 Young Joon Park
Young Joon Park Heung Kyu Lee
Heung Kyu Lee- 1Graduate School of Medical Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea
- 2KAIST Institute for Health Science and Technology, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, South Korea
The skin and orogenital mucosae, which constitute complex protective barriers against infection and injuries, are not only the first to come into contact with pathogens but are also colonized by a set of microorganisms that are essential to maintain a healthy physiological environment. Using 16S ribosomal RNA metagenomic sequencing, scientists recognized that the microorganism colonization has greater diversity and variability than previously assumed. These microorganisms, such as commensal bacteria, affect the host’s immune response against pathogens and modulate chronic inflammatory responses. Previously, a single pathogen was thought to cause a single disease, but current evidence suggests that dysbiosis of the tissue microbiota may underlie the disease status. Dysbiosis results in aberrant immune responses at the surface and furthermore, affects the systemic immune response. Hence, understanding the initial interaction between the barrier surface immune system and local microorganisms is important for understanding the overall systemic effects of the immune response. In this review, we describe current evidence for the basis of the interactions between pathogens, microbiota, and immune cells on surface barriers and offer explanations for how these interactions may lead to chronic inflammatory disorders.
Introduction
The skin and mucosae constitute complex protective barriers against infection and injuries. Oral and genital mucosae differ from those of lung or gut as they are in constant contact with extrinsic stimulation, such as food, medication, and physical trauma. The skin and orogenital mucosae are similar with respect to function, as these barriers are not only the body’s first line of defense against pathogens but also hosts for a substantial number of commensals, including bacteria, fungi, and viruses. These organisms influence the immune response at the barrier sites and can lead to aberrant responses and chronic inflammation. Additionally, dysbiosis caused by specific bacteria species in the skin and orogenital mucosae is associated with tissue-specific chronic immune-mediated disease. The specific species responsible for this disease include Staphylococcus aureus on skin, Porphyromonas gingivalis on oral mucosa, and Gardnerella vaginalis on vaginal mucosa. The association between certain systemic diseases and changes in the microbiota of these barrier surfaces has just recently been reported, due to the fact that the microbiota in these areas is given less attention compared to the microorganisms residing in the gut. Understanding the interactions between the local commensals and the immune system at these barrier sites is important, however, as the initial interaction occurs at these sites and can lead to widespread disease. Thus, in this review, we describe the major immune mediators of interactions between the surface barrier tissue and local microorganisms with respect to the representative disease.
Skin Microbiota
Human skin, which comprises the body’s largest organ, is home to many commensals. Across the 1.8 m2 of the skin surface, one million bacteria reside on each square centimeter, yielding a total of more than 1010 bacterial cells on the human body (1). While the gut supports 1014 bacterial cells per healthy individual, the skin contains the second highest number and diversity of bacterial cells with as many as 40 different species per individual (2, 3). The difference between skin microbiota and that of the large intestine stems largely from the direct contact of the skin with the surrounding environment. The combination of the multiple niches with various pH and temperatures created by these interactions with the environment and the diversity of the epidermal compositions and dermal appendages of the skin leads to the differences and diversity of the resident bacteria. The initial microbial skin colonization depends on the delivery mode. Vaginal delivery results in babies with microbiota similar to that of mother’s vagina, whereas babies born via cesarean section acquire microbiota related to skin. During puberty, the skin microbiota goes through a major transition with domination of lipophilic bacteria, such as Propionibacterium and Corynebacterium.
The Skin Immune System
The immune system of the skin is composed of complex network of keratinocytes and immune cells, including the skin-specific antigen-presenting cells, the Langerhans cells. As the first line of defense against infection, the innate and adaptive immune systems are delicately controlled, and even a slight interference to the network can initiate an inflammatory response. The immune-microbial interaction is therefore quite important, despite the fact that the skin microbiota are not required for immune system organization (4). When in symbiosis, which is defined as a persistent balanced state between the skin and skin-resident microorganisms, a specific member of the microbiota acts to preserve barrier functions, but when the barrier is breeched by intrinsic or extrinsic factors, the very same member can initiate an immune response.
Skin Microbiota and Innate Immunity
Epidermal keratinocytes release antimicrobial peptides (AMPs), such as cathelicidins and β-defensins, which comprise the majority of these AMPs (Figure 1A). The AMPs, which are also produced by sebocytes, provide microbicidal activity against pathogens and may also trigger the inflammatory response. Some of these AMPs are actually controlled by the microbiota. For example, Propionibacterium species induce the expression of AMPs in human sebocytes (5).
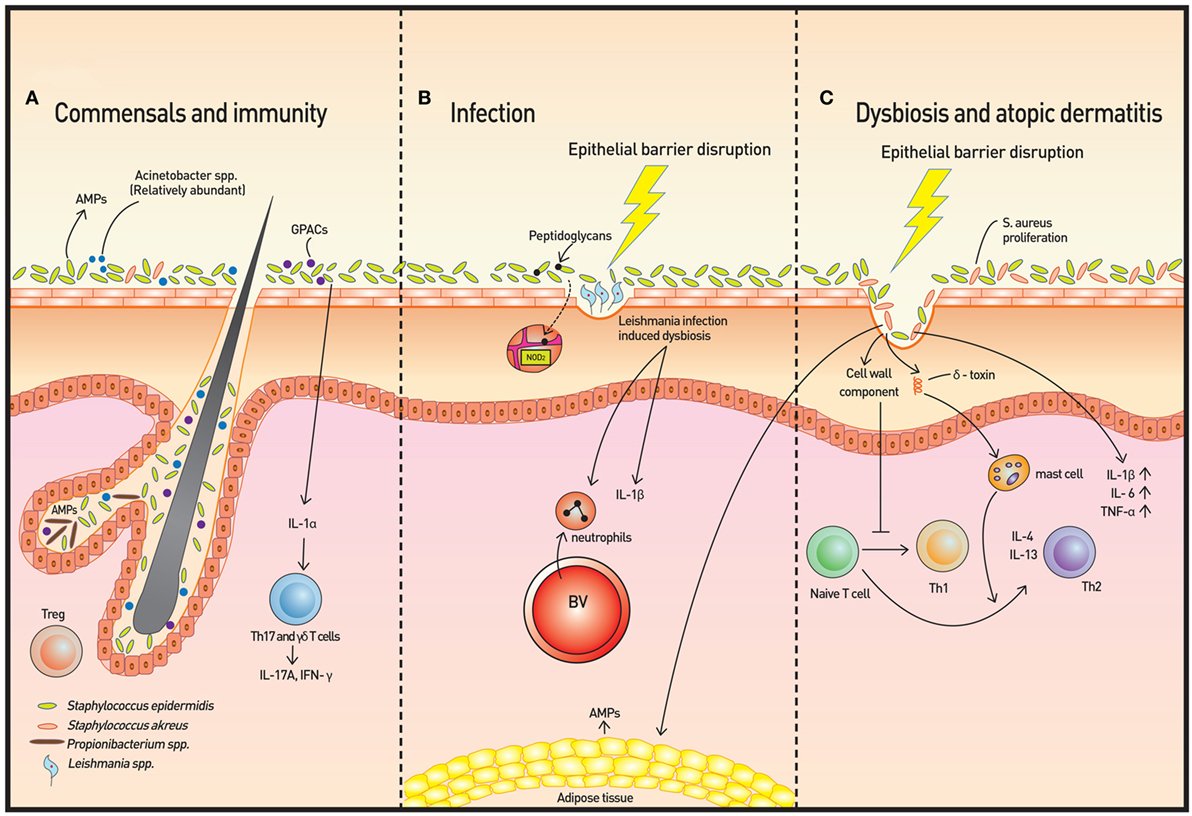
Figure 1. Skin microbiota and immunity. (A) The microbiome is more diverse in healthy skin. Staphylococcus epidermidis, Acinetobacter spp., and Gram-positive anaerobe cocci (GPAC) exhibit protective features against atopic disease. Keratinocytes and sebocytes release antimicrobial peptides (AMPs), and associations with skin commensals, such as Propionibacterium spp., have been demonstrated. Pattern recognition receptors (PRRs), such as nucleotide-binding oligomerization domain containing 2, recognize bacterial peptidoglycans to increase AMP production. Skin commensals also control the expression of interleukin (IL)-1, and increased IL-1 production is followed by IL-17A and subsequent interferon-γ production by dermal T helper 17 and γδ T cells. Regulatory T (Treg) cells reside primarily around fair follicles and interact with commensals within a specific time window to achieve immune tolerance. In addition to classical Foxp3+ Treg cells, Foxp3− Treg cells also interact with the bacterium Vitreoscilla filiformis to induce Foxp3− Treg cell differentiation. (B) The dysbiosis induced by Leishmaniasis infection is not only transmissible but also exacerbates skin inflammation via neutrophil recruitment and production of IL-1β. (C) In atopic dermatitis, Staphylococcus aureus proliferates and microbial diversity decreases concomitantly. Additionally, along with epithelial barrier disruption, proinflammatory cytokines are produced. Activation of T cells to Th2 cells occur via two mechanisms: degranulation of mast cells from δ-toxin and downregulation of IP-10 and other Th1 cell-recruiting chemokines.
In addition to microbiota regulation of AMPs, adipose tissue that interfaces with the skin may also contribute to the innate responses. In invasive S. aureus infection, local preadipocytes rapidly proliferate, leading to an expansion of the dermal fat layer, and simultaneously, cathelicidin is markedly increased in the adipose tissue and the infected skin (6). This response by the local adipose tissue provides both physical and immunologic antimicrobial defense.
Another response of the skin cells to bacterial pathogens is via pattern recognition receptors (PRRs). Nucleotide-binding oligomerization domain containing 2 (NOD2), an intracellular PRR, recognizes bacterial peptidoglycans of both Gram-positive and Gram-negative bacteria. Experimental manipulation of NOD2 led to bacterial dysbiosis with local changes in AMPs (7). Hence, although the specific mechanism has yet to be elucidated, NOD2-mediated immune surveillance appears to function in the immune response in association with the cutaneous microbiota.
Skin-resident microbes are also capable of regulating components of the complement system. Mice deficient in complement develop altered skin microbial communities (8). As inhibition of complement component C5a receptor in germ-free mice leads to decreased levels of AMPs and proinflammatory factors, the skin microbiome closely interacts with the complement system.
A recent study using a Leishmaniasis skin infection model demonstrated that human skin microbiota is altered following infection with Leishmania and that this dysbiosis is transmissible and capable of exacerbating skin inflammation via recruitment of neutrophils and production of interleukin (IL)-1β [Figure 1B (9)]. The skin microbiota also controls expression of IL-1α independently (4). The IL-1 cytokine is essential for the initiation and amplification of immune responses, and thus, the acute immune response is considered to be under the influence of host skin commensal interactions.
Skin Microbiota and Adaptive Immunity
Skin microbiota is capable of promoting both the innate and the adaptive immune responses to limit pathogen invasion and maintain homeostasis. Mice without adaptive immunity fail to control their skin microbiota, allowing pathogenic microbial invasion to occur (10). The adaptive response, however, does not occur alone and is an extension of the innate response. For example, increased IL-1 production is followed by production of IL-17 and interferon-γ (IFN-γ) from dermal T cells. Reciprocally, T helper 17 (Th17) cells along with IL-17A-producing γδ T cells are reduced in mice lacking IL-1R1 (4). Introduction of Staphylococcus epidermidis to germ-free mice restores the production of IL-17A, indicating that S. epidermidis as part of the skin commensal microbiome potently induces Th17 cells as well as other T cells that express IL-17A. Other human skin commensals, such as Corynebacterium pseudodiphtheriticum, Propionibacterium acnes, and Staphylococcus aureus, also increase the number of skin IL-17A+ cells and IFN-γ+ cells but do not induce as prominent a response as the introduction of S. epidermidis. In addition, S. epidermis is the only commensal species that increased the frequency of CD8+ T cells in the skin. These CD8+ T cells are capable of producing either IFN-γ or IL-17A and homed only to the epidermis. These commensal-specific IL-17A-producing T cells successfully enhance barrier immunity and limit pathogen invasion (11). Skin-resident Th17 cells that are affected by the skin microbiota are also independent of those from the gut microbiota, suggesting that Th17 cells of the barrier sites are regulated in a “compartmentalized” manner by local commensals (4).
In the skin of both mice and humans, Foxp3+ regulatory T (Treg) cells are present in the dermis, especially surrounding the hair follicles, where skin-resident microorganisms also reside (12). Occupation of these hair follicles by commensals may be coupled with a regulatory response by Treg cells to limit abnormal inflammatory response against them (13). The time window during which this commensal-specific tolerance is achieved may be as early as the neonatal period. Precolonization of neonatal mice with S. epidermidis suppresses skin inflammation upon S. epidermidis challenge. A wave of Foxp3+ Treg cell infiltration in the skin occurs in the second week in neonate mice, and this infiltration is accompanied by higher levels of CTLA-4 and ICOS, which are both critical mediators of immune tolerance (14). The skin of germ-free mice, however, has a high frequency of Foxp3+ Treg cells compared with the skin of specific pathogen-free (SPF) mice (4). Thus, the underlying mechanism of Tregs in controlling host-microorganism dialog has yet to be fully elucidated. Foxp3+ Treg cells are known for their function in promoting microbial persistence (15), but Foxp3− Treg cells may also be involved in establishing immune tolerance of certain microbiota. Vitreoscilla filiformis, a Gram-negative bacterium, induces dendritic cells to prime naïve T cells to type 1 Treg cells without Foxp3 expression. Furthermore, epicutaneous application of V. filiformis lysate induces IL-10high T cells and inhibits T-cell proliferation in NC/Nga mice that exhibit atopic dermatitis (AD)-like inflammation (16). Nonetheless, as Treg cells are not the sole inducers of immune tolerance, much remains to be revealed about the basis of tolerance of constitutive commensals.
Atopic Dermatitis
Atopic dermatitis is a prototype immune-mediated inflammatory skin disorder with prominent association with commensals. AD is considered to be an initiating stage of abnormal systemic Th2 response that progresses to allergic rhinitis and asthma. Some Staphylococcus spp., and in particular, S. aureus, have been found to be associated with AD and AD flares. Colonization of S. aureus on barrier-disrupted murine skin increases expression of IL-1β, IL-6, and TNF-α, demonstrating a pivotal role of S. aureus in promoting skin inflammation [Figure 1C (17)]. S. aureus colonization results in a Th2 immune response instead of a Th1 response despite the presence of its superantigen, which elicits a predominant Th1 cytokine profile, has been explained by two studies. First, δ-toxin released by S. aureus induces the degranulation of dermal mast cells and in turn, promotes the Th2 response (18). Second, components of the S. aureus cell wall downregulate IP-10 independent of IL-10; trigger activation of MAPK, p38, and ERK; and inhibit STAT1 signaling in monocytes, contributing to the abrogation of Th1 cell-recruiting chemokines (19). In contrast, S. epidermidis colonization provides innate immune signals to establish a functional threshold for adaptive immunity to facilitate pathogen control (20). As expected, overabundant colonization of S. aureus correlates with worsening AD in both mice and humans, but whether the loss of microbiome diversity leads to skin flares or skin flares result in reduced microbiotic diversity has not been illuminated (21). Dysbiosis with abundant Staphylococcus spp. and Corynebacterium spp., however, was observed in a genetically engineered murine model of AD. Also, administration of antibiotics to mice deficient in epidermal ADAM17 (Adam17ΔSox9) prevented eczematous lesion development, prevented an increase in skin-infiltrating T-cell numbers, and paradoxically, increased diversity of the skin microbiome. The study clearly showed that dysbiosis and overabundance of S. aureus contribute to AD-like lesions and are likely responsible for acute atopic flares in humans (22). The most recent reports also reveal a role for S. aureus in acute AD and new-onset pediatric AD, both of which show Th17 immune polarization. Epicutaneous S. aureus exposure induces skin inflammation by triggering IL-36R/MyD88 signaling and consequent IL-17 production from T cells (23, 24). The findings provide an explanation of increased IL-36α/γ transcripts and the increased number of Th17 cells in human AD skin (25, 26). On the other hand, skin commensals, including S. epidermidis, were found to inhibit S. aureus via production of AMPs that selectively kill S. aureus, and reintroduction of these commensals to human subjects decreased colonization by S. aureus, confirming the role of S. epidermis to prevent dysbiosis and initiation of inflammation (27). In addition to S. epidermidis, Acinetobacter species and Gram-positive anaerobe cocci (GPAC) may also play a protective role. Acinetobacter species were found to not only be more abundant in healthy subjects compared to atopic subjects but also to be associated with expression of anti-inflammatory molecules by peripheral blood mononuclear cells. Acinetobacter species induced strong Th1 and anti-inflammatory responses by immune cells and skin cells, suppressing allergic sensitization and lung inflammation in the skin (28). The abundance of GPAC was found to be low in human skin with a filaggrin deficiency, an important factor involved in AD pathogenesis. The cocci generated an inflammatory response distinct from that of S. aureus in vitro (29). These studies suggest that the loss of microbiome diversity is linked with a higher allergic response and subsequent inflammation.
Oral Microbiota
The oral microbiota is also an important part of the human microbiota. A minimum of 700 different species are present in the human oral cavity, inhabiting diverse locations such as the tongue, hard tissues, and dentures (Figure 2A) (30). Similar to the skin, the microbiota of the oral cavity differs substantially based on the ecological niche (31). One unique characteristic of the oral microbiota is that due to saliva, which washes the mucosa and soft tissue surfaces, only a non-pathogenic monolayer exists on these surfaces. On the contrary, biofilm develops on hard tissue surfaces. Biofilm is an aggregate of extremely heterogeneous bacteria and contains sufficient nutrients to sustain the metabolic needs of the microbiota (32). Dental plaque, which is a typical form of biofilm, is known for its potential to induce inflammation. Under certain circumstances, for example, a change in pH, oxygen tension, or host immune status, the resident microbiota of the biofilm can transform to a pathogenic population (33). Pathogenic colonization with or without mucosal epithelial disruption impairs host immune responses and may result in disease, such as gingivitis or periodontitis.
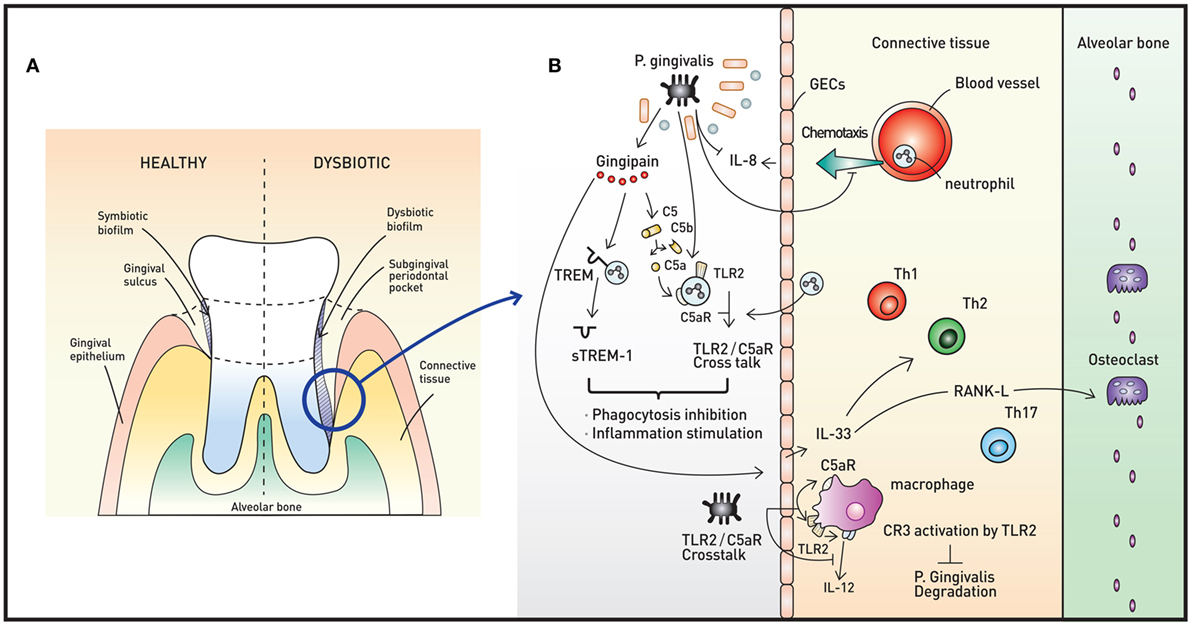
Figure 2. Oral microbiota and periodontal immunity. (A) The periodontium in healthy (left) and disease (right) states. When dysbiotic biofilm causes chronic inflammation of the gingiva, repeated swelling and inflammation results in deepening of the gingival sulcus, forming a subgingival periodontal pocket. The environment of the periodontal pocket intensifies the dysbiosis, and periodontal pathogens emerge. (B) A representative pathogen, Porphyromonas gingivalis, causes various immune responses that result in immune subversion and periodontitis. This organism inhibits secretion of IL-8 from gingival epithelial cells (GECs), interfering with the chemotaxis of neutrophils to the site of infection. Via the cysteine protease gingipain, P. gingivalis activates C5AR and toll-like receptor 2 (TLR2), triggering C5aR-TLR2 crosstalk. The crosstalk and cleavage of triggering receptor expressed on myeloid cells 1 (TREM-1) to soluble TREM-1 by gingipains result in simultaneous inhibition of phagocytosis and stimulation of inflammation. Gingipains also increase IL-33 production from GECs, downregulating antimicrobial peptides (AMPs) and increasing osteoclastogenecity. In macrophages, the C5aR-TLR2 crosstalk works to inhibit P. gingivalis degradation by suppressing production of nitric oxide and IL-12 without disruption of proinflammatory properties. The downregulation of IL-12 is also potentiated by binding of P. gingivalis to complement receptor 3.
The majority of bacteria in the oral cavity belong to five phyla: Actinobacteria, Bacteroidetes, Firmicutes, Fusobacterium, and Proteobacteria. As the microbial communities from different oral sites are distinct, the salivary microbiome is generally used as a representative microbiome (34). In a Japanese study on the salivary microbiome of 2,343 adults aged 40 years or older, the population could be divided in to two groups based on the composition of the microbiome. The group with more abundant Prevotella histicola, Prevotella melaninogenica, Veillonella parvula, Veillonella atypica, Streptococcus salivarius, and Streptococcus parasanguinis was associated with poorer oral hygiene and poorer general conditions, suggesting that the oral microbiota reflects both the local and the systemic immune status (35). Despite the characterization of the overall composition of the oral microbiome (36), studies on the immunologic role of each organism in the oral microbiome, notably bacteria without virulence factors, are remarkably scarce. The diversity of the oral microbiome based on spatial niches and constant temporal change due to its nature as an open system exposed to exogenous microbes may underlie the rarity of such studies, although a few studies have been completed. One example of such a study showed that bacteriocin produced by S. salivarius inhibits the growth of Gram-negative species associated with periodontitis and halitosis in vitro (37), in agreement with evidence that S. salivarius as a probiotic is beneficial to halitosis in vivo (38, 39).
Periodontal Microbiome, Protective Immunity, and Periodontitis
The gingival sulcus and periodontal pocket are the most-studied niches of microbial colonization in the oral mucosa. The crevice between the hard surface of the teeth and the gingiva harbors microbial communities that interact with the mucosal epithelial cells. Phylum Proteobacteria, particularly the gammaproteobacteriae of genus Acinetobacter, Haemophilus, and Moraxella, were most prevalent in healthy gingival sulci less than 4 mm deep. Periodontal pockets are formed when the attachment between the gingivae and teeth is lost. The space then can become colonized with anaerobic bacteria. The microbiota highly associated with pockets greater than 4 mm in depth include Spirochetes genus Treponema; Synergistetes genus Sinergistes; Bacteroidetes, such as genera Porphyromonas, Prevotella, and Tannerella; and Fusobacteria genera Fusobacterium and Leptotrichia (40, 41). Development of culture-independent techniques has expanded the range of associated species that were previously uncultivable or underappreciated. These species include Filifactor alocis, Peptostreptococcus stomatis, Prevotella denticola, Porpyromonas endodontalis, Anaeroglobus geminatus, and Eubacterium saphenum (40, 42).
Toll-like receptors (TLRs) and other PRRs act as immunologic sensors of the biofilm. The epithelial cells activated by microbe-associated molecular patterns (MAMPs) of the biofilm increase its proliferative rate, expression of adhesion molecules, IL-1β production, and production of AMPs, such as calprotectin (S100A8/A9) and defensins (27). Calprotectin from gingival epithelial cells (GECs) functions both extracellularly and intracellularly via incorporation into neutrophil extracellular traps and aiding innate intracellular immunity, respectively, providing resistance against pathogenic bacteria (31, 43).
In periodontal disease (also termed periodontitis), a chronic inflammatory disease that results in the destruction of bony apparatus, the components of the pattern recognition system, for example TLR2, initiate the sustained inflammation and ultimately induce the induction of bone loss in mice (44). Recently, the NOD1 cytoplasmic PRR was found to be responsible for the majority of periodontal bone destruction. Interestingly, monocolonization of germ-free animals with a single specific bacterium NI1060, which exhibits a greater than 60% homology with the coding sequences of Aggregatibacter actinomycetemcomitans, was sufficient to promote periodontal bone destruction in a NOD1-dependent manner (45). These findings demonstrate that a single bacterial component from a normal commensal microbiome can cause destructive disease through activation of a PRR in oral mucosa. Calprotectin is also abnormally activated in periodontitis and has been implicated in the pathogenesis of this disease (46).
T helper 17 cells have been implicated in mediating protective immunity as well as pathogenic inflammatory response at multiple barrier sites, including the oral cavity, skin, and gut. Gingival Th17 cells develop independently of commensal microbe colonization. As the frequencies of gingival Th17 cells are unchanged in germ-free mice compared to SPF mice, the oral microbiome does not appear to be a primary driver of gingival Th17 development, and this finding is in contrast to the situation in the skin and gastrointestinal tract. IL-6 expression in response to mechanical damage of epithelial cells instead is the major promoting factor for accumulation of Th17 cells and subsequent elevation of epithelial antimicrobial peptides and neutrophil chemo-attractants in the oral cavity (47). These data should be carefully interpreted, because it is evident that dysbiotic microbial communities aggravate periodontitis possibly in relation to accumulated Th17 cells (48–50). Mechanical damage may be critical for Th17 cell proliferation but also serves as an amplifier of the oral inflammatory response.
Porphryomonas gingivalis
Porphyromonas gingivalis, a member of the phylum Bacteroidetes, is a keystone oral pathogen that evades the immune system and alters local host responses. While this Gram-negative anaerobic rod-shaped bacterium mainly resides in biofilms and behave as a commensal, it also invades host cells, such as GECs in certain situations. This bacterium occupies its own replicative niche within autophagosomes (51). This microorganism also underlies the “dysbiosis hypothesis,” which surmises that P. gingivalis, even at low colonization levels, triggers changes in the amount and composition of the oral commensal microbiota, leading to inflammation and ultimately periodontal bone loss. Interestingly, the commensal microbiota and complement are both required for this P. gingivalis-induced bone loss, as P. gingivalis, although necessary, is not sufficient to induce inflammation (52). Thus, P. gingivalis exerts its pathogenic effects via its ability to induce dysbiotic microbial communities (Figure 2B). Manipulation of the host immune system (called immune subversion) by P. gingivalis, therefore, is essential and is therefore discussed in detail in this review.
P. gingivalis and Innate Immunity
As described above, PRRs, such as TLR2 and NOD1, are mediators of the innate immune response. P. gingivalis coactivates TLR2 and C5a receptor (C5aR) in neutrophils, resulting in crosstalk that leads to ubiquitination and proteasomal degradation of MyD88 and inhibits the host-protective antimicrobial response. Moreover, this C5aR-TLR2 crosstalk activates PI3K, which prevents phagocytosis through inhibition of RhoA activation and actin polymerization and stimulates an inflammatory response (53). Upon initiation of inflammation, GECs secrete the chemoattractant IL-8 to recruit neutrophils. P. gingivalis inhibits the secretion of IL-8 from GECs, lowering the number of neutrophils recruited to the site of inflammation. Another way that P. gingivalis inhibits the polymorphonuclear leukocyte function is through the binding of whole cells and lipopolysaccharides of the bacteria to adhesion molecules, interrupting leukocyte diapedesis. Other mechanisms to manipulate neutrophil activity include bacterial binding to fMLF receptor and PPAD-citrullinated C5a, resulting in inhibition of chemotaxis, and regulation of triggering receptor expressed on myeloid cells 1 (TREM-1) via both gingipain-dependent and -independent mechanisms, rendering the inflammatory response appropriate for the bacteria (54).
Gingipains are cysteine proteases that act as critical enzymes in periodontitis pathogenesis. In addition to cleavage of TREM-1 by Arg-gingipain, gingipain is responsible for proteasomal degradation of P. gingivalis and increased inflammation, as this enzyme cleaves C5 to C5a, leading to My88D degradation and PI3K activation (53). Gingipain is also associated with increased IL-33 production by GECs via PAR-2-p38/NF-κB signaling (55). IL-33 is an alarmin released in response to tissue damage, and increased IL-33 downregulates AMPs, such as LL-37 (56). Moreover, IL-33 induces receptor activator of NF-κB ligand, a crucial osteoclastogenic factor (57).
In macrophages, C5aR-TLR2 crosstalk by P. gingivalis suppresses production of nitric oxide and IL-12 simultaneously, inhibiting P. gingivalis killing but preserving the ability of these cells to elicit an inflammatory response (58, 59). P. gingivalis binds to complement receptor 3 (CR3) on macrophages to downregulate IL-12 selectively. This binding further allows P. gingivalis to persist intracellularly, as CR3 is not coupled to microbicidal mechanisms directly (60).
P. gingivalis and Adaptive Immunity
Macrophages engage microbes not only to eliminate them but also to work as antigen-presenting cells. Currently, the M1 subpopulation of macrophages is considered to be responsible for inflammatory function, and P. gingivalis clearly triggers M1-type-associated inflammatory pathways (61, 62). A recent study, however, indicated a novel M1 macrophage appears in response to P. gingivalis with the addition of IFN-γ, which significantly upregulated proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, and lowered production of chemokines related to T-cell recruitment (IL-12, IL-23, and CXCL10). These effects are in stark contrast to those induced by A. actinomycetemcomitans, another representative oral pathogen. Thus, P. gingivalis may have a unique capacity to alter the programmed course of the hyperinflammatory and T-cell immunomodulatory M1 macrophages (63).
Porphyromonas gingivalis appeared to drive dendritic cells, however, toward a Th2 antigen-presenting cell phenotype, at least in vitro (64). A Th2 cytokine-mediated inflammatory response was observed in association with P. gingivalis, and IL-33 was again identified recently using P. gingivalis gingipain-null mutant KDP cells. Of the various virulence factors, gingipain was entirely responsible for the IL-33 increase that was observed in human GECs (55). As evaluation of the protein levels in cells activated by P. gingivalis revealed no obvious Th1- or Th2-skewed profiles (65) and IL-1β and IL-6 were produced by CD4+ T cells (66), the specific response of Th cells induced by this bacterium remains to be fully delineated. Notably, P. gingivalis was posited to stimulate myeloid antigen-presenting cells to drive Th17 polarization, and inactive gingipains selectively generated a Th17 phenotype in an IL-6-dependent manner. Inhibition of IL-6 signaling in dendritic cells led to a significant depletion of the Th17 population without similar effects on other T-cell subsets. These studies unveiled the possibility that Th17 cells are involved in the pathogenesis of periodontitis and confirmed that IL-6 signaling is an attractive target for treatment of this disease (49).
Despite the paucity of in vivo evidence of P. gingivalis manipulation of the adaptive immune response, this organism should still be considered a keystone species and successful pathogen with the ability to modulate adaptive immunity, as P. gingivalis disables the overall host response while simultaneously enhancing the inflammatory response and the pathogenicity of a polymicrobial community.
Vaginal Microbiota
The human vaginal mucosa is home to abundant microflora. The environment is exposed to unique foreign substances, such as spermatozoa and sexually transmitted pathogens. The environment is also very versatile, constantly changing due to menstrual cycle changes of the female body. In addition, the composition of the microbial community is largely influenced by events such as pregnancy and menopause. The commensals of the vagina change throughout a woman’s life and function as a main component of the vaginal mucosal defense against pathogens.
Out of the ~200 bacterial species that reside in the vagina, Lactobacillus species predominantly colonize the vaginal tract (Figure 3A). Currently, more than 120 Lactobacillus species have been described and comprise more than 70% of resident bacteria in women. Lactobacilli are believed to contribute to the immunity in a healthy vagina. Lactobacilli produce significant amounts of lactic acid, ensuring the environment maintains a relatively low pH. Furthermore, these bacteria produce H2O2, which is presumed to contribute to protection of the vagina from pathogens, as H2O2-producing vaginal Lactobacillus spp. are more protective against bacterial vaginosis (BV) than those that do not produce H2O2 (67, 68).
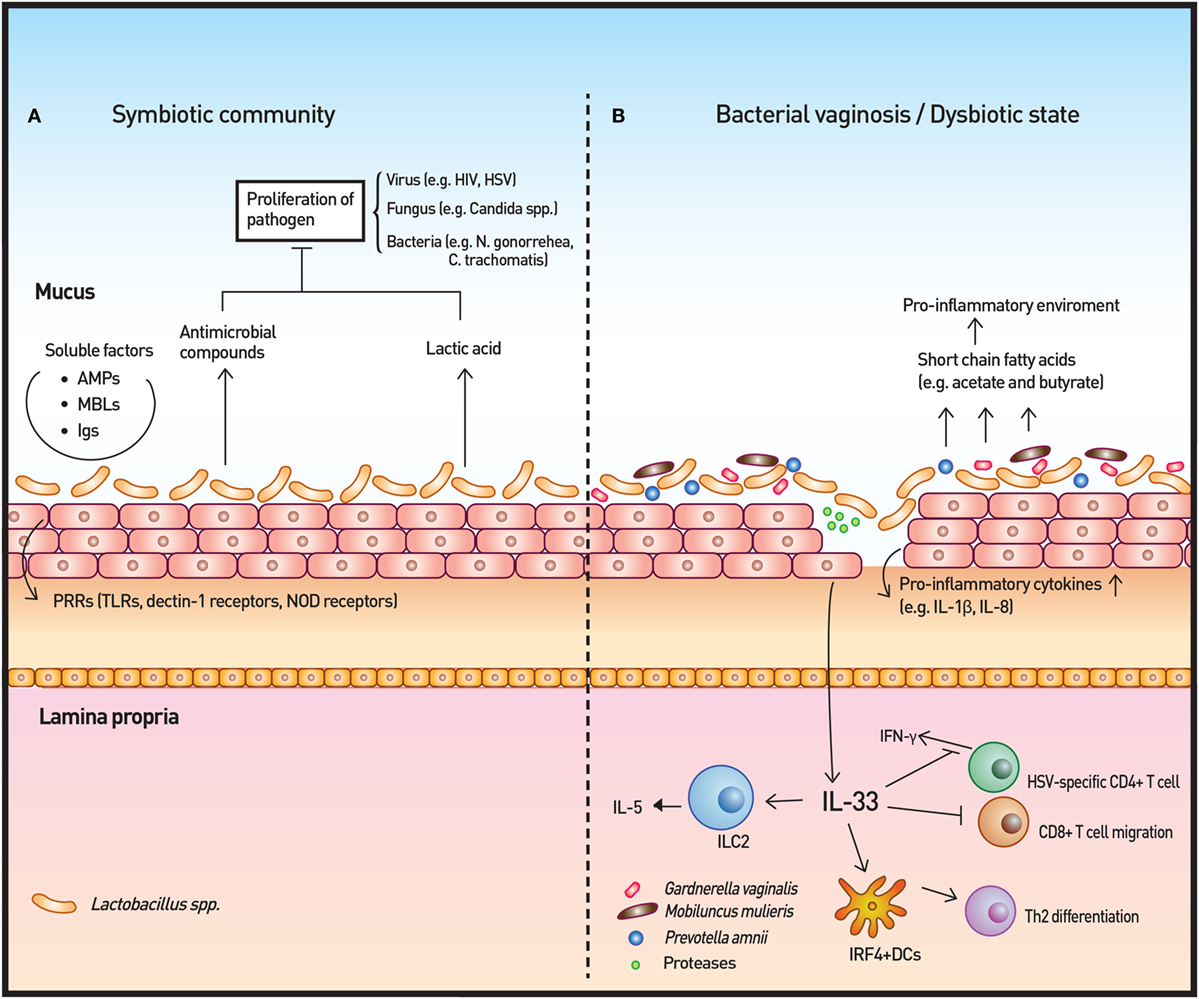
Figure 3. Vaginal microbiota and immunity. (A) The vaginal microbiota in a healthy individual is dominated by Lactobacillus spp. The Lactobacillus spp. produce lactic acid as well as antimicrobial compounds to control the growth of microbes. Other soluble factors, such as antimicrobial peptides (AMPs), mannose-binding lectins (MBLs), and immunoglobulins (Igs), contribute to the homeostatic immunity of the vaginal surface. In addition, the surveillance of commensals and pathogenic microbes is achieved by pattern recognition receptors (PRRs). (B) In cases of disrupted vaginal microbiota, such as bacterial vaginosis, community state type IV type microorganisms dominate to initiate an inflammatory response. Short-chain fatty acids produced by these microorganisms are likely to induce the production of proinflammatory cytokines. IL-33 has recently been identified as the key cytokine in association with antiviral immunity modulation by the vaginal microbiome. IL-33 is also responsible for the Th2-type immune response elicited by proteases that are secreted by pathogenic microbes.
The vaginal mucosa of a single woman is usually dominated by one or two species of Lactobacillus, and attempts to classify women by lactobacilli and other microbiota species have shown that ethnicity affects the microbiome. Caucasian women are dominated by L. iners, black and Hispanic women are dominated by L. genseni, and Asian women are dominated by L. crispatus. Similarly, reproductive-aged women have been classified to five community state types (CSTs). These types include women who harbored L. crispatus preferentially (CST I), women who were dominated by L. gasseri (CST II), and women who were dominated by L. iners (CST III). Furthermore, CST IV was initially defined as a complex mixture of bacteria, including Prevotella, without any Lactobacillus species dominance. Additionally, the presence of L. crispatus and/or L. iners without dominance was assigned as CST IV-A, and no or minimal lactobacilli was assigned as CST IV-B. The clinical significance of these groupings has yet to be fully explored, but once established, the vaginal microbiota is stable, despite frequent interruptions, such as sexual behavior (69, 70).
The vaginal microbiota is also influenced by gene polymorphisms, and the innate immune response to the microbiota in the female genital tract has been found to be associated with these genetic variants. Single-nucleotide polymorphisms that disrupt immune recognition are associated with increased susceptibility to disruption of vaginal microbiota and vaginal infections (71, 72). Especially, polymorphisms in IL-4, IL-10, TLR4, and TNF-α genes have been shown to induce aberrant responses to BV-associated bacteria and preterm delivery (72, 73). Of note, periodontal disease and BV are both influenced by gene polymorphisms and are both associated with preterm birth (74).
Vaginal Microbiota and Protective Immunity
Pattern recognition receptors, including TLRs, dectin-1 receptors, and NOD receptors, work as inspectors for MAMP on both commensal and pathogenic microbes (75, 76). Upon binding, PRRs initiate cytokine/chemokine signaling cascades for host defense. The cytokine/chemokines, including IL-1β, IL-6, and IL-8, recruit and activate various immune cells, such as macrophages, natural killer cells, and T cells. In vitro colonization of vaginal epithelial cell multilayers by Lactobacilli demonstrated that these bacteria do not elicit an inflammatory response in contrast to S. aureus. Rather, isolates of Lactobaclli, especially L. jensenii, tempered the induction of cytokines following TLR2/6 and 3 agonist treatment of the epithelial cells (77). Within the CST IV classification, Prevotella amnii, Mobiluncus mulieris, Sneathia amnii, and Sneathia sanguinegens were found to induce higher levels of inflammatory cytokines and chemokines, such as IL-1α/β and IL-8, relative to microbiota dominated by L. crispatus or L. iners. The study also showed significant increases of IL-1α/β and TNF-α during transition of communities from CST I to CST IV longitudinally, supporting the notion that the innate immune response is affected by vaginal bacterial community states (78).
Furthermore, although the number of vaginal antigen-presenting cells (VAPCs) does not vary with vaginal microbial populations, the CST IV VAPCs were more activated and mature and induced a marked response to LPS as well to IFN-γ and IL-1β. The study suggests that VAPCs are stimulated by contact with the resident flora to upregulate inflammatory cytokine/chemokine expression, resulting in a significant increase in vaginal CCR5+ CD4+ T cells (78). The increase of the accessible CD4 T-cell is important, as the vaginal mucosa is a restricted space for lymphocyte migration (79).
Soluble factors such as mannose-binding lectin (MBL), vaginal AMPs, and immunoglobulins (Igs) also contribute to vaginal defense (80). MBL binding to mannose, N-acetylglucosamine and fucose carbohydrate moieties present on microbial cell surfaces leads to cell lysis or recognition by immune cells (81). Women with MBL deficiency due to genetic polymorphism are more susceptible to recurrent vulvovaginal candidiasis (82). Defensins are a class of AMPs that act against various pathogens, including bacteria and viruses. The concentration of defensin three is associated with dysbiosis and BV during pregnancy (83). Human β-defensin-2 expression was associated with colonization by L. iners, Atopovium vaginae, and Prevotella bivia in ex vivo organotypic models of the vaginal epithelium and with L. jensenii but not G. vaginalis in vitro (71, 84). Natural Igs, such as IgG, present in vaginal mucus prevent viral infections, such as HSV, by forming multiple low-affinity bonds between the virus and mucus gel. A sufficient number of low-affinity bonds ensures that viruses are effectively trapped in mucus, thereby reducing the flux of infectious virions (85).
Lactobacilli dominance of vaginal microbiota has been shown to protect the host from other opportunistic microbial infections, including bacteria (e.g., Neisseria gonorrhoeae and Chlamydia trachomatis), viruses (e.g., HIV and HPV), and fungi [e.g., Candida albicans (86–90)]. The lactic acid and antimicrobial compounds produced by Lactobacilli are hypothesized to underlie such protection, and many studies are now focused on discovering the specific mechanisms (90, 91). Notably, of the Lactobacillus species, L. crispatus is most prominently involved in inhibition of the growth of other microbes and in suppression of cytokine production, whereas L. iners, despite its affiliation with Lactobacilli, shows conflicting results.
Bacterial Vaginosis
A disrupted vaginal microbiota may be related to various diseases but is directly connected with two pathogenic states, BV and aerobic vaginitis (AV) (Figure 3B). Both of these disease entities originate from the CST IV category. AV is mainly differentiated from BV by the presence of prominent inflammatory response associated with aerobes, such as group B Streptococcus, S. aureus, E. coli, and Enterococcus (80, 92). Despite prominent clinical symptoms and a higher risk of preterm labor and preterm birth than BV (93), the identity of AV as dysbiosis-induced inflammation is still controversial, as some believe that AV is primarily an immunologic disorder with secondary dysbiosis or a dermatological disease (94). Thus, we will focus our discussion on BV in this review.
Bacterial vaginosis is defined as a replacement of lactobacilli with characteristic groups of bacteria and subsequent change in vaginal fluid. Clinically, Amsel et al. proposed the first diagnostic criteria for BV: gray-white milky homogeneous discharge, vaginal fluid pH > 4.5, clue cells upon microscopy, and release of fishy odor on 10% potassium hydroxide solution (95). An alternative for diagnosis is to use a vaginal smear for Gram-staining of Lactobaccillus, Gardnerella, and other Gram-variable rods to yield a Nugent criteria (96). BV may or may not elicit overt inflammatory responses, and investigation of inflammatory cytokines in BV has led to inconsistent results (97).
Bacterial vaginosis has long been known to be associated with G. vaginalis; however, following the development of non-culture-based methods, it became evident that G. vaginalis is often present in the absence of BV, suggesting that colonization by this bacterium is not a precondition of BV. Furthermore, BV is not caused by transferring G. vaginalis to a healthy woman, but transfer of discharge from a woman with BV is enough to elicit BV. Hence, it is not a single type of bacteria, even at high numbers, that is important but a critical mixture of bacteria with potential pathogenic properties and a lack of favorable lactobacilli that is essential for the development of BV (98, 99).
BV-Altered Immune Response
Short-chain fatty acids (SCFAs) produced by commensal microbiota play an anti-inflammatory role in the gut (100); however, high concentrations (20 mM) of some SCFAs (acetate and butyrate) that are prevalent in BV induce PBMC production of the proinflammatory cytokines IL-8, TNF-α, and IL-1β at neutral pH, which resembles the pH of vagina. Lower levels of these SCFAs also significantly enhance TLR2 ligand- and TLR7 ligand-induced production of IL-8 and TNF-α in a time- and dose-dependent manner (101). The discordance between the effect of SCFAs in the gut and the effect of SCFAs in the vagina may be ascribed to inherent differences in these organs, including cell-type, SCFA concentration, and pH (101, 102). In vitro studies using vaginal epithelial cells and BV-associated bacteria have shown a similar proinflammatory response to that elicited by BV-associated SCFAs (71, 77, 103). In addition, a number of studies have reported higher levels of proinflammatory cytokines, such as IL-1β and IL-8, in women with BV than in controls with normal Nugent scores (104, 105). Taken together, these studies indicate that these metabolites may contribute to BV and the sub-clinical inflammation present in women with BV.
With respect to altered T-cell responses, CD4+, CD4+ CCR5+, and CD4+ CD69+ T cells were found to be decreased in the cervixes of Kenyan sex workers following BV treatment (106). Gamma delta T cells in the endocervix were also decreased in women with BV (107). On the contrary, qPCR-confirmed presence of both L. crispatus and L. jensenii was associated with lower numbers of cervical CD3+ HLADR+ and CD3+ CD4+ CCR5+ cells in healthy Belgian women (108). These results suggest a connection between BV and T cells, although the specific details of this relationship warrant further research.
Another important consequence of BV-altered immunity is the increased risk of viral infection, including sexually transmitted viruses, such as HIV and HSV. Commensal Lactobacilli inhibit HIV-1 replication in human tissue ex vivo by medium acidification, lactic acid production, and direct viricidal effects (89). The proinflammatory cytokines and chemokines that are increased by BV-associated bacteria in vitro and are associated with BV in vivo enhance the risk of HIV transmission by directly stimulating HIV replication in latent viral reservoirs and by facilitating the trafficking and activation of CD4+ host cells, which are normally sparse in the cervicovaginal mucosa (88). A recent study highlighted the importance of IL-33 in antiviral immunity of vaginal mucosa. The study demonstrated that the innate immune responses, including type I IFN and proinflammatory cytokine production at infection sites as well as induction of virus-specific CD4 and CD8 T-cell responses in draining lymph nodes, were not impaired when vaginal dysbiosis was induced using oral antibiotics. IL-33 alone was able to suppress local antiviral immunity by blocking the migration of effector T cells to the vaginal tissue, thereby inhibiting IFN-γ production (109). The study solidified the relationship between dysbiosis and HSV infection and revealed the mechanisms through which dysbiosis modulates antiviral immunity. The researchers further investigated the T-cell response from bacteria- or protozoan-secreted proteases. Challenge with the prototype protease papain on vaginal mucosa induced Th2 immunity that was dependent on IL-33. Furthermore, dendritic cells that express interferon regulatory factor 4 were responsible for the induction of Th2 differentiation (110).
Concluding Remarks
Skin, oral, and genital mucosa function similarly as the initial barriers of host defense from pathogens; however, the interactions of these mucosae with microbes and the microbial rendering of these environments are unique, which can also be described as “compartmentalized” (4). Still, researchers should not overlook the possibility of connections between the microbiota of different tissues. Recently, ectopic colonization of oral bacteria in the intestine was found to induce gut inflammation (111). Diseases, such as Behçet’s disease, which affects orogenitalia, gut, and skin, exhibit distinct salivary microbiota as well as gut microbiota (100, 112). Thus, despite the absence of continuity, it remains feasible that the microbiota of one tissue affects another tissue.
The interaction between the major pathogenic microbes and subsequent chronic inflammatory immune response sheds light on the diverse mechanisms of the host response against dysbiosis and chronic systemic disorders. In addition, these microbes with pathogenic potential can become more than commensals, and thus, illuminating the key metabolic alterations responsible for their expansion will increase our understanding of tissue-specific pathology.
In vivo studies describing the host adaptive immunity and its association with compartmentalized microbiota outside of the gut remain scarce, likely due to the difficulty of such research. Skin and orogenital mucosa are profound reservoirs of T cells, especially tissue-resident memory T (TRM) cells (12, 113–115), which are cells recently identified as lymphocytes that are distinct from recirculating central and effector memory T cells (116). Thus, studies that illustrate the relationship between the microbiota and TRM cells are very likely to increase our understanding of the continuous interaction between our body and microbes. Moreover, novel strategies targeting the host manipulation of microbes with immune cells can also be developed via information we learn from further studies on microbiota and the human immune system.
Author Contributions
YJ performed literature research and wrote the review. HK conceived the idea for the review, provided insightful discussion when necessary and edited the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the members of the Laboratory of Host Defenses for helpful advice and discussions.
Funding
This study was supported by the National Research Foundation of Korea (NRF-2016R1A2B2015028 and NRF-2015M3D6A1065121) and KAIST Future Systems Healthcare Project funded by the Ministry of Science and ICT of Korea. Young Joon Park is a recipient of Global PhD fellowship supported by the National Research Foundation of Korea (NRF-2017H1A2A1045742).
References
1. Grice EA, Kong HH, Renaud G, Young AC, NISC Comparative Sequencing ProgramBouffard GG, et al. A diversity profile of the human skin microbiota. Genome Res (2008) 18(7):1043–50. doi:10.1101/gr.075549.107
2. Belkaid Y, Segre JA. Dialogue between skin microbiota and immunity. Science (2014) 346(6212):954–9. doi:10.1126/science.1260144
3. Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annu Rev Immunol (2012) 30:759–95. doi:10.1146/annurev-immunol-020711-074937
4. Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, et al. Compartmentalized control of skin immunity by resident commensals. Science (2012) 337(6098):1115–9. doi:10.1126/science.1225152
5. Nagy I, Pivarcsi A, Kis K, Koreck A, Bodai L, McDowell A, et al. Propionibacterium acnes and lipopolysaccharide induce the expression of antimicrobial peptides and proinflammatory cytokines/chemokines in human sebocytes. Microbes Infect (2006) 8(8):2195–205. doi:10.1016/j.micinf.2006.04.001
6. Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, et al. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science (2015) 347(6217):67–71. doi:10.1126/science.1260972
7. Williams H, Crompton RA, Thomason HA, Campbell L, Singh G, McBain AJ, et al. Cutaneous NOD2 expression regulates the skin microbiome and wound healing in a murine model. J Invest Dermatol (2017) 137(11):2427–36. doi:10.1016/j.jid.2017.05.029
8. Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci U S A (2013) 110(37):15061–6. doi:10.1073/pnas.1307855110
9. Gimblet C, Meisel JS, Loesche MA, Cole SD, Horwinski J, Novais FO, et al. Cutaneous leishmaniasis induces a transmissible dysbiotic skin microbiota that promotes skin inflammation. Cell Host Microbe (2017) 22(1):13–24.e4. doi:10.1016/j.chom.2017.06.006
10. Shen W, Li W, Hixon JA, Bouladoux N, Belkaid Y, Dzutzev A, et al. Adaptive immunity to murine skin commensals. Proc Natl Acad Sci U S A (2014) 111(29):E2977–86. doi:10.1073/pnas.1401820111
11. Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature (2015) 520(7545):104–8. doi:10.1038/nature14052
12. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest (2014) 124(3):1027–36. doi:10.1172/JCI72932
13. Belkaid Y, Tamoutounour S. The influence of skin microorganisms on cutaneous immunity. Nat Rev Immunol (2016) 16(6):353–66. doi:10.1038/nri.2016.48
14. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity (2015) 43(5):1011–21. doi:10.1016/j.immuni.2015.10.016
15. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature (2002) 420(6915):502–7. doi:10.1038/nature01152
16. Volz T, Skabytska Y, Guenova E, Chen KM, Frick JS, Kirschning CJ, et al. Nonpathogenic bacteria alleviating atopic dermatitis inflammation induce IL-10-producing dendritic cells and regulatory Tr1 cells. J Invest Dermatol (2014) 134(1):96–104. doi:10.1038/jid.2013.291
17. Wanke I, Skabytska Y, Kraft B, Peschel A, Biedermann T, Schittek B. Staphylococcus aureus skin colonization is promoted by barrier disruption and leads to local inflammation. Exp Dermatol (2013) 22(2):153–5. doi:10.1111/exd.12083
18. Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature (2013) 503(7476):397–401. doi:10.1038/nature12655
19. Li Z, Levast B, Madrenas J. Staphylococcus aureus downregulates IP-10 production and prevents Th1 cell recruitment. J Immunol (2017) 198(5):1865–74. doi:10.4049/jimmunol.1601336
20. Biedermann T, Skabytska Y, Kaesler S, Volz T. Regulation of T cell immunity in atopic dermatitis by microbes: the Yin and Yang of cutaneous inflammation. Front Immunol (2015) 6:353. doi:10.3389/fimmu.2015.00353
21. Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res (2012) 22(5):850–9. doi:10.1101/gr.131029.111
22. Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity (2015) 42(4):756–66. doi:10.1016/j.immuni.2015.03.014
23. Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe (2017) 22(5):653–66.e5. doi:10.1016/j.chom.2017.10.006
24. Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus aureus virulent PSMalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe (2017) 22(5):667–77.e5. doi:10.1016/j.chom.2017.10.008
25. Suarez-Farinas M, Ungar B, Correa da Rosa J, Ewald DA, Rozenblit M, Gonzalez J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol (2015) 135(5):1218–27. doi:10.1016/j.jaci.2015.03.003
26. Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol (2008) 128(11):2625–30. doi:10.1038/jid.2008.111
27. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med (2017) 9(378). doi:10.1126/scitranslmed.aah4680
28. Fyhrquist N, Ruokolainen L, Suomalainen A, Lehtimaki S, Veckman V, Vendelin J, et al. Acinetobacter species in the skin microbiota protect against allergic sensitization and inflammation. J Allergy Clin Immunol (2014) 134(6):1301–9.e11. doi:10.1016/j.jaci.2014.07.059
29. Zeeuwen PL, Ederveen TH, van der Krieken DA, Niehues H, Boekhorst J, Kezic S, et al. Gram-positive anaerobe cocci are underrepresented in the microbiome of filaggrin-deficient human skin. J Allergy Clin Immunol (2017) 139(4):1368–71. doi:10.1016/j.jaci.2016.09.017
30. Arweiler NB, Netuschil L. The oral microbiota. Adv Exp Med Biol (2016) 902:45–60. doi:10.1007/978-3-319-31248-4_4
31. Costalonga M, Herzberg MC. The oral microbiome and the immunobiology of periodontal disease and caries. Immunol Lett (2014) 162(2 Pt A):22–38. doi:10.1016/j.imlet.2014.08.017
32. Listgarten MA. Microorganisms and dental implants. J Periodontol (1999) 70(2):220–2. doi:10.1902/jop.1999.70.2.220
33. Socransky SS, Haffajee AD. Periodontal microbial ecology. Periodontol 2000 (2005) 38:135–87. doi:10.1111/j.1600-0757.2005.00107.x
34. Hall MW, Singh N, Ng KF, Lam DK, Goldberg MB, Tenenbaum HC, et al. Inter-personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. NPJ Biofilms Microbiomes (2017) 3:2. doi:10.1038/s41522-016-0011-0
35. Takeshita T, Kageyama S, Furuta M, Tsuboi H, Takeuchi K, Shibata Y, et al. Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Sci Rep (2016) 6:22164. doi:10.1038/srep22164
36. Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al. The human oral microbiome. J Bacteriol (2010) 192(19):5002–17. doi:10.1128/JB.00542-10
37. Wescombe PA, Heng NC, Burton JP, Chilcott CN, Tagg JR. Streptococcal bacteriocins and the case for Streptococcus salivarius as model oral probiotics. Future Microbiol (2009) 4(7):819–35. doi:10.2217/fmb.09.61
38. Burton JP, Wescombe PA, Moore CJ, Chilcott CN, Tagg JR. Safety assessment of the oral cavity probiotic Streptococcus salivarius K12. Appl Environ Microbiol (2006) 72(4):3050–3. doi:10.1128/AEM.72.4.3050-3053.2006
39. Burton JP, Chilcott CN, Moore CJ, Speiser G, Tagg JR. A preliminary study of the effect of probiotic Streptococcus salivarius K12 on oral malodour parameters. J Appl Microbiol (2006) 100(4):754–64. doi:10.1111/j.1365-2672.2006.02837.x
40. Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, et al. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J (2012) 6(6):1176–85. doi:10.1038/ismej.2011.191
41. Vartoukian SR, Palmer RM, Wade WG. Diversity and morphology of members of the phylum “synergistetes” in periodontal health and disease. Appl Environ Microbiol (2009) 75(11):3777–86. doi:10.1128/AEM.02763-08
42. Kumar PS, Griffen AL, Barton JA, Paster BJ, Moeschberger ML, Leys EJ. New bacterial species associated with chronic periodontitis. J Dent Res (2003) 82(5):338–44. doi:10.1177/154405910308200503
43. Ross KF, Herzberg MC. Calprotectin expression by gingival epithelial cells. Infect Immun (2001) 69(5):3248–54. doi:10.1128/IAI.69.5.3248-3254.2001
44. Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol (2006) 177(12):8296–300. doi:10.4049/jimmunol.177.12.8296
45. Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, et al. Induction of bone loss by pathobiont-mediated NOD1 signaling in the oral cavity. Cell Host Microbe (2013) 13(5):595–601. doi:10.1016/j.chom.2013.04.005
46. Hiroshima Y, Sakamoto E, Yoshida K, Abe K, Naruishi K, Yamamoto T, et al. Advanced glycation end-products and Porphyromonas gingivalis lipopolysaccharide increase calprotectin expression in human gingival epithelial cells. J Cell Biochem (2018) 119(2):1591–603. doi:10.1002/jcb.26319
47. Dutzan N, Abusleme L, Bridgeman H, Greenwell-Wild T, Zangerle-Murray T, Fife ME, et al. On-going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity (2017) 46(1):133–47. doi:10.1016/j.immuni.2016.12.010
48. Lamont RJ, Hajishengallis G. Polymicrobial synergy and dysbiosis in inflammatory disease. Trends Mol Med (2015) 21(3):172–83. doi:10.1016/j.molmed.2014.11.004
49. Glowczyk I, Wong A, Potempa B, Babyak O, Lech M, Lamont RJ, et al. Inactive gingipains from P. gingivalis selectively skews T cells toward a Th17 phenotype in an IL-6 dependent manner. Front Cell Infect Microbiol (2017) 7:140. doi:10.3389/fcimb.2017.00140
50. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med (2014) 6(229):229ra40. doi:10.1126/scitranslmed.3007696
51. Mysak J, Podzimek S, Sommerova P, Lyuya-Mi Y, Bartova J, Janatova T, et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J Immunol Res (2014) 2014:476068. doi:10.1155/2014/476068
52. Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe (2011) 10(5):497–506. doi:10.1016/j.chom.2011.10.006
53. Maekawa T, Krauss JL, Abe T, Jotwani R, Triantafilou M, Triantafilou K, et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe (2014) 15(6):768–78. doi:10.1016/j.chom.2014.05.012
54. Uriarte SM, Edmisson JS, Jimenez-Flores E. Human neutrophils and oral microbiota: a constant tug-of-war between a harmonious and a discordant coexistence. Immunol Rev (2016) 273(1):282–98. doi:10.1111/imr.12451
55. Tada H, Matsuyama T, Nishioka T, Hagiwara M, Kiyoura Y, Shimauchi H, et al. Porphyromonas gingivalis gingipain-dependently enhances IL-33 production in human gingival epithelial cells. PLoS One (2016) 11(4):e0152794. doi:10.1371/journal.pone.0152794
56. Tada H, Shimizu T, Matsushita K, Takada H. Porphyromonas gingivalis-induced IL-33 down-regulates hCAP-18/LL-37 production in human gingival epithelial cells. Biomed Res (2017) 38(3):167–73. doi:10.2220/biomedres.38.167
57. Laperine O, Cloitre A, Caillon J, Huck O, Bugueno IM, Pilet P, et al. Interleukin-33 and RANK-l interplay in the alveolar bone loss associated to periodontitis. PLoS One (2016) 11(12):e0168080. doi:10.1371/journal.pone.0168080
58. Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal (2010) 3(109):ra11. doi:10.1126/scisignal.2000697
59. Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol (2011) 186(2):869–77. doi:10.4049/jimmunol.1003252
60. Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol (2007) 179(4):2359–67. doi:10.4049/jimmunol.179.4.2359
61. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol (2014) 29(6):248–57. doi:10.1111/omi.12065
62. Ebersole JL, Dawson D III, Emecen-Huja P, Nagarajan R, Howard K, Grady ME, et al. The periodontal war: microbes and immunity. Periodontol 2000 (2017) 75(1):52–115. doi:10.1111/prd.12222
63. Huang CB, Alimova Y, Ebersole JL. Macrophage polarization in response to oral commensals and pathogens. Pathog Dis (2016) 74(3):1–10. doi:10.1093/femspd/ftw011
64. Jotwani R, Pulendran B, Agrawal S, Cutler CW. Human dendritic cells respond to Porphyromonas gingivalis LPS by promoting a Th2 effector response in vitro. Eur J Immunol (2003) 33(11):2980–6. doi:10.1002/eji.200324392
65. Gonzales JR, Groger S, Boedeker RH, Meyle J. Expression and secretion levels of Th1 and Th2 cytokines in patients with aggressive periodontitis. Clin Oral Investig (2012) 16(5):1463–73. doi:10.1007/s00784-011-0634-8
66. Gonzales JR, Groeger S, Johansson A, Meyle J. T helper cells from aggressive periodontitis patients produce higher levels of interleukin-1 beta and interleukin-6 in interaction with Porphyromonas gingivalis. Clin Oral Investig (2014) 18(7):1835–43. doi:10.1007/s00784-013-1162-5
67. Vallor AC, Antonio MA, Hawes SE, Hillier SL. Factors associated with acquisition of, or persistent colonization by, vaginal lactobacilli: role of hydrogen peroxide production. J Infect Dis (2001) 184(11):1431–6. doi:10.1086/324445
68. Hawes SE, Hillier SL, Benedetti J, Stevens CE, Koutsky LA, Wolner-Hanssen P, et al. Hydrogen peroxide-producing lactobacilli and acquisition of vaginal infections. J Infect Dis (1996) 174(5):1058–63. doi:10.1093/infdis/174.5.1058
69. Gajer P, Brotman RM, Bai G, Sakamoto J, Schutte UM, Zhong X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med (2012) 4(132):132ra52. doi:10.1126/scitranslmed.3003605
70. Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A (2011) 108(Suppl 1):4680–7. doi:10.1073/pnas.1002611107
71. Doerflinger SY, Throop AL, Herbst-Kralovetz MM. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J Infect Dis (2014) 209(12):1989–99. doi:10.1093/infdis/jiu004
72. Verstraelen H, Verhelst R, Nuytinck L, Roelens K, De Meester E, De Vos D, et al. Gene polymorphisms of toll-like and related recognition receptors in relation to the vaginal carriage of Gardnerella vaginalis and Atopobium vaginae. J Reprod Immunol (2009) 79(2):163–73. doi:10.1016/j.jri.2008.10.006
73. Jones NM, Holzman C, Friderici KH, Jernigan K, Chung H, Wirth J, et al. Interplay of cytokine polymorphisms and bacterial vaginosis in the etiology of preterm delivery. J Reprod Immunol (2010) 87(1–2):82–9. doi:10.1016/j.jri.2010.06.158
74. Sanu O, Lamont RF. Periodontal disease and bacterial vaginosis as genetic and environmental markers for the risk of spontaneous preterm labor and preterm birth. J Matern Fetal Neonatal Med (2011) 24(12):1476–85. doi:10.3109/14767058.2010.545930
75. Horne AW, Stock SJ, King AE. Innate immunity and disorders of the female reproductive tract. Reproduction (2008) 135(6):739–49. doi:10.1530/REP-07-0564
76. Usluogullari B, Gumus I, Gunduz E, Kaygusuz I, Simavli S, Acar M, et al. The role of human dectin-1 Y238X gene polymorphism in recurrent vulvovaginal candidiasis infections. Mol Biol Rep (2014) 41(10):6763–8. doi:10.1007/s11033-014-3562-2
77. Rose WA II, McGowin CL, Spagnuolo RA, Eaves-Pyles TD, Popov VL, Pyles RB. Commensal bacteria modulate innate immune responses of vaginal epithelial cell multilayer cultures. PLoS One (2012) 7(3):e32728. doi:10.1371/journal.pone.0032728
78. Anahtar MN, Byrne EH, Doherty KE, Bowman BA, Yamamoto HS, Soumillon M, et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity (2015) 42(5):965–76. doi:10.1016/j.immuni.2015.04.019
79. Shin H, Iwasaki A. Tissue-resident memory T cells. Immunol Rev (2013) 255(1):165–81. doi:10.1111/imr.12087
80. Smith SB, Ravel J. The vaginal microbiota, host defence and reproductive physiology. J Physiol (2017) 595(2):451–63. doi:10.1113/JP271694
81. Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol (2003) 40(7):423–9. doi:10.1016/S0161-5890(03)00155-X
82. Babula O, Lazdane G, Kroica J, Ledger WJ, Witkin SS. Relation between recurrent vulvovaginal candidiasis, vaginal concentrations of mannose-binding lectin, and a mannose-binding lectin gene polymorphism in Latvian women. Clin Infect Dis (2003) 37(5):733–7. doi:10.1086/377234
83. Khan FF, Carpenter D, Mitchell L, Mansouri O, Black HA, Tyson J, et al. Accurate measurement of gene copy number for human alpha-defensin DEFA1A3. BMC Genomics (2013) 14:719. doi:10.1186/1471-2164-14-719
84. Valore EV, Wiley DJ, Ganz T. Reversible deficiency of antimicrobial polypeptides in bacterial vaginosis. Infect Immun (2006) 74(10):5693–702. doi:10.1128/IAI.00524-06
85. Wang YY, Kannan A, Nunn KL, Murphy MA, Subramani DB, Moench T, et al. IgG in cervicovaginal mucus traps HSV and prevents vaginal herpes infections. Mucosal Immunol (2014) 7(5):1036–44. doi:10.1038/mi.2013.120
86. Breshears LM, Edwards VL, Ravel J, Peterson ML. Lactobacillus crispatus inhibits growth of Gardnerella vaginalis and Neisseria gonorrhoeae on a porcine vaginal mucosa model. BMC Microbiol (2015) 15:276. doi:10.1186/s12866-015-0608-0
87. Mastromarino P, Di Pietro M, Schiavoni G, Nardis C, Gentile M, Sessa R. Effects of vaginal lactobacilli in Chlamydia trachomatis infection. Int J Med Microbiol (2014) 304(5–6):654–61. doi:10.1016/j.ijmm.2014.04.006
88. Buve A, Jespers V, Crucitti T, Fichorova RN. The vaginal microbiota and susceptibility to HIV. AIDS (2014) 28(16):2333–44. doi:10.1097/QAD.0000000000000432
89. Nahui Palomino RA, Zicari S, Vanpouille C, Vitali B, Margolis L. Vaginal Lactobacillus inhibits HIV-1 replication in human tissues ex vivo. Front Microbiol (2017) 8:906. doi:10.3389/fmicb.2017.00906
90. Wang S, Wang Q, Yang E, Yan L, Li T, Zhuang H. Antimicrobial compounds produced by vaginal Lactobacillus crispatus are able to strongly inhibit Candida albicans growth, hyphal formation and regulate virulence-related gene expressions. Front Microbiol (2017) 8:564. doi:10.3389/fmicb.2017.00564
91. Hearps AC, Tyssen D, Srbinovski D, Bayigga L, Diaz DJD, Aldunate M, et al. Vaginal lactic acid elicits an anti-inflammatory response from human cervicovaginal epithelial cells and inhibits production of pro-inflammatory mediators associated with HIV acquisition. Mucosal Immunol (2017) 10(6):1480–90. doi:10.1038/mi.2017.27
92. Donders GG, Vereecken A, Bosmans E, Dekeersmaecker A, Salembier G, Spitz B. Definition of a type of abnormal vaginal flora that is distinct from bacterial vaginosis: aerobic vaginitis. BJOG (2002) 109(1):34–43. doi:10.1111/j.1471-0528.2002.00432.x
93. Donders G, Bellen G, Rezeberga D. Aerobic vaginitis in pregnancy. BJOG (2011) 118(10):1163–70. doi:10.1111/j.1471-0528.2011.03020.x
94. Edwards L. Dermatologic causes of vaginitis: a clinical review. Dermatol Clin (2010) 28(4):727–35. doi:10.1016/j.det.2010.07.004
95. Amsel R, Totten PA, Spiegel CA, Chen KC, Eschenbach D, Holmes KK. Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am J Med (1983) 74(1):14–22. doi:10.1016/0002-9343(83)91112-9
96. Nugent RP, Krohn MA, Hillier SL. Reliability of diagnosing bacterial vaginosis is improved by a standardized method of Gram stain interpretation. J Clin Microbiol (1991) 29(2):297–301.
97. Mitchell C, Marrazzo J. Bacterial vaginosis and the cervicovaginal immune response. Am J Reprod Immunol (2014) 71(6):555–63. doi:10.1111/aji.12264
98. Lamont RF, Sobel JD, Akins RA, Hassan SS, Chaiworapongsa T, Kusanovic JP, et al. The vaginal microbiome: new information about genital tract flora using molecular based techniques. BJOG (2011) 118(5):533–49. doi:10.1111/j.1471-0528.2010.02840.x
99. Mendling W. Vaginal microbiota. Adv Exp Med Biol (2016) 902:83–93. doi:10.1007/978-3-319-31248-4_6
100. Consolandi C, Turroni S, Emmi G, Severgnini M, Fiori J, Peano C, et al. Behcet’s syndrome patients exhibit specific microbiome signature. Autoimmun Rev (2015) 14(4):269–76. doi:10.1016/j.autrev.2014.11.009
101. Mirmonsef P, Zariffard MR, Gilbert D, Makinde H, Landay AL, Spear GT. Short-chain fatty acids induce pro-inflammatory cytokine production alone and in combination with toll-like receptor ligands. Am J Reprod Immunol (2012) 67(5):391–400. doi:10.1111/j.1600-0897.2011.01089.x
102. Aldunate M, Srbinovski D, Hearps AC, Latham CF, Ramsland PA, Gugasyan R, et al. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front Physiol (2015) 6:164. doi:10.3389/fphys.2015.00164
103. Eade CR, Diaz C, Wood MP, Anastos K, Patterson BK, Gupta P, et al. Identification and characterization of bacterial vaginosis-associated pathogens using a comprehensive cervical-vaginal epithelial coculture assay. PLoS One (2012) 7(11):e50106. doi:10.1371/journal.pone.0050106
104. Thurman AR, Kimble T, Herold B, Mesquita PM, Fichorova RN, Dawood HY, et al. Bacterial vaginosis and subclinical markers of genital tract inflammation and mucosal immunity. AIDS Res Hum Retroviruses (2015) 31(11):1139–52. doi:10.1089/aid.2015.0006
105. Fichorova RN, Lai JJ, Schwartz JL, Weiner DH, Mauck CK, Callahan MM. Baseline variation and associations between subject characteristics and five cytokine biomarkers of vaginal safety among healthy non-pregnant women in microbicide trials. Cytokine (2011) 55(1):134–40. doi:10.1016/j.cyto.2011.03.016
106. Rebbapragada A, Howe K, Wachihi C, Pettengell C, Sunderji S, Huibner S, et al. Bacterial vaginosis in HIV-infected women induces reversible alterations in the cervical immune environment. J Acquir Immune Defic Syndr (2008) 49(5):520–2. doi:10.1097/QAI.0b013e318189a7ca
107. Alcaide ML, Strbo N, Romero L, Jones DL, Rodriguez VJ, Arheart K, et al. Bacterial vaginosis is associated with loss of gamma delta T cells in the female reproductive tract in women in the Miami Women Interagency HIV Study (WIHS): a cross sectional study. PLoS One (2016) 11(4):e0153045. doi:10.1371/journal.pone.0153045
108. Kyongo JK, Jespers V, Goovaerts O, Michiels J, Menten J, Fichorova RN, et al. Searching for lower female genital tract soluble and cellular biomarkers: defining levels and predictors in a cohort of healthy Caucasian women. PLoS One (2012) 7(8):e43951. doi:10.1371/journal.pone.0043951
109. Oh JE, Kim BC, Chang DH, Kwon M, Lee SY, Kang D, et al. Dysbiosis-induced IL-33 contributes to impaired antiviral immunity in the genital mucosa. Proc Natl Acad Sci U S A (2016) 113(6):E762–71. doi:10.1073/pnas.1518589113
110. Oh JE, Oh DS, Jung HE, Lee HK. A mechanism for the induction of type 2 immune responses by a protease allergen in the genital tract. Proc Natl Acad Sci U S A (2017) 114(7):E1188–95. doi:10.1073/pnas.1612997114
111. Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science (2017) 358(6361):359–65. doi:10.1126/science.aan4526
112. Coit P, Mumcu G, Ture-Ozdemir F, Unal AU, Alpar U, Bostanci N, et al. Sequencing of 16S rRNA reveals a distinct salivary microbiome signature in Behcet’s disease. Clin Immunol (2016) 169:28–35. doi:10.1016/j.clim.2016.06.002
113. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol (2013) 31:137–61. doi:10.1146/annurev-immunol-032712-095954
114. Park JY, Chung H, Choi Y, Park JH. Phenotype and tissue residency of lymphocytes in the murine oral mucosa. Front Immunol (2017) 8:250. doi:10.3389/fimmu.2017.00250
115. Posavad CM, Zhao L, Dong L, Jin L, Stevens CE, Magaret AS, et al. Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal Immunol (2017) 10(5):1259–69. doi:10.1038/mi.2016.118
Keywords: immune response, microbiota, skin mucosa, orogenital mucosa, inflammation
Citation: Park YJ and Lee HK (2018) The Role of Skin and Orogenital Microbiota in Protective Immunity and Chronic Immune-Mediated Inflammatory Disease. Front. Immunol. 8:1955. doi: 10.3389/fimmu.2017.01955
Received: 08 September 2017; Accepted: 19 December 2017;
Published: 10 January 2018
Edited by:
Yves Renaudineau, University of Western Brittany, FranceReviewed by:
Mario M. D’Elios, University of Florence, ItalySandip Chakraborty, College of Veterinary Sciences and Animal Husbandry, India
Copyright: © 2018 Park and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heung Kyu Lee, aGV1bmdreXUubGVlQGthaXN0LmFjLmty