 Wim Adriaensen
Wim Adriaensen Thomas P. C. Dorlo
Thomas P. C. Dorlo Guido Vanham
Guido Vanham Luc Kestens4
Luc Kestens4 Paul M. Kaye
Paul M. Kaye Johan van Griensven
Johan van Griensven- 1Unit of HIV and Neglected Tropical Diseases, Department of Clinical Sciences, Institute of Tropical Medicine, Antwerp, Belgium
- 2Department of Pharmacy and Pharmacology, Antoni van Leeuwenhoek Hospital, Netherlands Cancer Institute, Amsterdam, Netherlands
- 3Unit of Virology, Department of Biomedical Sciences, Institute of Tropical Medicine, Antwerp, Belgium
- 4Unit of Immunology, Department of Biomedical Sciences, Institute of Tropical Medicine, Antwerp, Belgium
- 5Centre for Immunology and Infection, Department of Biology, Hull York Medical School, University of York, Heslington, York, United Kingdom
Patients with visceral leishmaniasis (VL)–human immunodeficiency virus (HIV) coinfection experience increased drug toxicity and treatment failure rates compared to VL patients, with more frequent VL relapse and death. In the era of VL elimination strategies, HIV coinfection is progressively becoming a key challenge, because HIV-coinfected patients respond poorly to conventional VL treatment and play an important role in parasite transmission. With limited chemotherapeutic options and a paucity of novel anti-parasitic drugs, new interventions that target host immunity may offer an effective alternative. In this review, we first summarize current views on how VL immunopathology is significantly affected by HIV coinfection. We then review current clinical and promising preclinical immunomodulatory interventions in the field of VL and discuss how these may operate in the context of a concurrent HIV infection. Caveats are formulated as these interventions may unpredictably impact the delicate balance between boosting of beneficial VL-specific responses and deleterious immune activation/hyperinflammation, activation of latent provirus or increased HIV-susceptibility of target cells. Evidence is lacking to prioritize a target molecule and a more detailed account of the immunological status induced by the coinfection as well as surrogate markers of cure and protection are still required. We do, however, argue that virologically suppressed VL patients with a recovered immune system, in whom effective antiretroviral therapy alone is not able to restore protective immunity, can be considered a relevant target group for an immunomodulatory intervention. Finally, we provide perspectives on the translation of novel theories on synergistic immune cell cross-talk into an effective treatment strategy for VL–HIV-coinfected patients.
Introduction
Visceral leishmaniasis (VL), also called kala-azar, is a vector-borne protozoan infection caused by species of the Leishmania donovani complex, which mainly targets tissue macrophages of systemic organs, such as spleen, liver, and bone marrow (1). Characteristics of the disease include chronic fever, hepatosplenomegaly, and pancytopenia (1). Untreated, overt disease is universally lethal (1). Zoonotic VL, with dogs as the main reservoir, is mainly prevalent in the Mediterranean basin and in South America, and is caused by Leishmania infantum. Anthroponotic VL is prevalent on the Indian subcontinent and in East Africa and is typically caused by L. donovani (2). According to the recent World Health Organization (WHO) report, VL is endemic in 75 countries with an estimated 50,000–90,000 new cases occurring each year (3). Ninety percent of the global disease burden occurs in just six countries: India, Bangladesh, Sudan, South Sudan, Brazil, and Ethiopia (3).
Chemotherapy is currently the sole form of treatment in clinical practice. The pentavalent antimonial (SbV) compounds [sodium stibogluconate (SSG) commercialized as Pentostam®; meglumine antimoniate commercialized as Glucantime®] have been the cornerstone of first-line treatment of VL over the last 70 years. However, these compounds are far from optimal due to severe toxicity and the emergence of antimonial resistance on the Indian subcontinent (1, 4). Newer drugs that are increasingly used include paromomycin, miltefosine, pentamidine, and conventional and liposomal amphotericin B. All these drugs have several important disadvantages as shown in Table 1. While various combination therapy regimens designed to overcome some of the shortcomings are highly efficacious in India, disappointing findings on some combination regimens have been recently reported in East Africa (5–10). As of today, no comparative studies have been conducted to explain this geographical difference, but parasite genetic diversity and host immune phenotypes are assumed as key factors. Novel chemotherapeutic drugs are in the initial development pipeline and are, therefore, unlikely to be widely available within the next few years. Nevertheless, over 90–95% of immunocompetent patients display a good clinical response to currently recommended conventional treatment regimens, with treatment unresponsiveness, death or severe toxicity observed in less than 5–10% of patients (11). Less than 5% of immunocompetent individuals who initially cure develop a relapse, most commonly within 6–12 months after treatment (5). Treatment outcomes, however, vary substantially between different geographic regions and depend on the drug(s) used, drug exposure, parasite susceptibility to the drug, severity of disease, host immunity, and the presence of coinfections (11–13).
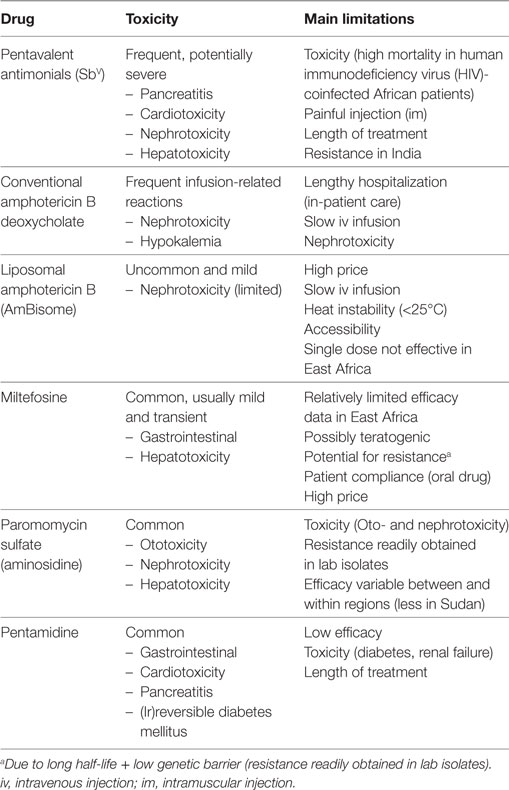
Table 1. The main drugs currently used for treatment of visceral leishmaniasis (VL), adapted from Ref. (5).
Emerging Challenge of VL–HIV Coinfection
Human immunodeficiency virus (HIV) has been identified as one of the emerging challenges facing the control of VL (14). The immunological status of HIV-infected patients is particularly favorable for the multiplication of Leishmania parasites. HIV coinfection substantially increases the risk of progression from asymptomatic Leishmania infection to active disease (14, 15). On the other hand, VL accelerates HIV disease progression towards acquired immunodeficiency syndrome (AIDS) and could induce expression of latent proviruses (14). HIV has fueled the re-emergence of VL in Southern Europe and Brazil, where up to 70% of VL cases are associated with HIV infection (7). The problem is currently particularly severe in areas such as Northern Ethiopia, where up to 30% of all VL patients are coinfected with HIV (16). Since 2001, 35 countries have reported between 2 and 30% of VL cases as coinfected with HIV, but these percentages are most probably underestimations (14). Because the disease affects the most poor and most neglected patients within an already neglected disease population, under-reporting in most endemic areas is common due to a lack of facilities to diagnose one or both of the diseases and to poor reporting systems. Importantly, VL–HIV-coinfected patients are also often considered super-spreaders of VL and, thus, pose a major threat to current elimination strategies (17).
Since 1996, combined antiretroviral treatment (cART), comprising three antiretroviral drugs, constitutes the cornerstone of HIV treatment. The treatment options continue to expand with new drugs and co-formulations; by the end of 2016, there were 40 antiretroviral drugs from six different classes approved by the Food and Drug Administration. In most resource-constrained settings, the standardized WHO guidelines are used for ART, which currently recommends a combination of tenofovir, lamivudine, and efavirenz as first-line treatment. WHO recommended first-line regimens have been found highly effective in resource-constrained settings (18). The main aim of cART is sustainable suppression of HIV replication, and with good adherence, this can generally be achieved, leading to a close to normal life expectancy (19).
Visceral leishmaniasis is one of the AIDS-defining conditions, requiring anti-leishmanial treatment and cART irrespective of CD4+ T cell count (7). Although there are limited in vitro data suggesting that HIV-1 protease inhibitors and possibly some other antiretroviral drugs might directly exert inhibitory effects on Leishmania, there is insufficient evidence for their clinical use against VL, and standard ART regimens are currently recommended in VL–HIV coinfection (5). In low income countries, this is provided by standardized first- and second-line regimens in a public health approach (20, 21).
Increased toxicity and parasitologically confirmed treatment failures (up to 30%) were observed in VL–HIV-coinfected patients treated with SbV, with case fatality rates up to 24% (14, 16, 22). While liposomal amphotericin B was consistently found to have excellent tolerability, VL cure rates in HIV-coinfected individuals have been rather disappointing in East Africa. For example, at a total dose of 30 mg/kg, around 16% of primary VL and 56% of VL relapse cases demonstrate parasitological failure in northern Ethiopia (16). WHO now proposes a total dose of 40 mg/kg (7, 23, 24). Experience with miltefosine in VL–HIV coinfection is limited, but suggests moderate efficacy and an acceptable toxicity profile (22, 25–28). To date, only one clinical trial in HIV-coinfected patients has been conducted with miltefosine, with 18% of patients displaying initial parasitological treatment failure and 25% relapsing, although deaths were excluded (22). The role of combination therapy in VL–HIV coinfection is currently under exploration in clinical trials in India and East Africa.
While in Europe widespread use of cART has resulted in a pronounced (i.e., 60%) reduction in the incidence of VL–HIV coinfection, relapse in coinfected subjects remains substantial at up to 60% after 1 year (14, 29, 30) and secondary prophylaxis has only a partial effect (31). In a pentamidine secondary prophylaxis trial in Ethiopia, the relapse-free survival rate at 2 years was only 58.3% (32). Even with access to all current chemotherapies, the prognosis in VL–HIV coinfection remains dire. Currently, it is believed that VL can only be effectively treated in HIV patients before profound immune deficiency has developed.
Visceral leishmaniasis–HIV coinfection has a number of unique clinical and immunological features. In contrast to many other HIV-associated opportunistic infections, CD4+ T cell reconstitution is severely delayed (even if virological suppression is reached) and the immune reconstitution inflammatory syndrome to a Leishmania infection after initiation of cART appears relatively rare, indicating a persistent suppression of host immunity (33, 34). Atypical clinical presentations can occur and amastigotes have been detected in tissues such as the intestine, where parasites are mostly undetectable in the immunocompetent host (14, 35). After clinical remission, parasitemia also appears to persist, at least intermittently (36). A chronic/intermittent course of VL lasting several years has been described, labeled as “active chronic visceral leishmaniasis” (36). Consequently, HIV-infected patients will develop multiple VL relapses and often become progressively more difficult to treat, ultimately leading to a stage of complete treatment unresponsiveness. Hence, there is an urgent need for innovative and effective alternative therapies against VL–HIV coinfection.
Promising Role of Immunomodulatory Therapy
It has become increasingly clear that the host immune response is a critical factor determining VL treatment response and control, acting in synergy with anti-leishmanial drugs (37). This implies that in immunosuppressed individuals, targeting parasites alone with conventional anti-leishmanial drugs but without enhancing the immune response might simply not be sufficient. This interaction between drugs and the immune system was first suggested in animal models of VL, where the efficacy of pentavalent antimony (Sbv) was lower after T cell depletion (38). This was probably related to the decreased cellular uptake of SbV into interferon-gamma (IFNγ) activated macrophages, where it is normally converted intracellularly into its active trivalent form (SbIII) (4). While this finding should be extrapolated with caution, this mechanism may explain the observations that immunocompromised patients with VL failed to respond to antimonial drugs.
Immunotherapy is defined as the use of biological molecules or pharmacological compounds to modulate immune responses directly or in combination with drugs. A combination of immunomodulatory and direct anti-parasitic drugs could enhance the efficacy of chemotherapy and even prevent drug resistance (39). On top of its successful use in treating several non-infectious disorders (e.g., cancer, rheumatoid arthritis, etc.), the use of immune-based combination therapy is increasingly being explored in infectious diseases, such as tuberculosis (40) and leprosy (41). Despite several candidates being in the drug development pipeline, there are no immunotherapeutic agents or vaccines against VL currently registered for human use in routine clinical practice due to multiple reasons (e.g., high costs of clinical trials, limited and remote patient populations, ineffectiveness, safety concerns) (42). Experimental immune-based approaches are also being explored in the domain of HIV, where many have reached Phase I and some Phase II clinical trials but as of today have failed to provide enough immune restoration, potent effectiveness, sustainable benefits, delay of clinical progression, or good safety profiles (40, 43–46). However, VL–HIV-coinfected patients are often excluded or neglected in such studies, although both individual patients as well as public health approaches in general could benefit from these interventions.
Here, we first summarize current views on how host immunity against VL is affected during HIV coinfection, and then discuss the potential of current immunomodulatory therapies against VL in the context of concurrent HIV infection (both human studies and promising experimental approaches, excluding prophylactic studies). In particular, key targets and potential caveats are emphasized to guide future research on immunomodulatory therapies against VL and support the inclusion of HIV-coinfected patients in clinical research.
Immunopathogenesis of VL–HIV Coinfection
Macrophages represent an important common reservoir for HIV and Leishmania and serve as vehicles that disseminate both virus and parasite throughout the host. In addition, both pathogens may interact with each other to exacerbate immune suppression (Figure 1). In fact, both pathogens severely alter the antigen processing and presentation capacities of dendritic cells and macrophages, and synergistically escape immune surveillance using an array of strategies yet to be fully understood (47).
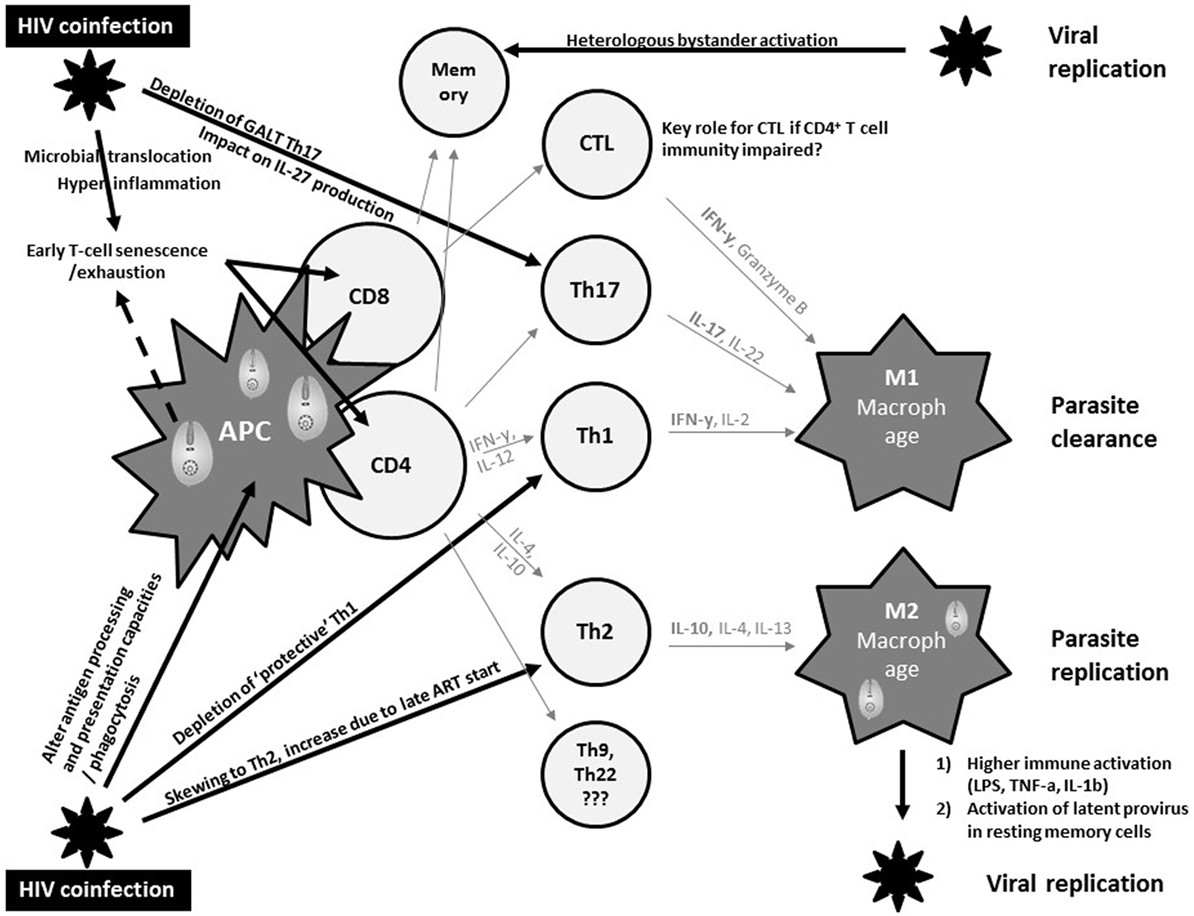
Figure 1. Current views on synergistic mechanisms in T cell immunity against visceral leishmaniasis (VL) due to human immunodeficiency virus (HIV) coinfection inciting persistent viral and parasite replication in VL–HIV-coinfected patients. APC, antigen-presenting cell; Th, T-helper; GALT, gut-associated lymphoid tissue; CTL, cytotoxic T cell; IL, interleukin; ART, antiretroviral therapy; IFN, interferon; LPS, lipopolysaccharide; TNF, tumor necrosis factor.
The control of VL in experimental models has been robustly associated with a strong T helper 1 (Th1) immune response, with large amounts of IL-2 and IFNγ (48) (Figure 1). In addition, a M2 polarization of macrophages has been associated with suppression of cell-mediated immunity, which confers susceptibility to intracellular infection. However, the immune mechanisms modulating VL in murine models or humans differ significantly. Human studies have shown a Th1/Th17 protective pattern with a somewhat different T cell functionality compared to experimental models, but lack comprehensive longitudinal data (49, 50). CD8+ T cells have also been shown to produce IFNγ that can contribute to VL control (51). The immunosuppressive effects of IL-10, and the regulatory role of other cytokines such as IL-27, have been implicated in the development of the different clinical pictures (50). Impaired neutrophil effector function has also been suggested to play a key role in the pathogenesis of VL (52). Partly due to the lack of good animal or in vitro models, it is currently unknown whether and how these protective and immunosuppressive patterns of VL are modulated by HIV and ART and how they define the pertinent clinical outcomes of VL–HIV patients.
Human immunodeficiency virus-1 causes a general profound impairment of cell-mediated immunity with low levels of CD4+ Th1 cells, the main protective cells in VL (Figure 1). HIV also skews the host immunity toward a Th2 response that only becomes affected at the later stages of the viral infection, potentially provoking parasite replication. Th17 cells are also associated with protection in VL, but are highly permissive to HIV infection. Their frequency is significantly and preferentially reduced in the gastrointestinal tract, even in patients with undetectable plasma viral load under ART (53). Depletion of Th17 cells from the gut-associated lymphoid tissue together with a series of immunopathological events occurring at the gastrointestinal tract mucosa leads to microbial translocation and consequently higher non-specific immune activation and hyper-inflammation (54). This microbial translocation has been postulated as one of the factors causing non-specific early T cell exhaustion and senescence (55), which may further weaken protective immunity toward VL. Likewise, VL was reported as an independent cause of increased non-specific immune activation, T cell senescence and the lack of immune recovery in virologically suppressed coinfected HIV patients (56, 57). In line with T cell exhaustion, chronic immune activation was recently associated with recurrent relapse of VL in HIV patients (58). Recent research in VL–HIV patients also suggested that weak antigen-specific functional responses or proliferation of T cells after in vitro stimulation was an important predictor of relapse (59). Despite the pivotal role of CD8+ T cells in viral and parasite clearance, their contribution in VL–HIV control and level of exhaustion remains unknown. Likewise, it is still unclear as to what impact Leishmania infection could have on the capacity of resting memory CD4+ T cells to act as a stable reservoir of latent HIV infection. What impact a spike in viral replication may have on anti-leishmanial immunity (e.g., by bystander activation of Leishmania-specific memory cells) also remains unknown (60, 61).
The consequences of infection by two immune-suppressive pathogens could, therefore, be a symbiotic and persistent incapacitation of the host’s immune system, favoring a state of immunological anergy, ultimately being fatal to the patient. A better understanding of the immune response against Leishmania infection in HIV-coinfected patients is crucial to establish a rational approach for immunomodulatory therapy.
Status of Immunotherapeutic Interventions in Human VL and Their Application in HIV Patients
Due to the lack of a protective role of anti-Leishmania antibodies in early studies, passive immunization was not further explored, while active immunization with immunomodulators and vaccine therapy was investigated (62). Early studies by Murray et al. (38, 63, 64) showed the therapeutic utility of interleukin-2 (IL-2), IL-12, interferon-gamma (IFNγ), and granulocyte–monocyte colony-stimulating factor (GM-CSF) in murine VL models. Although the Th1/Th2 dichotomy of immunity to VL is not fully upheld in humans, clinical immunotherapeutic studies on VL patients have been skewed toward Th1-associated cytokine-adjuvant therapy and are discussed below (see Table 2). For VL–HIV coinfection, only five published case reports using recombinant IFNγ, IL-2, and GM-CSF combined chemotherapy were found in literature (see Table 2).
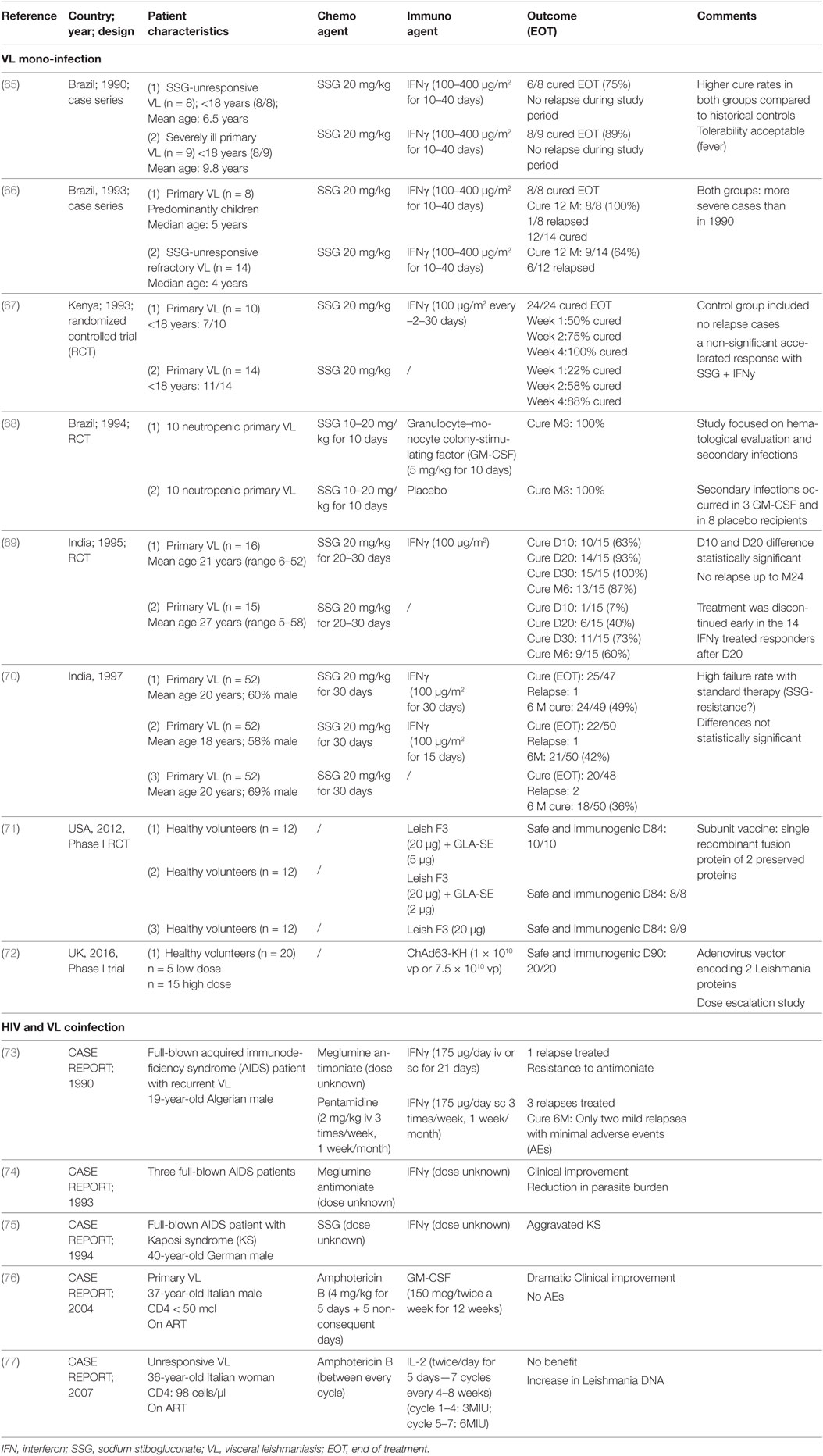
Table 2. Published clinical reports on the use of immuno(chemo)therapy against visceral leishmaniasis (VL) and VL–human immunodeficiency virus (HIV).
Interferon-γ
There has been limited success in small-scale clinical trials with combined therapy of IFNγ and SbV for treating VL. This combination therapy displayed stronger parasitological and clinical cure rates in VL patients (mainly children) from Brazil, Kenya and India compared with the drug alone, but these studies had several limitations (see Table 2 for details). In a subsequent larger randomized controlled trial (RCT) in India, these improved treatment outcomes could not be confirmed (70). Importantly, treatment response in this particular study was generally poor as drug resistance was emerging in that region.
There are a few case reports, mostly from the pre-ART era, providing information on whether IFNγ can be safely administered in VL–HIV patients (see Table 2), which is of relevance since IFNγ also has a vital but ambiguous role in the pathogenesis of HIV (78). IFNγ appeared to be fairly well tolerated but showed inconclusive results (73, 74, 79). In one old case report of a patient with VL–HIV coinfection, acceleration of Kaposi’s sarcoma has been reported (75). The therapeutic potential of IFNγ to treat HIV coinfections was supported by two Phase II trials, evaluating adjunctive IFNγ to improve treatment response to antifungals in HIV patients with cryptococcal meningitis (80, 81). However, in the early 1990s, a multicenter clinical trial of SSG plus IFNγ for VL in HIV-coinfected patients in Spain was suspended following an interim analysis indicating that there was an excess of severe secondary effects and no benefit over drug alone (79). The findings itself have never been published but suggested a limited value of IFNγ therapy for VL–HIV coinfection.
Granulocyte–Macrophage Colony-Stimulating Factor
Granulocyte–monocyte colony-stimulating factor can inhibit the intracellular replication of protozoa such as Leishmania. The justification to explore GM-CSF as immunotherapeutic agent stems from documented effects, such as monocyte mobilization, macrophage activation, the production of pro-inflammatory cytokines, and amelioration of neutropenia (63). GM-CSF combined with SbV was successfully explored in 20 neutropenic VL patients in Brazil. All responded well to VL treatment, neutropenia rapidly improved and secondary infections decreased (68) (Table 2). The authors did, however, not include a control arm, making it unclear whether the effect of GM-CSF, if any, could be due to the reversal of neutropenia (and might hence not apply in those without neutropenia) or whether other mechanisms were involved. On the other hand, in vitro studies have recently suggested that GM-CSF could contradictory promote Leishmania growth by inducing monocyte proliferation and induction of intracellular dNTP production (82), but whether this would also occur in humans remains unknown.
In terms of safety, several older clinical trials of GM-CSF administration in HIV patients indicated that it might accelerate HIV replication (83). By contrast, more recent RCTs have demonstrated benefits of using GM-CSF in virologically suppressed patients as an adjunct to conventional ART or therapeutic HIV vaccination (83, 84). This would argue against using GM-CSF in pre-ART patients, but might suggest it to be safe in those stable on ART. With regard to coinfections, some case reports were published on successful GM-CSF therapy of resistant-to-standard-therapy mycobacterial infection and pulmonary aspergillosis in HIV patients (85, 86). There is a single successful case report on immunotherapy targeting primary VL in an Italian AIDS patient, whereby human GM-CSF was combined with liposomal amphotericin B (Table 2) (76). Presently the evidence for beneficial effects of GM-CSF on HIV disease is limited, but GM-GSF adjuvant therapy could provide a potential value for treatment of neutropenic VL in stable ART patients.
Interleukin-2
Interleukin-2 induces clonal expansion of specific T cells; promotes natural killer and CD8+ T cell cytotoxicity, cytokine secretion by Th1, Th2, and Th17 cells; and modulates programmed cell death (42). Hence, IL-2 is necessary for the protection against Leishmania in immunodeficient mice, in which IL-2 restores the activity of SbV (38, 87). The impairment in IL-2 production is also one of the first functional defects described in untreated HIV-positive patients and its administration to boost the quantitative and/or qualitative CD4+ T cell restoration in HIV-infected patients has been evaluated in Phase I, II and III trials (42). These early results provided evidence that IL-2 therapy combined with existing cART has the potential to enhance quantitative and qualitative immune restoration, without triggering HIV replication, even when ART alone had failed to do so. However, restoring CD4+ T cell counts with IL-2 failed to show long-term clinical benefits in two large Phase III clinical trials, ESPRIT and SILCAAT (88). IL-2 recipients in the STALWART trial even experienced more opportunistic infections, death or grade 4 adverse events during IL-2 administration, than those not receiving IL-2 (89).
To date, no clinical trial for rIL-2 administration in VL patients has been reported. There has been one case report on the use of rIL-2 in a VL–HIV-coinfected patient failing to respond to anti-leishmanial and HIV treatment with low CD4 counts and incomplete HIV suppression despite ART use (77). This report indicated no benefit. Importantly, increased Leishmania parasitemia was observed at each rIL-2 cycle, which might have favored the progression of HIV infection and possibly explains the reported progressive decline in CD4 T cell count (77). In a BALB/c mouse model, IL-2 seemed to have a short protective effect against VL only at the priming phase, without any lasting benefit (90). Such a phase-specific effect could explain the lack of long-term clinical benefits. In general, the small therapeutic window, critical dosage with potential high toxicity and challenging treatment conditions suggest IL-2 is an unlikely candidate for boosting immunity in VL–HIV-coinfected patients.
Therapeutic Vaccines
Historically, leishmanization (inoculation with live parasites) was shown to have benefit for protection against re-infection with cutaneous leishmaniasis (CL) and this evidence has driven the search for an effective vaccine against VL (91). Besides prophylactic vaccine development, various approaches employing therapeutic vaccines have been tested experimentally and clinically; and currently resulted in three licensed vaccines for canine VL but none for human VL (92). Therapeutic immunization with a first generation vaccine of aluminum hydroxide precipitated autoclaved L. major (Alum-ALM)+Bacille Calmette–Guérin (BCG) was found clinically effective in CL, mucocutaneous leishmaniasis and persistent post-kala-azar dermal leishmaniasis (PKDL) cases, with studies progressing to Phase III clinical trials (93–99), but application to VL has not been reported (62). Similarly, LeishF1/F2 vaccine (alternatively called Leish-111f), a promising second-generation (i.e., recombinant protein) vaccine for CL, showed insufficient protection against VL in dogs (100). A modified version of these second generation vaccines, called LeishF3, which accommodated changes to enhance its efficacy against VL has been shown to be safe and immunogenic in a Phase I trial in healthy human volunteers, but therapeutic trials in patients have not been reported (Table 2) (71). A third-generation (i.e., DNA-based) adenovirus vaccine (ChAd63-KH) was designed to induce Leishmania-specific CD8+ T cells and aimed at therapeutic use in VL/PKDL patients. It was shown to be safe and immunogenic in healthy volunteers (72) and is currently in Phase II trial in persistent PKDL patients in Sudan.
A careful risk–benefit assessment needs to be made when considering therapeutic vaccination against VL in HIV patients, with depressed immunity. Safety concerns surely exist, but should not be overstated and should not impede evaluation of therapeutic VL vaccination studies in virally suppressed HIV patients as potential benefits can outweigh existing theoretical risks. In essence, these patients have a higher risk of developing VL and are most in need of an enhanced immune response upon VL development. Post-marketing trends suggest that routinely used inactivated (non-VL) vaccines have similar safety profiles among HIV-uninfected and HIV-infected persons on stable ART (101). Although data are still limited, HIV-infected individuals who are on ART with well-controlled HIV RNA levels and CD4+ T cell counts of >200 cells/μL (or ≥15%) may even receive indicated live-virus vaccines (101). In addition, modern post cART era studies did not indicate that vaccines are important triggers of HIV replication or disease progression (102). With regard to efficacy, a highly immunogenic vaccine will be needed, as well as detailed studies to define the optimal timing and dosing for vaccination among those with advanced disease. Despite the concerns of depressed immunity and sparse efficacy data for other types of vaccines, studies have clearly demonstrated the protective benefit of influenza and Streptococcus pneumoniae vaccinations even among advanced HIV patients. In summary, these data merit a concurrent evaluation of therapeutic VL vaccines in coinfected patients who are virologically suppressed at the time of VL presentation.
Promising Pipeline Immunomodulatory Molecules/Interventions
While both the pharmacokinetics and pharmacodynamics of a drug, but also the nature of drug–immune interactions in animals and humans may differ considerably, animal models may still provide new clues to potential approaches. Here, we selected the most promising molecules or interventions for their potential in an immunosuppressive environment of the coinfected individual and refer to recent review papers for a more extensive list (39, 44, 45, 92, 103). The formats discussed below are limited to active immunotherapy attempts including non-antigen-specific strategies such as cytokines that stimulate immunity or suppress the viral replication; antibodies that block negative regulatory pathways; and indirect immunomodulation (Figure 2). Antigen-specific strategies such as therapeutic vaccination and adoptive strategies such as cell therapy are also briefly discussed. Whether the molecules listed below could serve as putative targets for human immunotherapy remains to be demonstrated.
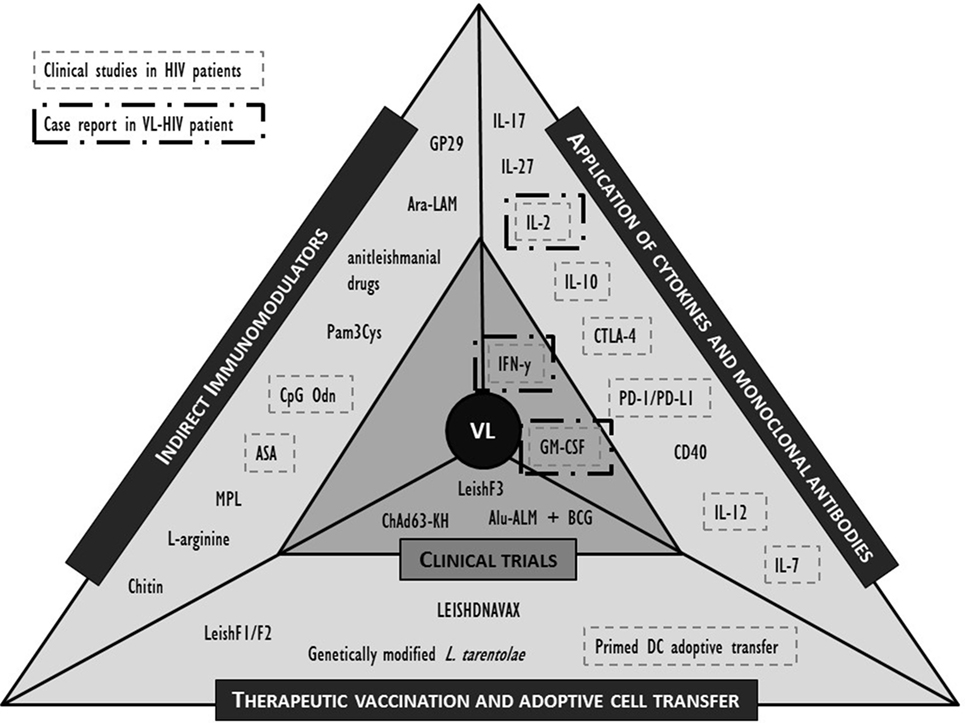
Figure 2. Overview of described clinical and preclinical immunomodulatory interventions in human visceral leishmaniasis (VL) and their application in (VL)-human immunodeficiency virus (HIV) (co)infection. IL, interleukin; IFN, interferon; PD-(L)1, programmed cell death-(ligand)1; GM-CSF, granulocyte–macrophage colony-stimulating factor; CTLA, cytotoxic T lymphocyte-associated molecule; CD, cluster of differentiation; BCG, Bacillus Calmette–Guérin; Alu-ALM, aluminum hydroxide precipitated autoclaved L. major; DC, dendritic cell; GP, Glycoprotein; Ara-LAM, arabinosylated lipoarabinomannan; Pam3Cys, synthetic bacterial lipopeptide; CpG Odn, CpG oligodeoxynucleotides; ASA, acetyl salicylic acid; MPL, monophosphoryl lipid.
Non-Antigen-Specific Strategies
The above listed clinical trials with cytokine-adjuvant chemotherapy were based on limited data from experimental models of VL conducted in the 1990s. Our knowledge of immune mechanisms has substantially expanded since then. For instance, IL-12, a pluripotent cytokine that plays a central role in the initiation/maintenance of Th1 responses and potentiates T cell IFNγ production, was shown to have similar effects as IFNγ in both CL and VL when injected in mice (64, 104) as well as dogs (105) and human PBMC from treated Sudanese VL patients (106). Likewise, IL-12 preconditioning of monkeys during acute SIV infection markedly delayed disease progression (107). While rhIL-12 administration was well tolerated and safe, no evidence of improvement in HIV antigen-specific immune response could be observed in a Phase I RCT (108). While this suggests that IL-12 therapy is unlikely to provide major benefits in the chronic phase of an HIV infection, it might still be valuable in the context of opportunistic infections that are best met with Th1-like effector immune responses. In line with this, rIL-12-adjuvant chemotherapy was successfully evaluated for patients with Kaposi’s sarcoma (109). In addition, it has been tested as part of a combination therapy for cryptosporidiosis in two AIDS patients that demonstrated signs of a brisk immune response and consequently symptomatic improvement, but with severe side effects that outweighed the clinical benefits (110). Data on the role of IL-12 as an immunotherapeutic agent or vaccine adjuvant for HIV coinfections could be promising and merits further research, although potential broad side effects due to its pluripotent role should be limited (e.g., tissue-targeted delivery, well-timed short boosting approach, etc.). Unfortunately, the incorporation of IL-12 into larger vaccine trials has lagged, largely due to the early setback in a renal carcinoma Phase II trial. However, the mechanisms underlying the severe acute toxicities that led to two deaths and 12 hospitalizations have been ascribed to an inappropriate dose and administration schedule (111).
Like IL-12, many chemokines or cytokines contributing to protection/pathogenesis of VL are regulated during HIV coinfection. For instance, Th17 cells are highly depleted from the gut in HIV-infected patients. Recent work in humans has, however, demonstrated the importance of IL-17 and IL-22 in protection against VL progression from asymptomatic infection to disease (49). In addition, elevated serum IL-27 concentrations were linked to severity of VL. IL-27 seems to regulate the Th1/Th17 profiles in a L. infantum mouse model of VL by suppressing the IL-17-induced neutrophil response (112). The IL-27–Th17–IL-17 axis, thus, seems to be strongly involved in resistance against VL and merits further therapeutic exploration, especially in HIV-coinfected patients with a Th-17-depleted immune response.
Despite the central role of IL-7 cytokine therapy in HIV patients in the past, this molecule has not been evaluated in VL–HIV-coinfected patients and remains under-investigated in experimental models of VL (113). IL-7, like IL-2, has a critical role in peripheral T cell homeostasis. IL-7 has, however, a more pleiotropic role and was shown to drive CD4+ T cell restoration in HIV patients, even when HIV replication is controlled. It is also able to promote Th1 responses, enhance memory T cell expansion (on top of naive T cell response), and increase CD8+ T cell counts and cytotoxicity in HIV patients (42). Moreover, damage to hepatocytes during full-blown VL may impair IL-7 production, as IL-7 is also produced by liver cells in response to inflammation (114). Recombinant IL-7 administration thus has the potential to safeguard the long-term survival of effector CD4+ T cells in response to persisting parasites in a VL–HIV coinfection. However, in the ERAMUNE 01 RCT, rIL-7 and dual ART intensification induced an amplification of the HIV reservoir in well-controlled HIV patients (115). The authors reasoned that this was the result of the expansion of central memory CD4+ T cells, carrying HIV DNA, thus limiting this IL-7 based strategy. In the context of VL–HIV coinfection, this strategy should only be considered if a pronounced clinical benefit to VL treatment outweighs its potential negative effects.
Blocking the action of immune-suppressive factors could prove more efficient as it might allow restoration of protective immunity in a more controlled manner. IL-10 correlates very well with the parasite load during VL infection. Moreover, in animals, IL-10 blockade (by means of anti-IL-10R or anti-IL-10 monoclonal antibody) has been proven successful in lowering parasite burden when combined with conventional treatment in multiple studies in mice (116, 117). These effects were confirmed in cultures of splenocytes or PBMCs from Indian and Sudanese VL patients (106, 118). However, in immunodeficient mice treated with anti-IL-10R monoclonal antibody, Murray et al. were not only able to show an acceleration of SbV-associated killing, but also reported a >10-fold SbV dose-sparing effect (119). Despite the clinical and experimental data suggesting IL-10 as a key target in the immunopathogenesis of VL, a clinical trial using a monoclonal antibody against IL-10 failed to start following the decision of the company to stop its production (NCT01437020, clinicaltrials.gov).
Increased serum IL-10 concentrations are also observed in HIV-infected patients with disease progression, in contrast to non-progressing patients where levels were stable (120). In addition, ART has a clear downregulating effect on IL-10. On the other hand, increasing evidence suggests that IL-10 impacts many aspects of HIV pathogenesis, including the regulation of HIV-specific CD4+ and CD8+ T cell functions, as well as modulation of HIV replication in PBMC subsets. Genetic polymorphisms in the IL-10 gene promoter that lead to decreased IL-10 expression have been associated with more rapid disease progression in late stages of HIV infection, suggesting that the anti-inflammatory effects of IL-10 may be solely protective in the setting of chronic immune activation and blocking IL-10 function would only make sense in an acute setting (121). When considering VL–HIV coinfection, these data would advocate the blocking of excessive IL-10 levels during the acute stage of VL in HIV patients (in particular pre-ART patients) to allow a beneficial acute response which should however be time limited to retain the beneficial role of IL-10 in controlling side damage of chronic HIV and parasitic infections. To reduce the unwanted side effects due to blockage of normal, and beneficial, biological activities, novel IL-10 signaling inhibitors with for instance shorter half-lives are first needed (122).
The concept of immune exhaustion and senescence as a stepwise and progressive loss of T cell function and proliferative potential, respectively, and evolving to complete T cell unresponsiveness has been robustly discussed in the context of HIV infection (123). The driving force is believed to be chronic antigen exposure and consequently extensive non-specific immune activation. Increased immune activation in patients on long-term suppressive cART has been associated with increased mortality, the occurrence of non-AIDS-defining conditions, and a poorer recovery in CD4+ T cell count (124, 125). Similarly, increased levels of programmed death-ligand 1 (PD-L1) expression on monocytes, B cells, and T cells from untreated HIV patients correlated directly with plasma viral load and inversely with CD4+ T cell count (126). This mechanism could partly explain the disappointing long-term effects of IL-2 therapy in HIV patients, as IL-2 was recently shown to upregulate the PD1–PD-L1/L2 pathway (127). While the causative factors of immune exhaustion or senescence are not completely understood, chronic immune activation, residual HIV-replication, and coinfections are likely main drivers of this process. Recent studies have also focused on the role of this process in the context of VL and other parasitic infections, showing an accelerated T cell senescence during VL infection (128). Likewise, a parasite-induced T cell anergy has been proposed (128). Hence, a modulatory approach to reverse this process or temporarily breaking the regulatory feedback loop using antibody therapies targeting PD-1, CTLA-4 or its ligands could prove efficient in coinfected individuals with a potential double-driven T cell unresponsiveness. Such an approach to reverse the reported T cell unresponsiveness has proved very effective in experimental VL (57, 129–131). In SIV-infected rhesus macaques, anti-PD-1 (in the absence of ART) was shown to enhance virus-specific CD8+ T cell activity, to reduce viral load, and to prolong survival (46). Similarly, anti-PD-L1 antibody therapy showed promising in a recent Phase I RCT on 6 ART patients, arguing in favor of its potential use in virologically suppressed VL–HIV patients (132). Recently, the major HIV cell reservoir was shown to be composed of PD-1+ CD4+ memory T cells, suggesting an additional positive effect of anti-PD-1 therapy to combat the concomitant HIV infection (133).
Antigen-Specific and Adoptive Strategies
There are multiple studies in which diverse antigens and adjuvants showed promising results as immunoprophylactic or therapeutic tools in animal models of VL, recently summarized in a review by Jain and Jain (92). Apart from the current clinically explored strategies and the safety/efficacy concerns in HIV patients (see above), a promising approach would be to vaccinate with a non-pathogenic L. tarentolae strain, genetically modified to improve its immunogenic potential as a live vaccine (134). Likewise, a novel third generation T cell epitope-enriched DNA vaccine (LEISHDNAVAX) showed significant efficacy when co-administered with a single dose of AmBisome in L. donovani-infected mice (135). The vaccine is based on minimalistic immunogenically defined gene expression vectors encoding five conserved antigens developed for efficient induction of Th1 immune responses. This candidate vaccine has yet to enter clinical Phase I trials.
Another cutting-edge approach to induce antigen-specific T cell immunity is dendritic cell-based immunotherapy (103, 136). While macrophages are one destination of Leishmania parasites in the human host, dendritic cells can also harbor parasites, but in addition present antigen and regulate immune mechanism governing control or progression of infection. Adoptive transfer of dendritic cells primed with different kinds of Leishmania antigens has been shown very effective in murine VL, improving both cellular and humoral immunity (136). Compared to the modest efficacy of immune therapy and therapeutic vaccines against HIV infection, ex vivo generated dendritic cell therapeutic vaccines aimed at inducing effective HIV-specific immune responses have yielded the best results in this field (137). The outcomes of monocyte-derived dendritic cell-based therapeutic vaccines still needs optimization as functional cure was not reached and most patients needed to restart ART, but this method could provide a strong immunogenic window for concomitant VL-targeted therapy of coinfected individuals. Due to high costs and required state-of-the-art equipment, adoptive cell transfer therapy may prove difficult to implement in low-resource settings of disease endemic countries.
Indirect Strategies
An alternative approach is to indirectly stimulate host immunity to optimize protection against infection. Such indirect immunomodulators can be obtained by many different types of substances, including natural products that have immunomodulatory activity. Such immunomodulators, however, carry the risk of inducing excessive immunopathology and side effects. Many compounds have been evaluated in VL animal studies over the years, including CpG oligodeoxynucleotides, acetyl salicylic acid and l-arginine (103). Most of these molecules increase T cell activation through enhanced antigen presentation by costimulation-based therapy or acting on toll-like receptors (TLRs) (e.g., TLR4/GP29 or MPL; TLR2/Ara-LAM, or Pam3Cys). This could be particularly beneficial in HIV-coinfected patients, as TLR-agonists such as TLR7 or TLR9 agonists have shown reduction of viral DNA or the viral reservoir and enhancement of HIV-specific CD8+ T cell immunity in experimental and human HIV (138, 139). Whether such a multi-TLR targeting approach would benefit human VL–HIV patients remains unclear and merits further research.
In a similar manner, it has been suggested that TLR4 and TLR9, two TLRs contributing to the immune response against Leishmania infection, play a role in the anti-leishmanial mechanism of miltefosine (140). An alternative strategy could, thus, be to concurrently capitalize on the indirect immunological effects of the combined anti-leishmanial drug in a immuno-chemotherapeutic approach. The relevance and impact of these immunomodulatory actions of current anti-leishmanials in HIV-coinfected VL patients remains to be determined. Besides a direct mechanism of action, anti-leishmanials can increase nitric oxide and reactive oxygen species production due to activation of infected macrophages, leading to elimination of the parasite. This indirect activation of macrophages has been shown for amphotericin B (141), miltefosine (142), antimonials (143), and paromomycin (144). Induction of macrophage-derived cytokine release promoting a Th1 response (IL-2, IL-12, IFNγ) has been noted for all conventional anti-leishmanials such as amphotericin B (141, 145), miltefosine (142, 145), paromomycin (145), and SSG (143, 145), even though contradictory results have been reported, e.g., for miltefosine (146). Related to this, miltefosine restored IFNγ responsiveness in Leishmania-infected macrophages (142). Another immunostimulatory property contributing to anti-leishmanial activity is a drug-induced increase in macrophage membrane fluidity, ameliorating defects in antigen presentation and enhancing T cell stimulation. This has been shown after exposure of infected host cells to higher concentrations of miltefosine, paromomycin, and SSG (145). For both antimonials (147) and miltefosine (148), it has been shown that they increase the phagocytic capacity of monocytes and macrophages. There are currently no data available whether all these effects are clinically relevant in terms of short-term treatment response, relapse, final cure, and the risk of development of PKDL. Despite the current lack of data on clinical relevance, these background effects should be taken into consideration in future combined immuno-chemotherapeutic strategies to incite an effective synergistic effect. The general lack of response to anti-leishmanial treatment in HIV-coinfected patients and the relevance of concomitant cART for the efficacy of current anti-leishmanials possibly indicate that these indirect effects are not negligible for a therapeutic response.
Perspectives
Despite the growing research in immunotherapy against VL (partly reviewed above), no immunotherapeutic approach has yet been licensed for use in human VL. HIV-coinfected patient groups, in particular, are often excluded from the above described clinical intervention studies due to the presumed hazards and challenging logistics. Although a vulnerable population, we would argue that VL–HIV patients should be considered as a relevant target group for an immunomodulatory approach against VL due to an intensified defect in T cell immunity, dependence of current anti-leishmanial drugs on the latter, inadequate treatment outcomes, and higher chronicity of the parasitic infection with frequent relapse. In addition, HIV-targeted immunomodulatory approaches, despite their drawbacks to achieve long-term functional cure in HIV patients, might find a temporarily window of opportunity in opportunistic coinfections such as VL, where cART alone is not able to restore protective immunity. The challenge, however, of immunomodulatory therapy in VL–HIV-coinfected patients is boosting effective VL-specific T cell responses while avoiding activation of latent provirus and inappropriate immune activation (in virologically suppressed ART patients) or HIV recrudescence and increased HIV-susceptibility of target cells (in unstable HIV/AIDS patients). Clinical trials are a necessity to study treatment effects, due to the lack of good animal or in vitro models mimicking VL–HIV coinfection.
In Figure 2, we summarized the discussed interventions against VL and highlighted those that have also been clinically evaluated in the context of HIV. Evidence is lacking to prioritize a target molecule, but attempts at immunotherapy in VL–HIV patients should best be performed in ART patients with a recovered immune system. Appropriate adjuvants can be included to enhance the efficacy of the response, but caution should be taken to avoid excessive and broad immune activation. The following perspectives are best taken into consideration when designing or evaluating an immunomodulatory approach in VL–HIV-coinfected patients.
Combination Strategies
As current anti-leishmanial drugs are highly dependent on host immunity, it is recommended to potentiate chemotherapeutic agents with various immunomodulators in HIV-coinfected patients. While the increment in immunocompetent patients could be potentially low, HIV-coinfected patients are probably in more need of a boost in effective T cell immunity against VL to decrease the high mortality and treatment failure rates typically observed in coinfected patients.
The current clinically explored techniques of single cytokine-adjuvant therapy in VL have the inherent danger of a very pluripotent effect in HIV-coinfected patients, due to the intricacies of cytokine networks, and may unpredictably impact the delicate balance between beneficial VL-specific responses and deleterious immune activation. Future therapeutic use of broad immunomodulators will most likely lead to unwanted side effects in coinfected patients until a system-level understanding of their mode of action is available and thus a more selective and well-timed approach can be performed (149). However, they could potentially prove valuable as a well-timed adjuvant in a more targeted immunomodulatory approach.
The other clinically explored strategy in VL is therapeutic vaccination. However, as T cell senescence and exhaustion could have occurred by persistent HIV replication, further stimulating effector-memory T cells could be futile or even harmful in VL–HIV patients. Perhaps a concurrent strategy to reverse this T cell exhaustion (e.g., anti-PD-1 therapy) could increase vaccine efficacy. It remains to be seen whether VL–based therapeutic vaccines deployed in HIV-coinfected patients are safe and whether a strong enough response can be induced against VL. In severely CD4+ depleted patients in particular, a concurrent need may be to first encourage immune reconstitution before vaccination. Combination strategies of diverse immunomodulators and drugs will, thus, be crucial in these patients to reach an effective treatment, perhaps with a more individualized approach.
Stratification
Among patients with tuberculous meningitis, different inflammatory patterns governed by host genetics are recognized, converging on dysregulated levels of TNF. At one end of the extreme, a hyper inflammatory phenotype was shown to benefit from steroid administration; at the other end, where inflammation is inadequate, other immunomodulatory interventions would be required (150). In a similar manner, subgroup analyses in HIV-associated cryptococcal meningitis suggested that the greatest benefit of a short-course IFNγ adjuvant therapy was gained among patients with a lack of Cryptococcus-specific IFNγ/TNF CD4+ T cell responses (151). In most settings, VL–HIV-coinfected individuals will also be (severely) malnourished upon VL diagnosis, and micro- and macro-nutrient deficiency can have profound immunological effects. These alterations could critically affect the efficacy of any immunomodulatory interventions, yet may also provide opportunities for complementary interventions. We, therefore, argue that there is a need to assess immune risk profiles based on functional T cell assays, RNA signatures, and other parameters that identify patients that are more likely to benefit from immune adjuvant therapy, across the heterogeneous group of VL–HIV patients.
Timing
Visceral leishmaniasis–HIV coinfection is a dynamic process with diverse stages of infection and regardless of choice of immunomodulatory intervention, timing will be critical to success. For instance, high IL-17 levels appeared protective for early VL progression, but its role is still debatable in chronic infection. The optimal timing of immunotherapy among HIV-coinfected adults in regard to HIV stage and receipt of antiretroviral therapy also remain important unanswered questions. Most benefit is probably to be gained in early stages of HIV infection as well as in under-therapy suppressed patients, who are able to effectively respond to immunomodulators. Therefore, we would argue for a primary evaluation of novel approaches in stable ART patients that have a somewhat reconstituted CD4+ T cell immunity and suppressed viral load, including frequent monitoring of blips in viral load and CD4+ T cell count. It remains to be investigated whether HIV patients with a severe suppression in T cell immunity are also able to respond to immune stimulators or whether virological suppression first has to be prioritized to enable T cell responsiveness.
Targeted Strategies
The delivery system is also an important part of an immune-based strategy and implementation of various novel approaches based on liposomes, electroporation, dendrimers, carbon nanotubes, etc., can boost efficacy (92). For instance, as an alternative for broad cytokine adjuvants, more effective and tolerable approaches are being explored like encapsulation in micro or nanoparticles, restricting the delivery to APCs and/or the co-delivery with another immunomodulatory molecule via transducing vectors. Similar techniques such as microRNA or small interference RNA-based therapy could be explored, but these novel drugs will be most likely unaffordable in most countries where the disease is endemic.
Accesibility
The target population is largely living in very rural and/or poor areas, where a highly controlled clinical trial setting can be challenging and costly to implement. It will be imperative to strengthen human and infrastructural capacity in disease endemic areas to ensure a sustainable base for immunotherapeutic research and to assess safety and efficacy of novel interventions. Moreover, designed therapeutics should become affordable and accessible to the patient population, suggesting innovative low-resource-demanding methods ideally without the need of a cold chain.
Conclusion
To advance the development of immunomodulatory approaches for VL–HIV coinfection, a more detailed account of the immunological status induced by the coinfection and surrogate markers of cure and protection are still required, as a forerunner to inclusion of such patients in clinical intervention studies. The main limitation for comprehensive immunological research is, however, the need for human samples of longitudinal studies and trials in (often very remote) low-resource settings. With more research aimed at discovering key synergistic pathways of immune cell cross-talk and renewed efforts to translate these findings into effective treatment modalities that target Leishmania without promoting HIV replication, the goal of improved patient outcome and clinical management of this neglected population may be achievable.
Author Contributions
WA, JG, GV, LK, TD, and PK wrote and conceived the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks goes to Mariana Abreu de Andrade for her contribution in screening the literature.
Funding
WA is personally supported by a “Fonds Wetenschappelijk Onderzoek—Vlaanderen” fellowship. TD is personally supported by a ZonMw/Netherlands Organisation for Scientific Research (NWO) Veni fellowship, project no. 91617140. PK is supported by a Wellcome Trust Senior Investigator Award (#104726).
References
1. van Griensven J, Diro E. Visceral leishmaniasis. Infect Dis Clin North Am (2012) 26(2):309–22. doi:10.1016/j.idc.2012.03.005
2. Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol (2014) 6:147–54. doi:10.2147/CLEP.S44267
3. World Health Organization. Leishmaniasis in high-burden countries: an epidemiological update based on data reported in 2014. Wkly Epidemiol Rec (2016) 91(22):287–96.
4. Haldar AK, Sen P, Roy S. Use of antimony in the treatment of leishmaniasis: current status and future directions. Mol Biol Int (2011) 2011:571242. doi:10.4061/2011/571242
5. van Griensven J, Balasegaram M, Meheus F, Alvar J, Lynen L, Boelaert M. Combination therapy for visceral leishmaniasis. Lancet Infect Dis (2010) 10(3):184–94. doi:10.1016/S1473-3099(10)70011-6
6. Musa A, Khalil E, Hailu A, Olobo J, Balasegaram M, Omollo R, et al. Sodium stibogluconate (SSG) & paromomycin combination compared to SSG for visceral leishmaniasis in East Africa: a randomised controlled trial. PLoS Negl Trop Dis (2012) 6(6):e1674. doi:10.1371/journal.pntd.0001674
7. WHO. Control of the Leishmaniasis. Report of a Meeting of the WHO Expert Committee on the Control of Leishmaniases. Geneva (2010). WHO Technical Report Series 9492010.
8. Atia AM, Mumina A, Tayler-Smith K, Boulle P, Alcoba G, Elhag MS, et al. Sodium stibogluconate and paromomycin for treating visceral leishmaniasis under routine conditions in eastern Sudan. Trop Med Int Health (2015) 20(12):1674–84. doi:10.1111/tmi.12603
9. Khalil EA, Weldegebreal T, Younis BM, Omollo R, Musa AM, Hailu W, et al. Safety and efficacy of single dose versus multiple doses of AmBisome for treatment of visceral leishmaniasis in eastern Africa: a randomised trial. PLoS Negl Trop Dis (2014) 8(1):e2613. doi:10.1371/journal.pntd.0002613
10. Wasunna M, Njenga S, Balasegaram M, Alexander N, Omollo R, Edwards T, et al. Efficacy and safety of AmBisome in combination with sodium stibogluconate or miltefosine and miltefosine monotherapy for African visceral leishmaniasis: phase II randomized trial. PLoS Negl Trop Dis (2016) 10(9):e0004880. doi:10.1371/journal.pntd.0004880
11. Alvar J, Croft S, Olliaro P. Chemotherapy in the treatment and control of leishmaniasis. Adv Parasitol (2006) 61:223–74. doi:10.1016/S0065-308X(05)61006-8
12. Dorlo TP, Rijal S, Ostyn B, de Vries PJ, Singh R, Bhattarai N, et al. Failure of miltefosine in visceral leishmaniasis is associated with low drug exposure. J Infect Dis (2014) 210(1):146–53. doi:10.1093/infdis/jiu039
13. Dorlo TPC, Kip AE, Younis BM, Ellis SJ, Alves F, Beijnen JH, et al. Visceral leishmaniasis relapse hazard is linked to reduced miltefosine exposure in patients from Eastern Africa: a population pharmacokinetic/pharmacodynamic study. J Antimicrob Chemother (2017) 72(11):3131–314. doi:10.1093/jac/dkx283
14. Alvar J, Aparicio P, Aseffa A, Den Boer M, Canavate C, Dedet JP, et al. The relationship between leishmaniasis and AIDS: the second 10 years. Clin Microbiol Rev (2008) 21(2):334–59, table of contents. doi:10.1128/CMR.00061-07
15. van Griensven J, Carrillo E, Lopez-Velez R, Lynen L, Moreno J. Leishmaniasis in immunosuppressed individuals. Clin Microbiol Infect (2014) 20(4):286–99. doi:10.1111/1469-0691.12556
16. Diro E, Lynen L, Ritmeijer K, Boelaert M, Hailu A, van Griensven J. Visceral leishmaniasis and HIV coinfection in East Africa. PLoS Negl Trop Dis (2014) 8(6):e2869. doi:10.1371/journal.pntd.0002869
17. Stein RA. Super-spreaders in infectious diseases. Int J Infect Dis (2011) 15(8):e510–3. doi:10.1016/j.ijid.2010.06.020
18. World Health Organization. Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection – Recommendations for a Public Health Approach. 2nd ed. (2016). Available from: http://apps.who.int/iris/bitstream/10665/208825/1/9789241549684_eng.pdf?ua=1
19. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents Living with HIV. Unites States of America: Department of Health and Human Services (DHHS) (2013). Available from: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf
20. van Griensven J, Diro E, Lopez-Velez R, Boelaert M, Lynen L, Zijlstra E, et al. HIV-1 protease inhibitors for treatment of visceral leishmaniasis in HIV-co-infected individuals. Lancet Infect Dis (2013) 13(3):251–9. doi:10.1016/S1473-3099(12)70348-1
21. Araujo CA, Araujo AA, Batista CL, Oliveira MA, Oliveira V, Lino Junior RS, et al. Morphological alterations and growth inhibition of Leishmania (L.)amazonensis promastigotes exposed to zidovudine (AZT). Parasitol Res (2011) 108(3):547–51. doi:10.1007/s00436-010-2096-3
22. Ritmeijer K, Dejenie A, Assefa Y, Hundie TB, Mesure J, Boots G, et al. A comparison of miltefosine and sodium stibogluconate for treatment of visceral leishmaniasis in an Ethiopian population with high prevalence of HIV infection. Clin Infect Dis (2006) 43(3):357–64. doi:10.1086/505217
23. Murray HW. Leishmaniasis in the United States: treatment in 2012. Am J Trop Med Hyg (2012) 86(3):434–40. doi:10.4269/ajtmh.2012.11-0682
24. Meyerhoff A. U.S. Food and Drug Administration approval of AmBisome (liposomal amphotericin B) for treatment of visceral leishmaniasis. Clin Infect Dis (1999) 28(1):42–8; discussion 9–51. doi:10.1086/515085
25. Sindermann H, Engel KR, Fischer C, Bommer W, Miltefosine Compassionate Use Program. Oral miltefosine for leishmaniasis in immunocompromised patients: compassionate use in 39 patients with HIV infection. Clin Infect Dis (2004) 39(10):1520–3. doi:10.1086/425359
26. Marques N, Sa R, Coelho F, Oliveira J, Saraiva Da Cunha J, Melico-Silvestre A. Miltefosine for visceral leishmaniasis relapse treatment and secondary prophylaxis in HIV-infected patients. Scand J Infect Dis (2008) 40(6–7):523–6. doi:10.1080/00365540701787800
27. Troya J, Casquero A, Refoyo E, Fernandez-Guerrero ML, Gorgolas M. Long term failure of miltefosine in the treatment of refractory visceral leishmaniasis in AIDS patients. Scand J Infect Dis (2008) 40(1):78–80. doi:10.1080/00365540701466215
28. Mahajan R, Das P, Isaakidis P, Sunyoto T, Sagili KD, Lima MA, et al. Combination treatment for visceral leishmaniasis patients coinfected with human immunodeficiency virus in India. Clin Infect Dis (2015) 61(8):1255–62. doi:10.1093/cid/civ530
29. ter Horst R, Collin SM, Ritmeijer K, Bogale A, Davidson RN. Concordant HIV infection and visceral leishmaniasis in Ethiopia: the influence of antiretroviral treatment and other factors on outcome. Clin Infect Dis (2008) 46(11):1702–9. doi:10.1086/587899
30. Cota GF, de Sousa MR, de Mendonca AL, Patrocinio A, Assuncao LS, de Faria SR, et al. Leishmania-HIV co-infection: clinical presentation and outcomes in an Urban Area in Brazil. PLoS Negl Trop Dis (2014) 8(4):e2816. doi:10.1371/journal.pntd.0002816
31. Diro E, Ritmeijer K, Boelaert M, Alves F, Mohammed R, Abongomera C, et al. Use of pentamidine as secondary prophylaxis to prevent visceral leishmaniasis relapse in HIV infected patients, the first twelve months of a prospective cohort study. PLoS Negl Trop Dis (2015) 9(10):e0004087. doi:10.1371/journal.pntd.0004087
32. Diro E, Ritmeijer K, Boelaert M, Alves F, Mohammed R, Abongomera C, et al. Long-term clinical outcomes in visceral leishmaniasis-HIV co-infected patients during and after pentamidine secondary prophylaxis in Ethiopia: a single-arm clinical trial Authors and affiliations. Clin Infect Dis (2017). doi:10.1093/cid/cix807
33. Lawn SD, Wilkinson RJ. Immune reconstitution disease associated with parasitic infections following antiretroviral treatment. Parasite Immunol (2006) 28(11):625–33. doi:10.1111/j.1365-3024.2006.00900.x
34. Badaro R, Goncalves LO, Gois LL, Maia ZP, Benson C, Grassi MF. Leishmaniasis as a manifestation of immune reconstitution inflammatory syndrome (IRIS) in HIV-infected patients: a literature review. J Int Assoc Provid AIDS Care (2015) 14(5):402–7. doi:10.1177/2325957414555225
35. Diro E, van Griensven J, Mohammed R, Colebunders R, Asefa M, Hailu A, et al. Atypical manifestations of visceral leishmaniasis in patients with HIV in north Ethiopia: a gap in guidelines for the management of opportunistic infections in resource poor settings. Lancet Infect Dis (2015) 15(1):122–9. doi:10.1016/S1473-3099(14)70833-3
36. Bourgeois N, Bastien P, Reynes J, Makinson A, Rouanet I, Lachaud L. ‘Active chronic visceral leishmaniasis’ in HIV-1-infected patients demonstrated by biological and clinical long-term follow-up of 10 patients. HIV Med (2010) 11(10):670–3. doi:10.1111/j.1468-1293.2010.00846.x
37. Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev (2006) 19(1):111–26. doi:10.1128/CMR.19.1.111-126.2006
38. Murray HW, Oca MJ, Granger AM, Schreiber RD. Requirement for T cells and effect of lymphokines in successful chemotherapy for an intracellular infection. Experimental visceral leishmaniasis. J Clin Invest (1989) 83(4):1253–7. doi:10.1172/JCI114009
39. Roatt BM, Aguiar-Soares RD, Coura-Vital W, Ker HG, Moreira N, Vitoriano-Souza J, et al. Immunotherapy and immunochemotherapy in visceral leishmaniasis: promising treatments for this neglected disease. Front Immunol (2014) 5:272. doi:10.3389/fimmu.2014.00272
40. Abate G, Hoft DF. Immunotherapy for tuberculosis: future prospects. Immunotargets Ther (2016) 5:37–45. doi:10.2147/ITT.S81892
41. Richardus JH, Oskam L. Protecting people against leprosy: chemoprophylaxis and immunoprophylaxis. Clin Dermatol (2015) 33(1):19–25. doi:10.1016/j.clindermatol.2014.07.009
42. Carcelain G, Autran B. Immune interventions in HIV infection. Immunol Rev (2013) 254(1):355–71. doi:10.1111/imr.12083
43. Bryceson A. A policy for leishmaniasis with respect to the prevention and control of drug resistance. Trop Med Int Health (2001) 6(11):928–34. doi:10.1046/j.1365-3156.2001.00795.x
44. Dalton JE, Kaye PM. Immunomodulators: use in combined therapy against leishmaniasis. Expert Rev Anti Infect Ther (2010) 8(7):739–42. doi:10.1586/eri.10.64
45. Singh OP, Sundar S. Immunotherapy and targeted therapies in treatment of visceral leishmaniasis: current status and future prospects. Front Immunol (2014) 5:296. doi:10.3389/fimmu.2014.00296
46. Vanham G, Van Gulck E. Can immunotherapy be useful as a “functional cure” for infection with human immunodeficiency virus-1? Retrovirology (2012) 9:72. doi:10.1186/1742-4690-9-72
47. Okwor I, Uzonna JE. The immunology of Leishmania/HIV co-infection. Immunol Res (2013) 56(1):163–71. doi:10.1007/s12026-013-8389-8
48. Gupta G, Oghumu S, Satoskar AR. Mechanisms of immune evasion in leishmaniasis. Adv Appl Microbiol (2013) 82:155–84. doi:10.1016/B978-0-12-407679-2.00005-3
49. Pitta MG, Romano A, Cabantous S, Henri S, Hammad A, Kouriba B, et al. IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J Clin Invest (2009) 119(8):2379–87. doi:10.1172/JCI38813
50. Rodrigues V, Cordeiro-da-Silva A, Laforge M, Silvestre R, Estaquier J. Regulation of immunity during visceral Leishmania infection. Parasit Vectors (2016) 9:118. doi:10.1186/s13071-016-1412-x
51. Gautam S, Kumar R, Singh N, Singh AK, Rai M, Sacks D, et al. CD8 T cell exhaustion in human visceral leishmaniasis. J Infect Dis (2014) 209(2):290–9. doi:10.1093/infdis/jit401
52. Yizengaw E, Getahun M, Tajebe F, Cruz Cervera E, Adem E, Mesfin G, et al. Visceral leishmaniasis patients display altered composition and maturity of neutrophils as well as impaired neutrophil effector functions. Front Immunol (2016) 7:517. doi:10.3389/fimmu.2016.00517
53. Gosselin A, Monteiro P, Chomont N, Diaz-Griffero F, Said EA, Fonseca S, et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T cells are highly permissive to HIV-1 infection. J Immunol (2010) 184(3):1604–16. doi:10.4049/jimmunol.0903058
54. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
55. Desai S, Landay A. Early immune senescence in HIV disease. Curr HIV/AIDS Rep (2010) 7(1):4–10. doi:10.1007/s11904-009-0038-4
56. Casado JL, Abad-Fernandez M, Moreno S, Perez-Elias MJ, Moreno A, Bernardino JI, et al. Visceral leishmaniasis as an independent cause of high immune activation, T-cell senescence, and lack of immune recovery in virologically suppressed HIV-1-coinfected patients. HIV Med (2015) 16(4):240–8. doi:10.1111/hiv.12206
57. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed death 1-mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J Immunol (2013) 191(11):5542–50. doi:10.4049/jimmunol.1301810
58. Silva-Freitas ML, Cota GF, Machado-de-Assis TS, Giacoia-Gripp C, Rabello A, Da-Cruz AM, et al. Immune activation and bacterial translocation: a link between impaired immune recovery and frequent visceral leishmaniasis relapses in HIV-infected patients. PLoS One (2016) 11(12):e0167512. doi:10.1371/journal.pone.0167512
59. Castro A, Carrillo E, San Martin JV, Botana L, Molina L, Matia B, et al. Lymphoproliferative response after stimulation with soluble Leishmania antigen (SLA) as a predictor of visceral leishmaniasis (VL) relapse in HIV+ patients. Acta Trop (2016) 164:345–51. doi:10.1016/j.actatropica.2016.09.026
60. Gois LL, Mehta S, Rodrigues MZ, Schooley RT, Badaro R, Grassi MF. Decreased memory T-cell response and function in human immunodeficiency virus-infected patients with tegumentary leishmaniasis. Mem Inst Oswaldo Cruz (2014) 109(1):9–14. doi:10.1590/0074-0276130174
61. Murray AJ, Kwon KJ, Farber DL, Siliciano RF. The latent reservoir for HIV-1: how immunologic memory and clonal expansion contribute to HIV-1 persistence. J Immunol (2016) 197(2):407–17. doi:10.4049/jimmunol.1600343
62. Khamesipour A. Therapeutic vaccines for leishmaniasis. Expert Opin Biol Ther (2014) 14(11):1641–9. doi:10.1517/14712598.2014.945415
63. Murray HW, Cervia JS, Hariprashad J, Taylor AP, Stoeckle MY, Hockman H. Effect of granulocyte-macrophage colony-stimulating factor in experimental visceral leishmaniasis. J Clin Invest (1995) 95(3):1183–92. doi:10.1172/JCI117767
64. Murray HW, Hariprashad J. Interleukin 12 is effective treatment for an established systemic intracellular infection: experimental visceral leishmaniasis. J Exp Med (1995) 181(1):387–91. doi:10.1084/jem.181.1.387
65. Badaro R, Falcoff E, Badaro FS, Carvalho EM, Pedral-Sampaio D, Barral A, et al. Treatment of visceral leishmaniasis with pentavalent antimony and interferon gamma. N Engl J Med (1990) 322(1):16–21. doi:10.1056/NEJM199001043220104
66. Badaro R, Johnson WD Jr. The role of interferon-gamma in the treatment of visceral and diffuse cutaneous leishmaniasis. J Infect Dis (1993) 167(Suppl 1):S13–7. doi:10.1093/infdis/167.Supplement_1.S13
67. Squires KE, Rosenkaimer F, Sherwood JA, Forni AL, Were JB, Murray HW. Immunochemotherapy for visceral leishmaniasis: a controlled pilot trial of antimony versus antimony plus interferon-gamma. Am J Trop Med Hyg (1993) 48(5):666–9. doi:10.4269/ajtmh.1993.48.666
68. Badaro R, Nascimento C, Carvalho JS, Badaro F, Russo D, Ho JL, et al. Recombinant human granulocyte-macrophage colony-stimulating factor reverses neutropenia and reduces secondary infections in visceral leishmaniasis. J Infect Dis (1994) 170(2):413–8. doi:10.1093/infdis/170.2.413
69. Sundar S, Rosenkaimer F, Lesser ML, Murray HW. Immunochemotherapy for a systemic intracellular infection: accelerated response using interferon-gamma in visceral leishmaniasis. J Infect Dis (1995) 171(4):992–6. doi:10.1093/infdis/171.4.992
70. Sundar S, Singh VP, Sharma S, Makharia MK, Murray HW. Response to interferon-gamma plus pentavalent antimony in Indian visceral leishmaniasis. J Infect Dis (1997) 176(4):1117–9. doi:10.1086/516526
71. Coler RN, Duthie MS, Hofmeyer KA, Guderian J, Jayashankar L, Vergara J, et al. From mouse to man: safety, immunogenicity and efficacy of a candidate leishmaniasis vaccine LEISH-F3+GLA-SE. Clin Transl Immunol (2015) 4(4):e35. doi:10.1038/cti.2015.6
72. Osman M, Mistry A, Keding A, Gabe R, Cook E, Forrester S, et al. A third generation vaccine for human visceral leishmaniasis and post kala azar dermal leishmaniasis: first-in-human trial of ChAd63-KH. PLoS Negl Trop Dis (2017) 11(5):e0005527. doi:10.1371/journal.pntd.0005527
73. Lortholary O, Mechali D, Christiaens D, Gougerot Pocidalo M, Brandely M, Babinet P. Interferon-gamma associated with conventional therapy for recurrent visceral leishmaniasis in a patient with AIDS. Rev Infect Dis (1990) 12(2):370–1. doi:10.1093/clinids/12.2.370
74. de Gorgolas M, Castrillo JM, Fernandez Guerrero ML. Visceral leishmaniasis in patients with AIDS: report of three cases treated with pentavalent antimony and interferon-gamma. Clin Infect Dis (1993) 17(1):56–8. doi:10.1093/clinids/17.1.56
75. Albrecht H, Stellbrink HJ, Gross G, Berg B, Helmchen U, Mensing H. Treatment of atypical leishmaniasis with interferon gamma resulting in progression of Kaposi’s sarcoma in an AIDS patient. Clin Investig (1994) 72(12):1041–7. doi:10.1007/BF00577752
76. Mastroianni A. Liposomal amphotericin B and rHuGM-CSF for treatment of visceral leishmaniasis in AIDS. Infez Med (2004) 12(3):197–204.
77. Bossolasco S, Nozza S, Gaiera G, Bestetti A, Lazzarin A, Cinque P. Lack of immune recovery in HIV/Leishmania co-infection treated with human recombinant IL-2. AIDS (2007) 21(9):1223–5. doi:10.1097/QAD.0b013e32810c8d27
78. Roff SR, Noon-Song EN, Yamamoto JK. The significance of interferon-gamma in HIV-1 pathogenesis, therapy, and prophylaxis. Front Immunol (2014) 4:498. doi:10.3389/fimmu.2013.00498
79. Laguna F. Treatment of leishmaniasis in HIV-positive patients. Ann Trop Med Parasitol (2003) 97(Suppl 1):135–42. doi:10.1179/000349803225002606
80. Jarvis JN, Meintjes G, Rebe K, Williams GN, Bicanic T, Williams A, et al. Adjunctive interferon-gamma immunotherapy for the treatment of HIV-associated cryptococcal meningitis: a randomized controlled trial. AIDS (2012) 26(9):1105–13. doi:10.1097/QAD.0b013e3283536a93
81. Pappas PG, Bustamante B, Ticona E, Hamill RJ, Johnson PC, Reboli A, et al. Recombinant interferon-gamma 1b as adjunctive therapy for AIDS-related acute cryptococcal meningitis. J Infect Dis (2004) 189(12):2185–91. doi:10.1086/420829
82. Mock DJ, Hollenbaugh JA, Daddacha W, Overstreet MG, Lazarski CA, Fowell DJ, et al. Leishmania induces survival, proliferation and elevated cellular dNTP levels in human monocytes promoting acceleration of HIV co-infection. PLoS Pathog (2012) 8(4):e1002635. doi:10.1371/journal.ppat.1002635
83. Brown PA, Angel JB. Granulocyte-macrophage colony-stimulating factor as an immune-based therapy in HIV infection. J Immune Based Ther Vaccines (2005) 3(1):3. doi:10.1186/1476-8518-3-3
84. Leth S, Schleimann MH, Nissen SK, Hojen JF, Olesen R, Graversen ME, et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV (2016) 3(10):e463–72. doi:10.1016/S2352-3018(16)30055-8
85. Bandera A, Trabattoni D, Ferrario G, Cesari M, Franzetti F, Clerici M, et al. Interferon-gamma and granulocyte-macrophage colony stimulating factor therapy in three patients with pulmonary aspergillosis. Infection (2008) 36(4):368–73. doi:10.1007/s15010-008-7378-7
86. de Silva TI, Cope A, Goepel J, Greig JM. The use of adjuvant granulocyte-macrophage colony-stimulating factor in HIV-related disseminated atypical mycobacterial infection. J Infect (2007) 54(4):e207–10. doi:10.1016/j.jinf.2006.11.005
87. Murray HW, Miralles GD, Stoeckle MY, McDermott DF. Role and effect of IL-2 in experimental visceral leishmaniasis. J Immunol (1993) 151(2):929–38.
88. Group I-ES, Committee SS, Abrams D, Levy Y, Losso MH, Babiker A, et al. Interleukin-2 therapy in patients with HIV infection. N Engl J Med (2009) 361(16):1548–59. doi:10.1056/NEJMoa0903175
89. Markowitz N, Lopardo G, Wentworth D, Gey D, Babiker A, Fox L, et al. Long-term effects of intermittent IL-2 in HIV infection: extended follow-up of the INSIGHT STALWART study. PLoS One (2012) 7(10):e47506. doi:10.1371/journal.pone.0047506
90. Bodas M, Jain N, Awasthi A, Martin S, Penke Loka RK, Dandekar D, et al. Inhibition of IL-2 induced IL-10 production as a principle of phase-specific immunotherapy. J Immunol (2006) 177(7):4636–43. doi:10.4049/jimmunol.177.7.4636
91. Khamesipour A, Dowlati Y, Asilian A, Hashemi-Fesharki R, Javadi A, Noazin S, et al. Leishmanization: use of an old method for evaluation of candidate vaccines against leishmaniasis. Vaccine (2005) 23(28):3642–8. doi:10.1016/j.vaccine.2005.02.015
92. Jain K, Jain NK. Vaccines for visceral leishmaniasis: a review. J Immunol Methods (2015) 422:1–12. doi:10.1016/j.jim.2015.03.017
93. Musa AM, Noazin S, Khalil EA, Modabber F. Immunological stimulation for the treatment of leishmaniasis: a modality worthy of serious consideration. Trans R Soc Trop Med Hyg (2010) 104(1):1–2. doi:10.1016/j.trstmh.2009.07.026
94. Musa AM, Khalil EA, Mahgoub FA, Elgawi SH, Modabber F, Elkadaru AE, et al. Immunochemotherapy of persistent post-kala-azar dermal leishmaniasis: a novel approach to treatment. Trans R Soc Trop Med Hyg (2008) 102(1):58–63. doi:10.1016/j.trstmh.2007.08.006
95. Badaro R, Lobo I, Munos A, Netto EM, Modabber F, Campos-Neto A, et al. Immunotherapy for drug-refractory mucosal leishmaniasis. J Infect Dis (2006) 194(8):1151–9. doi:10.1086/507708
96. Convit J, Ulrich M, Zerpa O, Borges R, Aranzazu N, Valera M, et al. Immunotherapy of American cutaneous leishmaniasis in Venezuela during the period 1990–99. Trans R Soc Trop Med Hyg (2003) 97(4):469–72. doi:10.1016/S0035-9203(03)90093-9
97. Convit J, Ulrich M, Polegre MA, Avila A, Rodriguez N, Mazzedo MI, et al. Therapy of Venezuelan patients with severe mucocutaneous or early lesions of diffuse cutaneous leishmaniasis with a vaccine containing pasteurized Leishmania promastigotes and bacillus Calmette-Guerin: preliminary report. Mem Inst Oswaldo Cruz (2004) 99(1):57–62. doi:10.1590/S0074-02762004000100010
98. Mayrink W, Botelho AC, Magalhaes PA, Batista SM, Lima Ade O, Genaro O, et al. Immunotherapy, immunochemotherapy and chemotherapy for American cutaneous leishmaniasis treatment. Rev Soc Bras Med Trop (2006) 39(1):14–21. doi:10.1590/S0037-86822006000100003
99. Machado-Pinto J, Pinto J, da Costa CA, Genaro O, Marques MJ, Modabber F, et al. Immunochemotherapy for cutaneous leishmaniasis: a controlled trial using killed Leishmania (Leishmania) amazonensis vaccine plus antimonial. Int J Dermatol (2002) 41(2):73–8. doi:10.1046/j.1365-4362.2002.01336.x
100. Trigo J, Abbehusen M, Netto EM, Nakatani M, Pedral-Sampaio G, de Jesus RS, et al. Treatment of canine visceral leishmaniasis by the vaccine Leish-111f+MPL-SE. Vaccine (2010) 28(19):3333–40. doi:10.1016/j.vaccine.2010.02.089
101. Crum-Cianflone NF, Wallace MR. Vaccination in HIV-infected adults. AIDS Patient Care STDS (2014) 28(8):397–410. doi:10.1089/apc.2014.0121
102. Rubin LG, Levin MJ, Ljungman P, Davies EG, Avery R, Tomblyn M, et al. 2013 IDSA clinical practice guideline for vaccination of the immunocompromised host. Clin Infect Dis (2014) 58(3):e44–100. doi:10.1093/cid/cit684
103. Taslimi Y, Zahedifard F, Rafati S. Leishmaniasis and various immunotherapeutic approaches. Parasitology (2016) 15:1–11. doi:10.1017/S003118201600216X
104. Murray HW, Montelibano C, Peterson R, Sypek JP. Interleukin-12 regulates the response to chemotherapy in experimental visceral leishmaniasis. J Infect Dis (2000) 182(5):1497–502. doi:10.1086/315890
105. Strauss-Ayali D, Baneth G, Shor S, Okano F, Jaffe CL. Interleukin-12 augments a Th1-type immune response manifested as lymphocyte proliferation and interferon gamma production in Leishmania infantum-infected dogs. Int J Parasitol (2005) 35(1):63–73. doi:10.1016/j.ijpara.2004.10.015
106. Ghalib HW, Whittle JA, Kubin M, Hashim FA, el-Hassan AM, Grabstein KH, et al. IL-12 enhances Th1-type responses in human Leishmania donovani infections. J Immunol (1995) 154(9):4623–9.
107. Villinger F, Ansari AA. Role of IL-12 in HIV infection and vaccine. Eur Cytokine Netw (2010) 21(3):215–8. doi:10.1684/ecn.2010.0206
108. Jacobson MA, Spritzler J, Landay A, Chan E, Katzenstein D, Schock B, et al. A phase I, placebo-controlled trial of multi-dose recombinant human interleukin-12 in patients with HIV infection. AIDS (2002) 16(8):1147–54. doi:10.1097/00002030-200205240-00008
109. Little RF, Aleman K, Kumar P, Wyvill KM, Pluda JM, Read-Connole E, et al. Phase 2 study of pegylated liposomal doxorubicin in combination with interleukin-12 for AIDS-related Kaposi sarcoma. Blood (2007) 110(13):4165–71. doi:10.1182/blood-2007-06-097568
110. Okhuysen PC, Chappell CL, Lewis DE, Robinson P, White AC Jr. Treatment of chronic cryptosporidiosis in AIDS with rIL-12 induces an immune response associated with improvement but severe side-effects. AIDS (2005) 19(12):1333–4. doi:10.1097/01.aids.0000180110.03759.e3
111. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood (1997) 90(7):2541–8.
112. Quirino GF, Nascimento MS, Davoli-Ferreira M, Sacramento LA, Lima MH, Almeida RP, et al. Interleukin-27 (IL-27) mediates susceptibility to visceral leishmaniasis by suppressing the IL-17-neutrophil response. Infect Immun (2016) 84(8):2289–98. doi:10.1128/IAI.00283-16
113. Gessner A, Vieth M, Will A, Schroppel K, Rollinghoff M. Interleukin-7 enhances antimicrobial activity against Leishmania major in murine macrophages. Infect Immun (1993) 61(9):4008–12.
114. Sawa Y, Arima Y, Ogura H, Kitabayashi C, Jiang JJ, Fukushima T, et al. Hepatic interleukin-7 expression regulates T cell responses. Immunity (2009) 30(3):447–57. doi:10.1016/j.immuni.2009.01.007
115. Katlama C, Lambert-Niclot S, Assoumou L, Papagno L, Lecardonnel F, Zoorob R, et al. Treatment intensification followed by interleukin-7 reactivates HIV without reducing total HIV DNA: a randomized trial. AIDS (2016) 30(2):221–30. doi:10.1097/QAD.0000000000000894
116. Bhattacharjee S, Gupta G, Bhattacharya P, Adhikari A, Majumdar SB, Majumdar S. Anti-iL-10 mAb protection against experimental visceral leishmaniasis via induction of Th1 cytokines and nitric oxide. Indian J Exp Biol (2009) 47(6):489–97.
117. Murray HW, Moreira AL, Lu CM, DeVecchio JL, Matsuhashi M, Ma X, et al. Determinants of response to interleukin-10 receptor blockade immunotherapy in experimental visceral leishmaniasis. J Infect Dis (2003) 188(3):458–64. doi:10.1086/376510
118. Faleiro RJ, Kumar R, Bunn PT, Singh N, Chauhan SB, Sheel M, et al. Combined immune therapy for the treatment of visceral leishmaniasis. PLoS Negl Trop Dis (2016) 10(2):e0004415. doi:10.1371/journal.pntd.0004415
119. Murray HW. Interleukin 10 receptor blockade – pentavalent antimony treatment in experimental visceral leishmaniasis. Acta Trop (2005) 93(3):295–301. doi:10.1016/j.actatropica.2004.11.008
120. Stylianou E, Aukrust P, Kvale D, Muller F, Froland SS. IL-10 in HIV infection: increasing serum IL-10 levels with disease progression – down-regulatory effect of potent anti-retroviral therapy. Clin Exp Immunol (1999) 116(1):115–20. doi:10.1046/j.1365-2249.1999.00865.x
121. Kwon DS, Kaufmann DE. Protective and detrimental roles of IL-10 in HIV pathogenesis. Eur Cytokine Netw (2010) 21(3):208–14. doi:10.1684/ecn.2010.0201
122. Ni GY, Wang TF, Walton S, Zhu B, Chen S, Wu XL, et al. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol (2015) 293(2):126–9. doi:10.1016/j.cellimm.2014.12.012
124. Paiardini M, Muller-Trutwin M. HIV-associated chronic immune activation. Immunol Rev (2013) 254(1):78–101. doi:10.1111/imr.12079
125. Okoye AA, Picker LJ. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev (2013) 254(1):54–64. doi:10.1111/imr.12066
126. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443(7109):350–4. doi:10.1038/nature05115
127. Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O’Shea MA, et al. The common gamma-chain cytokines IL-2, IL-7, IL-15, and IL-21 induce the expression of programmed death-1 and its ligands. J Immunol (2008) 181(10):6738–46. doi:10.4049/jimmunol.181.10.6738
128. Rodrigues V, Cordeiro-da-Silva A, Laforge M, Ouaissi A, Akharid K, Silvestre R, et al. Impairment of T cell function in parasitic infections. PLoS Negl Trop Dis (2014) 8(2):e2567. doi:10.1371/journal.pntd.0002567
129. Murray HW, Lu CM, Brooks EB, Fichtl RE, DeVecchio JL, Heinzel FP. Modulation of T-cell costimulation as immunotherapy or immunochemotherapy in experimental visceral leishmaniasis. Infect Immun (2003) 71(11):6453–62. doi:10.1128/IAI.71.11.6453-6462.2003
130. Chiku VM, Silva KL, de Almeida BF, Venturin GL, Leal AA, de Martini CC, et al. PD-1 function in apoptosis of T lymphocytes in canine visceral leishmaniasis. Immunobiology (2016) 221(8):879–88. doi:10.1016/j.imbio.2016.03.007
131. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stager S. B7-H1 blockade increases survival of dysfunctional CD8(+) T cells and confers protection against Leishmania donovani infections. PLoS Pathog (2009) 5(5):e1000431. doi:10.1371/journal.ppat.1000431
132. Gay CL, Bosch RJ, Ritz J, Hataye JM, Aga E, Tressler RL, et al. Clinical trial of the anti-PD-L1 antibody BMS-936559 in HIV-1 infected participants on suppressive antiretroviral therapy. J Infect Dis (2017) 215(11):1725–33. doi:10.1093/infdis/jix191
133. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med (2016) 22(7):754–61. doi:10.1038/nm.4113
134. Gannavaram S, Dey R, Avishek K, Selvapandiyan A, Salotra P, Nakhasi HL. Biomarkers of safety and immune protection for genetically modified live attenuated Leishmania vaccines against visceral leishmaniasis – discovery and implications. Front Immunol (2014) 5:241. doi:10.3389/fimmu.2014.00241
135. Seifert K, Juhls C, Salguero FJ, Croft SL. Sequential chemoimmunotherapy of experimental visceral leishmaniasis using a single low dose of liposomal amphotericin B and a novel DNA vaccine candidate. Antimicrob Agents Chemother (2015) 59(9):5819–23. doi:10.1128/AAC.00273-15
136. Bagirova M, Allahverdiyev AM, Abamor ES, Ullah I, Cosar G, Aydogdu M, et al. Overview of dendritic cell-based vaccine development for leishmaniasis. Parasite Immunol (2016) 38(11):651–62. doi:10.1111/pim.12360
137. Garcia F, Plana M, Climent N, Leon A, Gatell JM, Gallart T. Dendritic cell based vaccines for HIV infection: the way ahead. Hum Vaccin Immunother (2013) 9(11):2445–52. doi:10.4161/hv.25876
138. Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur J Immunol (2004) 34(5):1433–40. doi:10.1002/eji.200324021
139. Vieillard V, Gharakhanian S, Lucar O, Katlama C, Launay O, Autran B, et al. Perspectives for immunotherapy: which applications might achieve an HIV functional cure? Oncotarget (2016) 7(25):38946–58. doi:10.18632/oncotarget.7793
140. Mukherjee AK, Gupta G, Adhikari A, Majumder S, Kar Mahapatra S, Bhattacharyya Majumdar S, et al. Miltefosine triggers a strong proinflammatory cytokine response during visceral leishmaniasis: role of TLR4 and TLR9. Int Immunopharmacol (2012) 12(4):565–72. doi:10.1016/j.intimp.2012.02.002
141. Mukherjee AK, Gupta G, Bhattacharjee S, Guha SK, Majumder S, Adhikari A, et al. Amphotericin B regulates the host immune response in visceral leishmaniasis: reciprocal regulation of protein kinase C isoforms. J Infect (2010) 61(2):173–84. doi:10.1016/j.jinf.2010.05.003
142. Wadhone P, Maiti M, Agarwal R, Kamat V, Martin S, Saha B. Miltefosine promotes IFN-gamma-dominated anti-leishmanial immune response. J Immunol (2009) 182(11):7146–54. doi:10.4049/jimmunol.0803859
143. Mookerjee Basu J, Mookerjee A, Sen P, Bhaumik S, Sen P, Banerjee S, et al. Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother (2006) 50(5):1788–97. doi:10.1128/AAC.50.5.1788-1797.2006
144. Kulshrestha A, Singh R, Kumar D, Negi NS, Salotra P. Antimony-resistant clinical isolates of Leishmania donovani are susceptible to paromomycin and sitamaquine. Antimicrob Agents Chemother (2011) 55(6):2916–21. doi:10.1128/AAC.00812-10
145. Ghosh M, Roy K, Roy S. Immunomodulatory effects of antileishmanial drugs. J Antimicrob Chemother (2013) 68(12):2834–8. doi:10.1093/jac/dkt262
146. Griewank K, Gazeau C, Eichhorn A, von Stebut E. Miltefosine efficiently eliminates Leishmania major amastigotes from infected murine dendritic cells without altering their immune functions. Antimicrob Agents Chemother (2010) 54(2):652–9. doi:10.1128/AAC.01014-09
147. Muniz-Junqueira MI, de Paula-Coelho VN. Meglumine antimonate directly increases phagocytosis, superoxide anion and TNF-alpha production, but only via TNF-alpha it indirectly increases nitric oxide production by phagocytes of healthy individuals, in vitro. Int Immunopharmacol (2008) 8(12):1633–8. doi:10.1016/j.intimp.2008.07.011
148. Sane SA, Shakya N, Haq W, Gupta S. CpG oligodeoxynucleotide augments the antileishmanial activity of miltefosine against experimental visceral leishmaniasis. J Antimicrob Chemother (2010) 65(7):1448–54. doi:10.1093/jac/dkq164
149. Clerici M. Beyond IL-17: new cytokines in the pathogenesis of HIV infection. Curr Opin HIV AIDS (2010) 5(2):184–8. doi:10.1097/COH.0b013e328335c23c
150. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell (2012) 148(3):434–46. doi:10.1016/j.cell.2011.12.023
151. Jarvis JN, Casazza JP, Stone HH, Meintjes G, Lawn SD, Levitz SM, et al. The phenotype of the Cryptococcus-specific CD4+ memory T-cell response is associated with disease severity and outcome in HIV-associated cryptococcal meningitis. J Infect Dis (2013) 207(12):1817–28. doi:10.1093/infdis/jit099
Keywords: visceral leishmaniasis, kala-azar, human immunodeficiency virus, immunotherapy, immunomodulation, coinfection, immunity, vaccination
Citation: Adriaensen W, Dorlo TPC, Vanham G, Kestens L, Kaye PM and van Griensven J (2018) Immunomodulatory Therapy of Visceral Leishmaniasis in Human Immunodeficiency Virus-Coinfected Patients. Front. Immunol. 8:1943. doi: 10.3389/fimmu.2017.01943
Received: 10 October 2017; Accepted: 18 December 2017;
Published: 12 January 2018
Edited by:
Nahid Ali, Indian Institute of Chemical Biology, IndiaReviewed by:
Adrian John Frederick Luty, Institut de recherche pour le développement (IRD), FranceHenry Muriuki Kariithi, International Atomic Energy Agency, Austria
Copyright: © 2018 Adriaensen, Dorlo, Vanham, Kestens, Kaye and van Griensven. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wim Adriaensen, d2FkcmlhZW5zZW5AaXRnLmJl