 Pappanaicken R. Kumaresan
Pappanaicken R. Kumaresan Thiago Aparecido da Silva
Thiago Aparecido da Silva Dimitrios P. Kontoyiannis
Dimitrios P. Kontoyiannis- 1Department of Pediatrics, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 2Department of Infectious Diseases, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Invasive fungal infections (IFIs) cause high rates of morbidity and mortality in immunocompromised patients. Pattern-recognition receptors present on the surfaces of innate immune cells recognize fungal pathogens and activate the first line of defense against fungal infection. The second line of defense is the adaptive immune system which involves mainly CD4+ T cells, while CD8+ T cells also play a role. CD8+ T cell-based vaccines designed to prevent IFIs are currently being investigated in clinical trials, their use could play an especially important role in acquired immune deficiency syndrome patients. So far, none of the vaccines used to treat IFI have been approved by the FDA. Here, we review current and future antifungal immunotherapy strategies involving CD8+ T cells. We highlight recent advances in the use of T cells engineered using a Sleeping Beauty vector to treat IFIs. Recent clinical trials using chimeric antigen receptor (CAR) T-cell therapy to treat patients with leukemia have shown very promising results. We hypothesized that CAR T cells could also be used to control IFI. Therefore, we designed a CAR that targets β-glucan, a sugar molecule found in most of the fungal cell walls, using the extracellular domain of Dectin-1, which binds to β-glucan. Mice treated with D-CAR+ T cells displayed reductions in hyphal growth of Aspergillus compared to the untreated group. Patients suffering from IFIs due to primary immunodeficiency, secondary immunodeficiency (e.g., HIV), or hematopoietic transplant patients may benefit from bioengineered CAR T cell therapy.
Introduction
Opportunistic invasive fungal infections (IFIs) are a major threat to the immunocompromised individual; neutropenia is a major risk factor for these infections (1, 2). Patients who require prolonged immunosuppressive therapy, for example, those who have undergone solid organ transplantation or hematopoietic stem-cell transplantation (HSCT) and those who have severe autoimmune diseases are also highly susceptible to IFIs (3–7). Other risk factors include long-term stays in an intensive care unit, the use of indwelling catheters, chemotherapy, or broad-spectrum antibiotics. The main causative agents of IFI are Aspergillus spp., Candida spp., and Cryptococcus spp. The incidence of IFI is increasing worldwide (2, 8, 9) (Table 1), and the worldwide crude mortality rate of invasive aspergillosis and invasive candidiasis has been estimated to be 0.4 deaths per 100,000 people. However, mortality rates associated with IFIs in immunocompromised patients are considerably higher, reaching 60–85% for invasive aspergillosis. The emergence of fungal strains that are resistant to currently available antifungal drugs such as polyenes, triazoles, and echinocandins poses a dangerous problem (10) and immune-based treatments are giving new hope to combat these deadly fungal infections (11–14).
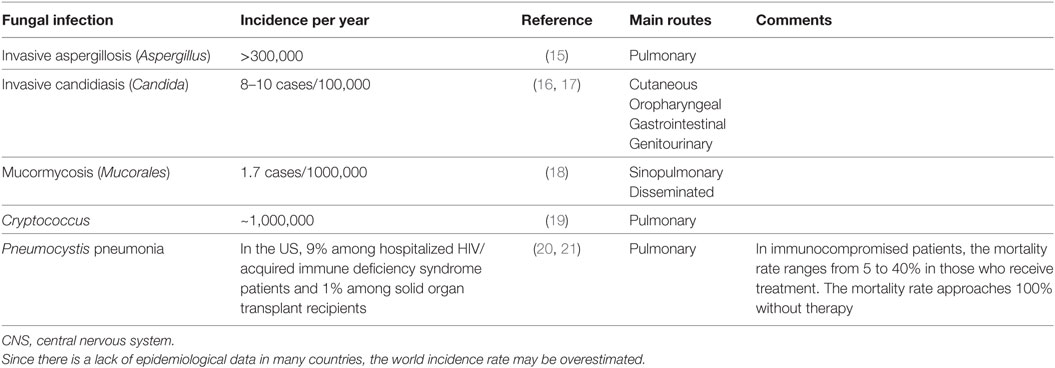
Table 1. Incidence and patterns of fungal infections worldwide.
The host response to fungal infection depends on several factors, including the host immune status, site of infection, fungal morphotype (yeast or hyphae), cell wall complexity, and virulence traits, such as the production of fungal exotoxins (22–25). The routes of various fungal infections are listed in Table 1; the majority occurs via the sinopulmonary and gastrointestinal routes (22). The host immune response to fungal infection occurs in a coordinated way via both innate and adaptive immune cells. Innate effector cells, mainly macrophages and neutrophils, are the first line of defense against inhaled fungal spores (11, 26). As a result, most initial fungal encounters go unnoticed (27). Pattern-recognition receptors (PRRs) are a family of receptors that is composed of the C-type lectin receptors (CLRs), toll-like receptors (TLRs), Nod-like receptors, and other receptors that initiate immune responses against invading fungal pathogens. Cellular expression and signaling mechanism of the PRRs have been reviewed previously (28–30).
Most of the sugars present on the fungal cell wall are recognized by the receptors from the CLR family, underscoring the constant vigil of the host innate immune system against invading fungal pathogens (28, 31–33). CLRs recognize the various carbohydrate glycoprotein components of the fungal cell wall, such as β-glucan or α-mannan, which trigger downstream signaling cascades that are essential for inducing protective immunity against fungi (34–37). When the fungal insult cannot be quickly controlled, adaptive immune cells, mainly CD4+ T cells, activate other cellular responses and antibody production. Adaptive immune cells produce cytokines to activate B cells, which in turn secrete antibodies against fungal antigens and activate the release of antimicrobial peptides from endothelial cells. Recent comprehensive reviews have already detailed the mechanisms of CD4+ T cells and surveyed current immunotherapeutic strategies to control fungal diseases (12, 38, 39). Despite having intact innate immune systems, patients with acquired immune deficiency syndrome (AIDS) are highly susceptible to fungal infections, highlighting the importance of the adaptive immune system. When CD4+ T cell counts are low, as in patients with AIDS, CD8+ T cells have a heightened role in controlling fungal infections (40). In this review, we focus on the functional role of CD8+ T cells in the immune response to fungal infections. We then discuss a new method of combating fungal infections, engineering T cells with the “Sleeping Beauty” (SB) vector system.
Current and Future Strategies to Control Fungal Infections
Drug Therapy
Antifungal drugs have had only modest success in reducing the high mortality rates associated with IFIs. In large part, this is because diagnosis of fungal infection and identification of the responsible organism is often delayed, leading to a delay in the administration of directed antifungal therapy. The use of available antifungal drugs is also restricted by their route of administration, spectrum of activity, and bioavailability in target tissues such as the brain (41). Additional issues include toxicity, undesirable drug interactions, and drug resistance. Use of the triazoles, for example, is limited by their interactions with statins, corticosteroids, and other drugs (42).
Despite tremendous improvements in the response rates of aspergillosis to modern antifungal agents, fatality rates of 40% are common in contemporary real life cohorts of unselected patients with leukemia and transplant recipients (43). The high rate of mortality following A. fumigatus infection is a result of the suboptimal diagnostic tools available, leading to late diagnosis. Other factors include rising Aspergillus resistance, and even more importantly, the relative ineffectiveness of existing antifungal drugs against established Aspergillus infections (44).
The development of effective and safe immune enhancement therapies is a major unmet need. Some patients with candidiasis struggle with poor outcomes, although this is less common in the era of widespread azole prophylaxis given to high-risk patients. Randomized controlled studies typically exclude high-risk immunosuppressed patients by use of their inclusion criteria (45).
Immunotherapy
Innate Immune Cells
Immunotherapy, which comprises cell-based therapies, such as the adoptive transfer of T cells, dendritic cells (DCs), or neutrophils, and other humoral approaches, such as antibodies and recombinant pentraxins, is a viable option for control of IFIs. Among immunocompetent individuals, the innate immunity efficiently prevents and clears IFIs (26). Alveolar macrophages are the first line of fungal defense; they recognize, phagocytize, and destroy fungal spores (46). Neutrophils also play a key role in killing fungal hyphae. They eliminate fungal hyphae by inducing an oxidative burst and by forming neutrophil extracellular traps (NETs) (47). Neutrophils utilize NETs to trap the invading pathogens by releasing chromatin fibers to form a meshwork adorned with cytoplasmic granules containing the antimicrobial enzymes myeloperoxidase, cathepsin G and neutrophil elastase that destroy trapped pathogens. The whole process is called NETosis (48).
To date, immunotherapeutic strategies to combat IFIs have primarily focused on augmenting the number of granulocytes, since these cells are known to have fungicidal activity. Granulocyte-focused immunotherapies include granulocyte transfusions (49), infusion of growth factors [granulocyte colony-stimulating factor (G-CSF), or granulocyte macrophage colony-stimulating factor (GM-CSF)] to increase granulocyte numbers (50), and the administration of cytokines such as interferon (IFN)-γ (51) and/or interleukin (IL)-15, the latter of which promotes the production of IL-8 (52), to augment phagocytic and cytotoxic function. However, the reconstitution of granulocytes is hampered by an inability to numerically expand large numbers of cells ex vivo. Moreover, after infusion, reconstituted granulocytes exhibit poor persistence owing to increased apoptosis, weak potency, and a propensity to become trapped in the pulmonary vasculature (53).
Natural Killer (NK) Cells
Natural killer cells are another type of innate immune cell reported to be involved in controlling fungal infections. NK cells make up from 5 to 15% of the peripheral blood mononuclear cells (PBMCs) of healthy individuals; the NK cell population is made up of CD56+ and CD3− cells (54). NK cells are activated when signals from activating receptors outweigh signals from inhibitory receptors, leading to cytotoxicity directed against tumor cells and virus-infected cells. NK cells also recognize infectious fungal pathogens, including A. fumigatus, C. albicans, C. neoformans, and Mucorales species (31, 55–58). Recently, CD56 has been identified as a PRR that can bind directly to both germ tubes and hyphae of Aspergillus fumigatus (59). Upon recognition, NK cells either induce lysis of these pathogens by secreting perforin and granulysin or trigger activation of other immune cells by releasing IFN-γ (60). Fungal pathogen-specific NK cell receptors and their mechanism of action has been reviewed (61, 62).
Dendritic Cells
Dendritic cells are professional antigen presenting cells (APCs) that can recognize and phagocytize fungal conidia and hyphae through PRRs and degrade them by fusing with lysosome vesicles (63). PRRs activate DCs to secrete cytokines, such as IL-12, IL-6, IL-4, and IL-1β that induce T-cell differentiation in the lymph nodes. During A. fumigatus infection, pulmonary DCs secrete IL-12 upon exposure to conidia, while IL-4 and IL-10 are secreted after exposure to hyphae. Therefore, IL-12 signaling generates a T helper (Th) 1 cell response, while IL-4 and IL-10 signaling generates a Th2 response. DCs also secrete tumor necrosis factor (TNF)-α and the chemokine CXCL8 which recruit neutrophils to the infection site (64).
In conventional DCs, β-glucan-induced Dectin-1-mediated signaling promotes secretion of the cytokines IL-2 and IL-23. The release of IL-23 induces Th17 differentiation but it is tightly regulated by IL-2 (65, 66). These data suggest that DCs direct naïve T cells to mature into functional T-cell sub types by secreting specific cytokines in the microenvironment based upon stimuli received by PRRs from different forms of fungi.
CD4+ T Cells
Even though DCs help to reduce the fungal burden to some extent through fusion with lysosome vesicles, the major function of DCs is to present fungal antigens to naive T-cells. DCs present processed antigens via major histocompatibility complex (MHC) class I or class II molecules and interact with naive T cells through formation of an immunological synapse. T cells are broadly classified into helper CD4+ T cells and cytotoxic CD8+ T cells. In fungal infections, both CD4+ and CD8+ T cells participate in the elimination of fungal pathogens (67, 68). On the basis of their function and cytokine secretion profile, CD4+ T cells are classified into several subsets: Th1, Th2, Th9, Th17, Th22, regulatory T cells, and follicular helper T cells. The activity of CD4+ T cells against fungal infection in immunocompetent individuals has been very well characterized. The most important CD4+ T cells in the antifungal immune response are the Th1 and Th17 helper T cells. After priming by DCs, CD4+ T cells differentiate into Th1 and Th17 helper T cells. Th1 helper T cells secrete the cytokines IFN-γ and TNF-α which activate innate immune cells, such as neutrophils, macrophages, DCs, and inflammatory monocytes, to fight against invading fungi and bacteria (12, 27). The cytokines secreted by Th1 cells also activate B cells, leading to the secretion of antigen-specific antibodies against fungi. IL-17 secreted by Th17 cells controls fungal infection by mobilizing neutrophils and protecting mucosal body sites by inducing epithelial cells to secrete defensin (69). IL-17 deficiency has been shown to enhance susceptibility to Candida albicans infections at mucosal sites (70).
CD8+ T Cells
Like CD4+ T cells, CD8+ T cells also have sub types, namely Tc1, Tc2, and Tc17 (Figure 1). APCs, mainly DCs, cross-present fungal antigens to CD8+ T cells. CD8+ T cells can be primed to recognize fungi by utilizing a “cross-presentation” and “cross-priming” approach, in which exogenous or fungal antigens are presented on MHC-I molecules (71). DCs internalize exogenous fungal products by CLRs and scavenger receptors for processing and presenting to MHC-I, and this process is called cross-presentation. Along with cross-presentation, some of the CLRs, for example, Dectin-1 activates DCs via Syk kinase signaling to produce IL-12, which favors Tc1 differentiation (72). Curdlan has been demonstrated to stimulate the Dectin-1-syk-CARD pathway, producing IL-23 to boost the differentiation of Th17 cells (73).
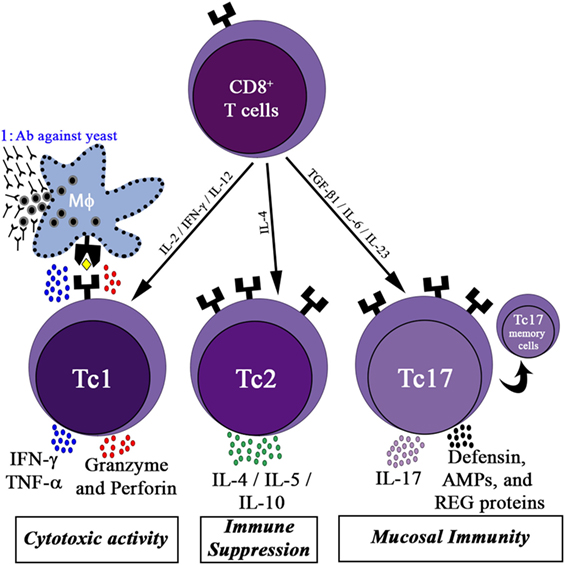
Figure 1. CD8+ T cells activity in the immune response. Differentiation of CD8+ T cells into three functional subsets: the cytotoxic cells (Tc1) cells, producing high levels of interferon (IFN)-γ, tumor necrosis factor (TNF)-α, granzyme, and perforin, which contribute to the killing of yeast infected host cells; Tc1 kills fungal infected macrophages and allows the participation of humoral immunity (marked as 1); Tc2 cells, release high amounts of interleukin (IL)-4 and IL-10, promoting immune suppression; Tc17 cells secrete IL-17, which activates mucosal immunity by inducing epithelial cells to secrete defensin, antimicrobial peptides (AMPs), and regenerating proteins (REG). Some of the activated Tc17 cells may differentiate into memory Tc17 cells.
Upon recognition of fungal peptides presented by APCs, CD8+ T cells differentiate into Tc1 cells and Tc17 cells (CD8+ T cells that secrete IL-17A), depending on the cytokines present in the environment. Several reports highlighted the role of Tc1 and Tc17 cells in protecting humans from fungal infection (74, 75). Tc1 cells act indirectly by secreting cytokines such as IFN-γ, TNF-α, and GM-CSF to activate innate immune cells such as neutrophils and macrophages involved in antifungal defense. Furthermore, Tc1 cells directly kill unresponsive fungal-infected macrophages by secreting cytotoxic factors such as perforin, granulysin, and granzyme K (76). DCs uptake fungal breakdown products from apoptotic macrophages by endocytosis to cross prime CD8+ Tc1 cells. CD8+ Tc17 cells, like CD4+ Th17 cells, secrete IL-17A cytokines to activate epithelial cells (mucosal immunity) to secrete antimicrobial products such as defensin to fight against fungal infections (Figure 1).
The Tc1 and Tc17 subtypes can be divided into effector T cells and effector memory T cells on the basis of their expression of surface receptors. The cell-surface markers used for phenotypical characterization of Tc1 and Tc17 cells are shown in Table 2. Tc1 cells that express C–X–C motif chemokine receptor 3 migrate to the lungs during pulmonary infections, such as pneumocystis (77). Tc17 cells have increased levels of effector memory phenotype markers on the cell surface (CD62Llo and CD27it/lo) as compared with Tc1 cells, suggesting that Tc17 cells may play a role in preserving long-term antifungal immunity in the host. Cytokines secreted by CD8+ Tc1 and Tc17 cells boost the innate immune system as well as the mucosal immune system to give protection from IFI (Figure 1).
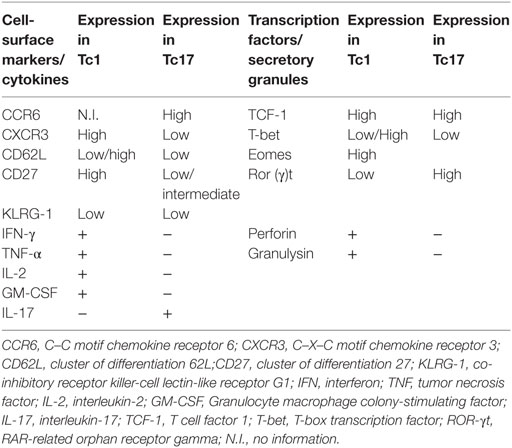
Table 2. Phenotypic characterization of CD8+ Tc1 and Tc17 cell subtypes.
CD8+ T cells share many cellular functional mechanisms with NK cells, such as releasing cytolytic granules and cytokines. As indicated above, in the absence of CD4+ T cells, CD8+ T cells also play a major role in controlling fungal infection. Immunotherapy could take advantage of several properties of CD8+ T cells, they can kill pathogen infected cells, be propagated in large numbers ex vivo, be genetically modified to recognize particular antigens, and contribute to immunologic memory.
The Role of CD8+ T Cells in the Antifungal Immune Response
The immune response to fungi elicited by CD8+ T cells can broadly classified into two processes: (1) T-cell receptor (TCR) mediated and (2) TLR and scavenger receptor mediated.
TCR-Mediated CD8+ T Cell Activation
Vaccines are a promising avenue for the treatment and prevention of IFIs (78–84) mediated through TCR receptors. The vaccine candidates developed against fungal antigens typically activate CD4+ T cells and Th17 cells. Several highly immunogenic and protective vaccine formulations for candidiasis are currently undergoing clinical trials (84, 85). It is worth noting that vaccination against fungi has mainly focused on yeast pathogens, such as Candida spp. and Cryptococcus spp. (80), and endemic mycoses that infect immunocompetent individuals, such as Coccidioides spp. (86), Blastomyces spp., and Histoplasma spp. It remains to be seen whether similar strategies will be as effective against opportunistic fungi, such as Aspergillus. In preclinical studies, vaccination using both crude and recombinant Aspergillus antigens improved the survival of immunocompromised mice following inhalation and intravenous administration of Aspergillus fumigatus (87).
Direct killing by CD8+ T cells has not been widely explored in the development of an immunotherapy against fungi, even though studies demonstrated the essential role of the CD8+ T-cell response in controlling fungal infections after vaccination (74, 75, 88–92) (Table 3). However, the presence of Aspergillus-specific CD8+ T cells has been shown in both mice and humans (93–96). Moreover, Mucorales (97) and Fusarium-specific T-cells (98) were reported in hematologic patients with IFI. Type I CD8+ T-cells (Tc1) were shown to provide protection against pneumocystis in mice (99). Preclinical studies demonstrated that the direct effect of CD8+ T cell-mediated cytotoxic activity and TNF-α and IFN-γ production were necessary to clear infected macrophages containing H. capsulatum (76), and provided full protection against coccidioidomycosis (88, 89). The activation of CD8+ T cells also contributed a protective response during Cryptococcus neoformans infection; involvement of Type 1 CD8+ T (Tc1) cells was triggered through immunization with the cytosolic proteins of the pathogens (90). Moreover, CD8+ T-cells secrete IL-17A to give protection against lethal fungal diseases, such as Blasotomyces dermatitidis and Histoplasma capsulatum, by supporting neutrophil activity (74) (Figure 1).
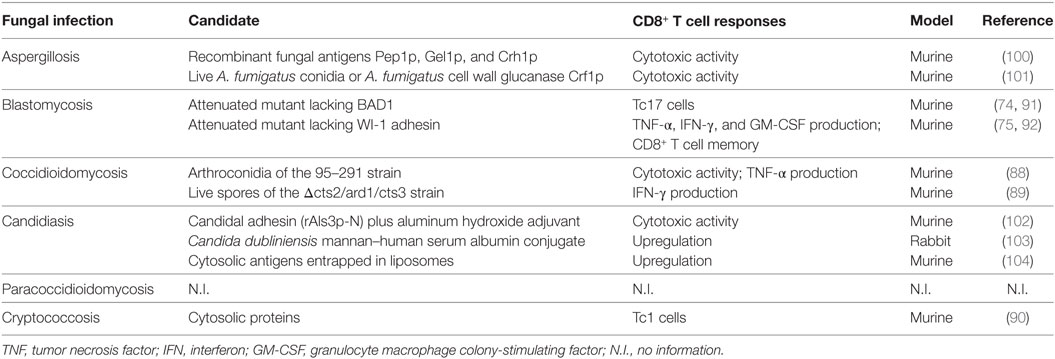
Table 3. Fungal vaccine candidates and their CD8+ T-cell mechanisms of action.
However, there are limitations for vaccine therapy. Currently, no FDA-approved vaccines are available to prevent the major opportunistic fungal infections, specifically candidiasis, aspergillosis, and cryptococcosis. Several reasons underlie the paucity of viable candidates. First, these infections are relatively uncommon, compared to viral and bacterial infections and typically occur in severely ill patients. Thus, finding sizable niche patient population who can benefit from a cost-effective vaccine strategy is difficult and not an area of priority for development by the pharmaceutical industry. Second, high-risk patients have pleiotropic and ever-evolving defects in both innate and adaptive immunity. As responses to fungi depend on both arms of the immune response, and because such responses are complex, depending on the site of infection (mucosal vs systemic infection) and the type of fungus (e.g., Candida or Cryptococcus vs a mold), much more groundwork needs to be done to decipher the key elements of a successful vaccine. In addition, there are questions regarding the efficacy and feasibility of using a vaccine in immunocompromised patients, since they are incapable of mounting a complete immune response (105).
TLR-Mediated CD8+ T Cell Activation
Toll-like receptors of the innate immune system play a major role in recognizing fungal cell wall carbohydrates, cell wall breakdown products, RNA, and DNA (13, 106–108) and thereby activate immune cells. One possible mechanism TLRs use to augment T-cell activation is when DCs activate fungal-specific CD8+ T cells by cross-presenting fungal antigens. TLR3 plays a crucial role in this process by sensing fungal RNA derived from necrotic cells and activating CD8+ memory T cells along with DCs. Indeed, TLR3−/− mice are more susceptible to Aspergillus infection than are control mice (101), and people with mutations in key TLR3 and TLR4 signaling components are susceptible to various fungal infections (109–111).
T-Cell Activation Mediated by Scavenger Receptors and Other Receptors
The scavenger receptor proteins are a highly heterogeneous set of proteins expressed on the cell surface that are involved in the uptake of modified low-density lipoproteins and a variety of microbes. One of the scavenger receptors, CD5, has been shown to bind β-glucan, a fungal cell wall sugar moiety, as well as many strains of yeast cells (112). CD5 is expressed on T cells and a small subset of mature B cells, where it associates with antigen receptors. Upon stimulation with zymosan (a protein-sugar moiety derived from the yeast cell wall), a CD5-transfected cell line produces IL-8, suggesting that CD5 has a pro-inflammatory role in fungal infection (112).
Besides TLRs, T cells have other receptors such as CD23 and CD56 for direct recognition of fungal antigens. CD23 is an inducible low-affinity receptor for immunoglobulin (Ig)E (113). It can recognize both β-glucan and α-mannan sugar moieties and thereby targets both yeast and hyphae forms of Candida (114), and upon activation, it upregulates nitric oxide production to destroy invading Candida. C-Jun N-terminal kinases (JNK1) activation suppresses the expression of CD23, which increases susceptibility of fungal infection. This was verified in JNK1 KO mice which showed resistance to Candida infection when compared to control mice (114). CD56 is a NK cell receptor that has been shown to bind to Aspergillus hyphae in a concentration-dependent manner. Blocking of CD56 reduced fungal-mediated NK cell activation (59). Activated T-cells expresses high levels of CD56 and its expression level directly correlates with T-cell effector functions (115). However, additional studies are warranted to verify that the CD56 mediated CD8+ T cells are activated during fungal infection.
Adoptive T-Cell Therapy
Over the years, several immunotherapies have been used to treat fungal infection. One such immunotherapy, adoptive T-cell therapy (ACT), is a promising therapeutic strategy not only for cancer but also for treating viral and fungal infections (38, 116–118). ACT involves the isolation and ex vivo expansion of autologous T cells in an antigen-specific manner; these expanded T cells are later infused into the patient. ACT has been shown to be effective in controlling viral infections, such as cytomegalovirus (119) and fungal infections, such as Aspergillus in HSCT patients (120). Immunocompromised patients, especially patients undergoing allogeneic HSCT, are highly susceptible to IFIs (7). The mortality rate from IFI in this patient population remains unacceptably high, partly due to the long-lasting immunosuppression in patients after HSCT (3, 121). Most IFIs in these patients occur after engraftment of the innate immune system, which suggests that adaptive cellular immunity plays a major role in controlling IFIs. In fact, adoptive transfer of CD4+ Th1 cells elicited significant protection against invasive aspergillosis in haploidentical HSCT settings (120). These findings have generated a growing interest in restoring adaptive immunity against fungal pathogens by infusing donor-derived antifungal T cells and in various ex vivo methods of propagating clinical-grade Aspergillus-specific T cells (38, 122). Recently, the FDA-approved chimeric antigen receptor (CAR) T-cell therapy to treat B-cell malignancies. CAR T cell technology can be applied to redirect T cell specificity to target fungal pathogens.
Three approaches are used to redirect T-cell specificity against a particular antigen (123).
1. Gene modification with antigen-specific TCRs in which the α and β chains of the TCR are cloned from tumor-associated antigen-specific T-cell clones (124, 125).
2. Gene modification using natural receptors other than TCRs, such as the Dectin-1 receptor (126).
3. Introduction of a CAR that recognizes tumor-associated antigens through a single-chain variable region (scFv) derived from the corresponding monoclonal antibody (127). Currently, there are no reports in the literature of scFv-derived CAR T-cells targeting fungal antigens. Therefore, in the current review, it was added for comparison analysis with D-CAR+ T cells (Figure 2).
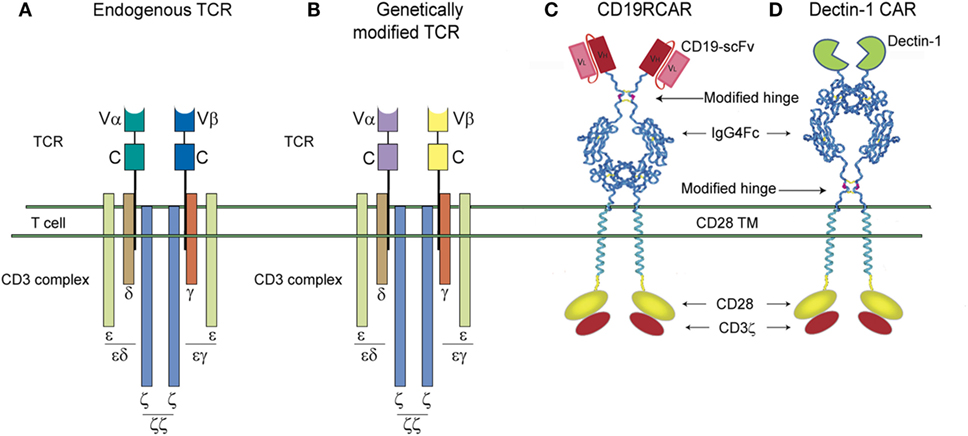
Figure 2. Schematic representations of the T-cell receptor (TCR) complex and second-generation single-chain variable region (scFv)-chimeric antigen receptor (CAR) and D-CAR. (A) Unmodified endogenous TCR complex and (B) genetically modified TCR complex. The α and β chains are highlighted in different colors. (C) The CD19R-CAR derived from a scFv region of a CD19 antigen-specific mouse monoclonal antibody and (D) the D-CAR+ derived from an extracellular domain of the Dectin-1 receptor. CD19R-CAR and D-CARs shown here have the same signaling domains, derived from costimulatory molecules, such as CD28 and CD3-ζ.
Gene Modification Using Pathogen-Specific TCRs
T-cell receptors are found on the surface of T cells as heterodimers of α and β chains and they recognize antigens presented by the MHC receptors of the APCs. For ACT, genes of tumor antigen-specific TCRs are isolated from patients and engineered into T cells using a viral or non-viral-based vector system (128, 129). These T cells are expanded ex vivo to generate large numbers for infusion. Improvements in vector design have recently increased the efficiency of this approach, and the avidity of the TCR has been improved by substituting amino acids in its complementarity determining region and introducing cysteines to form disulfide bonds, thereby preventing α- and β-chain mispairing. TCR-mediated T-cell responses to fungal antigens have been documented in both in vitro and in vivo studies (130, 131).
In colon cancer studies, TCR-driven ACT was effective in reducing tumor volume (132), but a high incidence of toxicity was reported, especially when high-avidity TCRs were used (133). Moreover, TCR-specific therapy is MHC restricted; if tumor cells lose antigen expression by downregulating MHC, they can evade the T cell attack (134). Hence, TCR-specific T-cell recognition is restricted to a single type of MHC molecule that presents the targeted antigen (135). In order to circumvent this problem, CAR-based therapy was developed. With CAR-based T-cell therapy, the tumor recognition of the CAR is not dependent on MHC (136).
Engineered CAR T-Cell Therapy
Engineered T-cell therapy involving the introduction of a CAR, which recognizes tumor-associated antigens through its scFv, is derived from the corresponding monoclonal antibody. CAR-based therapy involves the genetically engineered fusion of a variable light chain and a variable heavy chain that are specific for a cell-surface antigen and are tailored to produce an activating signal to host immune cells upon antigen engagement (137). CAR-based T-cell therapy has several key advantages. First, a CAR-based approach can be used in all tumor conditions expressing the antigen and is not MHC restricted. Second, tumor cells have no protection against CAR-based immunoediting. Finally, a varied range of tumor antigens can be targeted using this system, including glycoproteins and lipids (138, 139).
The CAR has been structurally refined over three generations of development. CAR’s structure consists of four elements: an antigen-targeting domain, an extracellular linker/spacer, a membrane-spanning (transmembrane) domain, and an intracellular signaling domain. The antigen-specific domain is usually derived from the scFv of the monoclonal antibody targeting the antigen. The linker makes the CAR flexible so that it can reach the antigen. A mutated IgG derived Fc sequence incapable of activating innate immune cells is commonly used because of its stability in expressing the CAR on the cell surface (140). In first-generation CARs, the transmembrane domain used CD4 or CD8, while CD28 was used in second-generation CARs (141). The intracellular signaling domain of second-generation CARs used CD3-ζ along with the costimulatory signaling domain CD28 (142). Tumor clearance and persistence is better in second- and third-generation CAR+ T cells than in the first generation (140, 141, 143, 144).
SB, a Non-Viral-Based Vector
Several vector systems have been used to introduce the CAR transgene into T cells. Of these, mammalian transposon/transposase-based vectors produce the most robust integration, have low immunogenicity, and allow for easy manipulation of plasmids. Multiple vectors have been studied in mammalian systems, including the SB transposon (derived from the fish, Tanichthys albonubes), the PiggyBac element (from the moth, Trichoplusia ni), Frog Prince (from the frog, Rana pipiens), Himar1 (from the horn fly, Haematobia irritans), Tol2 (from the fish, Oryzias latipes), and Passport (from the flatfish, Pleuronectes platessa) (145, 146).
Among all of the elements with activity in mammalian cells, the SB transposon is one of the most widely studied for use in gene transfer (147, 148). The SB transposase was derived by combining inactive transposase sequences from the genome of salmonid fish and then reversing the termination codon to activate transposase activity. A typical SB vector consists of 230-base pair (bp) regions containing long inverted and direct repeats (IR/DR) flanking the target gene sequence. These IR/DR sites bind with SB transposase to transfer the target gene to the host genome. In addition to the IR/DR sites, SB transposase also contains a DNA-recognition site, a nuclear localization signal, and a catalytic domain. Gene transfer using the SB transposon/transposase involves a cut-and-paste mechanism. The SB transposase protein is translated and accumulated in the cytoplasm, which is then imported into the nucleus using the nuclear localization signal. The SB transposase protein binds to the IR/DR sequence of the transposon, causing DNA breaks around the gene of interest.
The integration site of the gene cut from the SB transposon in the T cell genome depends on the presence of a TA dinucleotide site, DNA flexibility, and proximity of the donor and receiver (local hopping). More than 25% of integrations occur within a 200-bp region between the donor and receiver sites of the gene, and more than 75% of integrations are located in a single chromosome. CD19R-CAR T cells were developed using the SB vector and successfully used in clinical trials to treat acute myeloid leukemia and chronic lymphocytic leukemia (127, 149).
Dectin-1 CAR T-Cells to Target β-Glucan-Expressing Fungi
We modified this prototypical CAR to recognize carbohydrates by utilizing the pattern-recognition properties of Dectin-1 (126, 150, 151). It is specific for β-glucan, a glucose polymer consisting of β-1, 3-glucan and β-1, 6-glucan that is expressed on the cell wall of all known fungi (152–155). We hypothesized that the extracellular portion of Dectin-1 could be adapted as the specificity domain for a CAR (D-CAR) on T cells to redirect their specificity to β-glucan expressing fungi such as Aspergillus. Using the extracellular domain of Dectin-1, we engineered a CAR with specificity for the fungal cell wall sugar moiety β-glucan. This CAR was fused in frame to a modified human IgG4 hinge/Fc stalk (156), CD28 transmembrane domain, and a combination of CD28 and CD3ζ intracellular domains. This design is similar to that of our second-generation CD19-specific CAR (designated CD19RCD28), which is currently being employed in clinical trials to treat B-cell leukemia (157) (Figure 2).
Since this was the first time that a PRR was adapted to redirect T-cell specificity, we employed multiple assays [cell viability assay (XTT), cytokine production, upregulation of CD107a, and microscopy] to compare the ability of D-CAR+ T cells to target germinating Aspergillus hyphae with that of CD19-specific T cells. All of these assays demonstrated that the D-CAR activated the cytolytic machinery of the genetically modified T cells and probably their perforin/granzyme pathway as well (126). The production of IFN-γ by the D-CAR+ T cells may further augment innate immunity to IFIs. Treatment with recombinant IFN-γ or IFN-γ derived from CD4+ helper T cells or NK cells has been shown to augment anti-Aspergillus activity (55, 122). It remains to be determined whether the D-CAR-dependent production of IFN-γ contributes directly to the clearance of fungal infections or whether this activity works indirectly through the activation of granulocytes. Other cytokines, such as IL-17, may also participate in antifungal immunity. Reports indicate that IL-17 can activate neutrophils in a similar manner to IFN-γ (158), though increased levels of IL-17 are associated with mortality (153, 159–162) (Figure 3).
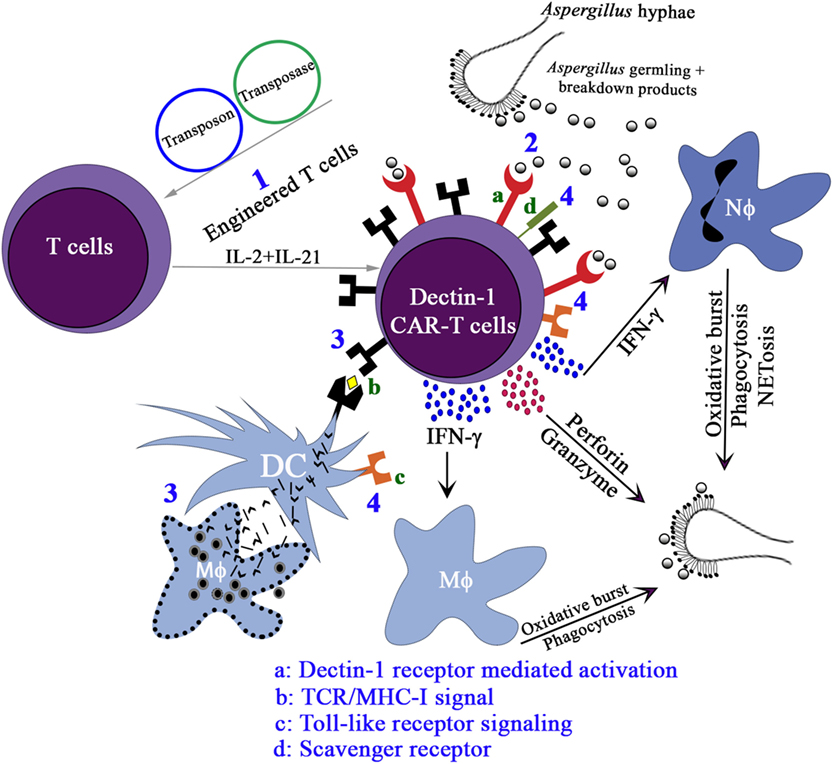
Figure 3. The proposed activation pathways of Engineered T cells. (1) Expression of D-CAR+ using Sleeping Beauty system, namely, D-CAR+ T cells; (2) the β-glucan expressing Aspergillus germlings are recognized by D-CAR+-T cells and induce the production of interferon (IFN)-γ, which favors the microbicidal activity of macrophages and neutrophils. Activated D-CAR+-T cells also secrete granzyme and perforin to degrade fungal cell walls. (3) The activation of the D-CAR+-T cells can also occur by cross-presentation of dendritic cells (DCs) and recognition by specific T-cell receptor (TCR), and (4) direct interaction of fungal breakdown products with toll-like receptors and scavenger receptor-ligands.
The use of combination therapies may supplement the antifungal efficacy of D-CAR+ T cells. For example, in Aspergillus pre-exposed to Caspofungin, β-glucan residues in the cell wall were unmasked, enhancing antifungal activity mediated by neutrophils (163). In HSCT patients, innate immune cells are present in the blood within 2 weeks of the stem-cell transplant, whereas it takes, on average, 7–12 months for NK, B, and T cells to be produced. Most IFIs occur during this period because no cellular immune system exists to support the innate immune system (7, 164). One clinical application for the add-back of donor-derived D-CAR+-T cells after allogeneic HSCT is to provide protection from IFIs by the recognition of β-glucan moieties present in all opportunistic fungi.
Bioengineered Dual CAR T Cells to Target B-Cell Leukemia and IFI
CD19-specific CAR T-cells have been used successfully to treat acute lymphoblastic leukemia (ALL) by eliminating both malignant and normal B-cells, since CD19 is also expressed on normal cells. However, total elimination of B-cells resulted in B-cell aplasia as a side effect (165). Therefore, patients undergoing CAR T cell therapy are typically given intravenous Ig to control bacterial and fungal infections. Also, high incidences of IFI are found in patients diagnosed with pediatric ALL (166, 167). These patients will gain additional benefits if CAR T cells can be engineered to destroy both IFI and tumor cells. With this goal in mind, we developed a novel gene therapy approach using dual CAR T cells to prevent IFIs such as Aspergillus and Candida and also treat B-cell leukemia. To target fungal infections, we adapted the PRR Dectin-1 to activate T cells via chimeric CD28 and CD3-ζ upon binding with β-1,3-gucan carbohydrate present in the fungal cell wall. The D-CAR+ T cells exhibited specificity for β-1,3-gucan and led to damage to fungal hyphae and inhibition of hyphal growth of Aspergillus and Candida upon testing in both in vitro and mouse models. To target B-cell leukemia, we adapted chimeric CD19R-CD28-CD3-ζ T-cells that are currently being used in clinical trials (149). The D-CAR+ T cells do not kill the yeast form of Candida so there should not be any reactions to normal commensals that live in the gut microbiota. Also, D-CAR T cells can control Aspergillus infections in the presence of immunosuppressive drugs at physiological concentrations (168). Thus, we propose utilizing the clinically appealing dual CAR T cells to control both leukemia and IFIs.
Future Directions for CAR T-Cell Therapy
The recent breakthroughs in bioengineered CAR T-cell therapy for cancer have opened up new horizons for targeting infectious disease-causing organisms, such as viruses, bacteria, and fungi. This approach promises to be especially useful in immunocompromised patients or those requiring long-term immunosuppressive drug therapy, such as solid organ transplant recipients. CAR T-cell therapy offers not only an immediate cure of the disease, but also long-term benefits because memory CAR T cells will protect the host from future attack by foreign invaders. This therapy will also give new hope to patients suffering from drug-resistant IFIs such as aspergillosis. An advantage of D-CAR T cell therapy is that it can be used with antifungal therapy such as Caspofungin and Amphotericin-B, thereby reducing drug-related toxicity such as nephrotoxicity associated with Amphotericin-B.
Several factors limit the immediate clinical applications of CAR T-cell therapy. Cytokine storm and neurotoxicity are the major side effects of CAR T-cell therapy and the good news is that now clinicians are successfully addressing these symptoms (169). Since D-CAR+-T cells are activated by the β-glucan sugar moiety which is not present in the mammalian system, off-target related toxicities may be minimal. At present, we cannot rule out the possibility of other toxicities such as macrophage activation syndrome or GvHD that are observed in CAR T therapy to treat cancers (170).
At present, CAR T-cell therapy is a personalized therapy; more CAR T-cell manufacturing centers are needed to produce clinical-grade T cells in a cost-effective way. The therapeutic success of any form of ACT depends on infusing sufficient numbers of T cells that lack replicative senescence and terminal differentiation and have the desired specificity (171). The CAR T-cell therapy used in current clinical trials requires the use of a Good Manufacturing Practice (GMP)-compliant facility to generate the T cells; it takes 2–4 weeks to propagate enough CAR T cells for infusion into the patient. However, the length of time T cells spend in culture, especially if they are propagated under non-physiological conditions, may erode the quality of the product despite increasing its quantity. Thus, a technique to generate T cells that can be harvested from peripheral blood, minimally manipulated, and infused within a few days of collection is appealing. Pharmaceutical and biotechnology companies are actively evaluating methods for generating CAR T cells in less than a week by automating the cell culture process. Automation has immediate appeal, as it avoids the expense and risk of contamination associated with prolonged culture and reduces human labor-associated error. Rapid production may in fact improve the therapeutic potential of the manufactured T cells by allowing them to avoid the replication senescence and terminal differentiation that causes them to lose in vivo persistence.
Approaches to generating T cells in compliance with GMPs are based on the ex vivo use of reagents to identify antigen-specific T cells. One approach is to use fluorescence-labeled or paramagnetic-labeled probes that bind TCRs to identify T cells with the desired specificity. The labeled T cells are subsequently subjected to fluorescence-activated cell sorting or magnetic selection to generate a homogeneously tagged product that can be immediately infused upon meeting release criteria (172, 173). The success of this approach is measured in terms of the time needed to identify antigen-specific T cells and the specificity of the harvested product. In another approach, antigen-specific T cells are isolated from donor PBMCs using a cytokine-capture system. In this process, donor PBMCs are incubated with a peptide antigen for 4 h; the activated T cells secrete IFN-γ, which is captured by a magnetic bead-conjugated bi-specific antibody. One arm of the bi-specific antibody is specific to IFN-γ, and the other arm is specific for the cell-surface CD45. T cells that secrete IFN-γ are then separated by passing them through a column. However, this approach is limited by the number of antigen-specific T cells in the donor. If more than one donor is available, prescreening of T cells (obtained from potential donors by simple venipuncture) for antigen-specific secretion of IFN-γ will determine the most suitable donor.
Despite these limitations, adoptive transfer of viral-antigen-specific T cells that have been modified for minimal manipulation and immediate infusion has been successful in clinical trials (174). TCR sequences can be identified from these antigen-specific T cells and can be used to generate TCR CAR T cells (175). Some clinical applications, such as infusion of allogeneic antigen-specific T cells after HSCT, are not possible because the initial donor may be unavailable or anonymous. In these cases, however, potential recipients may benefit from infusion of “captured” T cells from third-party donors that can recognize antigens via a human leukocyte antigen molecule shared by the recipient and the donor. These “off-the-shelf” T cells could be premanufactured and cryopreserved for infusion on demand. This approach might be better for prophylaxis in high-risk patients than for treatment in patients with recalcitrant IFIs. A precedent for this approach was reported, in which third-party Epstein–Barr virus-specific T cells and multivirus-specific T cells were infused (119, 176, 177). This approach could be adapted to treat or prevent IFIs.
Conclusion
Adoptive T-cell therapy could play a key role in controlling IFI. The GMP grade protocols for isolation of fungal-specific T cells are well characterized. Fungal-specific CD8+ T-cells protect the host by activating the host innate immune system (Tc1 mediated) and mucosal immune system (Tc17 mediated) against IFI. For direct control, D-CAR+ T-cells have been developed by fusing the extra cellular domain of Dectin-1 and cytoplasmic domains of CD3 and CD28 receptors. It can target various fungi, such as Aspergillus and Candida (126), and such treatment is highly warranted to combat IFI infections in immunocompromised patients. We have also developed Bi-specific CARs to target both B-cell malignancies and IFI by expressing CD19R-CAR and D-CAR in the same T-cell. The high costs involved with providing CAR T-cell therapy may prohibit many patients from receiving this potentially life-saving therapy, especially those located in the developing world, where fungal infections are highly prevalent. To reduce the manufacturing costs, off-the-shelf products are being developed which can be adapted for treating IFI in near future.
Author Contributions
DPK wrote the introduction and back ground. TAS wrote CD8+ T cell vaccines and PRK wrote engineered T-cells and Dectin1-CAR T cells. All authors have equally contributed for the tables. TAS and PRK contributed equally for figures.
Conflict of Interest Statement
Some of the technology described was advanced to clinic through research conducted at MD Anderson Cancer Center by PK and DK. A patent application based on research reported in this manuscript has been filed.
Acknowledgments
The authors thank Dr. Paul Hauser in the Pediatric Department and Dr. Amy Ninetto, Scientific Editor, Department of Scientific Publications for their assistance with proofreading this article.
Funding
National Institute of Allergy and Infectious Diseases grants R21 (AI127381-01), R33 (CA116127), P01 (CA148600); Burroughs Wellcome Fund; Cancer Prevention and Research Institute of Texas; CLL Global Research Foundation; DARPA (Defense Sciences Office); Department of Defense; Estate of Noelan L. Bibler; Gillson Longenbaugh Foundation; Harry T. Mangurian, Jr., Fund for Leukemia Immunotherapy; Institute of Personalized Cancer Therapy; Leukemia and Lymphoma Society; Lymphoma Research Foundation; MD Anderson Cancer Center’s Sister Institution Network Fund; Miller Foundation; Mr. Herb Simons; Mr. and Mrs. Joe H. Scales; Mr. Thomas Scott; National Foundation for Cancer Research; Pediatric Cancer Research Foundation; William Lawrence and Blanche Hughes Children’s Foundation. DK acknowledges the Texas 4000 Endowed Professorship for Cancer Research. Visiting scientist salary, Thiago Aparecido da Silva, was supported by funds received from Fundação de Amparo a Pesquisa do Estado de São Paulo (2016/23044-1).
References
1. Perfect JR, Pfaller MA, Ostrosky-Zeichner L, Kontoyiannis DP. Invasive mycoses: evolving challenges and opportunities in antifungal therapy (multimedia activity). Am J Med (2011) 124:S2–3. doi:10.1016/j.amjmed.2011.08.001
2. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol (2010) 36:1–53. doi:10.3109/10408410903241444
3. Neofytos D, Horn D, Anaissie E, Steinbach W, Olyaei A, Fishman J, et al. Epidemiology and outcome of invasive fungal infection in adult hematopoietic stem cell transplant recipients: analysis of multicenter prospective antifungal therapy (PATH) alliance registry. Clin Infect Dis (2009) 48:265–73. doi:10.1086/595846
4. Wirk B, Wingard JR. Current approaches in antifungal prophylaxis in high risk hematologic malignancy and hematopoietic stem cell transplant patients. Mycopathologia (2009) 168:299–311. doi:10.1007/s11046-009-9188-6
5. Person AK, Kontoyiannis DP, Alexander BD. Fungal infections in transplant and oncology patients. Infect Dis Clin North Am (2010) 24:439–59. doi:10.1016/j.idc.2010.01.002
6. Martino R, Subira M, Rovira M, Solano C, Vazquez L, Sanz GF, et al. Invasive fungal infections after allogeneic peripheral blood stem cell transplantation: incidence and risk factors in 395 patients. Br J Haematol (2002) 116:475–82. doi:10.1046/j.1365-2141.2002.03259.x
7. Kontoyiannis DP, Marr KA, Park BJ, Alexander BD, Anaissie EJ, Walsh TJ, et al. Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: overview of the transplant-associated infection surveillance network (TRANSNET) database. Clin Infect Dis (2010) 50:1091–100. doi:10.1086/651263
8. Groll AH, Schrey D, Tragiannidis A, Bochennek K, Lehrnbecher T. Invasive aspergillosis in children and adolescents. Curr Pharm Des (2013) 19:3545–68. doi:10.2174/13816128113199990311
9. Tragiannidis A, Roilides E, Walsh TJ, Groll AH. Invasive aspergillosis in children with acquired immunodeficiencies. Clin Infect Dis (2012) 54:258–67. doi:10.1093/cid/cir786
10. Kontoyiannis DP. Antifungal prophylaxis in hematopoietic stem cell transplant recipients: the unfinished tale of imperfect success. Bone Marrow Transplant (2011) 46:165–73. doi:10.1038/bmt.2010.256
11. Lionakis MS, Iliev ID, Hohl TM. Immunity against fungi. JCI Insight (2017) 2:93156. doi:10.1172/jci.insight.93156
12. Wuthrich M, Deepe GS, Klein B. Adaptive immunity to fungi. Annu Rev Immunol (2012) 30:115–48. doi:10.1146/annurev-immunol-020711-074958
14. Armstrong-James D, Brown GD, Netea MG, Zelante T, Gresnigt MS, van de Veerdonk FL, et al. Immunotherapeutic approaches to treatment of fungal diseases. Lancet Infect Dis (2017) 17:e393–e402. doi:10.1016/S1473-3099(17)30442-5
15. Global Action Found for Fugal Disease. Burden of Common Life-Threatening Fungal Infections. Global Action Found for Fugal Disease (2017). Available from: https://www.gaffi.org/why/fungal-disease-frequency/
16. Gudlaugsson O, Gillespie S, Lee K, Berg JV, Hu JF, Messer S, et al. Attributable mortality of nosocomial candidemia, revisited. Clin Infect Dis (2003) 37:1172–7. doi:10.1086/378745
17. Hajjeh RA, Sofair AN, Harrison LH, Lyon GM, Arthington-Skaggs BA, Mirza SA, et al. Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population-based active surveillance program. J Clin Microbiol (2004) 42:1519–27. doi:10.1128/JCM.42.4.1519-1527.2004
18. Gomes MZR, Lewis RE, Kontoyiannis DP. Mucormycosis caused by unusual mucormycetes, non-Rhizopus, -Mucor, and -Lichtheimia species. Clin Microbiol Rev (2011) 24:411. doi:10.1128/CMR.00056-10
19. Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TA. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS (2009) 23:525–30. doi:10.1097/QAD.0b013e328322ffac
20. Harris K, Maroun R, Chalhoub M, Elsayegh D. Unusual presentation of pneumocystis pneumonia in an immunocompetent patient diagnosed by open lung biopsy. Heart Lung Circ (2012) 21:221–4. doi:10.1016/j.hlc.2011.10.006
21. Harris JR, Marston BJ, Sangrujee N, DuPlessis D, Park B. Cost-effectiveness analysis of diagnostic options for pneumocystis pneumonia (PCP). PLoS One (2011) 6:e23158. doi:10.1371/journal.pone.0023158
22. Cottier F, Pavelka N. Complexity and dynamics of host-fungal interactions. Immunol Res (2012) 53:127–35. doi:10.1007/s12026-012-8265-y
23. Brunke S, Mogavero S, Kasper L, Hube B. Virulence factors in fungal pathogens of man. Curr Opin Microbiol (2016) 32:89–95. doi:10.1016/j.mib.2016.05.010
24. Gow NAR, Latge JP, Munro CA. The fungal cell wall: structure, biosynthesis, and function. Microbiol Spectr (2017) 5:1–25. doi:10.1128/microbiolspec.FUNK-0035-2016
25. Ibrahim AS. Host cell invasion in mucormycosis: role of iron. Curr Opin Microbiol (2011) 14:406–11. doi:10.1016/j.mib.2011.07.004
26. Espinosa V, Rivera A. First line of defense: innate cell-mediated control of pulmonary aspergillosis. Front Microbiol (2016) 7:272. doi:10.3389/fmicb.2016.00272
27. LeibundGut-Landmann S, Wuthrich M, Hohl TM. Immunity to fungi. Curr Opin Immunol (2012) 24:449–58. doi:10.1016/j.coi.2012.04.007
28. Plato A, Hardison SE, Brown GD. Pattern recognition receptors in antifungal immunity. Semin Immunopathol (2015) 37:97–106. doi:10.1007/s00281-014-0462-4
29. Claes AK, Zhou JY, Philpott DJ. NOD-like receptors: guardians of intestinal mucosal barriers. Physiology (Bethesda) (2015) 30:241–50. doi:10.1152/physiol.00025.2014
30. Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol (2009) 21:317–37. doi:10.1093/intimm/dxp017
31. Hoving JC, Wilson GJ, Brown GD. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol (2014) 16:185–94. doi:10.1111/cmi.12249
32. Underhill DM, Pearlman E. Immune interactions with pathogenic and commensal fungi: a two-way street. Immunity (2015) 43:845–58. doi:10.1016/j.immuni.2015.10.023
34. Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, et al. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med (2002) 196:407–12. doi:10.1084/jem.20020470
35. Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med (2009) 206:2037–51. doi:10.1084/jem.20082818
36. Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, et al. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity (2010) 32:681–91. doi:10.1016/j.immuni.2010.05.001
37. Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, Jiang YY, et al. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity (2013) 39:324–34. doi:10.1016/j.immuni.2013.05.017
38. Papadopoulou A, Kaloyannidis P, Yannaki E, Cruz CR. Adoptive transfer of Aspergillus-specific T cells as a novel anti-fungal therapy for hematopoietic stem cell transplant recipients: progress and challenges. Crit Rev Oncol Hematol (2016) 98:62–72. doi:10.1016/j.critrevonc.2015.10.005
39. Castellano-Gonzalez G, Clancy LE, Gottlieb D. Prospects for adoptive T-cell therapy for invasive fungal disease. Curr Opin Infect Dis (2017) 30(6):518–27. doi:10.1097/QCO.0000000000000403
40. van de Veerdonk FL, Netea MG. T-cell subsets and antifungal host defenses. Curr Fungal Infect Rep (2010) 4:238–43. doi:10.1007/s12281-010-0034-6
41. Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med (2014) 4:a019703. doi:10.1101/cshperspect.a019703
42. Vermeulen E, Lagrou K, Verweij PE. Azole resistance in Aspergillus fumigatus: a growing public health concern. Curr Opin Infect Dis (2013) 26:493–500. doi:10.1097/QCO.0000000000000005
43. Heo ST, Tatara AM, Jimenez-Ortigosa C, Jiang Y, Lewis RE, Tarrand J, et al. Changes in in vitro susceptibility patterns of Aspergillus to triazoles and correlation with aspergillosis outcome in a tertiary care cancer center, 1999–2015. Clin Infect Dis (2017) 65:216–25. doi:10.1093/cid/cix297
44. Kontoyiannis DP, Selleslag D, Mullane K, Cornely OA, Hope W, Lortholary O, et al. Impact of unresolved neutropenia in patients with neutropenia and invasive aspergillosis: a post hoc analysis of the SECURE trial. J Antimicrob Chemother (2017) 1–17. doi:10.1093/jac/dkx423
45. Kontoyiannis DP. Echinocandin-based initial therapy in fungemic patients with cancer: a focus on recent guidelines of the infectious diseases society of America. Clin Infect Dis (2009) 49:638–39; author reply 639–40. doi:10.1086/603585
46. Waldorf AR, Levitz SM, Diamond RD. In vivo bronchoalveolar macrophage defense against Rhizopus oryzae and Aspergillus fumigatus. J Infect Dis (1984) 150:752–60. doi:10.1093/infdis/150.5.752
47. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol (2012) 30:459–89. doi:10.1146/annurev-immunol-020711-074942
48. Kaplan MJ, Radic M. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol (2012) 189:2689–95. doi:10.4049/jimmunol.1201719
49. Mousset S, Hermann S, Klein SA, Bialleck H, Duchscherer M, Bomke B, et al. Prophylactic and interventional granulocyte transfusions in patients with haematological malignancies and life-threatening infections during neutropenia. Ann Hematol (2005) 84:734–41. doi:10.1007/s00277-005-1055-z
50. Grigull L, Beilken A, Schmid H, Kirschner P, Sykora KW, Linderkamp C, et al. Secondary prophylaxis of invasive fungal infections with combination antifungal therapy and G-CSF-mobilized granulocyte transfusions in three children with hematological malignancies. Support Care Cancer (2006) 14:783–6. doi:10.1007/s00520-005-0910-8
51. Gil-Lamaignere C, Winn RM, Simitsopoulou M, Maloukou A, Walsh TJ, Roilides E. Inteferon gamma and granulocyte-macrophage colony-stimulating factor augment the antifungal activity of human polymorphonuclear leukocytes against Scedosporium spp.: comparison with Aspergillus spp. Med Mycol (2005) 43:253–60. doi:10.1080/13693780412331271072
52. Winn RM, Gil-Lamaignere C, Roilides E, Simitsopoulou M, Lyman CA, Maloukou A, et al. Effects of interleukin-15 on antifungal responses of human polymorphonuclear leukocytes against Fusarium spp. and Scedosporium spp. Cytokine (2005) 31:1–8. doi:10.1016/j.cyto.2004.07.016
53. Valentini CG, Farina F, Pagano L, Teofili L. Granulocyte transfusions: a critical reappraisal. Biol Blood Marrow Transplant (2017) 23:2034–41. doi:10.1016/j.bbmt.2017.07.029
54. Robertson MJ, Ritz J. Biology and clinical relevance of human natural killer cells. Blood (1990) 76:2421–38.
55. Bouzani M, Ok M, McCormick A, Ebel F, Kurzai O, Morton CO, et al. Human NK cells display important antifungal activity against Aspergillus fumigatus, which is directly mediated by IFN-gamma release. J Immunol (2011) 187:1369–76. doi:10.4049/jimmunol.1003593
56. Schmidt S, Schneider A, Demir A, Lass-Florl C, Lehrnbecher T. Natural killer cell-mediated damage of clinical isolates of mucormycetes. Mycoses (2016) 59:34–8. doi:10.1111/myc.12431
57. Schmidt S, Tramsen L, Hanisch M, Latge JP, Huenecke S, Koehl U, et al. Human natural killer cells exhibit direct activity against Aspergillus fumigatus hyphae, but not against resting conidia. J Infect Dis (2011) 203:430–5. doi:10.1093/infdis/jiq062
58. Voigt J, Hunniger K, Bouzani M, Jacobsen ID, Barz D, Hube B, et al. Human natural killer cells acting as phagocytes against Candida albicans and mounting an inflammatory response that modulates neutrophil antifungal activity. J Infect Dis (2014) 209:616–26. doi:10.1093/infdis/jit574
59. Ziegler S, Weiss E, Schmitt AL, Schlegel J, Burgert A, Terpitz U, et al. CD56 is a pathogen recognition receptor on human natural killer cells. Sci Rep (2017) 7:6138. doi:10.1038/s41598-017-06238-4
60. Li SS, Kyei SK, Timm-McCann M, Ogbomo H, Jones GJ, Shi M, et al. The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe (2013) 14:387–97. doi:10.1016/j.chom.2013.09.007
61. Ogbomo H, Mody CH. Granule-dependent natural killer cell cytotoxicity to fungal pathogens. Front Immunol (2016) 7:692. doi:10.3389/fimmu.2016.00692
62. Schmidt S, Condorelli A, Koltze A, Lehrnbecher T. NK cells and their role in invasive mold infection. J Fungi (2017) 8:1623. doi:10.3389/fimmu.2017.01623
63. Fidan I, Kalkanci A, Yesilyurt E, Erdal B. In vitro effects of Candida albicans and Aspergillus fumigatus on dendritic cells and the role of beta glucan in this effect. Adv Clin Exp Med (2014) 23:17–24. doi:10.17219/acem/37016
64. Bozza S, Gaziano R, Spreca A, Bacci A, Montagnoli C, di Francesco P, et al. Dendritic cells transport conidia and hyphae of Aspergillus fumigatus from the airways to the draining lymph nodes and initiate disparate Th responses to the fungus. J Immunol (2002) 168:1362–71. doi:10.4049/jimmunol.168.3.1362
65. Thakur R, Anand R, Tiwari S, Singh AP, Tiwary BN, Shankar J. Cytokines induce effector T-helper cells during invasive aspergillosis; what we have learned about T-helper cells? Front Microbiol (2015) 6:429. doi:10.3389/fmicb.2015.00429
66. Zelante T, Wong AY, Ping TJ, Chen J, Sumatoh HR, Vigano E, et al. CD103(+) dendritic cells control Th17 cell function in the lung. Cell Rep (2015) 12:1789–801. doi:10.1016/j.celrep.2015.08.030
67. Cutler JE, Deepe GS Jr, Klein BS. Advances in combating fungal diseases: vaccines on the threshold. Nat Rev Microbiol (2007) 5:13–28. doi:10.1038/nrmicro1537
68. Chen K, Kolls JK. T cell-mediated host immune defenses in the lung. Annu Rev Immunol (2013) 31:605–33. doi:10.1146/annurev-immunol-032712-100019
69. Hernandez-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe (2012) 11:425–35. doi:10.1016/j.chom.2012.04.008
70. Huppler AR, Bishu S, Gaffen SL. Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthritis Res Ther (2012) 14:217. doi:10.1186/ar3893
71. Backer R, van Leeuwen F, Kraal G, den Haan JM. CD8-dendritic cells preferentially cross-present Saccharomyces cerevisiae antigens. Eur J Immunol (2008) 38:370–80. doi:10.1002/eji.200737647
72. Leibundgut-Landmann S, Osorio F, Brown GD, Reis e Sousa C. Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood (2008) 112:4971–80. doi:10.1182/blood-2008-05-158469
73. LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol (2007) 8:630–8. doi:10.1038/ni1460
74. Nanjappa SG, Heninger E, Wuthrich M, Gasper DJ, Klein BS. Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLoS Pathog (2012) 8:e1002771. doi:10.1371/journal.ppat.1002771
75. Nanjappa SG, Heninger E, Wuthrich M, Sullivan T, Klein B. Protective antifungal memory CD8(+) T cells are maintained in the absence of CD4(+) T cell help and cognate antigen in mice. J Clin Invest (2012) 122:987–99. doi:10.1172/JCI58762
76. Lin JS, Yang CW, Wang DW, Wu-Hsieh BA. Dendritic cells cross-present exogenous fungal antigens to stimulate a protective CD8 T cell response in infection by Histoplasma capsulatum. J Immunol (2005) 174:6282–91. doi:10.4049/jimmunol.174.10.6282
77. McAllister F, Ruan S, Steele C, Zheng M, McKinley L, Ulrich L, et al. CXCR3 and IFN protein-10 in pneumocystis pneumonia. J Immunol (2006) 177:1846–54. doi:10.4049/jimmunol.177.3.1846
78. Nanjappa SG, Klein BS. Vaccine immunity against fungal infections. Curr Opin Immunol (2014) 28:27–33. doi:10.1016/j.coi.2014.01.014
79. Reichard U, Herrmann S, Asif AR. Vaccination approaches against opportunistic fungal infections caused by Aspergillus fumigatus. Curr Protein Pept Sci (2014) 15:424–9. doi:10.2174/1389203715666140512122037
80. Chaturvedi AK, Wormley FL. Cryptococcus antigens and immune responses: implications for a vaccine. Expert Rev Vaccines (2013) 12:1261–72. doi:10.1586/14760584.2013.840094
81. Bocca AL, Amaral AC, Teixeira MM, Sato P, Shikanai-Yasuda MA, Soares Felipe MS. Paracoccidioidomycosis: eco-epidemiology, taxonomy and clinical and therapeutic issues. Future Microbiol (2013) 8:1177–91. doi:10.2217/fmb.13.68
82. Nguyen C, Barker BM, Hoover S, Nix DE, Ampel NM, Frelinger JA, et al. Recent advances in our understanding of the environmental, epidemiological, immunological, and clinical dimensions of coccidioidomycosis. Clin Microbiol Rev (2013) 26:505–25. doi:10.1128/CMR.00005-13
83. Levitz SM, Huang H, Ostroff GR, Specht CA. Exploiting fungal cell wall components in vaccines. Semin Immunopathol (2015) 37:199–207. doi:10.1007/s00281-014-0460-6
84. Edwards JE Jr. Fungal cell wall vaccines: an update. J Med Microbiol (2012) 61:895–903. doi:10.1099/jmm.0.041665-0
85. Cassone A, Casadevall A. Recent progress in vaccines against fungal diseases. Curr Opin Microbiol (2012) 15:427–33. doi:10.1016/j.mib.2012.04.004
86. Yoon HJ, Clemons KV. Vaccines against Coccidioides. Korean J Intern Med (2013) 28:403–7. doi:10.3904/kjim.2013.28.4.403
87. Ito JI, Lyons JM, Hong TB, Tamae D, Liu YK, Wilczynski SP, et al. Vaccinations with recombinant variants of Aspergillus fumigatus allergen Asp f 3 protect mice against invasive aspergillosis. Infect Immun (2006) 74:5075–84. doi:10.1128/IAI.00815-06
88. Fierer J, Waters C, Walls L. Both CD4+ and CD8+ T cells can mediate vaccine-induced protection against Coccidioides immitis infection in mice. J Infect Dis (2006) 193:1323–31. doi:10.1086/502972
89. Hung CY, Gonzalez A, Wuthrich M, Klein BS, Cole GT. Vaccine immunity to coccidioidomycosis occurs by early activation of three signal pathways of T helper cell response (Th1, Th2, and Th17). Infect Immun (2011) 79:4511–22. doi:10.1128/IAI.05726-11
90. Khan AA, Jabeen M, Chauhan A, Owais M. Vaccine potential of cytosolic proteins loaded fibrin microspheres of Cryptococcus neoformans in BALB/c mice. J Drug Target (2012) 20:453–66. doi:10.3109/1061186X.2012.685474
91. Nanjappa SG, Hernandez-Santos N, Galles K, Wuthrich M, Suresh M, Klein BS. Intrinsic MyD88-Akt1-mTOR signaling coordinates disparate Tc17 and Tc1 responses during vaccine immunity against fungal pneumonia. PLoS Pathog (2015) 11:e1005161. doi:10.1371/journal.ppat.1005161
92. Wuthrich M, Filutowicz HI, Warner T, Deepe GS Jr, Klein BS. Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune-deficient hosts. J Exp Med (2003) 197:1405–16. doi:10.1084/jem.20030109
93. Templeton SP, Buskirk AD, Law B, Green BJ, Beezhold DH. Role of germination in murine airway CD8+ T-cell responses to Aspergillus conidia. PLoS One (2011) 6:e18777. doi:10.1371/journal.pone.0018777
94. Chaudhary N, Staab JF, Marr KA. Healthy human T-cell responses to Aspergillus fumigatus antigens. PLoS One (2010) 5:e9036. doi:10.1371/journal.pone.0009036
95. Cui N, Wang H, Long Y, Liu D. CD8(+) T-cell counts: an early predictor of risk and mortality in critically ill immunocompromised patients with invasive pulmonary aspergillosis. Crit Care (2013) 17:R157. doi:10.1186/cc12836
96. Sun Z, Zhu P, Li L, Wan Z, Zhao Z, Li R. Adoptive immunity mediated by HLA-A*0201 restricted Asp f16 peptides-specific CD8+ T cells against Aspergillus fumigatus infection. Eur J Clin Microbiol Infect Dis (2012) 31:3089–96. doi:10.1007/s10096-012-1670-2
97. Potenza L, Vallerini D, Barozzi P, Riva G, Forghieri F, Zanetti E, et al. Mucorales-specific T cells emerge in the course of invasive mucormycosis and may be used as a surrogate diagnostic marker in high-risk patients. Blood (2011) 118:5416–9. doi:10.1182/blood-2011-07-366526
98. Vallerini D, Forghieri F, Lagreca I, Riva G, Barozzi P, Quadrelli C, et al. Detection of Fusarium-specific T cells in hematologic patients with invasive fusariosis. J Infect (2017) 74:314–8. doi:10.1016/j.jinf.2016.12.008
99. McAllister F, Steele C, Zheng M, Young E, Shellito JE, Marrero L, et al. T cytotoxic-1 CD8+ T cells are effector cells against pneumocystis in mice. J Immunol (2004) 172:1132–8. doi:10.4049/jimmunol.172.2.1132
100. De Luca A, Iannitti RG, Bozza S, Beau R, Casagrande A, D’Angelo C, et al. CD4(+) T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest (2012) 122:1816–31. doi:10.1172/JCI60862
101. Carvalho A, De Luca A, Bozza S, Cunha C, D’Angelo C, Moretti S, et al. TLR3 essentially promotes protective class I-restricted memory CD8(+) T-cell responses to Aspergillus fumigatus in hematopoietic transplanted patients. Blood (2012) 119:967–77. doi:10.1182/blood-2011-06-362582
102. Spellberg B, Ibrahim AS, Lin L, Avanesian V, Fu Y, Lipke P, et al. Antibody titer threshold predicts anti-candidal vaccine efficacy even though the mechanism of protection is induction of cell-mediated immunity. J Infect Dis (2008) 197:967–71. doi:10.1086/529204
103. Paulovicova E, Machova E, Tulinska J, Bystricky S. Cell and antibody mediated immunity induced by vaccination with novel Candida dubliniensis mannan immunogenic conjugate. Int Immunopharmacol (2007) 7:1325–33. doi:10.1016/j.intimp.2007.05.014
104. Chauhan A, Swaleha Z, Ahmad N, Farazuddin M, Vasco A, Abida M, et al. Escheriosome mediated cytosolic delivery of Candida albicans cytosolic proteins induces enhanced cytotoxic T lymphocyte response and protective immunity. Vaccine (2011) 29:5424–33. doi:10.1016/j.vaccine.2011.05.066
105. Hamad M. Universal fungal vaccines: could there be light at the end of the tunnel? Hum Vaccin Immunother (2012) 8:1758–63. doi:10.4161/hv.21838
106. Meier A, Kirschning CJ, Nikolaus T, Wagner H, Heesemann J, Ebel F. Toll-like receptor (TLR) 2 and TLR4 are essential for Aspergillus-induced activation of murine macrophages. Cell Microbiol (2003) 5:561–70. doi:10.1046/j.1462-5822.2003.00301.x
107. Shoham S, Huang C, Chen JM, Golenbock DT, Levitz SM. Toll-like receptor 4 mediates intracellular signaling without TNF-alpha release in response to Cryptococcus neoformans polysaccharide capsule. J Immunol (2001) 166:4620–6. doi:10.4049/jimmunol.166.7.4620
108. Calich VL, Pina A, Felonato M, Bernardino S, Costa TA, Loures FV. Toll-like receptors and fungal infections: the role of TLR2, TLR4 and MyD88 in paracoccidioidomycosis. FEMS Immunol Med Microbiol (2008) 53:1–7. doi:10.1111/j.1574-695X.2008.00378.x
109. Firinu D, Pisanu M, Piras B, Meleddu R, Lorrai MM, Manconi PE, et al. Genetic susceptibility to Candida infection: a new look at an old entity. Chin Med J (Engl) (2013) 126:378–81.
110. Nahum A, Dadi H, Bates A, Roifman CM. The biological significance of TLR3 variant, L412F, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun Rev (2012) 11:341–7. doi:10.1016/j.autrev.2011.10.007
111. Bochud PY, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, et al. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med (2008) 359:1766–77. doi:10.1056/NEJMoa0802629
112. Vera J, Fenutria R, Canadas O, Figueras M, Mota R, Sarrias MR, et al. The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc Natl Acad Sci U S A (2009) 106:1506–11. doi:10.1073/pnas.0805846106
113. Acharya M, Borland G, Edkins AL, Maclellan LM, Matheson J, Ozanne BW, et al. CD23/FcepsilonRII: molecular multi-tasking. Clin Exp Immunol (2010) 162:12–23. doi:10.1111/j.1365-2249.2010.04210.x
114. Zhao X, Guo Y, Jiang C, Chang Q, Zhang S, Luo T, et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat Med (2017) 23:337–46. doi:10.1038/nm.4260
115. Van Acker HH, Capsomidis A, Smits EL, Van Tendeloo VF. CD56 in the immune system: more than a marker for cytotoxicity? Front Immunol (2017) 8:892. doi:10.3389/fimmu.2017.00892
116. McLaughlin LP, Gottschalk S, Rooney CM, Bollard CM. EBV-directed T cell therapeutics for EBV-associated lymphomas. Methods Mol Biol (2017) 1532:255–65. doi:10.1007/978-1-4939-6655-4_19
117. Kumaresan P, Figliola M, Moyes JS, Huls MH, Tewari P, Shpall EJ, et al. Automated cell enrichment of cytomegalovirus-specific T cells for clinical applications using the cytokine-capture system. J Vis Exp (2015) 104:e52808. doi:10.3791/52808
118. Krishnamurthy J, Rabinovich BA, Mi T, Switzer KC, Olivares S, Maiti SN, et al. Genetic engineering of T cells to target HERV-K, an ancient retrovirus on melanoma. Clin Cancer Res (2015) 21:3241–51. doi:10.1158/1078-0432.CCR-14-3197
119. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol (2017) 35:3547–57. doi:10.1200/JCO.2017.73.0655
120. Perruccio K, Tosti A, Burchielli E, Topini F, Ruggeri L, Carotti A, et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood (2005) 106:4397–406. doi:10.1182/blood-2005-05-1775
121. Pagano L, Caira M, Nosari A, Van Lint MT, Candoni A, Offidani M, et al. Fungal infections in recipients of hematopoietic stem cell transplants: results of the SEIFEM B-2004 study – Sorveglianza Epidemiologica Infezioni Fungine Nelle Emopatie Maligne. Clin Infect Dis (2007) 45:1161–70. doi:10.1086/522189
122. Beck O, Topp MS, Koehl U, Roilides E, Simitsopoulou M, Hanisch M, et al. Generation of highly purified and functionally active human TH1 cells against Aspergillus fumigatus. Blood (2006) 107:2562–9. doi:10.1182/blood-2005-04-1660
123. Shi H, Liu L, Wang Z. Improving the efficacy and safety of engineered T cell therapy for cancer. Cancer Lett (2013) 328:191–7. doi:10.1016/j.canlet.2012.09.015
124. Stauss HJ, Morris EC, Abken H. Cancer gene therapy with T cell receptors and chimeric antigen receptors. Curr Opin Pharmacol (2015) 24:113–8. doi:10.1016/j.coph.2015.08.006
125. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med (2016) 22:26–36. doi:10.1038/nm.4015
126. Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci U S A (2014) 111:10660–5. doi:10.1073/pnas.1312789111
127. Singh H, Huls H, Kebriaei P, Cooper LJ. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol Rev (2014) 257:181–90. doi:10.1111/imr.12137
128. Dembic Z, Haas W, Weiss S, McCubrey J, Kiefer H, von Boehmer H, et al. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature (1986) 320:232–8. doi:10.1038/320232a0
129. Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther (2006) 13:151–9. doi:10.1016/j.ymthe.2005.07.688
130. Hohl TM. Immune responses to invasive aspergillosis: new understanding and therapeutic opportunities. Curr Opin Infect Dis (2017) 30:364–71. doi:10.1097/QCO.0000000000000381
131. Grazziutti M, Przepiorka D, Rex JH, Braunschweig I, Vadhan-Raj S, Savary CA. Dendritic cell-mediated stimulation of the in vitro lymphocyte response to Aspergillus. Bone Marrow Transplant (2001) 27:647–52. doi:10.1038/sj.bmt.1702832
132. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29:917–24. doi:10.1200/JCO.2010.32.2537
133. Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther (2011) 19:620–6. doi:10.1038/mt.2010.272
134. Garrido F, Ruiz-Cabello F, Cabrera T, Perez-Villar JJ, Lopez-Botet M, Duggan-Keen M, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today (1997) 18:89–95. doi:10.1016/S0167-5699(96)10075-X
135. Kershaw MH, Teng MW, Smyth MJ, Darcy PK. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol (2005) 5:928–40. doi:10.1038/nri1729
136. Eshhar Z. Tumor-specific T-bodies: towards clinical application. Cancer Immunol Immunother (1997) 45:131–6. doi:10.1007/s002620050415
137. Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun (1987) 149:960–8. doi:10.1016/0006-291X(87)90502-X
138. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev (2014) 257:107–26. doi:10.1111/imr.12131
139. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res (2017) 27:38–58. doi:10.1038/cr.2016.154
140. Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther (2010) 17:1206–13. doi:10.1038/gt.2010.91
141. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol (2010) 184:6938–49. doi:10.4049/jimmunol.0901766
142. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol (1998) 161:2791–7.
143. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121:1822–6. doi:10.1172/JCI46110
144. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi:10.1056/NEJMoa1103849
145. Hackett PB, Largaespada DA, Cooper LJN. A transposon and transposase system for human application. Mol Ther (2010) 18:674–83. doi:10.1038/mt.2010.2
146. Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping Beauty transposon-mediated gene therapy for prolonged expression. Adv Genet (2005) 54:189–232. doi:10.1016/S0065-2660(05)54009-4
147. Hudecek M, Izsvak Z, Johnen S, Renner M, Thumann G, Ivics Z. Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit Rev Biochem Mol Biol (2017) 52:355–80. doi:10.1080/10409238.2017.1304354
148. Kebriaei P, Izsvak Z, Narayanavari SA, Singh H, Ivics Z. Gene therapy with the Sleeping Beauty transposon system. Trends Genet (2017) 33:852–70. doi:10.1016/j.tig.2017.08.008
149. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest (2016) 126:3363–76. doi:10.1172/JCI86721
150. Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med (2003) 197:1119–24. doi:10.1084/jem.20021890
151. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol (2009) 10:203–13. doi:10.1038/ni.1692
152. Bowman SM, Free SJ. The structure and synthesis of the fungal cell wall. Bioessays (2006) 28:799–808. doi:10.1002/bies.20441
153. Werner JL, Metz AE, Horn D, Schoeb TR, Hewitt MM, Schwiebert LM, et al. Requisite role for the dectin-1 beta-glucan receptor in pulmonary defense against Aspergillus fumigatus. J Immunol (2009) 182:4938–46. doi:10.4049/jimmunol.0804250
154. Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, et al. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol (2002) 169:3876–82. doi:10.4049/jimmunol.169.7.3876
155. Willment JA, Marshall AS, Reid DM, Williams DL, Wong SY, Gordon S, et al. The human beta-glucan receptor is widely expressed and functionally equivalent to murine Dectin-1 on primary cells. Eur J Immunol (2005) 35:1539–47. doi:10.1002/eji.200425725
156. Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood (2003) 101:1637–44. doi:10.1182/blood-2002-07-1989
157. Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res (2008) 68:2961–71. doi:10.1158/0008-5472.CAN-07-5600
158. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28:454–67. doi:10.1016/j.immuni.2008.03.004
159. Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, et al. IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol (2007) 37:2695–706. doi:10.1002/eji.200737409
160. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell (2006) 126:1121–33. doi:10.1016/j.cell.2006.07.035
161. Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol (2007) 19:409–17. doi:10.1016/j.smim.2007.10.011
162. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity (2008) 28:29–39. doi:10.1016/j.immuni.2007.11.016
163. Lamaris GA, Lewis RE, Chamilos G, May GS, Safdar A, Walsh TJ, et al. Caspofungin-mediated beta-glucan unmasking and enhancement of human polymorphonuclear neutrophil activity against Aspergillus and non-Aspergillus hyphae. J Infect Dis (2008) 198:186–92. doi:10.1086/589305
164. Ogonek J, Kralj Juric M, Ghimire S, Varanasi PR, Holler E, Greinix H, et al. Immune reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol (2016) 7:507. doi:10.3389/fimmu.2016.00507
165. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics (2016) 3:16011. doi:10.1038/mto.2016.11
166. Pana ZD, Roilides E, Warris A, Groll AH, Zaoutis T. Epidemiology of invasive fungal disease in children. J Pediatric Infect Dis Soc (2017) 6:S3–11. doi:10.1093/jpids/pix046
167. Bochennek K, Hassler A, Perner C, Gilfert J, Schoning S, Klingebiel T, et al. Infectious complications in children with acute myeloid leukemia: decreased mortality in multicenter trial AML-BFM 2004. Blood Cancer J (2016) 6:e382. doi:10.1038/bcj.2015.110
168. Pappanaicken RK, Nathaniel A, Harjeet S, Simon O, Sourindra NM, Tiejuan M, et al. Bioengineered Dectin-1 CAR+ T cells to control invasive fungal infection. Inaugural International Cancer Immunotherapy Conference. New York (2016). Abstract A193.
169. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127:3321–30. doi:10.1182/blood-2016-04-703751
170. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T-cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol (2018) 15:47–62. doi:10.1038/nrclinonc.2017.148
171. DiGiusto D, Cooper L. Preparing clinical grade Ag-specific T cells for adoptive immunotherapy trials. Cytotherapy (2007) 9:613–29. doi:10.1080/14653240701650320
172. Yee C. Adoptive T-cell therapy for cancer: boutique therapy or treatment modality? Clin Cancer Res (2013) 19:4550–2. doi:10.1158/1078-0432.CCR-13-1367
173. Casati A, Varghaei-Nahvi A, Feldman SA, Assenmacher M, Rosenberg SA, Dudley ME, et al. Clinical-scale selection and viral transduction of human na < ve and central memory CD8(+) T cells for adoptive cell therapy of cancer patients. Cancer Immunol Immunother (2013) 62:1563–73. doi:10.1007/s00262-013-1459-x
174. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood (2013) 121:5113–23. doi:10.1182/blood-2013-02-486324
175. Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol (2011) 29:550–7. doi:10.1016/j.tibtech.2011.04.009
176. Cruz CRY, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood (2013) 122:2965–73. doi:10.1182/blood-2013-06-506741
Keywords: fungal infection, immunotherapy, chimeric antigen receptor, D-CAR+ T cells, cell therapy, Sleeping Beauty, CD8+ T cells, adoptive T cell therapy
Citation: Kumaresan PR, da Silva TA and Kontoyiannis DP (2018) Methods of Controlling Invasive Fungal Infections Using CD8+ T Cells. Front. Immunol. 8:1939. doi: 10.3389/fimmu.2017.01939
Received: 31 October 2017; Accepted: 15 December 2017;
Published: 08 January 2018
Edited by:
Steven Templeton, Indiana University School of Medicine, United StatesReviewed by:
Ana Serezani, Vanderbilt University Medical Center, United StatesAgostinho Carvalho, University of Minho, Portugal
Copyright: © 2018 Kumaresan, da Silva and Kontoyiannis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pappanaicken R. Kumaresan, cGt1bWFyZXNhbkBtZGFuZGVyc29uLm9yZw==;
Dimitrios P. Kontoyiannis, ZGtvbnRveWlAbWRhbmRlcnNvbi5vcmc=