose an extremely relevant source of soluble factors and adhesion molecules that promote tumor progression and escape from conventional treatments (254, 255). We have already referred in a separate section how clinical antibodies neutralize CKRs and their protumor activities in the TME. Below we review other approaches to disrupt the tumor niche, including: (i) neutralizing soluble survival/growth factors (mainly cytokines) or their associated receptors, all of them validated and valuable targets of antibody-based therapies of immunological disorders (256); (ii) blocking adhesion molecules that lodge tumor cells to their protective niche (257, 258); and (iii) blocking angiogenesis, an important process during development and vascular remodeling (259) that feeds tumor growth and progression (259, 260).
Soluble Factors and Associated Receptors
B-Cell Activating Factor (BAFF) and A Proliferation Inducing Ligand (APRIL)
These TNF-family members are produced as type II transmembrane proteins that are then proteolytically cleaved and released in soluble form (261). BAFF and APRIL are produced by a variety of hematopoietic and non hematopoietic cells including stromal microvascular endothelial cells and osteoclasts. Both factors share two receptors: transmembrane activator and cyclophilin ligand interactor (TACI) and B-cell maturation antigen (BCMA; CD269). Additionally, BAFF binds strongly to BAFF receptor (BAFF-R) (261). These receptors have distinct expression patterns and mediate separate functions. BAFF-R is absent on B-cell precursors but is gained on immature B-cells upon acquiring a functional BCR, which is critical for survival and maturation of immature B-cells. TACI is expressed on memory B-cells and is necessary for T-independent responses and promotion of class switch recombination in B-cells. Last, BCMA expression is restricted to plasmablasts and plasma cells and promotes their long-lived survival (261–263).
BAFF and APRIL are particularly relevant in MM, where BCMA and both soluble factors are augmented in samples from patients compared to healthy donors, and ligand–receptor interactions lead to increased survival of malignant cells (264–267). Moreover, higher concentrations of APRIL may promote resistance to lenalidomide, bortezomib and other standard-of-care drugs, and also may drive expression of programmed cell death ligand 1 PD-L1, interleukin (IL)-10, and TGFβ on BCMA+ tumor cells creating an immunosuppressive niche that favors tumor cells (266). In recent years, compelling evidence has suggested that neutralizing APRIL or BAFF could diminish MM cell survival, revert the immunosuppressive phenotype on BCMA+ cells and reduce resistance of malignant cells to treatment, regardless of the presence of protective stromal cells (266, 268–270). Tabalumab (LY2127399), a human IgG4 mAb that neutralizes membrane-bound and soluble BAFF, was entered in clinical trials after demonstrating both antitumor activity and osteoclastogenesis inhibition in xenograft models of MM (270). Results in two different studies showed limited efficacy of tabalumab in combination with standard-of-care drugs (NCT00689507, NCT01602224) (271, 272). In the near future, new molecules will burst into the field such as BION-1301, an anti-APRIL neutralizing mAb able to fully suppress in vitro APRIL-induced B-cell IgA and IgG class switching (273).
Other antibodies block BAFF-R, including VAY736 and belimumab, or are aimed to deliver payloads to tumor cells expressing BCMA as exemplified by two novel inmmunoconjugates in clinical studies: AMG 224 and GSK2857916 (Table 6). Notably, GSK2857916 acts through multiple mechanisms. It specifically blocks cell growth via G2/M arrest, induces caspase 3-dependent apoptosis in MM cells, and strongly inhibits colony formation by MM cells. Furthermore, GSK2857916 recruits macrophages and mediates ADCP of MM cells (274). Finally, BI836909 and JNJ-6400795 are the first MM cell-specific BiTEs in development, and both target BCMA/CD3 (275). Clinical studies to evaluate safety and efficacy of both BiTEs are still recruiting MM patients (NCT02514239 and NCT03145181).
Receptor Activator of Nuclear Factor Kappa-B Ligand (RANKL)
This soluble member of the TNF family is a key mediator in the pathogenesis of a broad range of skeletal diseases since it binds to RANK on preosteoclasts and mature osteoclasts which are involved in bone resorption (276). In particular, malignant plasma cells produce RANKL leading to an imbalance between bone formation and resorption, local bone lysis and the development of osteolytic lesions in MM patients (277, 278). Denosumab is a human IgG2 antibody that binds to the soluble and cell membrane-bound forms of RANKL thus preventing RANK-mediated differentiation, activation, and survival of osteoclasts. As a consequence, bone resorption and bone destruction are reduced (279). As expected, and based on its MOA, denosumab is not effective reducing tumor burden neither improving responses in MM nor B-NHL. But denosumab does inhibit RANKL regardless of previous exposure to bisphosphonates, standard-of-care drug in bone lessions (280) and delays the time to the first on-study skeletal-related event with similar results to zoledronic acid, another standard-of-care treatment (NCT00330759) (281). Similar to IgG1, denosumab is a big molecule that, contrary to bisphosphonates, is not cleared by the kidneys. Therefore, current investigation (NCT02833610) is evaluating whether denosumab could fill the unmet need for bone-targeted therapies in MM patients with renal insufficiency (approximately 25–50% of all patients) (282). Finally, denosumab has demonstrated efficacy at solving bisphosphonate-refractory hypercalcemia in hematological cancers (NCT00896454) (283). Despite the current US FDA-approved label for denosumab it does not include MM nor NHL, this situation may be reverted depending on the forthcoming results from these studies.
Interleukin-6
IL-6 is a pleiotropic cytokine with a critical role in the pathogenesis of MM and B-NHL by promoting tumor cell growth and interfering with chemotherapy drugs (284). Different mAbs against IL-6 or its soluble receptor IL-6R have been developed, with the two most promising being the chimeric siltuximab (CNTO 328) that neutralizes the cytokine, and tocilizumab that blocks the receptor (285). Siltuximab, was recently registered for multicentric Castleman’s disease and evaluated as a single agent or in combination with other agents in advanced MM and B-NHL (particularly CLL). Again, mAb-based strategies targeting soluble factors produce discouraging results in hematological malignancies. In addition, results are modest probably due to the complex interaction between malignant clones, inflammatory background and host response (NCT00412321, NCT00911859, NCT00401843) (286, 287). However, new investigations aim to uncover the application of siltuximab in the treatment of Waldenström macroglobulinemia and the early phase of smoldering MM (285). Moreover, one trial is exploring the utility of blocking the IL-6R in CLL (NCT02336048).
IL-3 Receptor Alpha Chain (IL-3Rα; CD123)
Interleukin-3 stimulates cell cycle progression in early hematopoietic progenitors and enhances the differentiation of various hematopoietic cells while inhibiting apoptosis (288). IL-3Rα is a novel molecular target that has emerged as a highly specific entity for CML, AML blasts, and LSCs (289–291). Notably, normal hematopoietic stem cells have limited expression of CD123 (289, 292). One of the first humanized anti-IL-3Rα antibodies tested in clinical trials was CSL360, a chimeric IgG1 molecule that achieved an improvement in blasts and LSCs percentage in bone marrow, but no clinical responses in high-risk rrAML (NCT00401739) (293). These results showed that the blockade of IL-3Rα alone was ineffective, leading to the development of second-generation molecules able to kill IL-3Rα-positive tumor cells by means of immune effector mechanisms. The one in most advanced stages is talacotuzumab (JNJ-56022473/CSL362), an Fc-engineered derivative from CSL360 which is undergoing phase II–III studies for rrAML (NCT02992860, NCT02472145). Talacotuzumab induces potent in vitro ADCC against IL-3Rα-expressing AML blasts/LSC and reduces leukemic cell growth in murine xenograft models of human AML (294). In addition, talacotuzumab inhibits IL-3–stimulated rescue of tyrosine kinases inhibitors (TKIs)-induced cell death, demonstrating that resistance to previous standard-of-care could be reverted (295). Actually, the combination of TKI therapy and talacotuzumab may eliminate leukemic cells in vivo more effectively than TKI treatment alone (296). Another second-generation mAb with similar MOA is XmAb14045 that will be tested early in a phase I study (NCT02730312). With different MOA, KHK2823 is a novel non-fucosylated fully human IgG1 mAb which mediates ADCC without inducing CDC. Its safety and efficacy is under evaluation in a phase I study (NCT02181699).
Other approaches targeting CD123 are based on bi-specific platforms. Examples are JNJ-63709178 (NCT02715011), a humanized CD123xCD3 DuoBody and flotetuzumab (NCT02152956), a CD123xCD3 bi-specific antibody-based molecular construct named dual affinity retargeting (DART) molecule. Both bispecific antibodies are effective in vitro and in vivo in preclinical settings (297). Nevertheless, recent evidence on human CD123-redirected T-cells (CAR-T123) shows some concerns regarding toxicity related to off-target events (298), suggesting the possibility that the same effects could be found with the redirection of T-cells by bi-specific antibodies. Finally, owing to CD123 internalization upon mAb binding, novel immunoconjugates aim to target CD123 (Table 6).
IL-2 Receptor Alpha Chain (IL-2Rα; CD25)
Commonly expressed by activated T- and B-cells, some thymocytes and myeloid precursors, IL2-Rα is also found in most of the malignacies corresponding to such lineages, particularly in ATL where IL2-Rα functions as the receptor for human T-cell leukemia virus 1 (HTLV-1) (299). Few mAbs have been developed for T-cell neoplasia. One of them, the chimeric IgG1 basiliximab selectively blocks IL-2Rα, thereby preventing IL-2-mediated activation of lymphocytes. Nevertheless, it lacked of activity in patients (299). Another anti-IL-2Rα is daclizumab, a humanized antibody which shows potential in T-cell disorders and HL, although its activity needs to be confirmed in a big cohort of patients (300, 301). A likely explanation for the modest results of anti-IL-2R therapy is related to the pharmacokinetics/pharmacodynamics of daclizumab. Indeed, a phase I–II study suggested that higher doses than previously used may be required to achieve clinical responses since high doses were needed to saturate targets in extravascular sites (301). Low activity in phase I–II was also documented for LMB-2, an immunotoxin comprised of the Fv of an anti-CD25 mAb connected to an exotoxin (302). The limited efficacy of naked or toxin-conjugated antibodies has led to the conjugation of basiliximab and daclizumab with radionuclides. Currently, both molecules are under evaluation. Interestingly, in HL daclizumab linked to radionuclides shows efficacy in patients with tumor cells expressing IL-2R, and in patients whose tumor cells lacking the receptor, suggesting off-target effects on accessory cells (303). Based on the same rationale, both basiliximab and daclizumab are being explored as adjuvant therapy to eliminate IL-2-Rα-positive Tregs in MM or to eliminate IL-2-Rα-positive naive T-cells to prevent the development of graft-versus host disease.
Type I Insulin-Like Growth Factor Receptor (IGF-1R; CD221)
This ubiquitously expressed tetramer binds insulin growth factor 1 (IGF-1) to activate multiple signaling pathways involved in cell growth, differentiation, migration, and cell survival (304). IGF-1R also mediates anchorage-independent growth and survival, and migration, thus facilitating tumor establishment and progression (305–307). IGF-1R has been widely studied in hematological tumors where a pathogenic role has been found, among others, for myeloid leukemias (308), and several B-cell malignancies (304, 309) but an exceptional role has been uncovered for MM (310). Therefore, the therapeutic potential of IGF-1R has been explored in MM with three different mAbs that directly block IGF-1R: dalotuzumab, AVE1642, and figitumumab. All of them prevent the binding of IGF-1 and the subsequent activation of PI3K/AKT signal transduction, nonetheless results derived from clinical studies were disappointing (310). In phase I studies, only dalotuzumab showed an evaluable antimyeloma activity (NCT00701103) (311). In contrast, AVE1642 (NCT01233895) and figitumumab (NCT01536145) did not result in significant improvement as single agents or in combination with standard-of-care drugs (312, 313). Hence, no further evaluation of these mAbs in MM patients is currently ongoing and the development of dalotuzumab in MM was consequently also discontinued. These discouraging results could be explained by the emergence of tumor cell independence from their microenvironment due to intraclonal heterogeneity, the involvement of other growth factors and the existence of hybrid receptors composed of IGF-1R and 2R that can be activated by all IGF ligands (314). Anti-IGF-1R mAbs are unable to neutralize these hybrid receptors. Moreover, it is thought that circulating IGF-1R can interact with the IGF-1R targeting antibodies and prevent their interaction with the IGF-1R on cancer cells (310).
Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF; CSF2)
This cytokine is a monomeric glycoprotein secreted by immune cells, endothelial cells and fibroblasts. Overall, GM-CSF participates in the development of innate immune cells since it stimulates stem cells to produce granulocytes and monocytes (315). GM-CSF is also involved in the pathogenesis of chronic myelomonocytic leukemia (CMML), where progenitor expansion through STAT5 signaling is mediated (316). Based on this, lenzilumab (KB003), a humanized antibody formerly developed for inflammation, is being investigated as single agent in CMML (NCT02546284) (317). In primary samples from CMML patients, lentizumab bound the cytokine and directly interrupted binding to its cognate receptor inducing apoptosis in vitro (318). However, the curative application in humans is uncertain as some CD34-positive subsets, including the LSCs, seem to be insensitive to GM-CSF signaling (318). Results from the ongoing clinical study will shed some light on the subject.
Hepatocyte Growth Factor (HGF)
This soluble factor is the only known ligand for the c-Met receptor tyrosine kinase that when bound to HGF activates key oncogenic signaling pathways that increase cell proliferation, survival, migration and invasion in several human cancers, including MM where HGF expression predicts poor prognosis (319). Ficlatuzumab (AV-299) is a potent HGF-neutralizing mAb able to interrupt HGF/c-Met interaction thus inhibiting c-Met-induced phosphorylation, cell proliferation, cell invasion and cell migration. With proven efficacy in solid tumors (320), a phase I study aimed to examine its efficacy in MM and NHL (NCT00725634), although preliminary results indicate that clinical effects are only seen in MM (321).
Adhesion Molecules
CD44
This cell-surface glycoprotein is a receptor for hyaluronic acid (HA), osteopontin, collagen, and matrix metalloproteases, which are typically found in the microenvironment of BM and lymphoid tissues (257). CD44 is particularly expressed by AML-LSCs and CLL cells which take advantage of HA-CD44 signaling to promote leukemic survival via PI3K/AKT and MAPK/ERK pathways (257, 322). Since AML-LSCs are more dependent on CD44 for their anchoring in the BM niche than their normal counterparts, CD44 is an exciting target to mobilize leukemic cells out of their protective niche (257). A novel humanized neutralizing mAb, RG7356 (RO5429083), has recently been evaluated alone or in combination with cytarabine in a phase I trial in rrAML patients (NCT01641250). RG7356 does not activate effector cells or complement. Very limited activity was observed in this study although the mAb was well tolerated (323). The ability of CD44 to complex with different partners, overcoming the neutralization mediated by the antibody, may explain this outcome. In CLL, the expression of CD44, in cooperation with VLA-4 and MMP9, helps in creating a protective TME within the lymphoid organs that circumvents spontaneous and drug-induced apoptosis in CLL cells (324). In preclinical models of CLL, RG7356 provoked apoptosis of CLL cells in a caspase-dependent manner and regardless of the presence of protective co-cultured stromal cells or HA, or even regardless of BCR signaling (325). These results indicate that RG7356 might have therapeutic activity in CLL patients.
Very Late Antigen 4 (VLA4; CD49d)
This molecule is the α-chain of the α4β1 integrin heterodimer which is normally expressed on monocytes and lymphocytes cell surface (326, 327). VLA-4 is involved in the firm adhesion step during the extravasation process, mediating the binding to fibronectin or to vascular cell adhesion molecule-1 (VCAM-1) located on the surface of endothelial cells (326, 327). In several hematological malignancies CD49d is considered as one of the main players at the TME as it mediates both cell–cell and cell–matrix interactions delivering prosurvival signals and protecting tumor cells from drug-induced damage (258, 328–330). Despite this, no anti-VLA4 antibodies are under development for blood cancers. In this context, the recombinant IgG4 anti-CD49d antibody natalizumab, which is an FDA-approved drug for relapsing multiple sclerosis, has demonstrated the potential to revert chemo-sensitivity and to inhibit both in vitro and in vivo adhesion of MM cells to non-cellular and cellular components of the TME. It also has the potential to arrest tumor growth in a xenograft model of MM (329). Unfortunately, a trial evaluating natalizumab as a single agent in MM patients was terminated due to low enrollment (NCT00675428). Natalizumab is also able to restore drug sensitivity in B-cell lymphomas (328) and primary ALL (330) providing the rationale for the clinical evaluation of natalizumab in many hematological tumors, preferably in combination with novel agents to enhance tumor cell cytotoxicity and improve patient outcome (329). Nevertheless, it should be taken into account that natalizumab treatment is associated with appearance of progressive multifocal leukoencephalopathy which may, in turn, dissuade this approach in hematological malignancies (331).
Angiogenesis
Vascular and Endothelial Growth Factor (VEGF)
This molecule is one of the most important mediators of neo-angiogenesis and tumor growth (332). Out of the five members of the VEGF family described to date, VEGF-A and its receptor VEGFR-2 are the main targets of current antiangiogenic agents (332). Bevacizumab is an IgG1 antibody that binds to all isoforms of VEGF-A preventing the interaction with its receptors and their subsequent activation (333). In solid tumor, this antibody promotes a regression of immature tumor vasculature, normalization of remaining tumor vasculature and inhibition of further tumor angiogenesis (334). In hematological malignancies, bevacizumab has been tested as a tool to solve resistances to previous chemotherapies. In myeloid malignancies, bevacizumab has not worked as monotherapy or combined with standard therapies (335). In CLL, preclinical studies demonstrated that bevacizumab was a proapoptotic and antiangiogenic drug (336, 337). Despite having no significant clinical activity as monotherapy (NCT00290810) (338), in combination with chemotherapy regimens the results had a better outcome (339). In FL, a phase II study (NCT00193492) revealed that a combination of rituximab with bevacizumab significantly extended progression free survival, although bevacizumab increased the toxicity as well (340). Encouraged by these works, bevacizumab has been used as adjuvant therapy in many other B-cell disorders, where addition of the anti-VEGF did not show improvement of the therapeutic responses (341, 342). It is tempting to assume that anti-VEGF in combination with other chemoimmunotherapies is a promising therapy for CLL and FL patients, but a close follow-up is recommended to ascertain the potential toxicities, including left ventricular dysfunction and heart failure, observed in many of the cited studies.
Endosialin (CD248; TEM1)
This glycoprotein is selectively expressed in vascular endothelial cells of malignant tumors (343). Targeting endosialin showed antitumor activity in different preclinical models where endosialin function was suppressed with the antiangiogenic antibody MORAB-004 (ontuxizumab) (343). Although the MOA of this drug is not completely understood, it was suggested that endosialin is removed from the cell surface by means of MORAB-004–mediated internalization. MORAB-004 could affect cellular signaling as well as protein–protein interactions that serve to communicate signals in the TME between tumor and stromal cells. Additional work is under way to further establish the exact mechanism of action of MORAB-004 (343). MORAB-004 antitumor activity has been observed in several phase I studies on solid tumors and phase II studies have recently started. Despite the role of endosialin in blood cancers is not fully understood, patients with different blood disorders have been enrolled in a phase I study (NCT01748721), although no results have been published (344).
Antibodies Targeting Immune Checkpoints
Antitumor innate and adaptive immune responses rely on cell-activation and cell-exhaustion balances which are regulated by stimulatory or inhibitory molecules most belonging to the B7-CD28 and the TNF-TNFR superfamilies (345). Due to their function modulating immune responses, these NLSAs are commonly known as “immune checkpoints.” Although checkpoint targeting with specific antibodies is a relatively new area (346), their accessibility to cell membranes and significance in regulating immune responses made them a very attractive therapy option, as exemplified by the plethora of novel agents that have been already approved or are under intensive studies in solid tumors. In Table 4, an account of the landscape of immune-checkpoint regulators in hematological malignancies is provided. Commonly, immune-checkpoints do not target tumor cells directly, but instead act on lymphocytes to boost their endogenous antitumor activity reversing tumor immune escape (347, 348). It is not our intention to review these types of molecules. Instead, in the next section, we will analyze a second-generation of immunomodulatory antibodies targeting receptors expressed in both tumor and immune cells. These antibodies are armed with a dual MOA combining direct tumoricidal properties with the ability to restore host antitumor immunity.
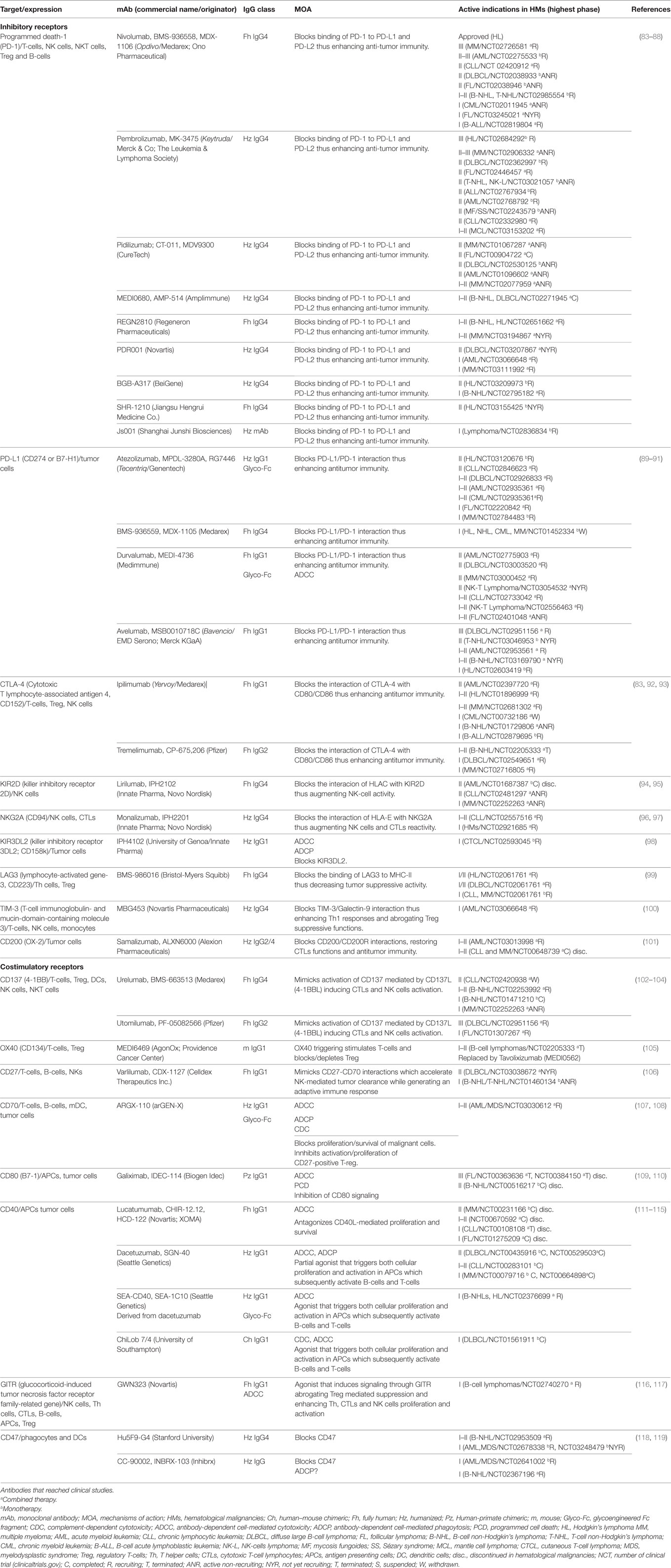
Table 4. Characteristics of antibodies directed to immune checkpoint receptors.
PD-L1 (B7-H1; CD274)
Programmed death-1 (PD-1) is an inhibitory receptor member of the B7 receptor family with a significant role in immune regulation (349). PD-1 is upregulated on activated T-cells, NK cells, NKT cells, and B-cells among other leukocytes (349–351). In many types of cancers, PD-1 engagement may represent one means by which tumors evade immune surveillance and clearance (349). Cancer cells express PD-L1, a PD-1 ligand that upon binding to PD-1 on tumor-infiltrating lymphocytes (TILs) leads to impairment of antitumor responses through multiple mechanisms including inhibition of T-cell activation and proliferation (352, 353) and increase in T-cell apoptosis (349, 354). In addition, PD-L1 drives the differentiation of naive CD4+ T-cells into induced Tregs, which also express PD-1 on their cell surface, and maintain their suppressive function (355). The end result of the PD1 axis activation is T-cell exhaustion or anergy, dampening effector T-cell functions and leading to immune tolerance.
This situation prompted the development of different mAbs to target either the receptor or the ligand with the goal of “releasing the brakes” on effector T-cells preventing suppression of the antitumor response and causing tumor cytotoxicity. Similar to antibodies targeting PD-1 (nivolumab and pembrolizumab are the most widely marketed anti-PD-1 antibodies), anti-PD-L1 antagonists aim to restore effector T-cell and NKs activities while abrogating intra-tumoral Treg-mediated suppression (83). In addition, some anti-PD-L1 are able to mediate ADCC and other Fc-mediated functions. Four PD-L1 mAbs have demonstrated clinical activity in several solid tumors including atezolizumab, durvalumab, avelumab, and MDX-1105 (BMS-936559) (356). Activity of the IgG4 MDX-1105 and the low-ADCC inducer, Fc-engineered, humanized, IgG1 atezolizumab rely on blocking PD-L1. In contrast, durvalumab and avelumab combine two MOA: blocking PD-L1/PD-1 interactions, and directly killing PD-L1-positive tumor cells (356). Clinical studies involving both molecules are recruiting patients or just initiating in different hematological malignancies. Some of the diseases that could be targeted by anti-PD-L1 double MOA are: HL, B-ALL, FL, or MM (357–359). Nonetheless, anti-PD1 targeting in MM had low efficacy and, most notably, established MM therapies such as IMiDs are able to reduce PD-L1 on MM cells and could interfere with the outcome (360).
CD40
This glycoprotein is a member of the TNFR superfamily that is principally expressed on APCs, but also on several tumors, such as B-cell lymphomas and carcinomas (111). Through the binding to its ligand CD40L (or CD154) on CD4+ T-cells, CD40 plays a key role in a broad variety of immune and inflammatory responses, including T-cell-dependent immunoglobulin class switching, memory B-cell development, germinal center formation, functional maturation of DC, and upregulation of macrophage cytotoxic function. To date, different anti-CD40 mAbs have been developed including three agonistic and one antagonistic which are being investigated in a range of lymphoid and solid tumors (111).
Lucatumumab (HCD122/CHIR12.12) is a fully human antibody that antagonizes CD40L-mediated proliferation and survival on CLL and MM cells, and triggers ADCC (111, 112, 361). Lucatumumab has overall modest activity as single agent or in combined regimens in multiple clinical studies on B-cell tumors, including HL (NCT00283101) (362), CLL (NCT00108108) (363), and MM (NCT00231166) (364). Similar to lucatumumab, the humanized IgG1 dacetuzumab (SGN-40) has tumoricidial activities in cultured NHL cells through ADCC, ADCP and direct apoptosis via caspase-3 activation. In contrast to lucatumumab, dacetuzumab does not prevent CD40/CD40L interaction, and behaves as a partial agonist by augmenting effector CTL responses (365, 366). The efficacy and safety of dacetuzumab as a single agent to treat rrMM, rrNHL, or rrCLL was investigated in three phase I studies, respectively (NCT 00079716, NCT00103779, NCT00283101), which demonstrated mild side effects but modest efficacy across the cancers tested (367–369). Nevertheless, combining dacetuzumab with chemotherapy and/or rituximab demonstrated synergistic activities in both preclinical and phase I clinical studies in rrDLBCL (NCT00529503, NCT00655837) (370–372).
Despite the limited activity of anti-CD40 antagonists, results with the partial agonist dacetuzumab and compelling evidence in preclinical models confirmed that CD40 agonists acting as CD40L could be a better venue to drive stronger antitumor responses (113, 373). Currently, two types of agonist anti-CD40 are available. The first type combines the activation of tumor-specific immune responses with a direct tumoricidal activity. In this group, we can include dacetuzumab along with the chimeric ChiLob7/4 and the human sugar-engineered SEA-CD40 antibodies (113–115, 365, 366). Upon binding to CD40, these drugs trigger both cellular proliferation and activation of APCs which activate innate and adaptive antitumor immunity (113, 373). In addition, these antibodies also directly kill CD40-expressing cancer cells through ADCC, and eventually inhibit proliferation and growth of CD40-expressing tumor cells. ChiLob7/4 has completed a phase I study in B-NHL showing a well-tolerated range of doses whereas SEA-CD40 is enrolling patients, at the time of writing, in a first phase I study in combination with pembrolizumab in solid tumors, B-NHLs and HL (NCT02376699).
A second type of CD40-directed antibodies triggers antitumor immune responses as sole MOA. Molecules such as APX005M, ADC-1013 or the IgG2 mAb CP-870,893 do not include FcR engagement as MOA and are being examined in solid tumors alone or in combination with immune checkpoint inhibitors (NCT03123783; NCT02482168; NCT02379741). Positive results may lead to the investigation to hematological malignancies. In both types, it is expected that antitumor efficacy highly depends on the CD40 status of the tumor infiltrate, mainly tumor-specific CTLs and possibly TAMs. Accordingly, the direct tumoricidal effects depend highly on the CD40 expression of the tumor.
Killer Cell Immunoglobulin-Like Receptor 3DL2 (KIR3DL2; CD158k)
This transmembrane glycoprotein belongs to the family of cell inhibitory receptors expressed by NK cells and subsets of CD8+ T-cells but not by most normal CD4+ T-cells. In contrast, KIR3DL2 expression is found in several CD4+ T malignancies, including SS, mycosis fungoides (MF) and anaplastic large cell lymphoma (ALCL) (98, 374). This receptor plays a dual role in the pathogenesis of these cancers: it acts as an inhibitory coreceptor which down-modulates CD3-dependent signaling events, and prevents programmed cell death. IPH-4102 is a humanized IgG1 antibody against KIR3DL2 whose potent antitumor properties ex vivo against SS and CTCL primary cells, and in vivo against KIR3DL2-positive tumor cells are achieved mainly through ADCC and ADCP. Only a minor contribution was attributed to the neutralization of the inhibitory receptor (98, 374). IPH-4102 advanced to phase I in 2015 (NCT02593045).
CD70
This member of the TNF family is a receptor transiently expressed on activated T- and B-cells and on mature DC (375). Its ligand is CD27, another costimulatory receptor found on the surface of T-cells, B-cell, and NKs (376). The interaction of both molecules accelerates NK-mediated tumor clearance while generating an adaptive immune response (377). For this reason these NSLAs are being investigated in oncoimmunology. Whereas anti-CD27 antibodies, such as Varilumab (CDX-1127), just boost innate and adaptive antitumor responses, anti-CD70 antibodies also target tumor cells inducing direct cell killing. CD70 is expressed in several hematological malignancies that activate NF-κB pathways leading to proliferation and survival of malignant cells (378–380). In addition, CD70 seems to be involved in the recruitment of CD27-positive Tregs to the TME thus allowing tumor evasion (381). Despite most anti-CD70 mAbs under development are immunoconjugates (Table 6), one classical antibody is under clinical evaluation. ARGX-110 is a defucosylated IgG1 mAb with several different MOA (107). By neutralizing CD70–CD27 interactions it deprives cell growth signaling in tumor cells while inhibiting the activation and proliferation of CD27-positive Tregs. In addition, ARGX-110 displays enhanced ADCC and ADCP while preserving a strong CDC (107). The first phase I study (NCT02759250) in patients with advanced solid tumors expressing CD70 provided evidence of good tolerability of ARGX-110 and antitumor activity at all dose levels (108). Currently, a phase I–II study is recruiting patients to evaluate ARGX-110 efficacy in AML (NCT03030612).
CD47 (Integrin-Associated Protein)
Phagocytosis is a complex process needed for programmed removal of apoptotic as well as IgG- or complement-opsonized cells that can be inhibited by the binding of the ubiquitous negative regulatory Ig receptor CD47 to the signal regulatory protein alpha (SIRPα), expressed on phagocytes and DCs (382, 383). CD47 was found to be universally expressed in human cancers where helps to prevent phagocytic elimination of tumor cells (118, 119). Notably, CD47 expression is preferentially found in AML-LSCs (384, 385) and negatively correlates with clinical outcome in AML, ALL, NHL, and MM (118, 119). The hypothesis that blocking CD47-SIRPα interactions would restore phagocytosis of tumor cells has been widely validated in primary human xenograft models treated with commercial and clinically developed anti-CD47 antibodies (118, 119, 384, 386–389). Based on this background, two novel anti-CD47 antibodies, Hu5F9-G4 and CC-900002, are being examined in several clinical studies (NCT02678338, NCT02953509, NCT02641002, NCT01410981, NCT02367196, NCT02663518), and many others have initiated clinical development, such as C47B222-(CHO).
CD47 is the first targeted receptor involved in phagocytosis, however, whether anti-CD47 MOA relies only in activation of immune cells or, in addition to immune cell activation, it strongly depends on Fc-mediated effector activities is a controversial issue. Based on preclinical investigations, it is assumed that these novel anti-CD47 mAbs impede CD47-SIRPα interactions leading to macrophage-mediated phagocytosis of B-NHL and AML cells, including LSC cells (118, 119). However, recent evidence suggests that the Fc region of the murine IgG1 (B6H12.2) and human IgG4 (Hu5F9-G4) are able to bind human and murine FcRs and mediate effector functions (118, 390, 391); hence, whether the therapeutic effect observed is due to solely blocking CD47 or to an opsonizing effect combined with CD47 blocking activity remains unclear. In this sense, antileukemic activity of the IgG1 antibody C47B222-(CHO) does not rely on CD47 neutralization but, on the contrary, depends on robust Fc effector functions such as ADCP and ADCC (118). Importantly, fusion proteins containing the high-affinity human SIPRα (known as CV1) only have antitumor activity when fused to IgG4, but not as SIRPα monomer (392, 393). Similarly, a SIRPα-Fc molecule known as TTI-621 demonstrated potent antileukemic activity as IgG1 Fc conjugate, but not with a Fc mutated to avoid effector functions (394). Furthermore, mice harboring inactivating mutations at the SIRPα cytoplasmic tail show similar growth and metastasis of implanted syngeneic melanoma tumor cells as wild-type mice, suggesting again that disruption of CD47-SIRPα alone does not yield an effective antitumor response (393).
Finally, another concern related to antibody-based CD47 therapy is tolerability. Except for C47B222-(CHO), anti-CD47 antibodies have been reported to cause platelet aggregation and red blood cell hemaglutination (118, 119). Therefore, it is not clear whether an optimal dosing strategy could be achieved that provides a therapeutic window with limited toxicity. Results from ongoing clinical studies will shed light on this issue.
Antibody-Drug and Antibody-Radionuclide Conjugates (ADCs AND ARCs)
ADCs, along with ARCs, comprise the largest group of the non-canonical antibody formats in clinical studies for hematological malignancies. The principles of ADC/ARC activity and the considerations for their development, including the choice of antibody, drug and radionuclide, are beyond the scope of this review and have been discussed elsewhere (3, 395, 396). However, as the most advanced ADCs in the clinic are directed to hematological indications, a brief account of this landscape with some examples of ADCs which target LSA and NLSAs are given in Tables 5 and 6, including the first ADCs developed and approved by the FDA: gemtuzumab ozogamicin, now discontinued, and brentuximab vedotin.
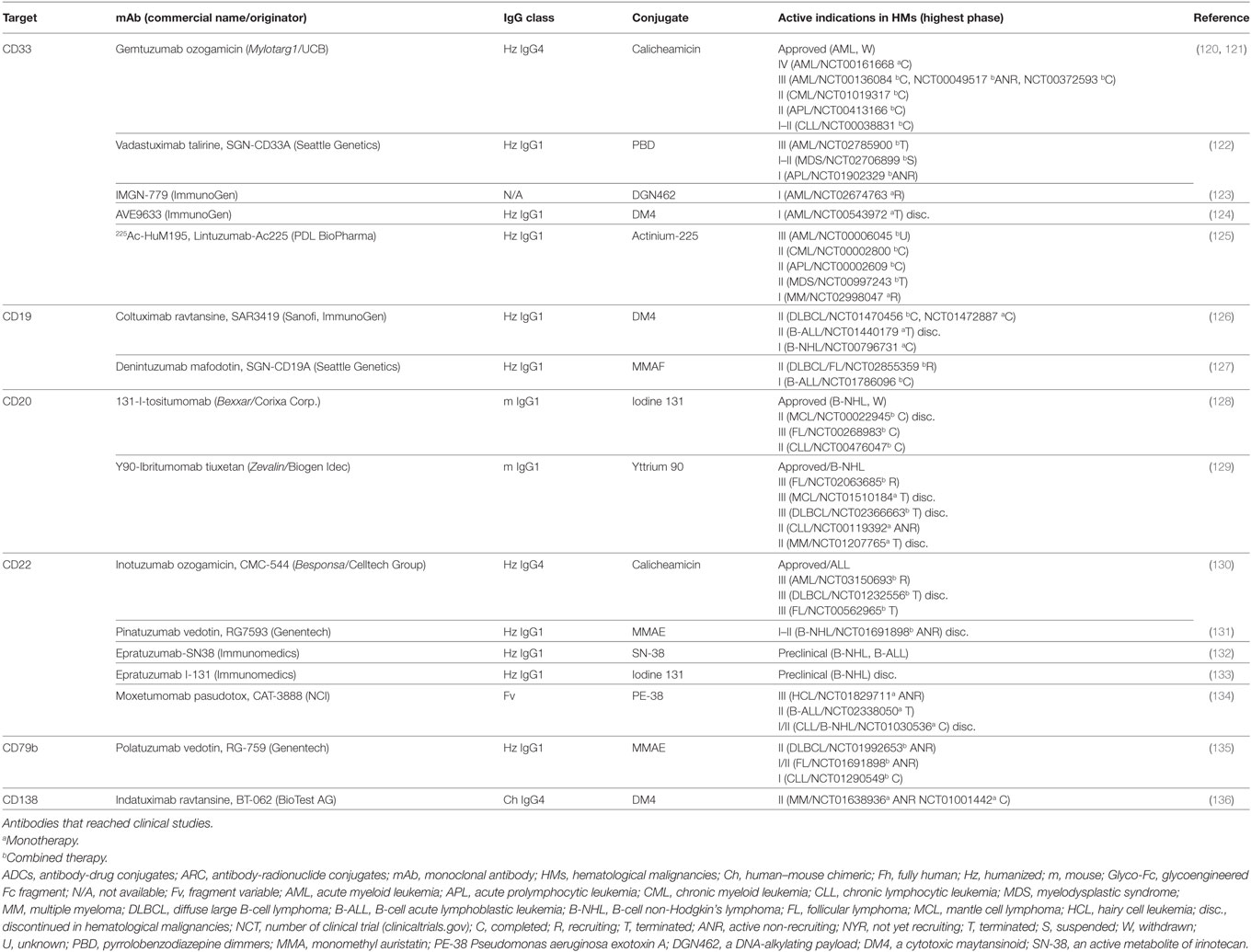
Table 5. Characteristics of ADCs and ARCs directed to LSAs.
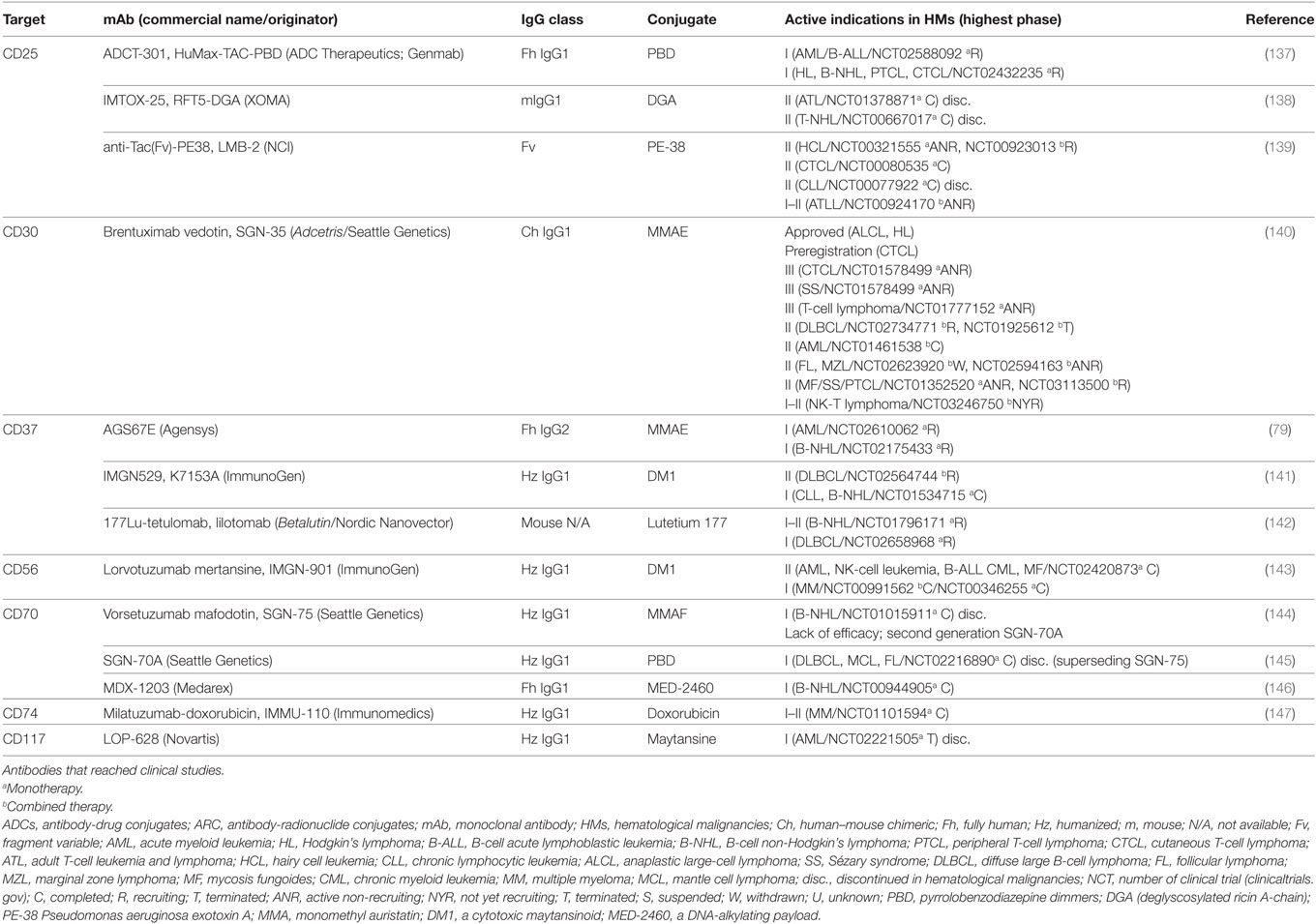
Table 6. Characteristics of ADCs and ARCs directed to NLSAs.
Conclusion
From rituximab, the first mAb approved in 1997 to treat cancer, to the recently approved daratumumab and elotuzumab to treat MM, several antibodies have changed the clinical practice and have transformed the therapeutic landscape of hematological malignancies. However, as we have shown, this number is extremely low compared with the total number of antibodies being studied in clinical trials. Today, most of the antibodies already approved target LSAs, whereas the majority of agents in development for hematological malignancies targets NLSAs. This situation reflects a paradigm shift in the criteria followed to select a target to develop as novel immune therapy in blood malignancies care. The impact of this change is still unknown in disorders where mAbs targeting LSAs were not used, or resulted to be ineffective, such as MM, AML, or T-NHL. In this respect, novel antibodies directly targeting NLSAs in MM or AML are a profound change compared with earlier treatment approaches. Remarkably, today most of the therapeutic antibodies under development target both disorders. Nonetheless, in other diseases, such as T-cell or NK-cells malignancies, the scenario is not so promising since few candidates are in clinical development, and many of them show lack of activity in these entities. For example, while patients with B-NHL may benefit from immune checkpoint drugs, it is likely that patients with T-lymphomas will not. However, efforts must be maintained. NLSAs constitute a bigger group of molecules than LSAs, and for this reason the number of antibodies showing successful results is likely to be higher.
Most of the antibodies reviewed in this work, used as single agent or in combined regimens, are not better than standard treatments. In some cases the selected NLSA is not appropriate. For instance, targeting soluble factors is a successful approach in immunological disorders that seems to be ineffective against hematological malignancies, likely due to the disseminated nature of these cancers. In other cases, the antibody under evaluation may not display all its potential or may not trigger the right MOA. In this respect, directed modifications both in the Fv and Fc may reverse the lack of activity. Moreover, combining antibodies with other drugs, without previous strong supporting evidence, may negatively affect the outcome in clinical settings. Unfavorable combination regimens may impair the main MOA of a single antibody, or even highly pretreated patients may harbor the unfavorable background themselves. To evaluate a novel antibody, the selection of patients must take into account several factors including sensitivity and tolerability to prior treatments, disease stage and cytogenetic profile. Finally, one of the most important issues in the evaluation of antibodies targeting NLSAs expressed in different disorders is the selection of the optimal dose, which is not usually based on biological criteria. As we have learnt from some antibodies targeting LSAs, benefits within a wide range of identical lineage disorders cannot be obtained using the same dosing schedule. The uncertainty over the optimal dosing schedule is even higher when a particular mAb is examined in diseases differing not only in maturation stages but most importantly in different lineages or even, in different tissues.
Far beyond these explanations, the abundance of novel targeted agents draw a promising and consolidated landscape in hematological malignancies. Hopefully, in the near future, clinicians may consider different standard treatments for a given disease as different treatments may be available and may be tailored for molecularly defined responsive groups. In the coming years, unmet medical needs in a wide variety of conditions should be reduced and patient choices for antibody therapeutics should increase in short- as well as long-term. The optimal integration of mAbs and other novel agents with current treatment strategies will require intelligent, rather than commercially driven, preclinical, and clinical design over the coming years.
Author Contributions
CC-M reviewed the literature, prepared the tables, and wrote the manuscript. AA-S and BS-C reviewed the literature and drafted the work. CM-C reviewed the literature and wrote the manuscript.
Conflict of Interest Statement
CM-C received research funding from BMS and IMMED.S.L. and has filed patents on targeting of the CCR7 chemokine receptor as cancer therapy. CC-M is an employee of IMMED. The remaining authors declare no conflict of interest.
Funding
The work in the author’s laboratories was supported by grants PI12/00494P and PI15/02085 from the Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III (ISCIII) to CMC, both cofinanced by FEDER funds from the EU. BS-C is financed by grant RTC-2015-3786-1 from the Spanish Ministry of Economy, Industry and Competitiveness, co-financed by FEDER funds from the EU, to Leonor Kremer (Operative program on Intelligent Growth 2014-2020).
References
1. Glassman PM, Balthasar JP. Mechanistic considerations for the use of monoclonal antibodies for cancer therapy. Cancer Biol Med (2014) 11:20–33. doi:10.7497/j.issn.2095-3941.2014.01.002
2. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer (2012) 12:278–87. doi:10.1038/nrc3236
3. Kim EG, Kim KM. Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol Ther (Seoul) (2015) 23:493–509. doi:10.4062/biomolther.2015.116
4. Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov (2009) 8:226–34. doi:10.1038/nrd2804
5. Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res (2008) 68:3863–72. doi:10.1158/0008-5472.CAN-07-6297
6. Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol (1980) 125:1678–85.
7. Drexler HG. Classification of acute myeloid leukemias – a comparison of FAB and immunophenotyping. Leukemia (1987) 1:697–705.
8. Lanier LL. Back to the future – defining NK cells and T cells. Eur J Immunol (2007) 37:1424–6. doi:10.1002/eji.200737418
9. Rossi JF. Targeted therapies in adult B-cell malignancies. Biomed Res Int (2015) 2015:217593. doi:10.1155/2015/217593
10. Oldham RK, Dillman RO. Monoclonal antibodies in cancer therapy: 25 years of progress. J Clin Oncol (2008) 26:1774–7. doi:10.1200/JCO.2007.15.7438
11. Maloney DG, Grillo-López AJ, Bodkin DJ, White CA, Liles TM, Royston I, et al. IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J Clin Oncol (1997) 15:3266–74. doi:10.1200/JCO.1997.15.10.3266
12. Maloney DG, Grillo-López AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, et al. IDEC-C2B8 (rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood (1997) 90:2188–95.
13. Gupta IV, Jewell RC. Ofatumumab, the first human anti-CD20 monoclonal antibody for the treatment of B cell hematologic malignancies. Ann N Y Acad Sci (2012) 1263:43–56. doi:10.1111/j.1749-6632.2012.06661.x
14. Teeling JL, French RR, Cragg MS, van den Brakel J, Pluyter M, Huang H, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood (2004) 104:1793–800. doi:10.1182/blood-2004-01-0039
15. Owen C, Stewart DA. Obinutuzumab for the treatment of lymphoproliferative disorders. Expert Opin Biol Ther (2012) 12:343–51. doi:10.1517/14712598.2012.657622
16. Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, Williams CD, Hellmann A, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol (2010) 28:1749–55. doi:10.1200/JCO.2009.25.3187
17. Stein R, Qu Z, Chen S, Rosario A, Shi V, Hayes M, et al. Characterization of a new humanized anti-CD20 monoclonal antibody, IMMU-106, and Its use in combination with the humanized anti-CD22 antibody, epratuzumab, for the therapy of non-Hodgkin’s lymphoma. Clin Cancer Res (2004) 10:2868–78. doi:10.1158/1078-0432.CCR-03-0493
18. Morschhauser F, Leonard JP, Fayad L, Coiffier B, Petillon MO, Coleman M, et al. Humanized anti-CD20 antibody, veltuzumab, in refractory/recurrent non-Hodgkin’s lymphoma: phase I/II results. J Clin Oncol (2009) 27:3346–53. doi:10.1200/JCO.2008.19.9117
19. Morschhauser F, Marlton P, Vitolo U, Lindén O, Seymour JF, Crump M, et al. Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann Oncol (2010) 21:1870–6. doi:10.1093/annonc/mdq027
20. Mössner E, Brünker P, Moser S, Püntener U, Schmidt C, Herter S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood (2010) 115:4393–402. doi:10.1182/blood-2009-06-225979
21. Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med (2014) 370:1101–10. doi:10.1056/NEJMoa1313984
22. Tobinai K, Ogura M, Kobayashi Y, Uchida T, Watanabe T, Oyama T, et al. Phase I study of LY2469298, an Fc-engineered humanized anti-CD20 antibody, in patients with relapsed or refractory follicular lymphoma. Cancer Sci (2011) 102:432–8. doi:10.1111/j.1349-7006.2010.01809.x
23. Sawas A, Farber CM, Schreeder MT, Khalil MY, Mahadevan D, Deng C, et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br J Haematol (2017) 177:243–53. doi:10.1111/bjh.14534
24. Horton HM, Bernett MJ, Pong E, Peipp M, Karki S, Chu SY, et al. Potent in vitro and in vivo activity of an Fc-engineered anti-CD19 monoclonal antibody against lymphoma and leukemia. Cancer Res (2008) 68:8049–57. doi:10.1158/0008-5472.CAN-08-2268
25. Leonard JP, Coleman M, Ketas JC, Chadburn A, Ely S, Furman RR, et al. Phase I/II trial of epratuzumab (humanized anti-CD22 antibody) in indolent non-Hodgkin’s lymphoma. J Clin Oncol (2003) 21:3051–9. doi:10.1200/JCO.2003.01.082
26. Pfeifer M, Zheng B, Erdmann T, Koeppen H, McCord R, Grau M, et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia (2015) 29:1578–86. doi:10.1038/leu.2015.48
27. Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, et al. A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood (2000) 95:2098–103.
28. Brandl C, Haas C, d’Argouges S, Fisch T, Kufer P, Brischwein K, et al. The effect of dexamethasone on polyclonal T cell activation and redirected target cell lysis as induced by a CD19/CD3-bispecific single-chain antibody construct. Cancer Immunol Immunother (2007) 56:1551–63. doi:10.1007/s00262-007-0298-z
29. Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol (2015) 16:57–66. doi:10.1016/S1470-2045(14)71170-2
30. Goebeler ME, Knop S, Viardot A, Kufer P, Topp MS, Einsele H, et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J Clin Oncol (2016) 34:1104–11. doi:10.1200/JCO.2014.59.1586
31. Ward E, Mittereder N, Kuta E, Sims GP, Bowen MA, Dall’Acqua W, et al. A glycoengineered anti-CD19 antibody with potent antibody-dependent cellular cytotoxicity activity in vitro and lymphoma growth inhibition in vivo. Br J Haematol (2011) 155:426–37. doi:10.1111/j.1365-2141.2011.08857.x
32. Woyach JA, Awan F, Flinn IW, Berdeja JG, Wiley E, Mansoor S, et al. A phase 1 trial of the Fc-engineered CD19 antibody XmAb5574 (MOR00208) demonstrates safety and preliminary efficacy in relapsed CLL. Blood (2014) 124:3553–60. doi:10.1182/blood-2014-08-593269
33. Cardarelli PM, Rao-Naik C, Chen S, Huang H, Pham A, Moldovan-Loomis MC, et al. A nonfucosylated human antibody to CD19 with potent B-cell depletive activity for therapy of B-cell malignancies. Cancer Immunol Immunother (2010) 59:257–65. doi:10.1007/s00262-009-0746-z
34. Cuesta-Mateos C, Loscertales J, Kreutzman A, Colom-Fernández B, Portero-Sáinz I, Pérez-Villar JJ, et al. Preclinical activity of anti-CCR7 immunotherapy in patients with high-risk chronic lymphocytic leukemia. Cancer Immunol Immunother (2015) 64:665–76. doi:10.1007/s00262-015-1670-z
35. Kapoor P, Greipp PT, Morice WG, Rajkumar SV, Witzig TE, Greipp PR. Anti-CD20 monoclonal antibody therapy in multiple myeloma. Br J Haematol (2008) 141:135–48. doi:10.1111/j.1365-2141.2008.07024.x
36. Gilleece MH, Dexter TM. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood (1993) 82:807–12.
37. Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Dyer MJ, et al. Levels of expression of CD52 in normal and leukemic B and T cells: correlation with in vivo therapeutic responses to Campath-1H. Leuk Res (1998) 22:185–91. doi:10.1016/S0145-2126(97)00158-6
38. Jabbour E, O’Brien S, Ravandi F, Kantarjian H. Monoclonal antibodies in acute lymphoblastic leukemia. Blood (2015) 125:4010–6. doi:10.1182/blood-2014-08-596403
39. Dearden C. How I treat prolymphocytic leukemia. Blood (2012) 120:538–51. doi:10.1182/blood-2012-01-380139
40. Osterborg A, Dyer MJ, Bunjes D, Pangalis GA, Bastion Y, Catovsky D, et al. Phase II multicenter study of human CD52 antibody in previously treated chronic lymphocytic leukemia. European Study Group of CAMPATH-1H Treatment in Chronic Lymphocytic Leukemia. J Clin Oncol (1997) 15:1567–74. doi:10.1200/JCO.1997.15.4.1567
41. Rai KR, Freter CE, Mercier RJ, Cooper MR, Mitchell BS, Stadtmauer EA, et al. Alemtuzumab in previously treated chronic lymphocytic leukemia patients who also had received fludarabine. J Clin Oncol (2002) 20:3891–7. doi:10.1200/JCO.2002.06.119
42. Stilgenbauer S, Dohner H. Campath-1H-induced complete remission of chronic lymphocytic leukemia despite p53 gene mutation and resistance to chemotherapy. N Engl J Med (2002) 347:452–3. doi:10.1056/NEJM200208083470619
43. Hu Y, Turner MJ, Shields J, Gale MS, Hutto E, Roberts BL, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology (2009) 128:260–70. doi:10.1111/j.1365-2567.2009.03115.x
44. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev (2016) 270:95–112. doi:10.1111/imr.12389
45. Wijdenes J, Vooijs WC, Clément C, Post J, Morard F, Vita N, et al. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br J Haematol (1996) 94:318–23. doi:10.1046/j.1365-2141.1996.d01-1811.x
46. Khagi Y, Mark TM. Potential role of daratumumab in the treatment of multiple myeloma. Onco Targets Ther (2014) 7:1095–100. doi:10.2147/OTT.S49480
47. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol (2011) 186:1840–8. doi:10.4049/jimmunol.1003032
48. van der Veer MS, de Weers M, van Kessel B, Bakker JM, Wittebol S, Parren PW, et al. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood Cancer J (2011) 1:e41. doi:10.1038/bcj.2011.42
49. Overdijk MB, Verploegen S, Bögels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs (2015) 7:311–21. doi:10.1080/19420862.2015.1007813
50. Overdijk MB, Jansen JH, Nederend M, Lammerts van Bueren JJ, Groen RW, Parren PW, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via Fcgamma receptor-mediated cross-linking. J Immunol (2016) 197:807–13. doi:10.4049/jimmunol.1501351
51. Krejcik J, Casneuf T, Nijhof IS, Verbist B, Bald J, Plesner T, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood (2016) 128:384–94. doi:10.1182/blood-2015-12-687749
52. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med (2015) 373:1207–19. doi:10.1056/NEJMoa1506348
53. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet (2016) 387:1551–60. doi:10.1016/S0140-6736(15)01120-4
54. Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375:1319–31. doi:10.1056/NEJMoa1607751
55. Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med (2016) 375:754–66. doi:10.1056/NEJMoa1606038
56. Chari A, Suvannasankha A, Fay JW, Arnulf B, Kaufman JL, Ifthikharuddin JJ, et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood (2017) 130(8):974–81. doi:10.1182/blood-2017-05-785246
57. Martin T, Hsu K, Strickland S, Glenn M, Mikhael J, Charpentier E, et al. A phase I trial of SAR650984, a CD38 monoclonal antibody, in relapsed or refractory multiple myeloma. J Clin Oncol (2014) 32(Suppl 5):abstract8532.
58. Martin T, Hsu K, Charpentier E, Vij R, Baz R, Benson D, et al. A phase Ib dose escalation trial of SAR650984 (anti-CD-38 mAb) in combination with lenalidomide and dexamethasone in relapsed/refractory multiple myeloma. J Clin Oncol (2014) 32(Suppl 5):abstract8512.
59. Raab M, Goldschmidt H, Agis H, Blau I, Einsele H, Engelhardt M, et al. A phase I/IIa study of the human anti-CD38 antibody MOR202 (MOR03087) in relapsed or refractory multiple myeloma (rrMM). J Clin Oncol (2015) 33(Suppl):abstract8574.
60. Raab M, Chatterjee M, Goldschmidt H, Agis H, Blau I, Einsele H, et al. MOR202 alone and in combination with pomalidomide or lenalidomide in relapsed or refractory multiple myeloma: data from clinically relevant cohorts from a phase I/IIa study. J Clin Oncol (2016) 34(Suppl):abstract8012.
61. Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallée F, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res (2014) 20:4574–83. doi:10.1158/1078-0432.CCR-14-0695
62. Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res (2008) 14:2775–84. doi:10.1158/1078-0432.CCR-07-4246
63. Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood (2008) 112:1329–37. doi:10.1182/blood-2007-08-107292
64. van Rhee F, Szmania SM, Dillon M, van Abbema AM, Li X, Stone MK, et al. Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther (2009) 8:2616–24. doi:10.1158/1535-7163.MCT-09-0483
65. Friend R, Bhutani M, Voorhees PM, Usmani SZ. Clinical potential of SLAMF7 antibodies – focus on elotuzumab in multiple myeloma. Drug Des Devel Ther (2017) 11:893–900. doi:10.2147/DDDT.S98053
66. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med (2015) 373:621–31. doi:10.1056/NEJMoa1505654
67. Collins SM, Bakan CE, Swartzel GD, Hofmeister CC, Efebera YA, Kwon H, et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother (2013) 62:1841–9. doi:10.1007/s00262-013-1493-8
68. Moreau P, Touzeau C. Elotuzumab for the treatment of multiple myeloma. Future Oncol (2014) 10:949–56. doi:10.2217/fon.14.56
69. Jakubowiak A, Offidani M, Pégourie B, De La Rubia J, Garderet L, Laribi K, et al. Randomized phase 2 study: elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood (2016) 127:2833–40. doi:10.1182/blood-2016-01-694604
70. Guo H, Cruz-Munoz ME, Wu N, Robbins M, Veillette A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol Cell Biol (2015) 35:41–51. doi:10.1128/MCB.01107-14
71. Perez-Quintero LA, Roncagalli R, Guo H, Latour S, Davidson D, Veillette A. EAT-2, a SAP-like adaptor, controls NK cell activation through phospholipase Cgamma, Ca++, and Erk, leading to granule polarization. J Exp Med (2014) 211:727–42. doi:10.1084/jem.20132038
72. Zonder JA, Mohrbacher AF, Singhal S, van Rhee F, Bensinger WI, Ding H, et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood (2012) 120:552–9. doi:10.1182/blood-2011-06-360552
73. Lonial S, Vij R, Harousseau JL, Facon T, Moreau P, Mazumder A, et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma. J Clin Oncol (2012) 30:1953–9. doi:10.1200/JCO.2011.37.2649
74. Richardson PG, Jagannath S, Moreau P, Jakubowiak AJ, Raab MS, Facon T, et al. Elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: final phase 2 results from the randomised, open-label, phase 1b-2 dose-escalation study. Lancet Haematol (2015) 2:e516–27. doi:10.1016/S2352-3026(15)00197-0
75. Jakubowiak AJ, Benson DM, Bensinger W, Siegel DS, Zimmerman TM, Mohrbacher A, et al. Phase I trial of anti-CS1 monoclonal antibody elotuzumab in combination with bortezomib in the treatment of relapsed/refractory multiple myeloma. J Clin Oncol (2012) 30:1960–5. doi:10.1200/JCO.2011.37.7069
76. Barrena S, Almeida J, Yunta M, López A, Fernández-Mosteirín N, Giralt M, et al. Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia (2005) 19:1376–83. doi:10.1038/sj.leu.2403822
77. Moore K, Cooper SA, Jones DB. Use of the monoclonal antibody WR17, identifying the CD37 gp40-45 Kd antigen complex, in the diagnosis of B-lymphoid malignancy. J Pathol (1987) 152:13–21. doi:10.1002/path.1711520103
78. Beckwith KA, Byrd JC, Muthusamy N. Tetraspanins as therapeutic targets in hematological malignancy: a concise review. Front Physiol (2015) 6:91. doi:10.3389/fphys.2015.00091
79. Pereira DS, Guevara CI, Jin L, Mbong N, Verlinsky A, Hsu SJ, et al. AGS67E, an anti-CD37 monomethyl auristatin E antibody-drug conjugate as a potential therapeutic for B/T-cell malignancies and AML: a new role for CD37 in AML. Mol Cancer Ther (2015) 14:1650–60. doi:10.1158/1535-7163.MCT-15-0067
80. Heider KH, Kiefer K, Zenz T, Volden M, Stilgenbauer S, Ostermann E, et al. A novel Fc-engineered monoclonal antibody to CD37 with enhanced ADCC and high proapoptotic activity for treatment of B-cell malignancies. Blood (2011) 118:4159–68. doi:10.1182/blood-2011-04-351932
81. Press OW, Eary JF, Badger CC, Martin PJ, Appelbaum FR, Levy R, et al. Treatment of refractory non-Hodgkin’s lymphoma with radiolabeled MB-1 (anti-CD37) antibody. J Clin Oncol (1989) 7:1027–38. doi:10.1200/JCO.1989.7.8.1027
82. Dahle J, Repetto-Llamazares AH, Mollatt CS, Melhus KB, Bruland OS, Kolstad A, et al. Evaluating antigen targeting and anti-tumor activity of a new anti-CD37 radioimmunoconjugate against non-Hodgkin’s lymphoma. Anticancer Res (2013) 33:85–95.
83. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol (2013) 94:25–39. doi:10.1189/jlb.1212621
84. Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) 372:311–9. doi:10.1056/NEJMoa1411087
85. Armand P, Shipp MA, Ribrag V, Michot JM, Zinzani PL, Kuruvilla J, et al. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J Clin Oncol (2016) 34(31):3733–9. doi:10.1200/JCO.2016.67.3467
86. Westin JR, Chu F, Zhang M, Fayad LE, Kwak LW, Fowler N, et al. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol (2014) 15:69–77. doi:10.1016/S1470-2045(13)70551-5
87. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14:3044–51. doi:10.1158/1078-0432.CCR-07-4079
88. Burova E, Hermann A, Waite J, Potocky T, Lai V, Hong S, et al. Characterization of the anti-PD-1 antibody REGN2810 and its antitumor activity in human PD-1 knock-in mice. Mol Cancer Ther (2017) 16:861–70. doi:10.1158/1535-7163.MCT-16-0665
89. Lee HT, Lee JY, Lim H, Lee SH, Moon YJ, Pyo HJ, et al. Molecular mechanism of PD-1/PD-L1 blockade via anti-PD-L1 antibodies atezolizumab and durvalumab. Sci Rep (2017) 7:5532. doi:10.1038/s41598-017-06002-8
90. Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis (2017) 215(11):1725–33. doi:10.1093/infdis/jix191
91. Kotsakis A, Georgoulias V. Avelumab, an anti-PD-L1 monoclonal antibody, shows activity in various tumour types. Lancet Oncol (2017) 18:556–7. doi:10.1016/S1470-2045(17)30227-9
92. Ansell SM, Hurvitz SA, Koenig PA, LaPlant BR, Kabat BF, Fernando D, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res (2009) 15:6446–53. doi:10.1158/1078-0432.CCR-09-1339
93. Camacho LH, Antonia S, Sosman J, Kirkwood JM, Gajewski TF, Redman B, et al. Phase I/II trial of tremelimumab in patients with metastatic melanoma. J Clin Oncol (2009) 27:1075–81. doi:10.1200/JCO.2008.19.2435
94. Benson DM Jr, Hofmeister CC, Padmanabhan S, Suvannasankha A, Jagannath S, Abonour R, et al. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood (2012) 120:4324–33. doi:10.1182/blood-2012-06-438028
95. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120:4317–23. doi:10.1182/blood-2012-06-437558
96. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology (2016) 5:e1226720. doi:10.1080/2162402X.2016.1226720
97. Ruggeri L, Urbani E, André P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica (2016) 101:626–33. doi:10.3324/haematol.2015.135301
98. Marie-Cardine A, Viaud N, Thonnart N, Joly R, Chanteux S, Gauthier L, et al. IPH4102, a humanized KIR3DL2 antibody with potent activity against cutaneous T-cell lymphoma. Cancer Res (2014) 74:6060–70. doi:10.1158/0008-5472.CAN-14-1456
99. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci (2016) 107:1193–7. doi:10.1111/cas.12986
100. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207:2187–94. doi:10.1084/jem.20100643
101. Kretz-Rommel A, Qin F, Dakappagari N, Cofiell R, Faas SJ, Bowdish KS. Blockade of CD200 in the presence or absence of antibody effector function: implications for anti-CD200 therapy. J Immunol (2008) 180:699–705. doi:10.4049/jimmunol.180.2.699
102. Segal NH, Logan TF, Hodi FS, McDermott D, Melero I, Hamid O, et al. Results from an integrated safety analysis of urelumab, an agonist anti-CD137 monoclonal antibody. Clin Cancer Res (2017) 23:1929–36. doi:10.1158/1078-0432.CCR-16-1272
103. Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med (1997) 3:682–5. doi:10.1038/nm0697-682
104. Fisher TS, Kamperschroer C, Oliphant T, Love VA, Lira PD, Doyonnas R, et al. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol Immunother (2012) 61:1721–33. doi:10.1007/s00262-012-1237-1
105. Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med (2009) 206:1103–16. doi:10.1084/jem.20082205
106. Burris HA, Infante JR, Ansell SM, Nemunaitis JJ, Weiss GR, Villalobos VM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-CD27 antibody, in patients with advanced solid tumors. J Clin Oncol (2017) 35:2028–36. doi:10.1200/JCO.2016.70.1508
107. Silence K, Dreier T, Moshir M, Ulrichts P, Gabriels SM, Saunders M, et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs (2014) 6:523–32. doi:10.4161/mabs.27398
108. Aftimos P, Rolfo C, Rottey S, Offner F, Bron D, Maerevoet M, et al. Phase 1 dose-escalation study of the anti-CD70 antibody ARGX-110 in advanced malignancies. Clin Cancer Res (2017) 2017:1078–432. doi:10.1158/1078-0432.CCR-17-0613
109. Bhat S, Czuczman MS. Galiximab: a review. Expert Opin Biol Ther (2010) 10:451–8. doi:10.1517/14712591003596318
110. Czuczman MS, Thall A, Witzig TE, Vose JM, Younes A, Emmanouilides C, et al. Phase I/II study of galiximab, an anti-CD80 antibody, for relapsed or refractory follicular lymphoma. J Clin Oncol (2005) 23:4390–8. doi:10.1200/JCO.2005.09.018
111. Remer M, White A, Glennie M, Al-Shamkhani A, Johnson P. The use of anti-CD40 mAb in cancer. Curr Top Microbiol Immunol (2017) 405:165–207. doi:10.1007/82_2014_427
112. Luqman M, Klabunde S, Lin K, Georgakis GV, Cherukuri A, Holash J, et al. The antileukemia activity of a human anti-CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood (2008) 112:711–20. doi:10.1182/blood-2007-04-084756
113. Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, et al. Clinical and biological effects of an agonist anti-CD40 antibody: a cancer research UK phase I study. Clin Cancer Res (2015) 21:1321–8. doi:10.1158/1078-0432.CCR-14-2355
114. Chowdhury F, Johnson PW, Glennie MJ, Williams AP. Ex vivo assays of dendritic cell activation and cytokine profiles as predictors of in vivo effects in an anti-human CD40 monoclonal antibody ChiLob 7/4 phase I trial. Cancer Immunol Res (2014) 2:229–40. doi:10.1158/2326-6066.CIR-13-0070
115. Gardai SJ, Epp A, Linares G, Westendorf L, Sutherland M, Neff-LaFord H, et al. SEA-CD40, a sugar engineered non-fucosylated anti-CD40 antibody with improved immune activating capabilities. [Abstract]. Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22. Philadelphia, PA: AACR (2015).
116. Aida K, Miyakawa R, Suzuki K, Narumi K, Udagawa T, Yamamoto Y, et al. Suppression of Tregs by anti-glucocorticoid induced TNF receptor antibody enhances the antitumor immunity of interferon-alpha gene therapy for pancreatic cancer. Cancer Sci (2014) 105:159–67. doi:10.1111/cas.12332
117. Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med (2005) 202:885–91. doi:10.1084/jem.20050940
118. Pietsch EC, Dong J, Cardoso R, Zhang X, Chin D, Hawkins R, et al. Anti-leukemic activity and tolerability of anti-human CD47 monoclonal antibodies. Blood Cancer J (2017) 7:e536. doi:10.1038/bcj.2017.7
119. Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS One (2015) 10:e0137345. doi:10.1371/journal.pone.0137345
120. Sievers EL, Larson RA, Stadtmauer EA, Estey E, Löwenberg B, Dombret H, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol (2001) 19:3244–54. doi:10.1200/JCO.2001.19.13.3244
121. Larson RA, Sievers EL, Stadtmauer EA, Löwenberg B, Estey EH, Dombret H, et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer (2005) 104:1442–52. doi:10.1002/cncr.21326
122. Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood (2013) 122:1455–63. doi:10.1182/blood-2013-03-491506
123. Whiteman KR, Noordhuis P, Walker R, Watkins K, Kovtun Y, Harvey L, et al. The antibody-drug conjugate (ADC) IMGN779 is highly active in vitro and in vivo against acute myeloid leukemia (AML) with FLT3-ITD mutations. Blood (2014) 124:2321.
124. Lapusan S, Vidriales MB, Thomas X, de Botton S, Vekhoff A, Tang R, et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Invest New Drugs (2012) 30:1121–31. doi:10.1007/s10637-011-9670-0
125. Miederer M, McDevitt MR, Sgouros G, Kramer K, Cheung NK, Scheinberg DA. Pharmacokinetics, dosimetry, and toxicity of the targetable atomic generator, 225Ac-HuM195, in nonhuman primates. J Nucl Med (2004) 45:129–37.
126. Younes A, Kim S, Romaguera J, Copeland A, Farial Sde C, Kwak LW, et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J Clin Oncol (2012) 30:2776–82. doi:10.1200/JCO.2011.39.4403
127. Borate U, Fathi AT, Shah BD, DeAngelo DJ, Silverman LB, Cooper TM, et al. A first-inhuman phase 1 study of the antibody-drug conjugate SGN-CD19A in relapsed or refractory B-lineage acute leukemia and highly aggressive lymphoma [abstract]. Blood (2013) 122(21):abstract1437.
128. Kaminski MS, Zelenetz AD, Press OW, Saleh M, Leonard J, Fehrenbacher L, et al. Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin’s lymphomas. J Clin Oncol (2001) 19:3918–28. doi:10.1200/JCO.2001.19.19.3918
129. Rizzieri D. Zevalin((R)) (ibritumomab tiuxetan): after more than a decade of treatment experience, what have we learned? Crit Rev Oncol Hematol (2016) 105:5–17. doi:10.1016/j.critrevonc.2016.07.008
130. Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med (2016) 375:740–53. doi:10.1056/NEJMoa1509277
131. Advani RH, Lebovic D, Chen A, Brunvand M, Goy A, Chang JE, et al. Phase I study of the anti-CD22 antibody-drug conjugate pinatuzumab vedotin with/without rituximab in patients with relapsed/refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res (2017) 23:1167–76. doi:10.1158/1078-0432.CCR-16-0772
132. Sharkey RM, Govindan SV, Cardillo TM, Goldenberg DM. Epratuzumab-SN-38: a new antibody-drug conjugate for the therapy of hematologic malignancies. Mol Cancer Ther (2012) 11:224–34. doi:10.1158/1535-7163.MCT-11-0632
133. Ghobrial I, Witzig T. Radioimmunotherapy: a new treatment modality for B-cell non-Hodgkin’s lymphoma. Oncology (Williston Park) (2004) 18:623–30; discussion 33–4, 37–8, 40.
134. Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol (2009) 27:2983–90. doi:10.1200/JCO.2008.20.2630
135. Palanca-Wessels MC, Czuczman M, Salles G, Assouline S, Sehn LH, Flinn I, et al. Safety and activity of the anti-CD79B antibody-drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study. Lancet Oncol (2015) 16:704–15. doi:10.1016/S1470-2045(15)70128-2
136. Schonfeld K, Zuber C, Pinkas J, Hader T, Bernoster K, Uherek C. Indatuximab ravtansine (BT062) combination treatment in multiple myeloma: pre-clinical studies. J Hematol Oncol (2017) 10:13. doi:10.1186/s13045-016-0380-0
137. Flynn MJ, Zammarchi F, Tyrer PC, Akarca AU, Janghra N, Britten CE, et al. ADCT-301, a pyrrolobenzodiazepine (PBD) dimer-containing antibody-drug conjugate (ADC) targeting CD25-expressing hematological malignancies. Mol Cancer Ther (2016) 15:2709–21. doi:10.1158/1535-7163.MCT-16-0233
138. Wayne AS, Fitzgerald DJ, Kreitman RJ, Pastan I. Immunotoxins for leukemia. Blood (2014) 123:2470–7. doi:10.1182/blood-2014-01-492256
139. Kreitman RJ, Wilson WH, White JD, Stetler-Stevenson M, Jaffe ES, Giardina S, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol (2000) 18:1622–36. doi:10.1200/JCO.2000.18.8.1622
140. Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med (2010) 363:1812–21. doi:10.1056/NEJMoa1002965
141. Beckwith KA, Frissora FW, Stefanovski MR, Towns WH, Cheney C, Mo X, et al. The CD37-targeted antibody-drug conjugate IMGN529 is highly active against human CLL and in a novel CD37 transgenic murine leukemia model. Leukemia (2014) 28:1501–10. doi:10.1038/leu.2014.32
142. Repetto-Llamazares AH, Larsen RH, Giusti AM, Riccardi E, Bruland ØS, Selbo PK, et al. 177Lu-DOTA-HH1, a novel anti-CD37 radio-immunoconjugate: a study of toxicity in nude mice. PLoS One (2014) 9:e103070. doi:10.1371/journal.pone.0103070
143. Ishitsuka K, Jimi S, Goldmacher VS, Ab O, Tamura K. Targeting CD56 by the maytansinoid immunoconjugate IMGN901 (huN901-DM1): a potential therapeutic modality implication against natural killer/T cell malignancy. Br J Haematol (2008) 141:129–31. doi:10.1111/j.1365-2141.2008.07000.x
144. Tannir NM, Forero-Torres A, Ramchandren R, Pal SK, Ansell SM, Infante JR, et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Invest New Drugs (2014) 32:1246–57. doi:10.1007/s10637-014-0151-0
145. McEarchern JA, Smith LM, McDonagh CF, Klussman K, Gordon KA, Morris-Tilden CA, et al. Preclinical characterization of SGN-70, a humanized antibody directed against CD70. Clin Cancer Res (2008) 14:7763–72. doi:10.1158/1078-0432.CCR-08-0493
146. Owonikoko TK, Hussain A, Stadler WM, Smith DC, Kluger H, Molina AM, et al. First-in-human multicenter phase I study of BMS-936561 (MDX-1203), an antibody-drug conjugate targeting CD70. Cancer Chemother Pharmacol (2016) 77:155–62. doi:10.1007/s00280-015-2909-2
147. Sapra P, Stein R, Pickett J, Qu Z, Govindan SV, Cardillo TM, et al. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin Cancer Res (2005) 11:5257–64. doi:10.1158/1078-0432.CCR-05-0204
148. Zhao X, Lapalombella R, Joshi T, Cheney C, Gowda A, Hayden-Ledbetter MS, et al. Targeting CD37-positive lymphoid malignancies with a novel engineered small modular immunopharmaceutical. Blood (2007) 110:2569–77. doi:10.1182/blood-2006-12-062927
149. Robak T, Hellmann A, Kloczko J, Loscertales J, Lech-Maranda E, Pagel JM, et al. Randomized phase 2 study of otlertuzumab and bendamustine versus bendamustine in patients with relapsed chronic lymphocytic leukaemia. Br J Haematol (2017) 176:618–28. doi:10.1111/bjh.14464
150. Lapalombella R, Yeh YY, Wang L, Ramanunni A, Rafiq S, Jha S, et al. Tetraspanin CD37 directly mediates transduction of survival and apoptotic signals. Cancer Cell (2012) 21:694–708. doi:10.1016/j.ccr.2012.03.040
151. Baum PR, Cerveny C, Gordon B, Nilsson C, Wiens J, Rafiq S, et al. Evaluation of the effect of TRU-016, an anti-CD37 directed SMIP in combination with other therapeutic drugs in models of non-Hodgkin’s lymphoma. J Clin Oncol (2009) 27:8571. doi:10.1200/jco.2009.27.15s.8571
152. Rafiq S, Siadak A, Butchar JP, Cheney C, Lozanski G, Jacob NK, et al. Glycovariant anti-CD37 monospecific protein therapeutic exhibits enhanced effector cell-mediated cytotoxicity against chronic and acute B cell malignancies. MAbs (2013) 5:723–35. doi:10.4161/mabs.25282
153. Byrd JC, Pagel JM, Awan FT, Forero A, Flinn IW, Deauna-Limayo DP, et al. A phase 1 study evaluating the safety and tolerability of otlertuzumab, an anti-CD37 mono-specific ADAPTIR therapeutic protein in chronic lymphocytic leukemia. Blood (2014) 123:1302–8. doi:10.1182/blood-2013-07-512137
154. Pagel JM, Spurgeon SE, Byrd JC, Forero A, Flinn IW, Deauna-Limayo DP, et al. Otlertuzumab (TRU-016), an anti-CD37 monospecific ADAPTIR() therapeutic protein, for relapsed or refractory NHL patients. Br J Haematol (2015) 168:38–45. doi:10.1111/bjh.13099
155. Deves R, Boyd CA. Surface antigen CD98(4F2): not a single membrane protein, but a family of proteins with multiple functions. J Membr Biol (2000) 173:165–77. doi:10.1007/s002320001017
156. Cantor JM, Ginsberg MH. CD98 at the crossroads of adaptive immunity and cancer. J Cell Sci (2012) 125:1373–82. doi:10.1242/jcs.096040
157. Mastroberardino L, Spindler B, Pfeiffer R, Skelly PJ, Loffing J, Shoemaker CB, et al. Amino-acid transport by heterodimers of 4F2hc/CD98 and members of a permease family. Nature (1998) 395:288–91. doi:10.1038/26246
158. Hayes GM, Chinn L, Cantor JM, Cairns B, Levashova Z, Tran H, et al. Antitumor activity of an anti-CD98 antibody. Int J Cancer (2015) 137:710–20. doi:10.1002/ijc.29415
159. Kaira K, Oriuchi N, Imai H, Shimizu K, Yanagitani N, Sunaga N, et al. L-type amino acid transporter 1 and CD98 expression in primary and metastatic sites of human neoplasms. Cancer Sci (2008) 99:2380–6. doi:10.1111/j.1349-7006.2008.00969.x
160. Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med (2003) 349:2483–94. doi:10.1056/NEJMoa030847
161. Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD Jr. Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood (2007) 109:2106–11. doi:10.1182/blood-2006-09-047712
162. Fulciniti M, Tassone P, Hideshima T, Vallet S, Nanjappa P, Ettenberg SA, et al. Anti-DKK1 mAb (BHQ880) as a potential therapeutic agent for multiple myeloma. Blood (2009) 114:371–9. doi:10.1182/blood-2008-11-191577
163. Pozzi S, Fulciniti M, Yan H, Vallet S, Eda H, Patel K, et al. In vivo and in vitro effects of a novel anti-Dkk1 neutralizing antibody in multiple myeloma. Bone (2013) 53:487–96. doi:10.1016/j.bone.2013.01.012
164. Munshi NC, Abonour R, Beck JT, Bensinger W, Facon T, Stockerl-Goldstein K, et al. Early evidence of anabolic bone activity of BHQ880, a fully human anti-DKK1 neutralizing antibody: results of a phase 2 study in previously untreated patients with smoldering multiple myeloma at risk of progression [abstract 331]. Presented at: ASH Annual Meeting; December 8–12, 2012. Atlanta, GA (2012).
165. Hernandez-Campo PM, Almeida J, Sanchez ML, Malvezzi M, Orfao A. Normal patterns of expression of glycosylphosphatidylinositol-anchored proteins on different subsets of peripheral blood cells: a frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry B Clin Cytom (2006) 70:71–81. doi:10.1002/cyto.b.20087
166. Hernandez-Campo PM, Almeida J, Matarraz S, de Santiago M, Sanchez ML, Orfao A. Quantitative analysis of the expression of glycosylphosphatidylinositol-anchored proteins during the maturation of different hematopoietic cell compartments of normal bone marrow. Cytometry B Clin Cytom (2007) 72:34–42. doi:10.1002/cyto.b.20143
167. Kaisho T, Ishikawa J, Oritani K, Inazawa J, Tomizawa H, Muraoka O, et al. BST-1, a surface molecule of bone marrow stromal cell lines that facilitates pre-B-cell growth. Proc Natl Acad Sci U S A (1994) 91:5325–9. doi:10.1073/pnas.91.12.5325
168. Krupka C, Lichtenegger FS, Köhnke T, Bögeholz J, Bücklein V, Roiss M, et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget (2017) 8:35707–17. doi:10.18632/oncotarget.16060
169. Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, Knop S, et al. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma – results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica (2015) 100:377–84. doi:10.3324/haematol.2014.117945
170. Casas C. GRP78 at the centre of the stage in cancer and neuroprotection. Front Neurosci (2017) 11:177. doi:10.3389/fnins.2017.00177
171. Rasche L, Duell J, Morgner C, Chatterjee M, Hensel F, Rosenwald A, et al. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS One (2013) 8:e63414. doi:10.1371/journal.pone.0063414
172. Rasche L, Menoret E, Dubljevic V, Menu E, Vanderkerken K, Lapa C, et al. A GRP78-directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res (2016) 22:4341–9. doi:10.1158/1078-0432.CCR-15-3111
173. Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science (1997) 277:818–21. doi:10.1126/science.277.5327.818
174. Tolcher AW, Mita M, Meropol NJ, von Mehren M, Patnaik A, Padavic K, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol (2007) 25:1390–5. doi:10.1200/JCO.2006.08.8898
175. Natoni A, MacFarlane M, Inoue S, Walewska R, Majid A, Knee D, et al. TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. Br J Haematol (2007) 139:568–77. doi:10.1111/j.1365-2141.2007.06852.x
176. Menoret E, Gomez-Bougie P, Geffroy-Luseau A, Daniels S, Moreau P, Le Gouill S, et al. Mcl-1L cleavage is involved in TRAIL-R1- and TRAIL-R2-mediated apoptosis induced by HGS-ETR1 and HGS-ETR2 human mAbs in myeloma cells. Blood (2006) 108:1346–52. doi:10.1182/blood-2005-12-007971
177. Pukac L, Kanakaraj P, Humphreys R, Alderson R, Bloom M, Sung C, et al. HGS-ETR1, a fully human TRAIL-receptor 1 monoclonal antibody, induces cell death in multiple tumour types in vitro and in vivo. Br J Cancer (2005) 92:1430–41. doi:10.1038/sj.bjc.6602487
178. Georgakis GV, Li Y, Humphreys R, Andreeff M, O’Brien S, Younes M, et al. Activity of selective fully human agonistic antibodies to the TRAIL death receptors TRAIL-R1 and TRAIL-R2 in primary and cultured lymphoma cells: induction of apoptosis and enhancement of doxorubicin- and bortezomib-induced cell death. Br J Haematol (2005) 130:501–10. doi:10.1111/j.1365-2141.2005.05656.x
179. Maddipatla S, Hernandez-Ilizaliturri FJ, Knight J, Czuczman MS. Augmented antitumor activity against B-cell lymphoma by a combination of monoclonal antibodies targeting TRAIL-R1 and CD20. Clin Cancer Res (2007) 13:4556–64. doi:10.1158/1078-0432.CCR-07-0680
180. Neeson PJ, Hsu AK, Chen YR, Halse HM, Loh J, Cordy R, et al. Induction of potent NK cell-dependent anti-myeloma cytotoxic T cells in response to combined mapatumumab and bortezomib. Oncoimmunology (2015) 4:e1038011. doi:10.1080/2162402X.2015.1038011
181. Younes A, Vose JM, Zelenetz AD, Smith MR, Burris HA, Ansell SM, et al. A phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br J Cancer (2010) 103:1783–7. doi:10.1038/sj.bjc.6605987
182. Katoh M. Molecular genetics and targeted therapy of WNT-related human diseases (review). Int J Mol Med (2017) 40:587–606. doi:10.3892/ijmm.2017.3071
183. Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, et al. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin Cancer Res (2008) 14:396–404. doi:10.1158/1078-0432.CCR-07-1823
184. Choi MY, Widhopf GF II, Wu CC, Cui B, Lao F, Sadarangani A, et al. Pre-clinical specificity and safety of UC-961, a first-in-class monoclonal antibody targeting ROR1. Clin Lymphoma Myeloma Leuk (2015) 15(Suppl):S167–9. doi:10.1016/j.clml.2015.02.010
185. Yu J, Chen L, Cui B, Wu C, Choi MY, Chen Y, et al. Cirmtuzumab inhibits Wnt5a-induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia (2017) 31:1333–9. doi:10.1038/leu.2016.368
186. Aghebati-Maleki L, Younesi V, Baradaran B, Abdolalizadeh J, Motallebnezhad M, Nickho H, et al. Antiproliferative and apoptotic effects of novel anti-ROR1 single-chain antibodies in hematological malignancies. SLAS Discov (2017) 22:408–17. doi:10.1177/2472555216689659
187. Previs RA, Coleman RL, Harris AL, Sood AK. Molecular pathways: translational and therapeutic implications of the Notch signaling pathway in cancer. Clin Cancer Res (2015) 21:955–61. doi:10.1158/1078-0432.CCR-14-0809
188. Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol (2006) 26:4642–51. doi:10.1128/MCB.01655-05
189. Weng AP, Ferrando AA, Lee W, Morris JP IV, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306:269–71. doi:10.1126/science.1102160
190. Aste-Amézaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One (2010) 5:e9094. doi:10.1371/journal.pone.0009094
191. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature (2010) 464:1052–7. doi:10.1038/nature08878
192. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature (2006) 444:1083–7. doi:10.1038/nature05313
193. Keane N, Freeman C, Swords R, Giles FJ. EPHA3 as a novel therapeutic target in the hematological malignancies. Expert Rev Hematol (2012) 5:325–40. doi:10.1586/ehm.12.19
194. Swords RT, Greenberg PL, Wei AH, Durrant S, Advani AS, Hertzberg MS, et al. KB004, a first in class monoclonal antibody targeting the receptor tyrosine kinase EphA3, in patients with advanced hematologic malignancies: results from a phase 1 study. Leuk Res (2016) 50:123–31. doi:10.1016/j.leukres.2016.09.012
195. Charmsaz S, Al-Ejeh F, Yeadon TM, Miller KJ, Smith FM, Stringer BW, et al. EphA3 as a target for antibody immunotherapy in acute lymphoblastic leukemia. Leukemia (2017) 31:1779–87. doi:10.1038/leu.2016.371
196. Vidovic D, Toral JI. Selective apoptosis of neoplastic cells by the HLA-DR-specific monoclonal antibody. Cancer Lett (1998) 128:127–35. doi:10.1016/S0304-3835(98)00034-2
197. Schweighofer CD, Tuchscherer A, Sperka S, Meyer T, Rattel B, Stein S, et al. Clinical safety and pharmacological profile of the HLA-DR antibody 1D09C3 in patients with B cell chronic lymphocytic leukemia and lymphoma: results from a phase I study. Cancer Immunol Immunother (2012) 61:2367–73. doi:10.1007/s00262-012-1362-x
198. Stein R, Qu Z, Chen S, Solis D, Hansen HJ, Goldenberg DM. Characterization of a humanized IgG4 anti-HLA-DR monoclonal antibody that lacks effector cell functions but retains direct antilymphoma activity and increases the potency of rituximab. Blood (2006) 108:2736–44. doi:10.1182/blood-2006-04-017921
199. Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol (2004) 22:891–928. doi:10.1146/annurev.immunol.22.012703.104543
200. Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS. Chemokines in tumor progression and metastasis. Oncotarget (2013) 4:2171–85. doi:10.18632/oncotarget.1426
201. Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol (2012) 33:496–504. doi:10.1016/j.it.2012.05.007
202. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13:23–35. doi:10.1016/j.ccr.2007.12.004
203. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med (2004) 10:942–9. doi:10.1038/nm1093
204. Villablanca EJ, Raccosta L, Zhou D, Fontana R, Maggioni D, Negro A, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat Med (2010) 16:98–105. doi:10.1038/nm.2074
205. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328:749–52. doi:10.1126/science.1185837
206. Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol (2015) 6:12. doi:10.3389/fimmu.2015.00012
207. Klarenbeek A, Maussang D, Blancheto C, Saunders M, vander Woning S, Smit M, et al. Targeting chemokines and chemokine receptors with antibodies. Drug Discov Today Technol (2012) 9:e227–314. doi:10.1016/j.ddtec.2012.09.006
208. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol (2015) 27:11–20. doi:10.1093/intimm/dxu079
209. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol (2007) 8:639–46. doi:10.1038/ni1467
210. Bonecchi R, Bianchi G, Bordignon PP, D’Ambrosio D, Lang R, Borsatti A, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med (1998) 187:129–34. doi:10.1084/jem.187.1.129
211. Campbell JJ, Haraldsen G, Pan J, Rottman J, Qin S, Ponath P, et al. The chemokine receptor CCR4 in vascular recognition by cutaneous but not intestinal memory T cells. Nature (1999) 400:776–80. doi:10.1038/23495
212. Ishida T, Iida S, Akatsuka Y, Ishii T, Miyazaki M, Komatsu H, et al. The CC chemokine receptor 4 as a novel specific molecular target for immunotherapy in adult T-cell leukemia/lymphoma. Clin Cancer Res (2004) 10:7529–39. doi:10.1158/1078-0432.CCR-04-0983
213. Ishida T, Inagaki H, Utsunomiya A, Takatsuka Y, Komatsu H, Iida S, et al. CXC chemokine receptor 3 and CC chemokine receptor 4 expression in T-cell and NK-cell lymphomas with special reference to clinicopathological significance for peripheral T-cell lymphoma, unspecified. Clin Cancer Res (2004) 10:5494–500. doi:10.1158/1078-0432.CCR-04-0371
214. Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. J Invest Dermatol (2002) 119:1405–10. doi:10.1046/j.1523-1747.2002.19610.x
215. Ishii T, Ishida T, Utsunomiya A, Inagaki A, Yano H, Komatsu H, et al. Defucosylated humanized anti-CCR4 monoclonal antibody KW-0761 as a novel immunotherapeutic agent for adult T-cell leukemia/lymphoma. Clin Cancer Res (2010) 16:1520–31. doi:10.1158/1078-0432.CCR-09-2697
216. Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, et al. Defucosylated chimeric anti-CC chemokine receptor 4 IgG1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T-cell leukemia and lymphoma. Cancer Res (2004) 64:2127–33. doi:10.1158/0008-5472.CAN-03-2068
217. Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem (2003) 278:3466–73. doi:10.1074/jbc.M210665200
218. Yamamoto K, Utsunomiya A, Tobinai K, Tsukasaki K, Uike N, Uozumi K, et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J Clin Oncol (2010) 28:1591–8. doi:10.1200/JCO.2009.25.3575
219. Tobinai K, Takahashi T, Akinaga S. Targeting chemokine receptor CCR4 in adult T-cell leukemia-lymphoma and other T-cell lymphomas. Curr Hematol Malig Rep (2012) 7:235–40. doi:10.1007/s11899-012-0124-3
220. Kanazawa T, Hiramatsu Y, Iwata S, Siddiquey M, Sato Y, Suzuki M, et al. Anti-CCR4 monoclonal antibody mogamulizumab for the treatment of EBV-associated T- and NK-cell lymphoproliferative diseases. Clin Cancer Res (2014) 20:5075–84. doi:10.1158/1078-0432.CCR-14-0580
221. Ishida T, Ishii T, Inagaki A, Yano H, Kusumoto S, Ri M, et al. The CCR4 as a novel-specific molecular target for immunotherapy in Hodgkin lymphoma. Leukemia (2006) 20:2162–8. doi:10.1038/sj.leu.2404415
222. Kuppers R. New insights in the biology of Hodgkin lymphoma. Hematology Am Soc Hematol Educ Program (2012) 2012:328–34. doi:10.1182/asheducation-2012.1.328
223. Ni X, Jorgensen JL, Goswami M, Challagundla P, Decker WK, Kim YH, et al. Reduction of regulatory T cells by mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and Sezary syndrome. Clin Cancer Res (2015) 21:274–85. doi:10.1158/1078-0432.CCR-14-0830
224. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A (2013) 110:17945–50. doi:10.1073/pnas.1316796110
225. Chang DK, Sui J, Geng S, Muvaffak A, Bai M, Fuhlbrigge RC, et al. Humanization of an anti-CCR4 antibody that kills cutaneous T-cell lymphoma cells and abrogates suppression by T-regulatory cells. Mol Cancer Ther (2012) 11:2451–61. doi:10.1158/1535-7163.MCT-12-0278
226. Chang DK, Peterson E, Sun J, Goudie C, Drapkin RI, Liu JF, et al. Anti-CCR4 monoclonal antibody enhances antitumor immunity by modulating tumor-infiltrating Tregs in an ovarian cancer xenograft humanized mouse model. Oncoimmunology (2015) 5:e1090075. doi:10.1080/2162402X.2015.1090075
227. Han T, Abdel-Motal UM, Chang DK, Sui J, Muvaffak A, Campbell J, et al. Human anti-CCR4 minibody gene transfer for the treatment of cutaneous T-cell lymphoma. PLoS One (2012) 7:e44455. doi:10.1371/journal.pone.0044455
228. Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res (2011) 17:2074–80. doi:10.1158/1078-0432.CCR-10-2636
229. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res (2010) 16:2927–31. doi:10.1158/1078-0432.CCR-09-2329
230. Balkwill F. The significance of cancer cell expression of the chemokine receptor CXCR4. Semin Cancer Biol (2004) 14:171–9. doi:10.1016/j.semcancer.2003.10.003
231. López-Giral S, Quintana NE, Cabrerizo M, Alfonso-Pérez M, Sala-Valdés M, De Soria VG, et al. Chemokine receptors that mediate B cell homing to secondary lymphoid tissues are highly expressed in B cell chronic lymphocytic leukemia and non-Hodgkin lymphomas with widespread nodular dissemination. J Leukoc Biol (2004) 76:462–71. doi:10.1189/jlb.1203652
232. Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, et al. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell (2015) 27:755–68. doi:10.1016/j.ccell.2015.05.002
233. Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood (1998) 91:4523–30.
234. Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood (2006) 107:1761–7. doi:10.1182/blood-2005-08-3182
235. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood (2009) 113:6215–24. doi:10.1182/blood-2008-05-158311
236. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity (2006) 25:977–88. doi:10.1016/j.immuni.2006.10.016
237. Chen Y, Jacamo R, Konopleva M, Garzon R, Croce C, Andreeff M. CXCR4 downregulation of let-7a drives chemoresistance in acute myeloid leukemia. J Clin Invest (2013) 123:2395–407. doi:10.1172/JCI66553
238. Reinholdt L, Laursen MB, Schmitz A, Bødker JS, Jakobsen LH, Bøgsted M, et al. The CXCR4 antagonist plerixafor enhances the effect of rituximab in diffuse large B-cell lymphoma cell lines. Biomark Res (2016) 4:12. doi:10.1186/s40364-016-0067-2
239. Sison EA, McIntyre E, Magoon D, Brown P. Dynamic chemotherapy-induced upregulation of CXCR4 expression: a mechanism of therapeutic resistance in pediatric AML. Mol Cancer Res (2013) 11:1004–16. doi:10.1158/1541-7786.MCR-13-0114
240. Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res (2013) 19:357–66. doi:10.1158/1078-0432.CCR-12-2333
241. Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai YT, Shi J, et al. CXCR4 regulates extra-medullary myeloma through epithelial-mesenchymal-transition-like transcriptional activation. Cell Rep (2015) 12:622–35. doi:10.1016/j.celrep.2015.06.059
242. Kashyap MK, Kumar D, Jones H, Amaya-Chanaga CI, Choi MY, Melo-Cardenas J, et al. Ulocuplumab (BMS-936564/ MDX1338): a fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget (2016) 7:2809–22. doi:10.18632/oncotarget.6465
243. Becker PS, Foran JM, Altman JK, Yacoub A, Castro JE, Sabbatini P, et al. Targeting the CXCR4 pathway: safety, tolerability and clinical activity of ulocuplumab (BMS-936564), an anti-CXCR4 antibody, in relapsed/refractory acute myeloid leukemia. Blood (2014) 124:386.
244. Peng SB, Zhang X, Paul D, Kays LM, Ye M, Vaillancourt P, et al. Inhibition of CXCR4 by LY2624587, a fully humanized anti-CXCR4 antibody induces apoptosis of hematologic malignancies. PLoS One (2016) 11:e0150585. doi:10.1371/journal.pone.0150585
245. Broussas M, Boute N, Akla B, Berger S, Beau-Larvor C, Champion T, et al. A new anti-CXCR4 antibody that blocks the CXCR4/SDF-1 axis and mobilizes effector cells. Mol Cancer Ther (2016) 15:1890–9. doi:10.1158/1535-7163.MCT-16-0041
246. Kashyap MK, Amaya-Chanaga CI, Kumar D, Simmons B, Huser N, Gu Y, et al. Targeting the CXCR4 pathway using a novel anti-CXCR4 IgG1 antibody (PF-06747143) in chronic lymphocytic leukemia. J Hematol Oncol (2017) 10:112. doi:10.1186/s13045-017-0435-x
247. Zhang Y, Saavedra E, Tang R, Gu Y, Lappin P, Trajkovic D, et al. Targeting primary acute myeloid leukemia with a new CXCR4 antagonist IgG1 antibody (PF-06747143). Sci Rep (2017) 7:7305. doi:10.1038/s41598-017-07848-8
248. Macanas-Pirard P, Quezada T, Navarrete L, Broekhuizen R, Leisewitz A, Nervi B, et al. The CCL2/CCR2 axis affects transmigration and proliferation but not resistance to chemotherapy of acute myeloid leukemia cells. PLoS One (2017) 12:e0168888. doi:10.1371/journal.pone.0168888
249. Moreaux J, Hose D, Kassambara A, Reme T, Moine P, Requirand G, et al. Osteoclast-gene expression profiling reveals osteoclast-derived CCR2 chemokines promoting myeloma cell migration. Blood (2011) 117:1280–90. doi:10.1182/blood-2010-04-279760
250. Somovilla-Crespo B, Alfonso-Pérez M, Cuesta-Mateos C, Carballo-de Dios C, Beltrán AE, Terrón F, et al. Anti-CCR7 therapy exerts a potent anti-tumor activity in a xenograft model of human mantle cell lymphoma. J Hematol Oncol (2013) 6:89. doi:10.1186/1756-8722-6-89
251. Chamorro S, Vela M, Franco-Villanueva A, Carramolino L, Gutiérrez J, Gómez L, et al. Antitumor effects of a monoclonal antibody to human CCR9 in leukemia cell xenografts. MAbs (2014) 6:1000–12. doi:10.4161/mabs.29063
252. Hoellenriegel J, Zboralski D, Maasch C, Rosin NY, Wierda WG, Keating MJ, et al. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood (2014) 123:1032–9. doi:10.1182/blood-2013-03-493924
253. Dudal S, Subramanian K, Flandre T, Law WS, Lowe PJ, Skerjanec A, et al. Integrated pharmacokinetic, pharmacodynamic and immunogenicity profiling of an anti-CCL21 monoclonal antibody in cynomolgus monkeys. MAbs (2015) 7:829–37. doi:10.1080/19420862.2015.1060384
254. Jourdan M, Cren M, Robert N, Bolloré K, Fest T, Duperray C, et al. IL-6 supports the generation of human long-lived plasma cells in combination with either APRIL or stromal cell-soluble factors. Leukemia (2014) 28:1647–56. doi:10.1038/leu.2014.61
255. Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program (2011) 2011:96–103. doi:10.1182/asheducation-2011.1.96
256. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol (2010) 10:301–16. doi:10.1038/nri2761
257. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med (2006) 12:1167–74. doi:10.1038/nm1483
258. Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood (2014) 123:2691–702. doi:10.1182/blood-2013-06-511527
259. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature (2011) 473:298–307. doi:10.1038/nature10144
260. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med (2011) 17:1359–70. doi:10.1038/nm.2537
261. Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol (2003) 21:231–64. doi:10.1146/annurev.immunol.21.120601.141152
262. Avery DT, Kalled SL, Ellyard JI, Ambrose C, Bixler SA, Thien M, et al. BAFF selectively enhances the survival of plasmablasts generated from human memory B cells. J Clin Invest (2003) 112:286–97. doi:10.1172/JCI18025
263. Vincent FB, Saulep-Easton D, Figgett WA, Fairfax KA, Mackay F. The BAFF/APRIL system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev (2013) 24:203–15. doi:10.1016/j.cytogfr.2013.04.003
264. Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood (2008) 111:2755–64. doi:10.1182/blood-2007-09-110858
265. O’Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med (2004) 199:91–8. doi:10.1084/jem.20031330
266. Tai YT, Acharya C, An G, Moschetta M, Zhong MY, Feng X, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood (2016) 127:3225–36. doi:10.1182/blood-2016-01-691162
267. Tai YT, Li XF, Breitkreutz I, Song W, Neri P, Catley L, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res (2006) 66:6675–82. doi:10.1158/0008-5472.CAN-06-0190
268. Ryan MC, Hering M, Peckham D, McDonagh CF, Brown L, Kim KM, et al. Antibody targeting of B-cell maturation antigen on malignant plasma cells. Mol Cancer Ther (2007) 6:3009–18. doi:10.1158/1535-7163.MCT-07-0464
269. Guadagnoli M, Kimberley FC, Phan U, Cameron K, Vink PM, Rodermond H, et al. Development and characterization of APRIL antagonistic monoclonal antibodies for treatment of B-cell lymphomas. Blood (2011) 117:6856–65. doi:10.1182/blood-2011-01-330852
270. Neri P, Kumar S, Fulciniti MT, Vallet S, Chhetri S, Mukherjee S, et al. Neutralizing B-cell activating factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin Cancer Res (2007) 13:5903–9. doi:10.1158/1078-0432.CCR-07-0753
271. Raje NS, Faber EA Jr, Richardson PG, Schiller G, Hohl RJ, Cohen AD, et al. Phase 1 study of tabalumab, a human anti-B-cell activating factor antibody, and bortezomib in patients with relapsed/refractory multiple myeloma. Clin Cancer Res (2016) 22:5688–95. doi:10.1158/1078-0432.CCR-16-0201
272. Raje NS, Moreau P, Terpos E, Benboubker L, Grzaśko N, Holstein SA, et al. Phase 2 study of tabalumab, a human anti-B-cell activating factor antibody, with bortezomib and dexamethasone in patients with previously treated multiple myeloma. Br J Haematol (2017) 176:783–95. doi:10.1111/bjh.14483
273. Dulos J, Driessen L, Guadagnoli M, Bertens A, Hulsik DL, Tai YT, et al. Development of a first in class APRIL fully blocking antibody BION-1301 for the treatment of multiple myeloma. [2645/4]. Presented at the 2017 AACR Annual Meeting, April 3, 2017. Washington, DC (2017).
274. Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood (2014) 123:3128–38. doi:10.1182/blood-2013-10-535088
275. Hipp S, Tai YT, Blanset D, Deegen P, Wahl J, Thomas O, et al. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia (2017) 31:1743–51. doi:10.1038/leu.2016.388
276. Cheng ML, Fong L. Effects of RANKL-targeted therapy in immunity and cancer. Front Oncol (2014) 3:329. doi:10.3389/fonc.2013.00329
277. Johnson DC, Weinhold N, Mitchell J, Chen B, Stephens OW, Försti A, et al. Genetic factors influencing the risk of multiple myeloma bone disease. Leukemia (2016) 30:883–8. doi:10.1038/leu.2015.342
278. Schmiedel BJ, Scheible CA, Nuebling T, Kopp HG, Wirths S, Azuma M, et al. RANKL expression, function, and therapeutic targeting in multiple myeloma and chronic lymphocytic leukemia. Cancer Res (2013) 73:683–94. doi:10.1158/0008-5472.CAN-12-2280
279. Kostenuik PJ, Nguyen HQ, McCabe J, Warmington KS, Kurahara C, Sun N, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res (2009) 24:182–95. doi:10.1359/jbmr.081112
280. Vij R, Horvath N, Spencer A, Taylor K, Vadhan-Raj S, Vescio R, et al. An open-label, phase 2 trial of denosumab in the treatment of relapsed or plateau-phase multiple myeloma. Am J Hematol (2009) 84:650–6. doi:10.1002/ajh.21509
281. Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol (2011) 29:1125–32. doi:10.1200/JCO.2010.31.3304
282. Knudsen LM, Hippe E, Hjorth M, Holmberg E, Westin J. Renal function in newly diagnosed multiple myeloma – a demographic study of 1353 patients. The Nordic Myeloma Study Group. Eur J Haematol (1994) 53:207–12. doi:10.1111/j.1600-0609.1994.tb00190.x
283. Hu MI, Glezerman IG, Leboulleux S, Insogna K, Gucalp R, Misiorowski W, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab (2014) 99:3144–52. doi:10.1210/jc.2014-1001
284. Voorzanger N, Touitou R, Garcia E, Delecluse HJ, Rousset F, Joab I, et al. Interleukin (IL)-10 and IL-6 are produced in vivo by non-Hodgkin’s lymphoma cells and act as cooperative growth factors. Cancer Res (1996) 56:5499–505.
285. Ferrario A, Merli M, Basilico C, Maffioli M, Passamonti F. Siltuximab and hematologic malignancies. A focus in non Hodgkin lymphoma. Expert Opin Investig Drugs (2017) 26:367–73. doi:10.1080/13543784.2017.1288213
286. San-Miguel J, Blade J, Shpilberg O, Grosicki S, Maloisel F, Min CK, et al. Phase 2 randomized study of bortezomib-melphalan-prednisone with or without siltuximab (anti-IL-6) in multiple myeloma. Blood (2014) 123:4136–42. doi:10.1182/blood-2013-12-546374
287. Orlowski RZ, Gercheva L, Williams C, Sutherland H, Robak T, Masszi T, et al. A phase 2, randomized, double-blind, placebo-controlled study of siltuximab (anti-IL-6 mAb) and bortezomib versus bortezomib alone in patients with relapsed or refractory multiple myeloma. Am J Hematol (2015) 90:42–9. doi:10.1002/ajh.23868
288. Bagley CJ, Woodcock JM, Stomski FC, Lopez AF. The structural and functional basis of cytokine receptor activation: lessons from the common beta subunit of the granulocyte-macrophage colony-stimulating factor, interleukin-3 (IL-3), and IL-5 receptors. Blood (1997) 89:1471–82.
289. Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia (2000) 14:1777–84. doi:10.1038/sj.leu.2401903
290. Muñoz L, Nomdedéu JF, López O, Carnicer MJ, Bellido M, Aventín A, et al. Interleukin-3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica (2001) 86:1261–9.
291. Hope KJ, Jin L, Dick JE. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat Immunol (2004) 5:738–43. doi:10.1038/ni1080
292. Ehninger A, Kramer M, Röllig C, Thiede C, Bornhäuser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J (2014) 4:e218. doi:10.1038/bcj.2014.39
293. He SZ, Busfield S, Ritchie DS, Hertzberg MS, Durrant S, Lewis ID, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymphoma (2015) 56:1406–15. doi:10.3109/10428194.2014.956316
294. Busfield SJ, Biondo M, Wong M, Ramshaw HS, Lee EM, Ghosh S, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia (2014) 28:2213–21. doi:10.1038/leu.2014.128
295. Chen CI, Koschmieder S, Kerstiens L, Schemionek M, Altvater B, Pscherer S, et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia (2012) 26:465–74. doi:10.1038/leu.2011.239
296. Nievergall E, Ramshaw HS, Yong AS, Biondo M, Busfield SJ, Vairo G, et al. Monoclonal antibody targeting of IL-3 receptor alpha with CSL362 effectively depletes CML progenitor and stem cells. Blood (2014) 123:1218–28. doi:10.1182/blood-2012-12-475194
297. Al-Hussaini M, Rettig MP, Ritchey JK, Karpova D, Uy GL, Eissenberg LG, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood (2016) 127:122–31. doi:10.1182/blood-2014-05-575704
298. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood (2014) 123:2343–54. doi:10.1182/blood-2013-09-529537
299. Flynn MJ, Hartley JA. The emerging role of anti-CD25 directed therapies as both immune modulators and targeted agents in cancer. Br J Haematol (2017) 2017:14770. doi:10.1111/bjh.14770
300. Linden O. Remission of a refractory, anaplastic large-cell lymphoma after treatment with daclizumab. N Engl J Med (2004) 351:1466–7. doi:10.1056/NEJM200409303511424
301. Berkowitz JL, Janik JE, Stewart DM, Jaffe ES, Stetler-Stevenson M, Shih JH, et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of daclizumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma. Clin Immunol (2014) 155:176–87. doi:10.1016/j.clim.2014.09.012
302. Chaudhary VK, Queen C, Junghans RP, Waldmann TA, FitzGerald DJ, Pastan I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature (1989) 339:394–7. doi:10.1038/339394a0
303. Janik JE, Morris JC, O’Mahony D, Pittaluga S, Jaffe ES, Redon CE, et al. 90Y-daclizumab, an anti-CD25 monoclonal antibody, provided responses in 50% of patients with relapsed Hodgkin’s lymphoma. Proc Natl Acad Sci U S A (2015) 112:13045–50. doi:10.1073/pnas.1516107112
304. Yaktapour N, Übelhart R, Schüler J, Aumann K, Dierks C, Burger M, et al. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood (2013) 122:1621–33. doi:10.1182/blood-2013-02-484386
305. Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol (1990) 10:464–73. doi:10.1128/MCB.10.2.464
306. Resnicoff M, Abraham D, Yutanawiboonchai W, Rotman HL, Kajstura J, Rubin R, et al. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res (1995) 55:2463–9.
307. Himmelmann B, Terry C, Dey BR, Lopaczynski W, Nissley P. Anchorage-independent growth of fibroblasts that express a truncated IGF-I receptor. Biochem Biophys Res Commun (2001) 286:472–7. doi:10.1006/bbrc.2001.5417
308. Chapuis N, Tamburini J, Cornillet-Lefebvre P, Gillot L, Bardet V, Willems L, et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica (2010) 95:415–23. doi:10.3324/haematol.2009.010785
309. Vishwamitra D, Shi P, Wilson D, Manshouri R, Vega F, Schlette EJ, et al. Expression and effects of inhibition of type I insulin-like growth factor receptor tyrosine kinase in mantle cell lymphoma. Haematologica (2011) 96:871–80. doi:10.3324/haematol.2010.031567
310. Bieghs L, Johnsen HE, Maes K, Menu E, Van Valckenborgh E, Overgaard MT, et al. The insulin-like growth factor system in multiple myeloma: diagnostic and therapeutic potential. Oncotarget (2016) 7:48732–52. doi:10.18632/oncotarget.8982
311. Scartozzi M, Bianconi M, Maccaroni E, Giampieri R, Berardi R, Cascinu S. Dalotuzumab, a recombinant humanized mAb targeted against IGFR1 for the treatment of cancer. Curr Opin Mol Ther (2010) 12:361–71.
312. Lacy MQ, Alsina M, Fonseca R, Paccagnella ML, Melvin CL, Yin D, et al. Phase I, pharmacokinetic and pharmacodynamic study of the anti-insulinlike growth factor type 1 Receptor monoclonal antibody CP-751,871 in patients with multiple myeloma. J Clin Oncol (2008) 26:3196–203. doi:10.1200/JCO.2007.15.9319
313. Moreau P, Cavallo F, Leleu X, Hulin C, Amiot M, Descamps G, et al. Phase I study of the anti insulin-like growth factor 1 receptor (IGF-1R) monoclonal antibody, AVE1642, as single agent and in combination with bortezomib in patients with relapsed multiple myeloma. Leukemia (2011) 25:872–4. doi:10.1038/leu.2011.4
314. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer (2012) 12:335–48. doi:10.1038/nrc3257
315. Francisco-Cruz A, Aguilar-Santelises M, Ramos-Espinosa O, Mata-Espinosa D, Marquina-Castillo B, Barrios-Payan J, et al. Granulocyte-macrophage colony-stimulating factor: not just another haematopoietic growth factor. Med Oncol (2014) 31:774. doi:10.1007/s12032-013-0774-6
316. Wang J, Liu Y, Li Z, Du J, Ryu MJ, Taylor PR, et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood (2010) 116:5991–6002. doi:10.1182/blood-2010-04-281527
317. Molfino NA, Kuna P, Leff JA, Oh CK, Singh D, Chernow M, et al. Phase 2, randomised placebo-controlled trial to evaluate the efficacy and safety of an anti-GM-CSF antibody (KB003) in patients with inadequately controlled asthma. BMJ Open (2016) 6:e007709. doi:10.1136/bmjopen-2015-007709
318. Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood (2013) 121:5068–77. doi:10.1182/blood-2012-10-460170
319. Pour L, Svachova H, Adam Z, Mikulkova Z, Buresova L, Kovarova L, et al. Pretreatment hepatocyte growth factor and thrombospondin-1 levels predict response to high-dose chemotherapy for multiple myeloma. Neoplasma (2010) 57:29–34. doi:10.4149/neo_2010_01_029
320. D’Arcangelo M, Cappuzzo F. Focus on the potential role of ficlatuzumab in the treatment of non-small cell lung cancer. Biologics (2013) 7:61–8. doi:10.2147/BTT.S28908
321. Patnaik A, Weiss GJ, Papadopoulos KP, Hofmeister CC, Tibes R, Tolcher A, et al. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br J Cancer (2014) 111:272–80. doi:10.1038/bjc.2014.290
322. Rossi D, Zucchetto A, Rossi FM, Capello D, Cerri M, Deambrogi C, et al. CD49d expression is an independent risk factor of progressive disease in early stage chronic lymphocytic leukemia. Haematologica (2008) 93:1575–9. doi:10.3324/haematol.13103
323. Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, et al. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget (2016) 7:32532–42. doi:10.18632/oncotarget.8687
324. Gutjahr JC, Greil R, Hartmann TN. The role of CD44 in the pathophysiology of chronic lymphocytic leukemia. Front Immunol (2015) 6:177. doi:10.3389/fimmu.2015.00177
325. Zhang S, Wu CC, Fecteau JF, Cui B, Chen L, Zhang L, et al. Targeting chronic lymphocytic leukemia cells with a humanized monoclonal antibody specific for CD44. Proc Natl Acad Sci U S A (2013) 110:6127–32. doi:10.1073/pnas.1221841110
326. Rommer PS, Dudesek A, Stuve O, Zettl UK. Monoclonal antibodies in treatment of multiple sclerosis. Clin Exp Immunol (2014) 175:373–84. doi:10.1111/cei.12197
327. Danylesko I, Beider K, Shimoni A, Nagler A. Novel strategies for immunotherapy in multiple myeloma: previous experience and future directions. Clin Dev Immunol (2012) 2012:753407. doi:10.1155/2012/753407
328. Dal Bo M, Tissino E, Benedetti D, Caldana C, Bomben R, Poeta GD, et al. Functional and clinical significance of the integrin alpha chain CD49d expression in chronic lymphocytic leukemia. Curr Cancer Drug Targets (2016) 16:659–68. doi:10.2174/1568009616666160809102219
329. Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, et al. The selective adhesion molecule inhibitor natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol (2011) 155:438–48. doi:10.1111/j.1365-2141.2011.08864.x
330. Hsieh YT, Gang EJ, Geng H, Park E, Huantes S, Chudziak D, et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood (2013) 121:1814–8. doi:10.1182/blood-2012-01-406272
331. Fontoura P. Monoclonal antibody therapy in multiple sclerosis: paradigm shifts and emerging challenges. MAbs (2010) 2:670–81. doi:10.4161/mabs.2.6.13270
333. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov (2004) 3:391–400. doi:10.1038/nrd1381
334. Panares RL, Garcia AA. Bevacizumab in the management of solid tumors. Expert Rev Anticancer Ther (2007) 7:433–45. doi:10.1586/14737140.7.4.433
335. Ossenkoppele GJ, Stussi G, Maertens J, van Montfort K, Biemond BJ, Breems D, et al. Addition of bevacizumab to chemotherapy in acute myeloid leukemia at older age: a randomized phase 2 trial of the Dutch-Belgian Cooperative Trial Group for Hemato-Oncology (HOVON) and the Swiss Group for Clinical Cancer Research (SAKK). Blood (2012) 120:4706–11. doi:10.1182/blood-2012-04-420596
336. Bogusz J, Majchrzak A, Medra A, Cebula-Obrzut B, Robak T, Smolewski P. Mechanisms of action of the anti-VEGF monoclonal antibody bevacizumab on chronic lymphocytic leukemia cells. Postepy Hig Med Dosw (Online) (2013) 67:107–18. doi:10.5604/17322693.1038349
337. Kay NE, Bone ND, Tschumper RC, Howell KH, Geyer SM, Dewald GW, et al. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia (2002) 16:911–9. doi:10.1038/sj.leu.2402467
338. Shanafelt T, Zent C, Byrd J, Erlichman C, Laplant B, Ghosh A, et al. Phase II trials of single-agent anti-VEGF therapy for patients with chronic lymphocytic leukemia. Leuk Lymphoma (2010) 51:2222–9. doi:10.3109/10428194.2010.524327
339. Kay NE, Strati P, LaPlant BR, Leis JF, Nikcevich D, Call TG, et al. A randomized phase II trial comparing chemoimmunotherapy with or without bevacizumab in previously untreated patients with chronic lymphocytic leukemia. Oncotarget (2016) 7:78269–80. doi:10.18632/oncotarget.13412
340. Hainsworth JD, Greco FA, Raefsky EL, Thompson DS, Lunin S, Reeves J Jr, et al. Rituximab with or without bevacizumab for the treatment of patients with relapsed follicular lymphoma. Clin Lymphoma Myeloma Leuk (2014) 14:277–83. doi:10.1016/j.clml.2014.02.010
341. Stopeck AT, Unger JM, Rimsza LM, LeBlanc M, Farnsworth B, Iannone M, et al. A phase 2 trial of standard-dose cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP) and rituximab plus bevacizumab for patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: SWOG 0515. Blood (2012) 120:1210–7. doi:10.1182/blood-2012-04-423079
342. Ruan J, Gregory SA, Christos P, Martin P, Furman RR, Coleman M, et al. Long-term follow-up of R-CHOP with bevacizumab as initial therapy for mantle cell lymphoma: clinical and correlative results. Clin Lymphoma Myeloma Leuk (2014) 14:107–13. doi:10.1016/j.clml.2013.10.002
343. Rybinski K, Imtiyaz HZ, Mittica B, Drozdowski B, Fulmer J, Furuuchi K, et al. Targeting endosialin/CD248 through antibody-mediated internalization results in impaired pericyte maturation and dysfunctional tumor microvasculature. Oncotarget (2015) 6:25429–40. doi:10.18632/oncotarget.4559
344. Diaz LA Jr, Coughlin CM, Weil SC, Fishel J, Gounder MM, Lawrence S, et al. A first-in-human phase I study of MORAb-004, a monoclonal antibody to endosialin in patients with advanced solid tumors. Clin Cancer Res (2015) 21:1281–8. doi:10.1158/1078-0432.CCR-14-1829
345. Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity (2016) 44:1069–78. doi:10.1016/j.immuni.2016.04.023
346. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science (2013) 342:1432–3. doi:10.1126/science.342.6165.1432
347. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer (2017) 81:116–29. doi:10.1016/j.ejca.2017.01.035
348. Kamta J, Chaar M, Ande A, Altomare DA, Ait-Oudhia S. Advancing cancer therapy with present and emerging immuno-oncology approaches. Front Oncol (2017) 7:64. doi:10.3389/fonc.2017.00064
349. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
350. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
351. Moll M, Kuylenstierna C, Gonzalez VD, Andersson SK, Bosnjak L, Sönnerborg A, et al. Severe functional impairment and elevated PD-1 expression in CD1d-restricted NKT cells retained during chronic HIV-1 infection. Eur J Immunol (2009) 39:902–11. doi:10.1002/eji.200838780
352. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi:10.1038/nature04444
353. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192:1027–34. doi:10.1084/jem.192.7.1027
354. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99:12293–7. doi:10.1073/pnas.192461099
355. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
356. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366:2455–65. doi:10.1056/NEJMoa1200694
357. Feucht J, Kayser S, Gorodezki D, Hamieh M, Döring M, Blaeschke F, et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget (2016) 7:76902–19. doi:10.18632/oncotarget.12357
358. Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature (2011) 471:377–81. doi:10.1038/nature09754
359. Atanackovic D, Luetkens T, Kroger N. Coinhibitory molecule PD-1 as a potential target for the immunotherapy of multiple myeloma. Leukemia (2014) 28:993–1000. doi:10.1038/leu.2013.310
360. Görgün G, Samur MK, Cowens KB, Paula S, Bianchi G, Anderson JE, et al. Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res (2015) 21:4607–18. doi:10.1158/1078-0432.CCR-15-0200
361. Tai YT, Li X, Tong X, Santos D, Otsuki T, Catley L, et al. Human anti-CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res (2005) 65:5898–906. doi:10.1158/0008-5472.CAN-04-4125
362. Fanale M, Assouline S, Kuruvilla J, Solal-Céligny P, Heo DS, Verhoef G, et al. Phase IA/II, multicentre, open-label study of the CD40 antagonistic monoclonal antibody lucatumumab in adult patients with advanced non-Hodgkin or Hodgkin lymphoma. Br J Haematol (2014) 164:258–65. doi:10.1111/bjh.12630
363. Byrd JC, Kipps TJ, Flinn IW, Cooper M, Odenike O, Bendiske J, et al. Phase I study of the anti-CD40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk Lymphoma (2012) 53:2136–42. doi:10.3109/10428194.2012.681655
364. Bensinger W, Maziarz RT, Jagannath S, Spencer A, Durrant S, Becker PS, et al. A phase 1 study of lucatumumab, a fully human anti-CD40 antagonist monoclonal antibody administered intravenously to patients with relapsed or refractory multiple myeloma. Br J Haematol (2012) 159:58–66. doi:10.1111/j.1365-2141.2012.09251.x
365. Law CL, Gordon KA, Collier J, Klussman K, McEarchern JA, Cerveny CG, et al. Preclinical antilymphoma activity of a humanized anti-CD40 monoclonal antibody, SGN-40. Cancer Res (2005) 65:8331–8. doi:10.1158/0008-5472.CAN-05-0095
366. Oflazoglu E, Stone IJ, Brown L, Gordon KA, van Rooijen N, Jonas M, et al. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J Cancer (2009) 100:113–7. doi:10.1038/sj.bjc.6604812
367. Advani R, Forero-Torres A, Furman RR, Rosenblatt JD, Younes A, Ren H, et al. Phase I study of the humanized anti-CD40 monoclonal antibody dacetuzumab in refractory or recurrent non-Hodgkin’s lymphoma. J Clin Oncol (2009) 27:4371–7. doi:10.1200/JCO.2008.21.3017
368. Hussein M, Berenson JR, Niesvizky R, Munshi N, Matous J, Sobecks R, et al. A phase I multidose study of dacetuzumab (SGN-40; humanized anti-CD40 monoclonal antibody) in patients with multiple myeloma. Haematologica (2010) 95:845–8. doi:10.3324/haematol.2009.008003
369. Furman RR, Forero-Torres A, Shustov A, Drachman JG. A phase I study of dacetuzumab (SGN-40, a humanized anti-CD40 monoclonal antibody) in patients with chronic lymphocytic leukemia. Leuk Lymphoma (2010) 51:228–35. doi:10.3109/10428190903440946
370. Lewis TS, McCormick RS, Emmerton K, Lau JT, Yu SF, McEarchern JA, et al. Distinct apoptotic signaling characteristics of the anti-CD40 monoclonal antibody dacetuzumab and rituximab produce enhanced antitumor activity in non-Hodgkin lymphoma. Clin Cancer Res (2011) 17:4672–81. doi:10.1158/1078-0432.CCR-11-0479
371. Fayad L, Ansell SM, Advani R, Coiffier B, Stuart R, Bartlett NL, et al. Dacetuzumab plus rituximab, ifosfamide, carboplatin and etoposide as salvage therapy for patients with diffuse large B-cell lymphoma relapsing after rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone: a randomized, double-blind, placebo-controlled phase 2b trial. Leuk Lymphoma (2011) 56:2569–78. doi:10.3109/10428194.2015.1007504
372. Forero-Torres A, Bartlett N, Beaven A, Myint H, Nasta S, Northfelt DW, et al. Pilot study of dacetuzumab in combination with rituximab and gemcitabine for relapsed or refractory diffuse large B-cell lymphoma. Leuk Lymphoma (2013) 54:277–83. doi:10.3109/10428194.2012.710328
373. French RR, Chan HT, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med (1999) 5:548–53. doi:10.1038/8426
374. Sicard H, Bonnafous C, Morel A, Bagot M, Bensussan A, Marie-Cardine A. A novel targeted immunotherapy for CTCL is on its way: anti-KIR3DL2 mAb IPH4102 is potent and safe in non-clinical studies. Oncoimmunology (2015) 4:e1022306. doi:10.1080/2162402X.2015.1022306
375. Tesselaar K, Xiao Y, Arens R, van Schijndel GM, Schuurhuis DH, Mebius RE, et al. Expression of the murine CD27 ligand CD70 in vitro and in vivo. J Immunol (2003) 170:33–40. doi:10.4049/jimmunol.170.1.33
376. Hendriks J, Gravestein LA, Tesselaar K, van Lier RA, Schumacher TN, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1:433–40. doi:10.1038/80877
377. Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H, et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol (2002) 3:83–90. doi:10.1038/ni746
378. Ranheim EA, Cantwell MJ, Kipps TJ. Expression of CD27 and its ligand, CD70, on chronic lymphocytic leukemia B cells. Blood (1995) 85:3556–65.
379. Nilsson A, de Milito A, Mowafi F, Winberg G, Björk O, Wolpert EZ, et al. Expression of CD27-CD70 on early B cell progenitors in the bone marrow: implication for diagnosis and therapy of childhood ALL. Exp Hematol (2005) 33:1500–7. doi:10.1016/j.exphem.2005.10.005
380. Ho AW, Hatjiharissi E, Ciccarelli BT, Branagan AR, Hunter ZR, Leleu X, et al. CD27-CD70 interactions in the pathogenesis of Waldenstrom macroglobulinemia. Blood (2008) 112:4683–9. doi:10.1182/blood-2007-04-084525
381. Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. CD70+ non-Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+CD25 T cells. Blood (2007) 110:2537–44. doi:10.1182/blood-2007-03-082578
382. Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis-Mortari A, Oldenborg PA, Iizuka K, et al. CD47 (integrin-associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med (2001) 194:541–9. doi:10.1084/jem.194.4.541
383. Oldenborg PA, Gresham HD, Lindberg FP. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J Exp Med (2001) 193:855–62. doi:10.1084/jem.193.7.855
384. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell (2009) 138:286–99. doi:10.1016/j.cell.2009.05.045
385. Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138:271–85. doi:10.1016/j.cell.2009.05.046
386. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell (2017) 142:699–713. doi:10.1016/j.cell.2010.07.044
387. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman-Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res (2011) 71:1374–84. doi:10.1158/0008-5472.CAN-10-2238
388. Chao MP, Tang C, Pachynski RK, Chin R, Majeti R, Weissman IL. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood (2011) 118:4890–901. doi:10.1182/blood-2011-02-338020
389. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia (2012) 26:2538–45. doi:10.1038/leu.2012.141
390. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi:10.1182/blood-2008-09-179754
391. Overdijk MB, Verploegen S, Ortiz Buijsse A, Vink T, Leusen JH, Bleeker WK, et al. Crosstalk between human IgG isotypes and murine effector cells. J Immunol (2012) 189:3430–8. doi:10.4049/jimmunol.1200356
392. Weiskopf K, Ring AM, Ho CC, Volkmer JP, Levin AM, Volkmer AK, et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science (2013) 341:88–91. doi:10.1126/science.1238856
393. Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, et al. CD47-signal regulatory protein-alpha (SIRPalpha) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci U S A (2011) 108:18342–7. doi:10.1073/pnas.1106550108
394. Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K, et al. TTI-621 (SIRPalphaFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res (2017) 23:1068–79. doi:10.1158/1078-0432.CCR-16-1700
395. Steiner M, Neri D. Antibody-radionuclide conjugates for cancer therapy: historical considerations and new trends. Clin Cancer Res (2011) 17:6406–16. doi:10.1158/1078-0432.CCR-11-0483
Keywords: monoclonal antibody, immunotherapy, hematological malignancies, non-lineage antigens, mechanism of action
Citation: Cuesta-Mateos C, Alcaraz-Serna A, Somovilla-Crespo B and Muñoz-Calleja C (2018) Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 8:1936. doi: 10.3389/fimmu.2017.01936
Received: 23 September 2017; Accepted: 15 December 2017;
Published: 17 January 2018
Edited by:
José Mordoh, Leloir Institute Foundation (FIL), ArgentinaReviewed by:
Alessandro Poggi, Ospedale Policlinico San Martino, ItalyBipulendu Jena, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2018 Cuesta-Mateos, Alcaraz-Serna, Somovilla-Crespo and Muñoz-Calleja. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Muñoz-Calleja, Y211bm96Y0BzYWx1ZC5tYWRyaWQub3Jn