 Helder Novais Bastos1,2,3†
Helder Novais Bastos1,2,3† Nuno S. Osório2,3†
Nuno S. Osório2,3† Sebastien Gagneux4,5
Sebastien Gagneux4,5 Iñaki Comas6,7*†
Iñaki Comas6,7*† Margarida Saraiva8,9*†
Margarida Saraiva8,9*†
- 1Department of Pneumology, Centro Hospitalar do São João, Porto, Portugal
- 2Life and Health Sciences Research Institute (ICVS), School of Medicine, University of Minho, Braga, Portugal
- 3ICVS/3B’s—PT Government Associate Laboratory, Braga, Portugal
- 4Department of Medical Parasitology and Infection Biology, Swiss Tropical and Public Health Institute, Basel, Switzerland
- 5University of Basel, Basel, Switzerland
- 6Institute of Biomedicine of Valencia (IBV-CSIC), Valencia, Spain
- 7CIBER of Epidemiology and Public Health (CIBERESP), Madrid, Spain
- 8i3S—Instituto de Investigação e Inovação em Saúde, University of Porto, Porto, Portugal
- 9Instituto de Biologia Molecular e Celular (IBMC), University of Porto, Porto, Portugal
The already enormous burden caused by tuberculosis (TB) will be further aggravated by the association of this disease with modern epidemics, as human immunodeficiency virus and diabetes. Furthermore, the increasingly aging population and the wider use of suppressive immune therapies hold the potential to enhance the incidence of TB. New preventive and therapeutic strategies based on recent advances on our understanding of TB are thus needed. In particular, understanding the intricate network of events modulating inflammation in TB will help to build more effective vaccines and host-directed therapies to stop TB. This review integrates the impact of host, pathogen, and extrinsic factors on inflammation and the almost scientifically unexplored complexity emerging from the interactions between these three factors. We highlight the exciting data showing a contribution of this troika for the clinical outcome of TB and the need of incorporating it when developing novel strategies to rewire the immune response in TB.
Introduction
According to current estimates, tuberculosis (TB) accounted for approximately 1.7 million deaths in 2016 and affected one-quarter of the world’s population in its latent form (1). TB is a heterogeneous disease, characterized by a continuous spectrum of infection, for which molecular and clinical biomarkers of progression are just starting to be unveiled. Several stages of latent TB infection (LTBI) exist and include subclinical forms of TB with an increased likelihood of progressing to active disease (2, 3). The clinical manifestation of the active disease is highly variable, with mild or extensive pulmonary involvement, extrapulmonary, or disseminated forms of TB. Many known TB precipitating factors, which either increase the susceptibility to TB or the risk of transition from LTBI to active TB, are connected with immune imbalances (4, 5). However, the molecular mechanisms governing the transitions along the TB spectrum remain unknown.
The immune condition of infected hosts is shaped by genetics, extrinsic factors altering the local microenvironment, and the heterogeneity of the infecting bacteria. This troika determines the threshold of the immune response generated during infection and possibly the disease outcome (Figure 1). Modulating these thresholds and uncovering the links between host, pathogen, and microenvironments should allow for the discovery of solid correlates of protection, molecular markers for disease prognosis, and the development of safe and effective host-directed therapies (HDTs) to TB. This review covers our current understanding of the impact of these elements on TB, their interactions, and how they may be further explored as a platform for developing new products and strategies against TB.
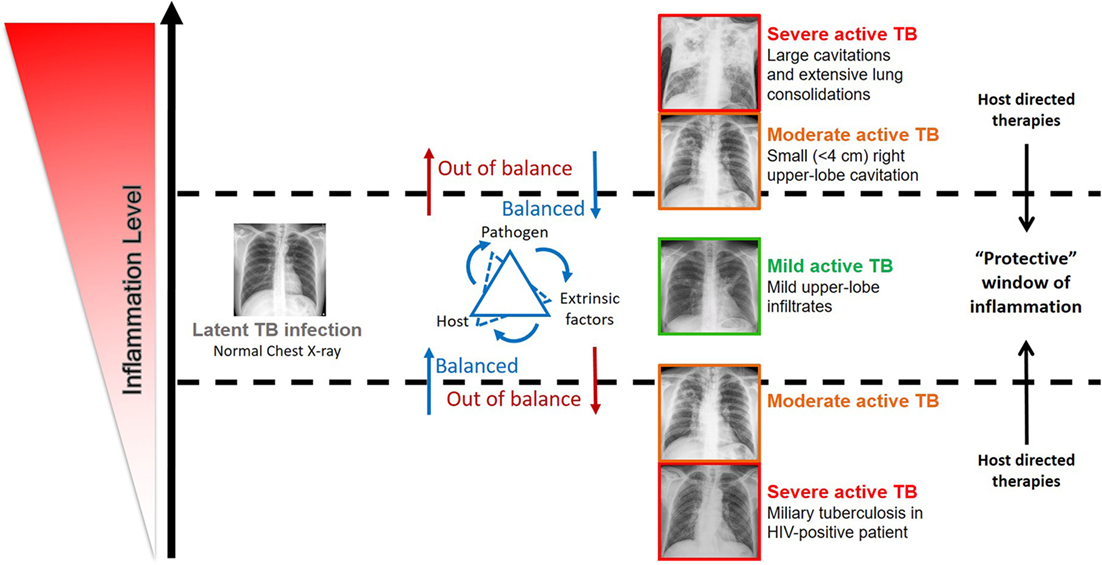
Figure 1. Impact of the inflammation level in the disease outcome in individuals infected by Mycobacterium tuberculosis complex bacteria. The spectrum of tuberculosis (TB) disease is strongly linked with the host immune status. The inflammation level results from the interaction of host, pathogen, and extrinsic factors. Very low and high inflammation levels often associated with severe active TB, while balanced immune responses associated with mild active TB, latent TB, and possibly TB clearance. Evidence supports that host-directed therapies (see Table 1) have the potential to successfully modulate inflammation and ameliorate disease outcome, by ensuring a protective immune response.
Host Genetics: Role in Immunomodulation
The association of single-gene inborn errors related to interferon (IFN)-γ immunity with severe childhood TB provides the clearest genetic basis for TB susceptibility (6, 7). These conditions, globally named Mendelian susceptibility to mycobacterial disease (MSMD) encompass a series of germline mutations in seven autosomal (IFNGR1, IFNGR2, IL12B, IL12RB1, STAT1, IRF8, and ISG15) and two X-linked (NEMO and CYBB) genes (6, 7). These defects are functionally and physiologically related, as they all result in an impairment of the CD4 T cell-mediated immunity and have provided decisive evidence on the critical protective role of the interleukin (IL)-12/IL-23/IFN-γ loop in TB (6, 7). This role was further confirmed in the context of secondary immunodeficiencies, such as in human immunodeficiency virus (HIV) infection, as discussed below.
Genetic association studies with adult patients showed more limited success than MSMD, and strikingly no consistent association of variants of genes from the IL-12/IFN-γ axis with TB susceptibility in adulthood was found. Instead, candidate-based studies found a number of genetic variants associated with TB susceptibility in humans (7–9). However, results remain inconsistent and have not been validated in different populations nor in genome-wide association studies (GWAS) (10–12). This most likely reflects the association of adult pulmonary TB with complex genetic traits, where the role of genetic–extrinsic factors and gene–gene interactions (epistasis) dominate over single polymorphisms on their own (13, 14). The identification of genetic risk factors is also likely masked by the experimental design, where important contributors, such as extrinsic factors, or pathogen variability are largely neglected. Furthermore, the full spectrum of TB has been mostly ignored in the group definition, with all phenotypes being analyzed together in two main study groups: active TB versus healthy controls/LTBI. Finally, combining and integrating genetic association studies with the investigation of the human epigenome will certainly lead to critical insights into the genetic basis of infection and clinical TB.
The Modulation of the Immune Response by Extrinsic Factors
The lack of clear association of human genotypes with TB susceptibility and the fact that progression from LTBI to pulmonary TB in adults usually reflects an impairment of host resistance due to non-genetic factors, highlighting the relevance of extrinsic factors in shaping the host immunity with an impact on TB outcome (Figure 2).
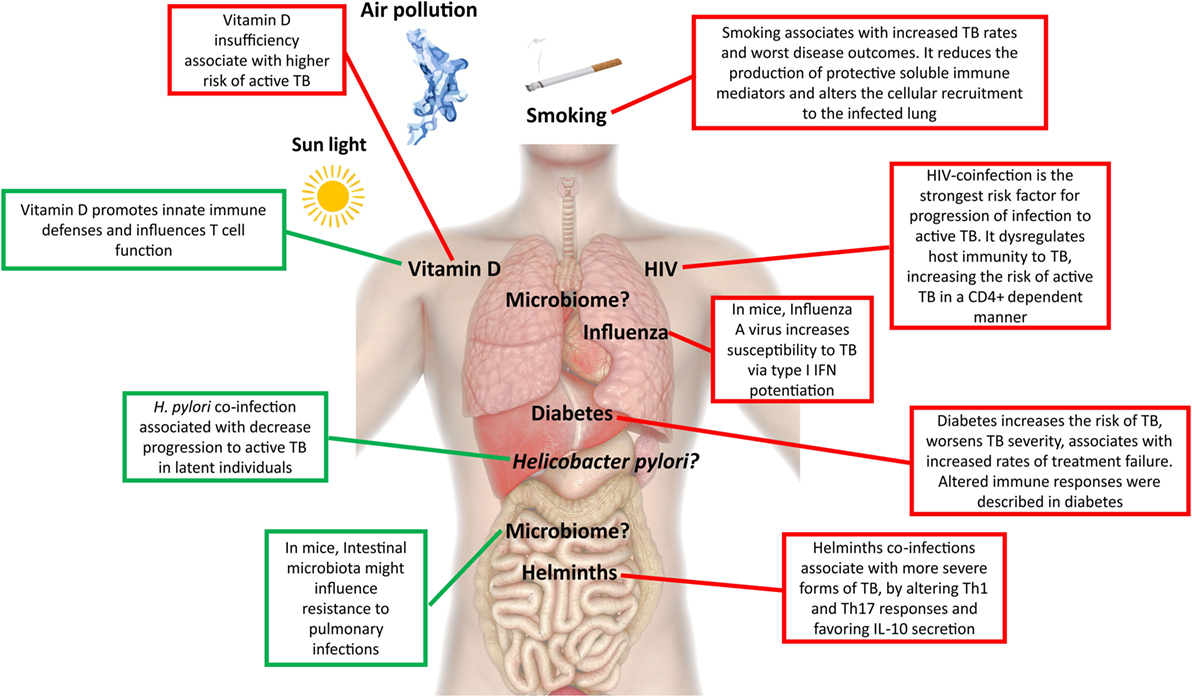
Figure 2. Extrinsic factors associated with active tuberculosis (TB). This figure depicts extrinsic factors associated with protection to active TB (green) or susceptibility/increased risk (red) for active TB development.
Coinfections
A major driver of the current TB epidemics has been the HIV syndemic, which dramatically decreases the host protective responses to TB in a CD4 T cell count-dependent manner (15), leading to an acceleration of both diseases. Because the risk of developing TB is largely increased in HIV-infected individuals even before CD4 T cell counts decrease (16), other factors must also play a role. Indeed, a detrimental alternative activation of macrophages, accompanied by less nitric oxide (NO) synthase expression and poorly formed granulomas was described in HIV–TB, which in turn downregulated the Mycobacterium tuberculosis DosR regulon (17), a set of genes known to be induced during anaerobic dormancy (18). Therefore, changes in the host immunity resulting from HIV coinfection remodel the bacterial physiology, further rewiring the host tissue microenvironment. Consequently, the pathogenesis of TB is different in HIV-coinfected individuals, resulting in the lack of complete cavitation and in a higher incidence of disseminated disease (19, 20). Other less studied coinfections with impact on TB include helminths, influenza, and Helicobacter pylori. In TB patients coinfected with helminths, a more advanced form of disease was reported (21), possibly related with decreased T helper (Th) 1 and Th17 cell responses and increased secretion of IL-10 (22, 23). In the case of influenza, the increased susceptibility to M. tuberculosis infection is likely mediated by type I IFN signaling (24). Contrastingly, infection with the ubiquitous bacteria H. pylori may help to avoid progression to active TB in latent individuals, due to enhanced IFN-γ and other Th1-like cytokine responses generated in response to H. pylori and that restrain M. tuberculosis (25). Another example of coinfection cross talk in TB comes from the mouse model, in which prior Helicobacter hepaticus colonization impaired the immune control of M. tuberculosis (26). It is interesting that of these three infections, only influenza is also a lung disease, thus indicating that distant events shape the lung microenvironment.
The Host Microbiome
Different mouse models of infection depleted of commensal gut microbiota after antibiotic treatment showed an increased risk of colonization by respiratory pathogens, such as Streptococcus pneumoniae (27), Staphylococcus aureus (28), and Klebsiella pneumoniae (29). Alterations in the gut microbiota also alter the susceptibility to TB (30), and an increase in the lung bacterial burden early post M. tuberculosis infection was reported in germ-free mice (31). Limited available data suggest that the gut microbiota of mice infected with M. tuberculosis is drastically reduced after initial infection (32). Gut diversity in M. tuberculosis-infected mice is recovered about the time that the adaptive immune system is onset, although significant differences in taxa composition remain in pre- and postinfection samples (32). In addition, experiment removal of the gut microbiota with antibiotic treatment leads to higher susceptibility to M. tuberculosis infection in mice (30). How the intestinal microbiota distally affects pulmonary immunity, and eventually the course of infection and disease, and how the gut–lung axis may impact TB await further research.
We are also still far from understanding the composition and the impact of changes in the oral–nasal cavity and lung microbiota on TB. M. tuberculosis establishes infection in the lower respiratory tract, and as such has to initially evade microbiota-activated macrophages of the upper respiratory tract (33). It remains to be seen if the presence or absence of certain microbial species in the upper respiratory airway generates a more permissive environment for the establishment of TB infection. If this is the case, restoring key players of the microbiota may help to fight invasion or even improve immunity, therapeutic possibilities not yet explored in TB infection.
Non-Communicable Comorbidities
Among non-communicable comorbidities, the presence of diabetes remains the major risk factor for TB. Owing to the dimension of the diabetes epidemics, its foreseen impact on the global numbers of TB cases is alarming. Diabetic patients have a three times higher risk of developing TB than healthy individuals (34). Diabetes also worsens disease severity (35), is a risk factor for death in TB patients (36, 37), and is associated with increased failure of standard TB treatment (35, 38, 39). Previous studies suggest that the interaction of M. tuberculosis with macrophages and dendritic cells (DCs) is impaired in the context of diabetes, leading to an initial hypo-inflammatory state (40–43). Once the infection is established, there is evidence for increased inflammation in TB/diabetes patients, as an augmented level of pro-inflammatory cytokines in the peripheral blood is measured, likely due to hyperactive T cell responses (41–43). It is possible that many of the alterations seen and their impact on TB are actually interconnected to changes in the composition of the human gut microbiota imposed by diabetes (44).
Subjects with chronic obstructive pulmonary disease (COPD) present greater risks for developing active TB (45, 46) and TB death as compared with TB patients without this comorbidity (36, 45). Altered immune responses likely underlie the mechanisms linking COPD and TB. COPD is characterized by a disruption of innate defense mechanisms in the airways, including decreased mucociliary clearance and impaired macrophage phagocytosis (47, 48). Furthermore, an accumulation of lung regulatory T cells and the increase of circulating IL-10 and TGF-β (49) were described in COPD patients. Thus, COPD limits the effector function of T cells in response to pathogens, which may explain the increased susceptibility to lower respiratory tract bacterial infections, including M. tuberculosis.
Environmental Factors
Smoking exposure is an independent risk factor for M. tuberculosis infection, progression to active disease, and for poor treatment outcomes (50–52). The underlying immunological mechanisms are just starting to be unveiled and include reduced production of tumor necrosis factor (TNF), IL-1β, and IFN-γ by in vitro infected alveolar macrophages (53), decreased number of DCs (54, 55), and compromised recruitment of IFN-γ-producing CD4 T cells to the lung, thus weakening the formation of granuloma (56). More recently, alveolar macrophages from smokers were found to exhibit lysosomal accumulations of tobacco smoke particulates, which impaired their migration toward M. tuberculosis-infected cells (57).
Malnutrition has also been associated with an increased risk of active TB, although it remains unclear whether the nutritional status is a cause or a consequence of the disease. A strong link between TB, malnutrition, and immune dysregulation is in place (58). This association is even worse in the framework of HIV infection (59). Nutritional status not only affects the function of several immune cells, including T cells, but additionally impacts the pharmacodynamics of the drugs (60). Less clear is the role of anemia, a common symptom, and prognosis marker of TB (36), particularly linked to iron deprivation, in facilitating or exacerbating TB disease. An emerging role is being recognized for hepcidin, a protein that regulates the homeostasis and cell type distribution of iron in the body and that plays a role on innate immune responses to mycobacterial infection (61, 62). In agreement, in epidemiological studies hepcidin levels have been positively correlated with increased risk of mortality in TB–HIV coinfection usually associated with more death-threatening manifestation of the disease, as extrapulmonary and miliary TB (63). Another nutrient, vitamin D, has been the focus of renewed attention by researchers. Historically, both vitamin D and exposure to sunlight, which endogenously promotes the conversion in the skin of 7-dehydrocholesterol into pre-vitamin D3, were used in the treatment of TB (64). Insufficiency of this molecule has been linked to higher risk of active TB (65, 66) and increased propensity for extrapulmonary involvement (67). The immunomodulatory role of vitamin D is well established leading to several changes in immune responses, including the induction of cathelicidin antimicrobial peptide, beta-defensin, and the promotion of authophagy and/or bacterial killing (68, 69). However, it is important to mention that despite several reports on the protective role for autophagy during M. tuberculosis infection (70), a recent study based on a genetic approach targeting multiple autophagy-related genes concluded that the cellular autophagic capacity did not correlate with the outcome of M. tuberculosis infection (71). Therefore, it is possible that the impact of autophagy during M. tuberculosis infection results from the use of in vitro models, thus calling for further in vivo studies when correlating protective mediators with induction of autophagy. Vitamin D levels have been shown to impact adaptive immune responses by influencing Th cell function and by promoting Tregs (72). It thus seems that vitamin D promotes the macrophage effector function, while at the same time keeping the immune response at check through its action on T cells. Several clinical trials have shown that supplementation of drug regimens with vitamin D does not improve TB outcomes in the general population (73), but it has been shown to accelerate sputum conversion in patients with the tt genotype of the Taql vitamin D receptor polymorphism (74). It remains to be seen if vitamin D can play a role in preventing infection or progression to active TB. It is possible that through the modulation of vitamin D, variations in sunlight may underlie some of the differences in TB incidence rate across the globe (65). This not only includes natural seasonal variations but also artificial variation due to human-associated activities, such as pollution.
M. tuberculosis Diversity and Immunomodulation: Paradigm Changing Evidence
How Variable Is M. tuberculosis?
Tuberculosis is caused by a group of phylogenetically closely related bacteria, collectively known as the M. tuberculosis complex (MTBC), now known to encompass seven main phylogenetic lineages of human-adapted bacteria (75). Within this complex, M. tuberculosis and Mycobacterium africanum (in West Africa) are responsible for the large majority of human cases of TB (76). The genetic diversity within the MTBC is higher than originally expected and can be observed at different evolutionary and geographical scales (Figure 3). Most of the diversity observed is likely due to genetic drift, i.e., stochastic variation of diversity due to limited population sizes, or to neutral variation with no impact on the fitness of the bacteria (77). Diversification of the initial infecting bacteria in subpopulations within a single patient has also been reported (78), part of it is likely due to antibiotic selection pressures (79, 80). Although still unclear, additional diversity maybe selected independently of antibiotic pressure (80). It is possible that some of the diversity within the host reflects the heterogeneous immune responses associated with different lung lesions (81). The complexity of the lung microstructure is now acknowledged, with the immune response being spatially separated even in single granulomas (82).
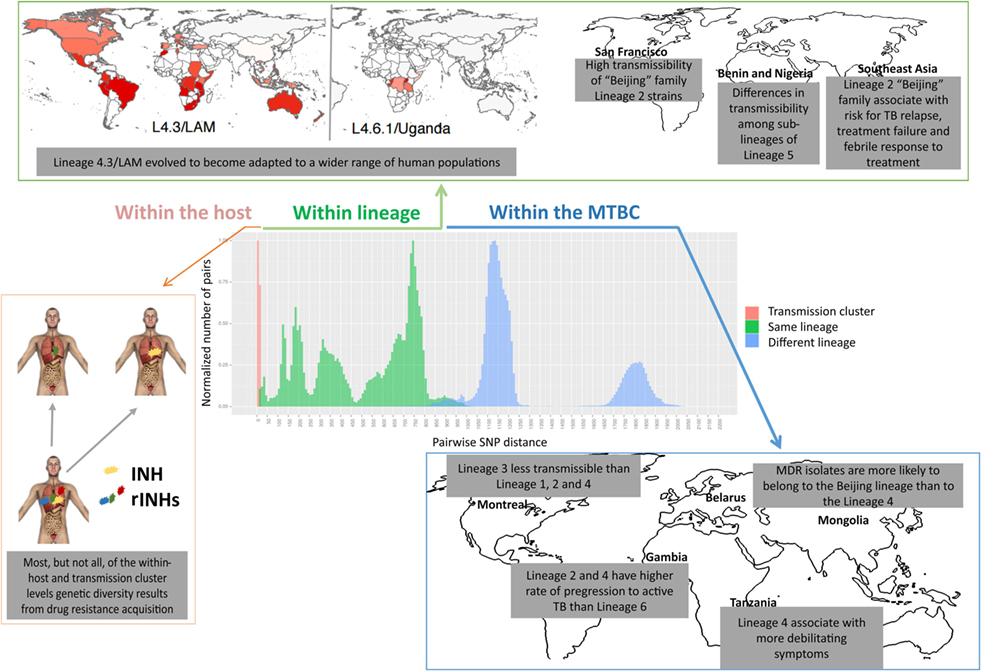
Figure 3. Levels of genetic diversity across the Mycobacterium tuberculosis complex (MTBC) and its epidemiological and clinical impact. There are different levels of diversity across the MTBC. Within a host or transmission chain, M. tuberculosis isolates typically differ in less than 25 single-nucleotide polymorphisms (SNPs). Diversity increases when comparing isolates within the same lineage (around 25–1,000 SNPs) or within different lineages of the MTBC (around 1,000–2,000 SNPs). This diversity impacts host/pathogen interactions, particularly the intensity and quality of the immune response and the clinical outcome, at the levels of drug acquisition, adaptation to different populations, transmissibility, or disease manifestation.
What Evolutionary Forces Shape MTBC Diversity?
In the long-term evolution of the pathogen, the main driving forces shaping MTBC diversity were a balance between genetic drift, during the global expansion of the bacteria and host-to-host transmission, and positive and purifying selection (77, 83). A long period of parallel evolution of the pathogen with different populations of modern humans following the out-of-Africa migrations (84) resulted in different bacterial lineages to prevail in different geographical regions of the world. Echoes of this type of interaction are still observed in cosmopolitan settings, where the phylogeny of the infecting MTBC isolates correlates with the patient’s ethnic origin (85, 86). A recent study showed that even within one specific MTBC lineage, i.e., Lineage 4 (or Euro-American), a phylogeographical structure can be found, with some sublineages being geographically restricted and others occurring globally (87).
Whether the immune system is also a driving force for M. tuberculosis diversity remains unclear. On one hand, extraordinary insights can be gained, as shown by the identification of single nucleotide polymorphisms (SNPs) in the two-component regulation system PhoP/PhoR impacting Lineage 6 and Mycobacterium bovis strains (88). On the other hand, while most pathogens have evolved to evade host immunity by antigenic variation, M. tuberculosis seems to apply a different strategy, thus questioning the role of T cells in driving M. tuberculosis evolution. Genome sequencing of M. tuberculosis showed that the known human T cell epitopes are evolutionarily hyperconserved, with the large majority of individual epitopes analyzed showing no amino acid change at all (89). It is interesting to note that epitopes appear to be significantly more conserved than the mean of the genome, and this result is robust to the number of strains analyzed (89, 90). Furthermore, these hyperconserved epitopes fall on genes that are not biased toward particular functional categories, but in which the major common theme is that they encode peptides that are recognized by the immune system (90). This hypothesis still awaits functional and experimental confirmation, but based on in silico analyses it is tempting to speculate that, despite the general conservation of the M. tuberculosis genome as compared with other bacteria, T cell responses may drive selective forces toward the hyperconservation of epitopes. The relevance of host T cell responses for the natural history of M. tuberculosis infection comes from findings in HIV+ patients, showing that less frequent cavitation, in a CD4 T cell-dependent frequency, associates with lower transmissibility (as inferred by looking at the number of household contacts infected by an HIV+ patient) (15). Therefore, it is conceivable that the T cell response that locally tries to eliminate the bacilli, which results in cavitation and thus in transmission. Ensuring conservation of T cell responses could thus be a mechanism explored by M. tuberculosis to ensure transmission. Nonetheless, outlier epitopes to the general rule of hyperconservation have been described both in previously known and newly identified antigenic regions (89, 91). Moreover, amino acid substitutions in these variable epitopes were shown to impact the host response with some patients responding only to the wild-type epitope variant and others only to the mutated forms (89). This is important, as the alteration of a single amino acid in an epitope can impact its affinity to a specific HLA molecule, thus influencing the T cell synapse and modulating the level and type of immune response elicited (92, 93). These variable epitopes could be potentially exploited as vaccine components to increase the protection provided by the existing TB vaccine.
Impact of Bacteria Genotypes on Immune Responses and Clinical Outcomes
Mycobacterium tuberculosis complex diversity impacts the host immune response, as certain clinical isolates are more potent than others in inducing the secretion of immune mediators by infected monocytes (94–99) and in experimental infections (100–104). The impact of bacterial diversity is not only reflected at the level of soluble immune mediators but also in the ability of virulent mycobacteria to inhibit apoptosis, while triggering necrosis of host macrophages to promote an innate delay in the initiation of adaptive immunity (105). Clearly, unbalancing the immune response is a strategy used by M. tuberculosis to increase its virulence, and this strategy might be modulated differently by diverse strains.
The realization of the MTBC diversity in all its extent has also led to multiple studies exploring the impact of this variation on the clinical outcome of TB (Figure 3). For example, Lineage 2 strains (which includes the Beijing family) have been repeatedly associated with treatment failure and relapse (106–108). One in vitro study further demonstrated that Lineage 2 may acquire drug resistance more rapidly than Lineage 4 (109), in line with reports from clinical settings where multidrug-resistant (MDR) isolates were more likely to belong to the Beijing lineage than to Lineage 4 (110, 111). High transmissibility of Beijing family Lineage 2 strains was shown in San Francisco (112), whereas Lineage 3 was reported to be less transmissible than Lineages 1, 2, and 4 in TB patients from Montreal, Canada (113), and striking differences in transmissibility among sublineages of Lineage 5 in Benin and Nigeria were reported (114). Lineages 2 and 4 were shown to have a higher rate of progression to active TB as compared with Lineage 6 in the Gambia (115). In addition, more debilitating symptoms (such as weight loss) have been associated with Lineage 4 strains in Tanzania (116). The same lineage was linked mainly to pulmonary TB (117), while Lineages 2 (117–119) and 3 (120) were reported to associate with extrapulmonary disease. Strikingly, within-host bacterial diversity also seems to contribute to disease manifestation. Indeed, the presence of distinct bacterial subpopulations is associated with poor clinical outcomes (121), resulting in differential resolution of granulomas (122), that may even contrast the overall trend of disease progression (123–125).
Bacteria and Host Genotype Interactions
There is emerging evidence showing an impact of host–pathogen genotype interactions in host immune responses, TB transmission, and disease presentation (76). This is the case of the association observed between the presence of the T597C allele of the toll-like receptor (TLR) 2 gene and susceptibility to disseminated disease, in a Vietnamese population upon infection by MTBC Lineage 2 (117). Three studies conducted in Ghana further support genotype–genotype interactions. First, the variant G57E of the mannose-binding Lectin (Protein C) 2 (MBL2) gene was associated with TB caused by M. africanum, but not by M. tuberculosis sensu stricto (126). Second, the variant 261TT of the immunity-related GTPase M (IRGM) gene was protective against TB caused by Lineage 4, but not for disease caused by other MTBC lineages (127). Third, M. africanum was significantly more common in TB patients belonging to the Ewe ethnic group, an association mainly driven by Lineage 5 (128, 129). In a South African population of mixed ancestry, an association between different HLA class I types and disease caused by different MTBC strain families was reported (130). Polymorphisms in the macrophage receptor with collagenous structure (MARCO) gene, a receptor involved in M. tuberculosis phagocytosis (131), preferentially associated with Lineage 2 over Lineage 1 or 4, implying that the host MARCO genotypes may interact with M. tuberculosis of the Lineage 2 genotype to increase susceptibility to TB (132). The first report of a M. tuberculosis lineage-based GWAS was recently published (133). In this study, an SNP on chromosome 1p13, near the CD53 gene, was specifically associated with non-Beijing lineage-infected old age onset cases (133). Altogether, these studies demonstrate that interactions between bacterial and human genetic loci exist and jointly influence clinical phenotypes. This evidence calls for the need of integrating the pathogen genotype in human genetic association studies, as well as in the study of TB immunity.
Rewiring the Immune Response in TB: The Need to Incorporate Diversity in Host, Pathogen, and Environment
Host-directed therapies are gaining momentum in the field of TB treatment (Table 1). HDTs aim at modulating host inflammation as a way to improve the efficacy of current treatments, while shortening the duration of these treatments, lowering toxicity and decreasing rates of resistance acquisition (134). A particular benefit of HDTs would be their application to treat MDR- and extensively drug resistant-TB, where antibiotics have limited effectiveness and immunopathological inflammation, tissue damage and high fatality are observed.
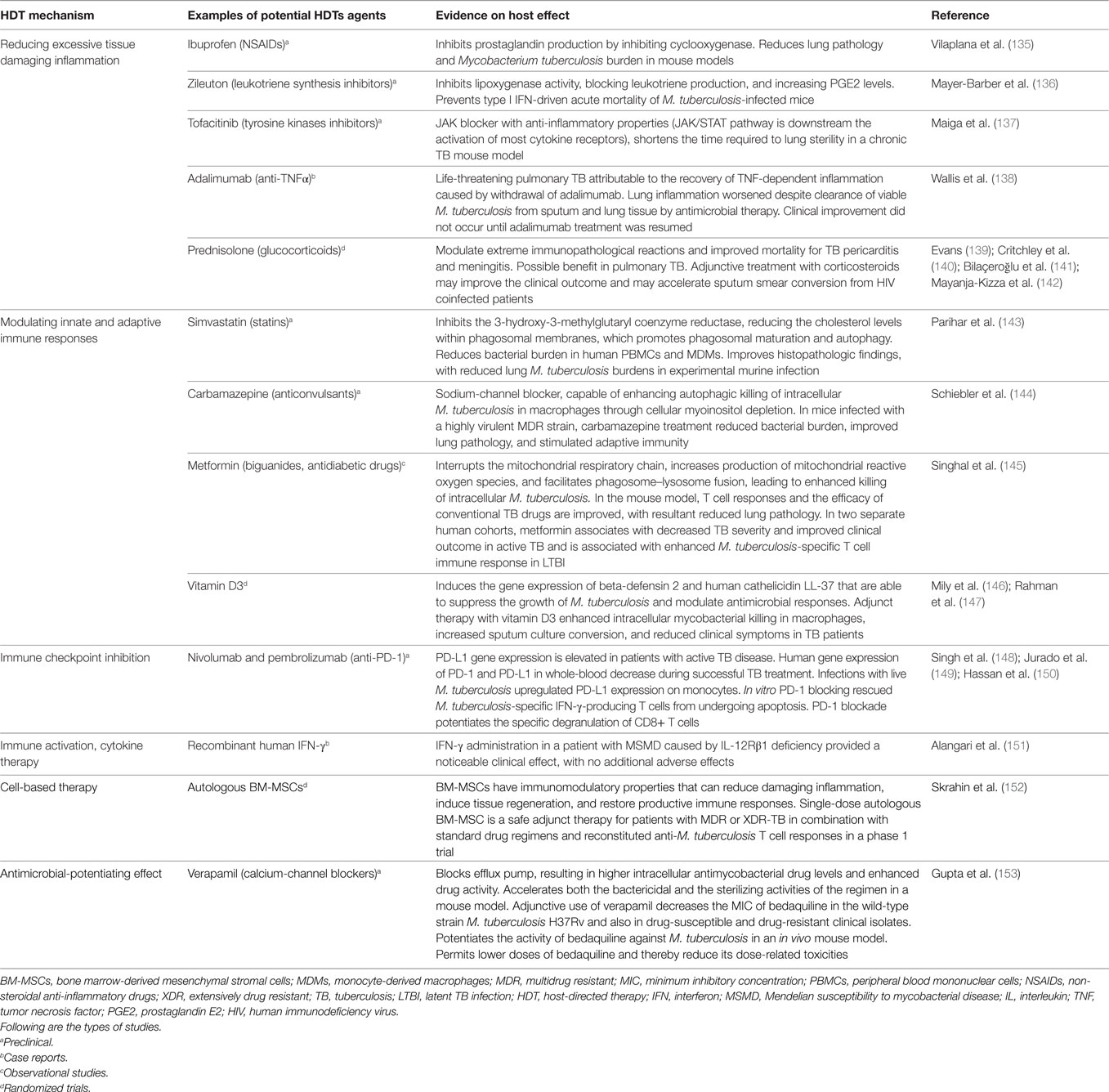
Table 1. Evidence supporting HDTs for TB.
Although HDTs take advantage of immunomodulatory mechanisms, they rarely account for the interaction between the diversity of the bacteria, the host, and the environment. However, in some clinical sites, indirect data suggest that successful HDTs have to be tailored toward these variables. For example, there is a large amount of data showing Lineage 2 strains as being low-cytokine inducers, but potent inducers of type I IFN (97, 98, 104, 154), which is at least in part mediated by the differential activation of TLR2 and TLR4 receptors (98). In this way, Lineage 2 strains may subvert antituberculous host defenses by inhibiting the enzyme inducible NO synthase, as well as IL-1β, IL-18, and IL-12p40, while inducing the immunosuppressive mediators IL-10 and IL-1 receptor antagonist (4, 155). In addition, there are data from TB patients in Vietnam showing an association between individuals with a T597C allele in the TLR2 gene and susceptibility to infection by the Lineage 2/Beijing genotype (117); and an association of individuals with an SNP in the leukotriene A4 hydrolase (LTA4H) promoter with an excess of inflammation and TB severity (156). How all the combinations of human/pathogen genotypes interact in Vietnam remains to be elucidated, but it will surely impact the outcome of HDTs in TB, as well as teaching important lessons. Furthermore, HDTs will need to deal with our partial understanding of what constitutes protective immunity to TB. For example, the role of type I IFNs is now being reconsidered. A recent study showed that in certain scenarios, namely, in the absence of IFN-γ, the induction of type I IFN is actually protective to the host, as it allows the control of the switch of M1 (effector) to M2 (detrimental) macrophages (157). The fact that type I IFN plays dynamic roles during infection (157, 158) may have implications in the use of eicosanoid modulators to enhance prostaglandin E2 levels and decrease the unfavorable type I IFN response (136). So, HDTs designed to modulate excessive type I IFN response, like the one associated with Lineage 2 strains, will need to consider host-specific characteristics, such as a high or low ability of differentiating IFN-γ responses, as well as the time of intervention.
Conclusion
The host immune status is tightly linked to the spectrum of TB infection and disease. However, the molecular determinants bridging inflammatory thresholds and TB outcomes remain elusive. Likely, this is due to the fact that what finally dictates disease outcomes and transmission is not a single factor, but the interacting (antagonistically or synergistically) action of multiple factors. Diversity in host, pathogen, and extrinsic factors needs to be studied in concert rather than individually, so that the full extent of the biological interplay underlying the immune response can be captured and modeled. In the long term, these models will lead the discovery of solid correlates of protection, biomarkers of prognosis, therapeutic targets, and more accurate epidemiology models. The great challenge is now to integrate this troika of interactions in the development of HDTs, at a personalized level, to develop sterilizing therapies. Most likely, we will be soon talking about personalized medicine to treat TB, by rewiring the immune response through host–pathogen–environment directed therapies. Only then will we be able to translate the many years of research devoted to the study of the protective immune response into real clinical applications, i.e., better vaccines, therapeutics and novel biomarkers of prognosis.
Author Contributions
All authors conceived and wrote the paper. SG, IC, and MS coordinated and completed the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
HN-B acknowledges the receipt of research scholarships from Bolsa D. Manuel de Mello and the Portuguese Society for Pneumology. NO acknowledges FCT IF/00474/2014. SG is funded by the European Research Council (grant number 309540-EVODRTB); the Swiss National Science Foundation (grant number 310030_166687) SystemsX.ch. IC lab is financed by Ministerio de Economía y Competitividad (Spanish Government) research grant SAF2013-43521-R, SAF2016-77346-R, and PROMETEO/2016/122 from Generalitat Valenciana and the European Research Council (ERC) (638553-TB-ACCELERATE). The MS lab is financed by FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operacional Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274). MS is an FCT Principal Investigator. Some graphical elements in the figures were designed by kjpargeter/Freepik. The funding agencies had no role in the design of the manuscript.
Abbreviations
CAMP, cathelicidin antimicrobial peptide; COPD, chronic obstructive pulmonary disease; DCs, dendritic cells; GWAS, genome-wide association studies; HDTs, host-directed therapies; HIV, human immunodeficiency virus; IFN, interferon; IL, interleukin; iNOS, inducible NO synthase; IRGM, immunity-related GTPase M; LTA4H, leukotriene A4 hydrolase; LTBI, latent tuberculosis infection; MARCO, macrophage receptor with collagenous structure; MBL2, mannose-binding lectin 2; MDR, multidrug resistant; MSMD, Mendelian susceptibility to mycobacterial disease; MTBC, Mycobacterium tuberculosis complex; NO, nitric oxide; PGE2, prostaglandin E2; SNP, single-nucleotide polymorphisms; TB, tuberculosis; Th, T helper; TLR, toll-like receptor; TNF, tumor necrosis factor; VDR, vitamin D receptor; XDR, extensively drug resistant.
References
2. Esmail H, Lai RP, Lesosky M, Wilkinson KA, Graham CM, Coussens AK, et al. Characterization of progressive HIV-associated tuberculosis using 2-deoxy-2-[18F]fluoro-d-glucose positron emission and computed tomography. Nat Med (2016) 22:1090–3. doi:10.1038/nm.4161
3. Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet (2016) 6736:1–11. doi:10.1016/S0140-6736(15)01316-1
4. O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi:10.1146/annurev-immunol-032712-095939
5. Barry CE, Boshoff H, Dartois V, Dick T, Ehrt S, Flynn J, et al. The spectrum of latent tuberculosis: rethinking the goals of prophylaxis. Nat Rev Microbiol (2009) 7:845–55. doi:10.1038/nrmicro2236
6. Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol (2014) 26:454–70. doi:10.1016/j.smim.2014.09.008
7. Abel L, El-Baghdadi J, Bousfiha AA, Casanova J-L, Schurr E. Human genetics of tuberculosis: a long and winding road. Philos Trans R Soc Lond B Biol Sci (2014) 369:20130428. doi:10.1098/rstb.2013.0428
8. Qu H-Q, Fisher-Hoch SP, McCormick JB. Knowledge gaining by human genetic studies on tuberculosis susceptibility. J Hum Genet (2011) 56:177–82. doi:10.1038/jhg.2010.164
9. Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun (2012) 80:3343–59. doi:10.1128/IAI.00443-12
10. Curtis J, Luo Y, Zenner HL, Cuchet-Lourenço D, Wu C, Lo K, et al. Susceptibility to tuberculosis is associated with variants in the ASAP1 gene encoding a regulator of dendritic cell migration. Nat Genet (2015) 47:523–7. doi:10.1038/ng.3248
11. Grant AV, Sabri A, Abid A, Abderrahmani Rhorfi I, Benkirane M, Souhi H, et al. A genome-wide association study of pulmonary tuberculosis in Morocco. Hum Genet (2016) 135:299–307. doi:10.1007/s00439-016-1633-2
12. Meyer CG, Thye T. Host genetic studies in adult pulmonary tuberculosis. Semin Immunol (2014) 26:445–53. doi:10.1016/j.smim.2014.09.005
13. de Wit E, van der Merwe L, van Helden PD, Hoal EG. Gene-gene interaction between tuberculosis candidate genes in a South African population. Mamm Genome (2011) 22:100–10. doi:10.1007/s00335-010-9280-8
14. Daya M, Van Der Merwe L, Van Helden PD, Möller M, Hoal EG. Investigating the role of gene-gene interactions in TB susceptibility. PLoS One (2015) 10:e0123970. doi:10.1371/journal.pone.0123970
15. Kwan C, Ernst JD. HIV and tuberculosis: a deadly human syndemic. Clin Microbiol Rev (2011) 24:351–76. doi:10.1128/CMR.00042-10
16. Sonnenberg P, Glynn JR, Fielding K, Murray J, Godfrey-Faussett P, Shearer S. How soon after infection with HIV does the risk of tuberculosis start to increase? A retrospective cohort study in South African gold miners. J Infect Dis (2005) 191:150–8. doi:10.1086/426827
17. Walter ND, De Jong BC, Garcia BJ, Dolganov GM, Worodria W, Byanyima P, et al. Adaptation of Mycobacterium tuberculosis to impaired host immunity in HIV-infected patients. J Infect Dis (2016) 214:1205–11. doi:10.1093/infdis/jiw364
18. Leistikow RL, Morton RA, Bartek IL, Frimpong I, Wagner K, Voskuil MI. The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J Bacteriol (2010) 192:1662–70. doi:10.1128/JB.00926-09
19. Diedrich CR, Flynn JAL. HIV-1/Mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect Immun (2011) 79:1407–17. doi:10.1128/IAI.01126-10
20. Deffur A, Mulder NJ, Wilkinson RJ. Co-infection with Mycobacterium tuberculosis and human immunodeficiency virus: an overview and motivation for systems approaches. Pathog Dis (2013) 69:101–13. doi:10.1111/2049-632X.12060
21. Resende CT, Hirsch CS, Toossi Z, Dietze R, Ribeiro-Rodrigues R. Intestinal helminth co-infection has a negative impact on both anti-Mycobacterium tuberculosis immunity and clinical response to tuberculosis therapy. Clin Exp Immunol (2007) 147:45–52. doi:10.1111/j.1365-2249.2006.03247.x
22. Chatterjee S, Nutman TB. Helminth-induced immune regulation: implications for immune responses to tuberculosis. PLoS Pathog (2015) 11:e1004582. doi:10.1371/journal.ppat.1004582
23. George PJ, Anuradha R, Kumar NP, Sridhar R, Banurekha VV, Nutman TB, et al. Helminth infections coincident with active pulmonary tuberculosis inhibit mono- and multifunctional CD4+ and CD8+ T cell responses in a process dependent on IL-10. PLoS Pathog (2014) 10:e1004375. doi:10.1371/journal.ppat.1004375
24. Redford PS, Mayer-Barber KD, McNab FW, Stavropoulos E, Wack A, Sher A, et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor-dependent pathway. J Infect Dis (2014) 209:270–4. doi:10.1093/infdis/jit424
25. Perry S, De Jong BC, Solnick JV, De La Luz Sanchez M, Yang S, Lin PL, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS One (2010) 5:e8804. doi:10.1371/journal.pone.0008804
26. Majlessi L, Sayes F, Bureau J-F, Pawlik A, Michel V, Jouvion G, et al. Colonization with Helicobacter is concomitant with modified Gut microbiota and drastic failure of the immune control of Mycobacterium tuberculosis. Mucosal Immunol (2017) 10:1178–89. doi:10.1038/mi.2016.140
27. Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJTH, de Boer JD, et al. The Gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut (2016) 65:575–83. doi:10.1136/gutjnl-2015-309728
28. Gauguet S, D’Ortona S, Ahnger-Pier K, Duan B, Surana NK, Lu R, et al. Intestinal microbiota of mice influences resistance to Staphylococcus aureus pneumonia. Infect Immun (2015) 83:4003–14. doi:10.1128/IAI.00037-15
29. Clarke TB. Early innate immunity to bacterial infection in the lung is regulated systemically by the commensal microbiota via nod-like receptor ligands. Infect Immun (2014) 82:4596–606. doi:10.1128/IAI.02212-14
30. Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, Agrewala JN. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol (2016) 7:529. doi:10.3389/fimmu.2016.00529
31. Huempfner HR, Deuschle KW. Experimental tuberculosis in germ-free and conventional mice. Am Rev Respir Dis (1966) 93:465–7. doi:10.1164/arrd.1966.93.3P1.465
32. Winglee K, Eloe-Fadrosh E, Gupta S, Guo H, Fraser C, Bishai W. Aerosol Mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS One (2014) 9:e97048. doi:10.1371/journal.pone.0097048
33. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature (2013) 505:218–22. doi:10.1038/nature12799
34. Jeon CY, Murray MB. Diabetes mellitus increases the risk of active tuberculosis: a systematic review of 13 observational studies. PLoS Med (2008) 5:e152. doi:10.1371/journal.pmed.0050152
35. Jiménez-Corona ME, Cruz-Hervert LP, García-García L, Ferreyra-Reyes L, Delgado-Sánchez G, Bobadilla-del-Valle M, et al. Association of diabetes and tuberculosis: impact on treatment and post-treatment outcomes. Thorax (2013) 68:214–20. doi:10.1136/thoraxjnl-2012-201756
36. Bastos HN, Osório NS, Castro AG, Ramos A, Carvalho T, Meira L, et al. A prediction rule to stratify mortality risk of patients with pulmonary tuberculosis. PLoS One (2016) 11:e0162797. doi:10.1371/journal.pone.0162797
37. Baker MA, Harries AD, Jeon CY, Hart JE, Kapur A, Lönnroth K, et al. The impact of diabetes on tuberculosis treatment outcomes: a systematic review. BMC Med (2011) 9:81. doi:10.1186/1741-7015-9-81
38. Alisjahbana B, Sahiratmadja E, Nelwan EJ, Purwa AM, Ahmad Y, Ottenhoff THM, et al. The effect of type 2 diabetes mellitus on the presentation and treatment response of pulmonary tuberculosis. Clin Infect Dis (2007) 45:428–35. doi:10.1086/519841
39. Viswanathan V, Vigneswari A, Selvan K, Satyavani K, Rajeswari R, Kapur A. Effect of diabetes on treatment outcome of smear-positive pulmonary tuberculosis – a report from South India. J Diabetes Complications (2014) 28:162–5. doi:10.1016/j.jdiacomp.2013.12.003
40. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology (2015) 144:171–85. doi:10.1111/imm.12394
41. Martinez N, Kornfeld H. Diabetes and immunity to tuberculosis. Eur J Immunol (2014) 44:617–26. doi:10.1002/eji.201344301
42. Restrepo BI, Schlesinger LS. Host-pathogen interactions in tuberculosis patients with type 2 diabetes mellitus. Tuberculosis (Edinb) (2013) 93(Suppl):S10–4. doi:10.1016/S1472-9792(13)70004-0
43. Ronacher K, Joosten SA, van Crevel R, Dockrell HM, Walzl G, Ottenhoff THM. Acquired immunodeficiencies and tuberculosis: focus on HIV/AIDS and diabetes mellitus. Immunol Rev (2015) 264:121–37. doi:10.1111/imr.12257
44. Larsen N, Vogensen FK, Van Den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One (2010) 5:e9085. doi:10.1371/journal.pone.0009085
45. Inghammar M, Ekbom A, Engström G, Ljungberg B, Romanus V, Löfdahl C-G, et al. COPD and the risk of tuberculosis – a population-based cohort study. PLoS One (2010) 5:e10138. doi:10.1371/journal.pone.0010138
46. Lee C-H, Lee M-C, Shu C-C, Lim C-S, Wang J-Y, Lee L-N, et al. Risk factors for pulmonary tuberculosis in patients with chronic obstructive airway disease in Taiwan: a nationwide cohort study. BMC Infect Dis (2013) 13:194. doi:10.1186/1471-2334-13-194
47. Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med (2008) 359:2355–65. doi:10.1056/NEJMra0800353
48. Taylor AE, Finney-Hayward TK, Quint JK, Thomas CMR, Tudhope SJ, Wedzicha JA, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J (2010) 35:1039–47. doi:10.1183/09031936.00036709
49. Bhat TA, Panzica L, Kalathil SG, Thanavala Y. Immune dysfunction in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc (2015) 12:S169–75. doi:10.1513/AnnalsATS.201503-126AW
50. Slama K, Chiang C-Y, Enarson DA, Hassmiller K, Fanning A, Gupta P, et al. Tobacco and tuberculosis: a qualitative systematic review and meta-analysis. Int J Tuberc Lung Dis (2007) 11:1049–61.
51. Lin H-H, Ezzati M, Murray M. Tobacco smoke, indoor air pollution and tuberculosis: a systematic review and meta-analysis. PLoS Med (2007) 4:e20. doi:10.1371/journal.pmed.0040020
52. Bates MN, Khalakdina A, Pai M, Chang L, Lessa F, Smith KR. Risk of tuberculosis from exposure to tobacco smoke: a systematic review and meta-analysis. Arch Intern Med (2007) 167:335–42. doi:10.1001/archinte.167.4.335
53. O’Leary SM, Coleman MM, Chew WM, Morrow C, McLaughlin AM, Gleeson LE, et al. Cigarette smoking impairs human pulmonary immunity to Mycobacterium tuberculosis. Am J Respir Crit Care Med (2014) 190:1430–6. doi:10.1164/rccm.201407-1385OC
54. Robbins CS, Dawe DE, Goncharova SI, Pouladi MA, Drannik AG, Swirski FK, et al. Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsiveness. Am J Respir Cell Mol Biol (2004) 30:202–11. doi:10.1165/rcmb.2003-0259OC
55. Givi ME, Folkerts G, Wagenaar GTM, Redegeld FA, Mortaz E. Cigarette smoke differentially modulates dendritic cell maturation and function in time. Respir Res (2015) 16:131. doi:10.1186/s12931-015-0291-6
56. Shaler CR, Horvath CN, McCormick S, Jeyanathan M, Khera A, Zganiacz A, et al. Continuous and discontinuous cigarette smoke exposure differentially affects protective Th1 immunity against pulmonary tuberculosis. PLoS One (2013) 8:e59185. doi:10.1371/journal.pone.0059185
57. Berg RD, Levitte S, O’Sullivan MP, O’Leary SM, Cambier CJ, Cameron J, et al. Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell (2016) 165:139–52. doi:10.1016/j.cell.2016.02.034
58. Cegielski JP, McMurray DN. The relationship between malnutrition and tuberculosis: evidence from studies in humans and experimental animals. Int J Tuberc Lung Dis (2004) 8:286–98.
59. Scalcini M, Occenac R, Manfreda J, Long R. Pulmonary tuberculosis, human immunodeficiency virus type-1 and malnutrition. Bull Int Union Tuberc Lung Dis (1991) 66:37–41.
60. Jeremiah K, Denti P, Chigutsa E, Faurholt-Jepsen D, PrayGod G, Range N, et al. Nutritional supplementation increases rifampin exposure among tuberculosis patients coinfected with HIV. Antimicrob Agents Chemother (2014) 58:3468–74. doi:10.1128/AAC.02307-13
61. Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood (2011) 118:4129–39. doi:10.1182/blood-2011-04-351957
62. Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol (2007) 82:934–45. doi:10.1189/jlb.0407216
63. Kerkhoff AD, Meintjes G, Burton R, Vogt M, Wood R, Lawn SD. Relationship between blood concentrations of hepcidin and anemia severity, mycobacterial burden, and mortality among patients with HIV-associated tuberculosis. J Infect Dis (2016) 213:61–70. doi:10.1093/infdis/jiv364
64. Martineau AR, Honecker FU, Wilkinson RJ, Griffiths CJ. Vitamin D in the treatment of pulmonary tuberculosis. J Steroid Biochem Mol Biol (2007) 103:793–8. doi:10.1016/j.jsbmb.2006.12.052
65. Martineau AR, Nhamoyebonde S, Oni T, Rangaka MX, Marais S, Bangani N, et al. Reciprocal seasonal variation in vitamin D status and tuberculosis notifications in Cape Town, South Africa. Proc Natl Acad Sci U S A (2011) 108:19013–7. doi:10.1073/pnas.1111825108
66. Kim JH, Park J-S, Cho Y-J, Yoon H-I, Song JH, Lee C-T, et al. Low serum 25-hydroxyvitamin D level: an independent risk factor for tuberculosis? Clin Nutr (2013) 33:1–6. doi:10.1016/j.clnu.2013.11.014
67. Pareek M, Innes J, Sridhar S, Grass L, Connell D, Woltmann G, et al. Vitamin D deficiency and TB disease phenotype. Thorax (2015) 70:1171–80. doi:10.1136/thoraxjnl-2014-206617
68. Coussens AK, Wilkinson RJ, Hanifa Y, Nikolayevskyy V, Elkington PT, Islam K, et al. Vitamin D accelerates resolution of inflammatory responses during tuberculosis treatment. Proc Natl Acad Sci U S A (2012) 109:15449–54. doi:10.1073/pnas.1200072109
69. Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog (2012) 8:e1002689. doi:10.1371/journal.ppat.1002689
70. Deretic V. Autophagy in tuberculosis. Cold Spring Harb Perspect Med (2014) 4:a018481. doi:10.1101/cshperspect.a018481
71. Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature (2015) 528:565–9. doi:10.1038/nature16451
72. Chambers ES, Hawrylowicz CM. The impact of vitamin D on regulatory T cells. Curr Allergy Asthma Rep (2011) 11:29–36. doi:10.1007/s11882-010-0161-8
73. Cegielski P, Vernon A. Tuberculosis and vitamin D: what’s the rest of the story? Lancet Infect Dis (2015) 15:489–90. doi:10.1016/S1473-3099(15)70163-5
74. Martineau AR, Timms PM, Bothamley GH, Hanifa Y, Islam K, Claxton AP, et al. High-dose vitamin D(3) during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet (2011) 377:242–50. doi:10.1016/S0140-6736(10)61889-2
75. Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A (2006) 103:2869–73. doi:10.1073/pnas.0511240103
76. Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev (2015) 264:6–24. doi:10.1111/imr.12264
77. Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol (2008) 6:e311. doi:10.1371/journal.pbio.0060311
78. Pérez-Lago L, Comas I, Navarro Y, González-Candelas F, Herranz M, Bouza E, et al. Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis (2014) 209:98–108. doi:10.1093/infdis/jit439
79. Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis (2012) 206:1724–33. doi:10.1093/infdis/jis601
80. Osório NS, Rodrigues F, Gagneux S, Pedrosa J, Pinto-Carbó M, Castro AG, et al. Evidence for diversifying selection in a set of Mycobacterium tuberculosis genes in response to antibiotic- and nonantibiotic-related pressure. Mol Biol Evol (2013) 30:1326–36. doi:10.1093/molbev/mst038
81. Liu Q, Via LE, Luo T, Liang L, Liu X, Wu S, et al. Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci Rep (2015) 5:17507. doi:10.1038/srep17507
82. Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med (2016) 22:531–8. doi:10.1038/nm.4073
83. Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, et al. The role of selection in shaping diversity of natural M. tuberculosis populations. PLoS Pathog (2013) 9:e1003543. doi:10.1371/journal.ppat.1003543
84. Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet (2013) 45:1176–82. doi:10.1038/ng.2744
85. Hirsh AE, Tsolaki AG, DeRiemer K, Feldman MW, Small PM. Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci U S A (2004) 101:4871–6. doi:10.1073/pnas.0305627101
86. Reed MB, Pichler VK, Mcintosh F, Mattia A, Fallow A, Masala S, et al. Major Mycobacterium tuberculosis lineages associate with patient country of origin. J Clin Microbiol (2009) 47:1119–28. doi:10.1128/JCM.02142-08
87. Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat Genet (2016) 48:1535–43. doi:10.1038/ng.3704
88. Gonzalo-Asensio J, Malaga W, Pawlik A, Astarie-Dequeker C, Passemar C, Moreau F, et al. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci U S A (2014) 111:11491–6. doi:10.1073/pnas.1406693111
89. Coscolla M, Copin R, Sutherland J, Gehre F, De Jong B, Owolabi O, et al. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host Microbe (2015) 18:538–48. doi:10.1016/j.chom.2015.10.008
90. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet (2010) 42:498–503. doi:10.1038/ng.590
91. Yruela I, Contreras-Moreira B, Magalhães C, Osório NS, Gonzalo-Asensio J. Mycobacterium tuberculosis complex exhibits lineage-specific variations affecting protein ductility and epitope recognition. Genome Biol Evol (2016) 8:3751–64. doi:10.1093/gbe/evw279
92. da Silva J. The evolutionary adaptation of HIV-1 to specific immunity. Curr HIV Res (2003) 1:363–71. doi:10.2174/1570162033485249
93. Copin R, Coscollá M, Efstathiadis E, Gagneux S, Ernst JD. Impact of in vitro evolution on antigenic diversity of Mycobacterium bovis bacillus Calmette-Guerin (BCG). Vaccine (2014) 32:5998–6004. doi:10.1016/j.vaccine.2014.07.113
94. Manca C, Tsenova L, Barry CE III, Bergtold A, Freeman S, Haslett PAJ, et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol (1999) 162:6740–6.
95. Sarkar R, Lenders L, Wilkinson KA, Wilkinson RJ, Nicol MP. Modern lineages of Mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLoS One (2012) 7:e43170. doi:10.1371/journal.pone.0043170
96. Krishnan N, Malaga W, Constant P, Caws M, Thi Hoang Chau T, Salmons J, et al. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS One (2011) 6:e23870. doi:10.1371/journal.pone.0023870
97. Wang C, Peyron P, Mestre O, Kaplan G, van Soolingen D, Gao Q, et al. Innate immune response to Mycobacterium tuberculosis Beijing and other genotypes. PLoS One (2010) 5:e13594. doi:10.1371/journal.pone.0013594
98. Carmona J, Cruz A, Moreira-Teixeira L, Sousa C, Sousa J, Osório NS, et al. Mycobacterium tuberculosis strains are differentially recognized by TLRs with an impact on the immune response. PLoS One (2013) 8:e67277. doi:10.1371/journal.pone.0067277
99. Portevin D, Gagneux S, Comas I, Young D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog (2011) 7:e1001307. doi:10.1371/journal.ppat.1001307
100. Aguilar LD, Hanekom M, Mata D, Gey Van Pittius NC, Van Helden PD, Warren RM, et al. Mycobacterium tuberculosis strains with the Beijing genotype demonstrate variability in virulence associated with transmission. Tuberculosis (2010) 90:319–25. doi:10.1016/j.tube.2010.08.004
101. Dormans J, Burger M, Aguilar D, Hernandez-Pando R, Kremer K, Roholl P, et al. Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin Exp Immunol (2004) 137:460–8. doi:10.1111/j.1365-2249.2004.02551.x
102. Dunn PL, North RJ. Virulence ranking of some Mycobacterium tuberculosis and Mycobacterium bovis strains according to their ability to multiply in the lungs, induce lung pathology, and cause mortality in mice. Infect Immun (1995) 63:3428–37.
103. López B, Aguilar D, Orozco H, Burger M, Espitia C, Ritacco V, et al. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol (2003) 133:30–7. doi:10.1046/j.1365-2249.2003.02171.x
104. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A (2001) 98:5752–7. doi:10.1073/pnas.091096998
105. Divangahi M, Behar SM, Remold H. Dying to live: how the death modality of the infected macrophage modulates immunity to tuberculosis. Adv Exp Med Biol (2013) 783:103–20. doi:10.1007/978-1-4614-6111-1_6
106. Parwati I, Alisjahbana B, Apriani L, Soetikno RD, Ottenhoff TH, van der Zanden AGM, et al. Mycobacterium tuberculosis Beijing genotype is an independent risk factor for tuberculosis treatment failure in Indonesia. J Infect Dis (2010) 201:553–7. doi:10.1086/650311
107. Lan NTN, Lien HTK, Tung LB, Borgdorff MW, Kremer K, Van Soolingen D. Mycobacterium tuberculosis Beijing genotype and risk for treatment failure and relapse, Vietnam. Emerg Infect Dis (2003) 9:1633–5. doi:10.3201/eid0912.030169
108. Sun Y-J, Lee a SG, Wong S-Y, Paton NI. Association of Mycobacterium tuberculosis Beijing genotype with tuberculosis relapse in Singapore. Epidemiol Infect (2006) 134:329–32. doi:10.1017/S095026880500525X
109. Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet (2013) 45:784–90. doi:10.1038/ng.2656
110. Wollenberg KR, Desjardins CA, Zalutskaya A, Slodovnikova V, Oler AJ, Quiñones M, et al. Whole-genome sequencing of Mycobacterium tuberculosis provides insight into the evolution and genetic composition of drug-resistant tuberculosis in Belarus. J Clin Microbiol (2017) 55:457–60. doi:10.1128/JCM.02116-16
111. Gurjav U, Erkhembayar B, Burneebaatar B, Narmandakh E, Tumenbayar O, Hill-Cawthorne GA, et al. Transmission of multi-drug resistant tuberculosis in Mongolia is driven by Beijing strains of Mycobacterium tuberculosis resistant to all first-line drugs. Tuberculosis (2016) 101:49–53. doi:10.1016/j.tube.2016.07.010
112. Kato-Maeda M, Kim EY, Flores L, Jarlsberg LG, Osmond D, Hopewell PC. Differences among sublineages of the East-Asian lineage of Mycobacterium tuberculosis in genotypic clustering. Int J Tuberc Lung Dis (2010) 14:538–44.
113. Albanna AS, Reed MB, Kotar KV, Fallow A, McIntosh FA, Behr MA, et al. Reduced transmissibility of East African Indian strains of Mycobacterium tuberculosis. PLoS One (2011) 6:e25075. doi:10.1371/journal.pone.0025075
114. Gehre F, Antonio M, Faïhun F, Odoun M, Uwizeye C, de Rijk P, et al. The first phylogeographic population structure and analysis of transmission dynamics of M. africanum West African 1-combining molecular data from Benin, Nigeria and Sierra Leone. PLoS One (2013) 8:e77000. doi:10.1371/journal.pone.0077000
115. de Jong BC, Hill PC, Aiken A, Awine T, Martin A, Adetifa IM, et al. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in the Gambia. J Infect Dis (2008) 198:1037–43. doi:10.1086/591504
116. Stavrum R, PrayGod G, Range N, Faurholt-Jepsen D, Jeremiah K, Faurholt-Jepsen M, et al. Increased level of acute phase reactants in patients infected with modern Mycobacterium tuberculosis genotypes in Mwanza, Tanzania. BMC Infect Dis (2014) 14:309. doi:10.1186/1471-2334-14-309
117. Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NTN, Thuong NTT, et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog (2008) 4:e1000034. doi:10.1371/journal.ppat.1000034
118. Kong Y, Cave MD, Zhang L, Foxman B, Marrs CF, Bates JH, et al. Association between Mycobacterium tuberculosis Beijing/W lineage strain infection and extrathoracic tuberculosis: insights from epidemiologic and clinical characterization of the three principal genetic groups of M. tuberculosis clinical isolates. J Clin Microbiol (2007) 45:409–14. doi:10.1128/JCM.01459-06
119. Kong Y, Cave MD, Yang D, Zhang L, Marrs CF, Foxman B, et al. Distribution of insertion- and deletion-associated genetic polymorphisms among four Mycobacterium tuberculosis phospholipase C genes and associations with extrathoracic tuberculosis: a population-based study. J Clin Microbiol (2005) 43:6048–53. doi:10.1128/JCM.43.12.6048-6053.2005
120. Lari N, Rindi L, Cristofani R, Rastogi N, Tortoli E, Garzelli C. Association of Mycobacterium tuberculosis complex isolates of BOVIS and Central Asian (CAS) genotypic lineages with extrapulmonary disease. Clin Microbiol Infect (2009) 15:538–43. doi:10.1111/j.1469-0691.2009.02712.x
121. Cohen T, Chindelevitch L, Misra R, Kempner ME, Galea J, Moodley P, et al. Within-host heterogeneity of Mycobacterium tuberculosis infection is associated with poor early treatment response a prospective cohort study. J Infect Dis (2016) 213:2005–11. doi:10.1093/infdis/jiw014
122. Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med (2013) 20:75–9. doi:10.1038/nm.3412
123. Via LE, Schimel D, Weiner DM, Dartois V, Dayao E, Cai Y, et al. Infection dynamics and response to chemotherapy in a rabbit model of tuberculosis using [18F]2-fluoro-deoxy-d-glucose positron emission tomography and computed tomography. Antimicrob Agents Chemother (2012) 56:4391–402. doi:10.1128/AAC.00531-12
124. Lin PL, Coleman T, Carney JPJ, Lopresti BJ, Tomko J, Fillmore D, et al. Radiologic responses in cynomolgus macaques for assessing tuberculosis chemotherapy regimens. Antimicrob Agents Chemother (2013) 57:4237–44. doi:10.1128/AAC.00277-13
125. Coleman MT, Maiello P, Tomko J, Frye LJ, Fillmore D, Janssen C, et al. Early changes by (18)Fluorodeoxyglucose positron emission tomography coregistered with computed tomography predict outcome after Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun (2014) 82:2400–4. doi:10.1128/IAI.01599-13
126. Thye T, Niemann S, Walter K, Homolka S, Intemann CD, Chinbuah MA, et al. Variant G57E of mannose binding lectin associated with protection against tuberculosis caused by Mycobacterium africanum but not by M. tuberculosis. PLoS One (2011) 6:e20908. doi:10.1371/journal.pone.0020908
127. Intemann CD, Thye T, Niemann S, Browne ENL, Chinbuah MA, Enimil A, et al. Autophagy gene variant IRGM-261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog (2009) 5:e1000577. doi:10.1371/journal.ppat.1000577
128. Asante-Poku A, Yeboah-Manu D, Otchere ID, Aboagye SY, Stucki D, Hattendorf J, et al. Mycobacterium africanum is associated with patient ethnicity in Ghana. PLoS Negl Trop Dis (2015) 9:e3370. doi:10.1371/journal.pntd.0003370
129. Asante-Poku A, Otchere ID, Osei-Wusu S, Sarpong E, Baddoo A, Forson A, et al. Molecular epidemiology of Mycobacterium africanum in Ghana. BMC Infect Dis (2016) 16:385. doi:10.1186/s12879-016-1725-6
130. Salie M, Van Der Merwe L, Möller M, Daya M, Van Der Spuy GD, Van Helden PD, et al. Associations between human leukocyte antigen class I variants and the Mycobacterium tuberculosis subtypes causing disease. J Infect Dis (2014) 209:216–23. doi:10.1093/infdis/jit443
131. Benard EL, Roobol SJ, Spaink HP, Meijer AH. Phagocytosis of mycobacteria by zebrafish macrophages is dependent on the scavenger receptor Marco, a key control factor of pro-inflammatory signalling. Dev Comp Immunol (2014) 47:223–33. doi:10.1016/j.dci.2014.07.022
132. Thuong NTT, Tram TTB, Dinh TD, Thai PVK, Heemskerk D, Bang ND, et al. MARCO variants are associated with phagocytosis, pulmonary tuberculosis susceptibility and Beijing lineage. Genes Immun (2016) 17:419–25. doi:10.1038/gene.2016.43
133. Omae Y, Toyo-oka L, Yanai H, Nedsuwan S, Wattanapokayakit S, Satproedprai N, et al. Pathogen lineage-based genome-wide association study identified CD53 as susceptible locus in tuberculosis. J Hum Genet (2017) 62(12):1015–22. doi:10.1038/jhg.2017.82
134. Zumla A, Chakaya J, Hoelscher M, Ntoumi F, Rustomjee R, Vilaplana C, et al. Towards host-directed therapies for tuberculosis. Nat Rev Drug Discov (2015) 14:511–2. doi:10.1038/nrd4696
135. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J Infect Dis (2013) 208:199–202. doi:10.1093/infdis/jit152
136. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature (2014) 511:99–103. doi:10.1038/nature13489
137. Maiga M, Ahidjo BA, Maiga MC, Cheung L, Pelly S, Lun S, et al. Efficacy of adjunctive tofacitinib therapy in mouse models of tuberculosis. EBioMedicine (2015) 2:868–73. doi:10.1016/j.ebiom.2015.07.014
138. Wallis RS, van Vuuren C, Potgieter S. Adalimumab treatment of life-threatening tuberculosis. Clin Infect Dis (2009) 48:1429–32. doi:10.1086/598504
139. Evans DJ. The use of adjunctive corticosteroids in the treatment of pericardial, pleural and meningeal tuberculosis: do they improve outcome? Respir Med (2008) 102:793–800. doi:10.1016/j.rmed.2008.01.018
140. Critchley JA, Young F, Orton L, Garner P. Corticosteroids for prevention of mortality in people with tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis (2013) 13:223–37. doi:10.1016/S1473-3099(12)70321-3
141. Bilaçeroglu S, Perim K, Büyükşirin M, Çelikten E. Prednisolone: a beneficial and safe adjunct to antituberculosis treatment? A randomized controlled trial. Int J Tuberc Lung Dis (1999) 3:47–54.
142. Mayanja-Kizza H, Jones-Lopez E, Okwera A, Wallis RS, Ellner JJ, Mugerwa RD, et al. Immunoadjuvant prednisolone therapy for HIV-associated tuberculosis: a phase 2 clinical trial in Uganda. J Infect Dis (2005) 191:856–65. doi:10.1086/427995
143. Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis (2014) 209:754–63. doi:10.1093/infdis/jit550
144. Schiebler M, Brown K, Hegyi K, Newton SM, Renna M, Hepburn L, et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol Med (2015) 7:127–39. doi:10.15252/emmm.201404137
145. Singhal A, Jie L, Kumar P, Hong GS, Leow MKS, Paleja B, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med (2014) 6:263ra159. doi:10.1126/scitranslmed.3009885
146. Mily A, Rekha RS, Kamal SMM, Arifuzzaman ASM, Rahim Z, Khan L, et al. Significant effects of oral phenylbutyrate and Vitamin D3 adjunctive therapy in pulmonary tuberculosis: a randomized controlled trial. PLoS One (2015) 10:1–25. doi:10.1371/journal.pone.0138340
147. Rahman S, Rehn A, Rahman J, Andersson J, Svensson M, Brighenti S. Pulmonary tuberculosis patients with a vitamin D deficiency demonstrate low local expression of the antimicrobial peptide LL-37 but enhanced FoxP3+ regulatory T cells and IgG-secreting cells. Clin Immunol (2015) 156:85–97. doi:10.1016/j.clim.2014.12.003
148. Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon γ-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis (2013) 208:603–15. doi:10.1093/infdis/jit206
149. Jurado JO, Alvarez IB, Pasquinelli V, Martínez GJ, Quiroga MF, Abbate E, et al. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol (2008) 181:116–25.
150. Hassan SS, Akram M, King EC, Dockrell HM, Cliff JM. PD-1, PD-L1 and PD-L2 gene expression on t-cells and natural killer cells declines in conjunction with a reduction in PD-1 protein during the intensive phase of tuberculosis treatment. PLoS One (2015) 10(9):e0137646. doi:10.1371/journal.pone.0137646
151. Alangari AA, Al-Zamil F, Al-Mazrou A, Al-Muhsen S, Boisson-Dupuis S, Awadallah S, et al. Treatment of disseminated mycobacterial infection with high-dose IFN-γ in a patient with IL-12Rβ1 deficiency. Clin Dev Immunol (2011) 2011:691956. doi:10.1155/2011/691956
152. Skrahin A, Ahmed RK, Ferrara G, Rane L, Poiret T, Isaikina Y, et al. Autologous mesenchymal stromal cell infusion as adjunct treatment in patients with multidrug and extensively drug-resistant tuberculosis: an open-label phase 1 safety trial. Lancet Respir Med (2014) 2:108–22. doi:10.1016/S2213-2600(13)70234-0
153. Gupta S, Tyagi S, Bishaia WR. Verapamil increases the bactericidal activity of bedaquiline against mycobacterium tuberculosis in a mouse model. Antimicrob Agents Chemother (2015) 59:673–6. doi:10.1128/AAC.04019-14
154. Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, et al. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res (2005) 25:694–701. doi:10.1089/jir.2005.25.694
155. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity (2011) 35:1023–34. doi:10.1016/j.immuni.2011.12.002
156. Tobin DM, Vary JC, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell (2010) 140:717–30. doi:10.1016/j.cell.2010.02.013
157. Moreira-Teixeira L, Sousa J, Mcnab FW, Torrado E, Cardoso F, Machado H, et al. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-g signaling. J Immunol (2016) 197:4714–26. doi:10.4049/jimmunol.1600584
Keywords: tuberculosis, genotypic diversity, immune phenotypes, severity of disease, inflammation, microenvironments
Citation: Bastos HN, Osório NS, Gagneux S, Comas I and Saraiva M (2018) The Troika Host–Pathogen–Extrinsic Factors in Tuberculosis: Modulating Inflammation and Clinical Outcomes. Front. Immunol. 8:1948. doi: 10.3389/fimmu.2017.01948
Received: 25 October 2017; Accepted: 18 December 2017;
Published: 09 January 2018
Edited by:
Olivier Garraud, Institut National de la Transfusion Sanguine, FranceReviewed by:
Marc Veldhoen, Instituto de Medicina Molecular (IMM), PortugalLaleh Majlessi, Institut Pasteur, France
Copyright: © 2018 Bastos, Osório, Gagneux, Comas and Saraiva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Iñaki Comas, aWNvbWFzQGlidi5jc2ljLmVz;
Margarida Saraiva, bWFyZ2FyaWRhLnNhcmFpdmFAaWJtYy51cC5wdA==
†These authors have contributed equally to this work.