 Rohan Ameratunga1,2*
Rohan Ameratunga1,2* See-Tarn Woon1
See-Tarn Woon1 Vanessa L. Bryant3,4Richard Steele1
Vanessa L. Bryant3,4Richard Steele1 Charlotte Slade3,5
Charlotte Slade3,5 Euphemia Yee Leung6
Euphemia Yee Leung6 Klaus Lehnert7
Klaus Lehnert7
- 1Department of Virology and Immunology, Auckland City Hospital, Auckland, New Zealand
- 2Department of Clinical Immunology, Auckland City Hospital, Auckland, New Zealand
- 3Department of Immunology, Walter and Eliza Hall Institute of Medical Research, Parkville, VIC, Australia
- 4Department of Medical Biology, University of Melbourne, Parkville, VIC, Australia
- 5Department of Allergy and Clinical Immunology, Royal Melbourne Hospital, Parkville, VIC, Australia
- 6Auckland Cancer Society Research Centre, University of Auckland, Auckland, New Zealand
- 7School of Biological Sciences, University of Auckland, Auckland, New Zealand
The existence of epistasis in humans was first predicted by Bateson in 1909. Epistasis describes the non-linear, synergistic interaction of two or more genetic loci, which can substantially modify disease severity or result in entirely new phenotypes. The concept has remained controversial in human genetics because of the lack of well-characterized examples. In humans, it is only possible to demonstrate epistasis if two or more genes are mutated. In most cases of epistasis, the mutated gene products are likely to be constituents of the same physiological pathway leading to severe disruption of a cellular function such as antibody production. We have recently described a digenic family, who carry mutations of TNFRSF13B/TACI as well as TCF3 genes. Both genes lie in tandem along the immunoglobulin isotype switching and secretion pathway. We have shown they interact in an epistatic way causing severe immunodeficiency and autoimmunity in the digenic proband. With the advent of next generation sequencing, it is likely other families with digenic inheritance will be identified. Since digenic inheritance does not always cause epistasis, we propose an epistasis index which may help quantify the effects of the two mutations. We also discuss the clinical implications of digenic inheritance and epistasis in humans with primary immunodeficiency disorders.
History of Epistasis
Epistasis was a term coined by Bateson in 1909 (1). Based on a series of experiments, Bateson predicted there would be interactions between two or more genetic loci to produce novel phenotypes. Bateson and Punnett undertook experiments on the genetics of flower color in peas and comb morphology in roosters. When white pea flowers were crossed, all of the F1 hybrid was purple, a new phenotype. When the purple F1 hybrids were crossed, the result was unexpected: a ratio of 9:7 purple to white colored flowers. This result could not be explained by Mendelian genetics (2).
Bateson and Punnett described a similar phenomenon in the comb morphology of roosters. They speculated there must be at least two genes, which were interacting in order to explain their observations. Bateson’s predictions were made long before the structure of DNA was elucidated and underlying biochemical pathways were understood. It is now known that there are two enzymes, which lie in tandem along the pathway for the production of the purple anthocyanin pigment (3). Homozygous null mutations of either enzyme result in white colored pea flowers.
Bateson and Punnett’s observations could not be explained at the time and the concept of epistasis became controversial. Sir Ronald Fisher was famously quoted “no epistasis on my watch.” Over time epistasis became a statistical concept (4). It is much easier to demonstrate interactions of multiple genes in a population than in a single individual (Figure 1).
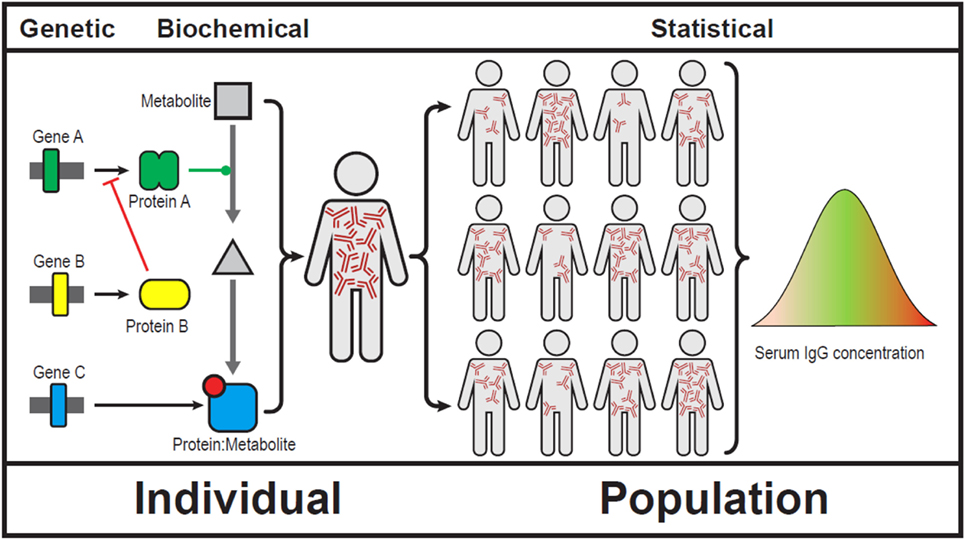
Figure 1. Showing epistasis at a genetic, biochemical, clinical, and population level (5). We use serum immunoglobulins to show gene–gene interactions can result in broad array of serum IgG levels. Note that IgG levels do not follow a Gaussian distribution in human populations.
Studies in Non-Mammalian Organisms and Laboratory Animals
Epistasis plays an important role in evolution. Multiple mutations may confer a survival advantage in a rapidly changing environment and therefore enhance reproductive fitness. A dramatic clinical example is the swift evolution of bacterial multidrug antibiotic resistance in intensive care units. In the face of extensive antibiotic usage, bacteria are under intense selection pressure to quickly evolve drug resistance to maintain reproductive fitness. Strains rapidly developing multiple antibiotic resistance from the epistatic interactions of two or more mutations (or acquiring resistance elements horizontally) are at a survival advantage (6).
In yeast, combinations of two different genetic variants can result in a phenotype that is different from each individual strain, i.e., qualitative epistasis (7). Subsequent work with iRNA screens to knock down gene function in Caenorhabditis elegans and Drosophila melanogaster has revealed that genetic networks are likely to be similarly affected in eukaryotic systems (8).
Epistasis can also be explored in laboratory animals (9). Single gene mutations can arise either spontaneously or may be induced by a variety of techniques including gene targeting or CRISPR-Cas9 gene editing technology. Cross-breeding allows the exploration of the phenotypic effects of digenic (or multigenic) disorders.
There are, however, caveats to such an approach. There may be substantial phenotypic differences between humans and mice carrying the same mutations (10). This could complicate interpretation of data in mice. We have also discussed problems utilizing CRISPR-Cas9 gene edited mice to replicate human genetic disease (11). Apart from the technical difficulties creating these mice, there is the potential for undetected off-target effects. This could compromise the interpretation of data.
Studies in Humans
Because of the lack of well-characterized examples, epistasis is not a commonly used term in human clinical genetics. In general, epistatic interactions occur along a single biochemical pathway where mutations can aggravate (negative epistasis), ameliorate (positive epistasis), or produce an entirely new phenotype. We suggest that positive and negative epistasis are grouped as quantitative epistasis while gene–gene interactions resulting in a new phenotype are termed qualitative epistasis. This simplified classification makes no presumptions on how digenic disorders are inherited or how the genes interact.
In the case of quantitative epistasis, there should be a clinical method for measuring the severity of the phenotype such as a disease-specific severity score. If clinical epistasis is to be demonstrated in an individual with digenic inheritance, the disease severity score should be much worse (or much better) compared with family members bearing only one mutation. The results could be quantified as an epistasis index (EI) as described below. If the biochemical basis of the clinical disorder is known, epistasis can potentially be quantified and confirmed in vitro. In an ideal model, clinical (phenotypic) epistasis should mirror relevant in vitro functional studies (biochemical epistasis) in family members (Table 1).

Table 1. Requirements for confirming epistasis in humans.
Unlike individuals bearing a single gene defect, we suggest that epistatic effects of digenic disorders can only be fully evaluated in an informative family (Table 1). Each of the mutations must have a demonstrable phenotype, even if mild. In order to demonstrate epistasis with confidence, both mutations should be present in at least one family member (the proband) and there must be individuals carrying only one of the mutations in the kindred (Table 1). Individuals with neither mutation serve as the wild-type control for the family.
Digenic Inheritance in Human Autoimmunity
Recently, a consanguineous Kuwaiti family was described with mutations of both the LRBA and NEIL3 genes (12). Three children bearing both mutations died at a young age from severe autoimmunity. The authors speculated that interaction of the two mutations may have contributed to the severe clinical manifestations. Interestingly, an unrelated Kuwaiti woman with the same homozygous NEIL3 deficiency had no clinical manifestations of autoimmunity. The frequency of NEIL3 mutations in the Kuwaiti population is approximately 2%. Because the three children had died, the authors did not have the opportunity to undertake detailed clinical or quantitative functional studies outlined in Table 1. Both mutations in this digenic disorder may have had a similar effect of predisposing to autoimmunity (category #5 of digenic disorders in Table 3).
Epistasis in Primary Immunodeficiency
Primary immunodeficiency disorders (PIDs) are rare genetic defects of immunity predisposing to infections, autoimmunity, allergies, and malignancy. To date, over 300 monogenic defects causing PIDs have been described (16). There are many clinical advantages in identifying the genetic basis of these conditions (Table 2).
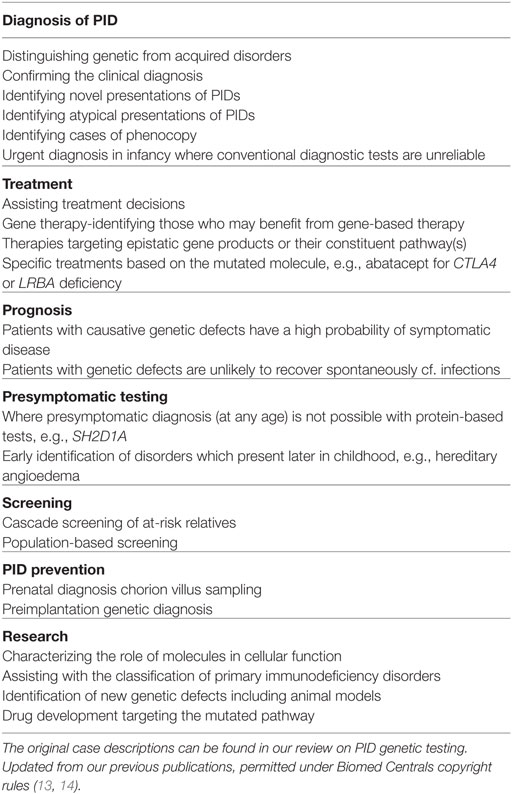
Table 2. Advantages of molecular analysis for primary immunodeficiency disorders (PIDs).
Common variable immunodeficiency disorders (CVID) are the most frequent symptomatic primary immunodeficiencies in humans and can present from infancy to late in life. CVID is characterized by late onset antibody failure (LOAF) leading to immune system failure (ISF). Over the last 5 years, an increasing number of CVID patients have been identified with monogenic disorders (17–20). Most of these patients were identified with the assistance of next generation sequencing (NGS) (21). Once identified, these patients are removed from umbrella diagnosis of CVID and are deemed to have a CVID-like disorder (22, 23).
We have recently shown that a digenic CVID-like disorder resulted from epistatic interactions of two mutated genes: transcription factor 3 (TCF3) and TNFRSF13B/TACI (21). The proband, who has both mutations is severely affected with multiple infections and she also meets the American College of Rheumatology criteria for systemic lupus erythematosus.
In this family, virtually all combinations of mutations were present allowing us to quantify the severity of disorder in order to investigate epistasis (Table 1; Figure 3). The clinical severity, measured by the clinical score for CVID, closely matched the pattern of the mutations (24). In vitro antibody production studies, which are the ultimate correlates of LOAF/ISF, mirrored the clinical phenotype of each family member (Figure 3). In vitro antibody production was most severely impaired in the digenic proband and milder defects were identified in other family members carrying a single mutation.
A consanguineous family was recently described, where the proband has mutations of the IFNAR1 and IFNGR2 genes. The patient was susceptible to mycobacterial, bacterial, and viral infections. There were no other siblings in the family. Given the autosomal recessive nature of the two mutations, both parents were carriers and were asymptomatic. In this case, it is likely both mutations had a similar function (category #5, Table 3) (25).

Table 3. Patterns of digenic inheritance, expanded from Gazzo et al. (15).
Patterns of Digenic Inheritance and EI
In a recent review, the authors suggested digenic disorders could be categorized according to the likely interactions of genes (Table 3) (15). An extended version is shown in Table 3 and it might be predicted that not all cases of digenic inheritance result in epistasis. Mutations of genes in unrelated pathways may not interact (category #6). In contrast, epistasis may occur in directly interacting genes as well as those functioning at different positions along the same pathway (categories #1 and #3). In order to further quantify this, we propose an EI. We suggest the following equation to identify non-linear effects of two or more mutations. Higher numbers indicate more severe disease (e.g., clinical score when computing CVID severity).
where M1 and M2 refer to scores of individuals bearing each mutation. When a lower number (e.g., in vitro antibody production) indicates greater severity, the equation can be inverted:
In either case, an EI value >1 indicates a synergistic effect consistent with negative epistasis. In the case of positive epistasis, the scores will be <1 indicating mitigation of effect. It is possible the EI is >1 for some in vitro parameters but not others. If there is no epistasis, the EI will be 1 indicating additive effects of the mutations.
We suggest computing epistasis indices independently, using the clinical disease severity score and the most informative biochemical marker to demonstrate epistasis, in this case in vitro antibody production. The EI can be applied to quantitative epistasis but not qualitative epistasis, where there is a completely different phenotype, such as a change in the color of pea flowers in F1 and F2 hybrids. As stated above, the EI cannot be computed from the digenic individual alone, in the absence of family members bearing each of the mutations.
In the case of the family described above, the calculated results are as follows:
Clinical EI (Based on the clinical score, Figure 3B) 34/(13 + 3) = 2.13
Biochemical EI (In vitro IgG production, nanogram per milliliter: Figure 3C) (749 + 647)/138 = 10.1
It should be noted that sequence variations in the absence of a phenotype (category #7, Table 3) do not necessarily indicate digenic or oligogenic inheritance. These sequences may be occurring in deeply intronic regions or they may be non-synonymous variants, with no effect on protein translation. In such situations, there may be no phenotype associated with these variants, making epistasis unlikely.
Discussion: Clinical Implications of Digenic Inheritance and Epistasis
The most important aspect of digenic inheritance is its clinical recognition. Failure to recognize digenic disorders could have catastrophic consequences for patients. It was previously thought mutations of TNFRSF13B/TACI caused CVID (26). More recently, TNFRSF13B/TACI mutations are perceived to have only mild effects on the phenotype (27) and mostly function as a risk allele for CVID and related disorders. Had this family been advised to undertake implantation genetic diagnosis (PGD)/chorion villus sampling (CVS) or amniocentesis based on the TNFRS13B/TACI mutation alone, serious consequences could have resulted from this error.
Even if recognized, digenic disorders will complicate diagnostic testing in the case of PGD. At this time, it seems unlikely both mutations could be reliably identified by single cell PCR for PGD. There is high probability of allele drop out, leading to a false-negative result. Given access to greater amounts of tissue and therefore DNA, CVS, and amniocentesis are more likely to reliably identify both mutations. We have indicated priority should be given to identifying the more damaging mutation (TCF3) for PGD or CVS/amniocentesis (13, 14).
Similarly, the more deleterious TCF3 mutation is likely to determine long-term prognosis and should be utilized when making therapeutic decisions such as administration of SCIG/IVIG. As seen in this and similar CVID-like disorders, it is likely most individuals bearing causative mutations will become symptomatic at some stage in their lives. In this family, the individuals heterozygous for the TNFRSF13B/TACI mutation alone are unlikely to require immunoglobulin replacement in the absence of the concomitant TCF3 mutation.
As in the example of flower color in peas and immunoglobulin production our family, it is likely future examples of digenic inheritance leading to epistasis will involve molecules along the same biochemical pathway (Table 3). In our family, one molecule was a cell surface receptor (TNFRSF13B/TACI) and the second more significant mutation (TCF3) was in a transcription factor (Figure 2). The TCF3 mutation can be considered a “hub” in the epistatic interaction of the two pathways (9). Being a transcription factor, TCF3 influences the expression and function of multiple molecules: activation-induced cystidine deaminase, I14-3-3, and immunoglobulin production (Figure 2) (28). Given its broad range of effects, it is not surprising the TCF3 is the critical mutation in this digenic family and the reason we consider it the epistatic hub. In future cases of epistasis, one molecule is likely to be a transcription factor or a regulatory molecule in a signaling pathway.
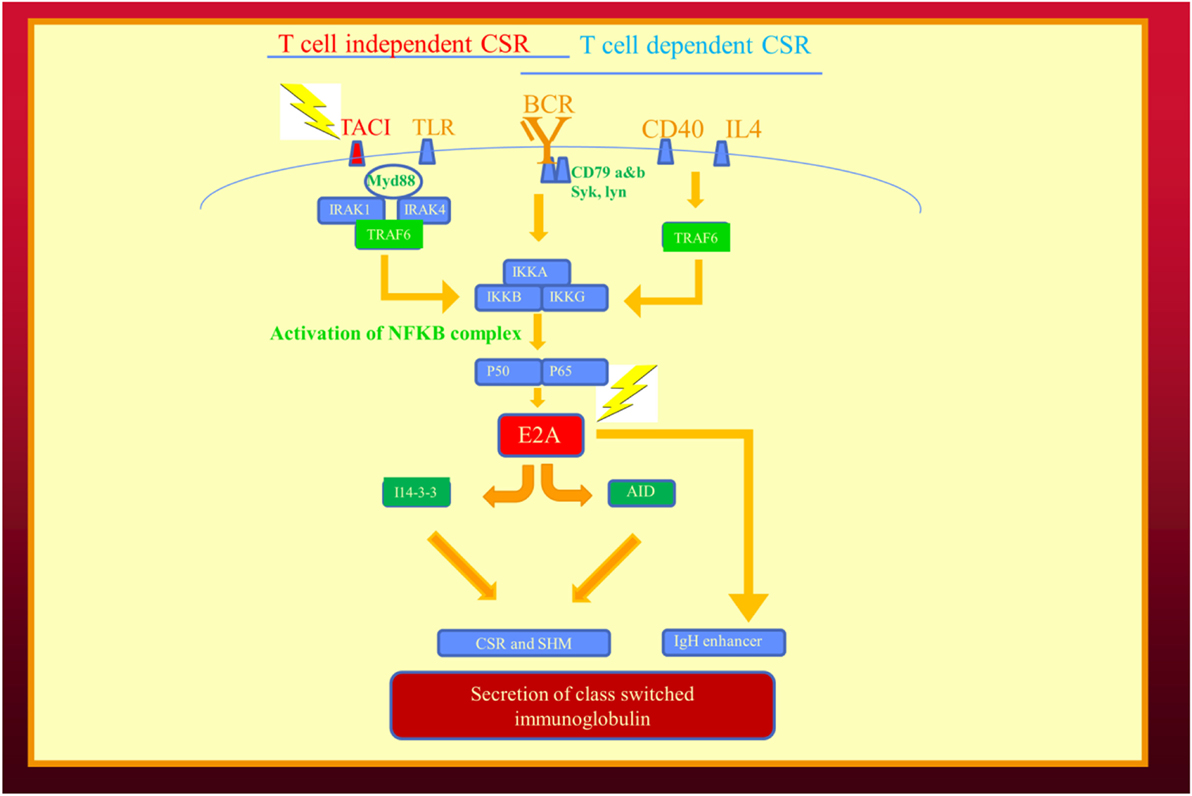
Figure 2. Epistatic interaction of mutated genes in the immunoglobulin isotype switching and secretion pathways from our recent publication describing human epistasis (21). TACI plays a critical role in T cell independent isotype class switching while E2A/(TCF3) affects both pathways. I14-3-3 is a scaffolding protein for activation induced cystidine deaminase. Mutations depicted by lightening. BCR, B cell receptor; CSR, immunoglobulin class switch recombination; SHM, somatic hypermutation; TLR, toll-like receptors.
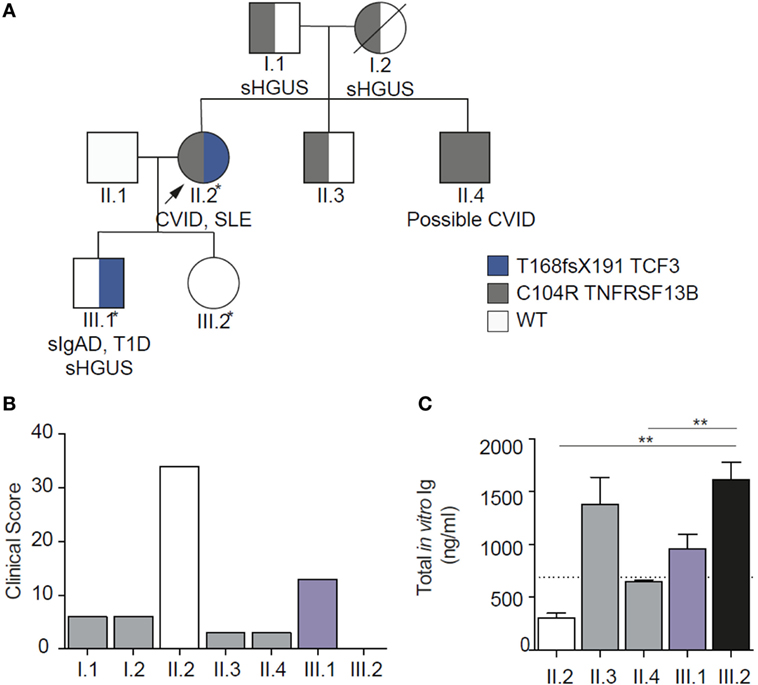
Figure 3. Family pedigree showing genetic, biochemical, and clinical epistasis. (A). Showing the digenic kindred, with inheritance of the two mutations. (B) Clinical epistasis shown with the clinical score. The proband carrying both mutations is much more severely affected than the sum of her symptomatic son [bearing the transcription factor 3 (TCF3) mutation] or any of the other individuals heterozygous for TACI mutation. Note that the clinical score for the unaffected daughter (III.2) is 0. (C) In vitro IgG production through the T cell independent pathway, showing severely impaired in vitro IgG production in the proband carrying both mutations. Diagram modified under the creative commons license (21).
Our findings in this family also justify the segregation of genes predisposing to CVID from those causing a CVID-like disorder (22, 23). We have included genes predisposing to CVID (TNFRSF13B/TACI, TNFRSF13C/BAFFR, TWEAK, and MSH5) in the Ameratunga et al. CVID diagnostic criteria primarily for patients enrolled in clinical research studies (23). As discussed previously, the prevalence of mutations in these predisposing genes is much higher than the lifetime estimates of CVID in the same population (29). We and others have also shown these mutations do not segregate with symptomatic family members (30). This is convincing evidence that these mutations cannot be causative in the absence of other mutated genes. In this digenic family, those individuals bearing the more severe mutation (TCF3) are classified as having CVID-like disorders, while those with TNFRSF13B/TACI mutation remain within the broad spectrum of CVID (31).
Our observation has shed new light on the role of TNFRSF13B/TACI mutations. In this family, it appears that the TNSRSF13B/TACI mutation has a modifying effect on the TCF3 mutation. The phenotype of the TCF3 mutation is exacerbated (negative epistasis) by the presence of the TNFRSF13B/TACI mutation in the proband (category #3, Table 3). We have previously advised against the routine sequencing of genes predisposing to CVID such as TNFRSF13B/TACI, TNFRSF13C/BAFFR, TWEAK, and MSH5 (22). Given the mild influence on phenotype, their role has not been clear. It now appears they may be playing an epistatic role, at least in some patients. Given their newly discovered role, there may be an argument for sequencing these genes, particularly if a more significant genetic defect has already been identified in a family.
This is the first example of digenic inheritance resulting in a CVID-like disorder as a result of epistasis (21). Both mutations are required for the complete phenotypic expression of the disorder in this proband. We have classified this digenic condition as a CVID-like disorder as the genetic basis is understood (22). Such digenic disorders will require a new classification of PIDs as neither mutation alone can cause the full phenotype on its own in this family (21). Similarly, they will require two OMIM gene annotations when identified. A recently curated digenic database will be very helpful in understanding these complex disorders (15). It will be important to document if clinical and biochemical epistasis has been confirmed in an informative digenic family.
A recent retrospective study suggested that up to 5% of patients undergoing NGS had a digenic disorder (32). Other examples of digenic inheritance leading to PIDs are likely to be discovered in the future. As in the family we have described, the initial clue indicating the presence of digenic inheritance will emerge from family studies showing lack of co-segregation between the pattern of symptoms and the inheritance of the initial candidate mutation. It will be important to ensure the phenotypic features of each family member can be satisfactorily explained by the functional effects of the initially identified mutation. If there is a discrepancy in the segregation analysis, there may be another unidentified mutation, as seen in our family. It is likely the first mutation will be identified by Sanger sequencing of a candidate gene (33) and the second mutation will be obtained by NGS-based gene discovery techniques.
In the absence of consanguinity, causal mutations are more likely inherited in an autosomal dominant manner, or arise de novo as seen in the New Zealand family described in our publications (21). This crucial information will inform the curation strategy in NGS, and the NGS data should be used to determine consanguinity in case this knowledge is not known to, or available from the family. The curation strategies for NGS will thus depend on the frequency of consanguinity in the population being surveyed. It will also be critical to understand the frequencies of candidate variants in population(s) representing the proband’s ethnic population. There is a serious risk of assigning disease significance to a common polymorphism in an under-surveyed ethnic group.
If none of the identified candidate genes are known to cause an immunodeficiency in a patient with PID, this will complicate evaluation of the digenic disorder. The role of each gene may have to be determined separately prior to attributing the epistatic effects of both mutations to the phenotype of the proband. There should be no discrepancy when the phenotypic and laboratory effects of each mutation are correlated with the clinical features of each member of the kindred. Within the limits discussed above, reproduction of the orthologous mutation in laboratory animals may support the existence of epistatic interactions between the two genes.
A computer program predicting epistasis in silico will greatly advance the investigation of digenic disorders and might assist in cases where the effects of genes are not completely understood (15). Such programs may attempt to quantify epistatic effects from network perturbation scores, score all pairwise contributions to known pathways, or simply identify pairs of genes acting in the same pathway. Particularly exciting are recent studies that compute tolerance to loss-of-function mutations for each gene from large-scale surveys of human genetic variation (34, 35). These scores may be combined with the approaches above to refine candidate pairs and prioritize follow-up experiments. For example, observing fewer loss-of-function mutations in TCF3 than statistically expected in 60,000 healthy individuals strongly suggests that humans are intolerant to this type of mutation in TCF3. In contrast, the same metric suggests that loss-of-function mutations in TNFRSF13B/TACI are better tolerated (27). The global genome sequencing efforts (now exceeding variants from 250,000 chromosomes) should allow a similar estimation of intolerance to loss-of-function mutations affecting gene pairs.
Investigation of this family has given us insight into previously unanswered questions in human genetics, which were first raised over a century ago. Apart from being a well-characterized example of human epistasis our observation offers a molecular explanation for variable penetrance and expressivity. It has long been speculated that individual genetic variation in modifying genes could be the basis of variable penetrance and expressivity. Other explanations are of course possible including environmental influences and epigenetic changes in methylation patterns leading to altered gene expression (36). Investigation of similar families will not only identify new PID disorders but will fundamentally change our understanding of human heredity.
Ethics Statement
All studies were approved by Auckland Hospital (3435), NZ Ministry of Health (MEC/06/10/134), and the Walter and Eliza Hall Institute (WEHI) Human Research Ethics Committee (HREC 10/02).
Author Contributions
RA conceptualized the article and wrote the first draft. S-TW contributed to molecular biology section in non-human models. VB contributed to the figure and text. Performed functional work in the original paper. RS contributed to conceptualization and helped edit the manuscript. CS contributed to original paper through functional assays. EL undertook practical work on the epistasis project and contributed to the text. KL played a major role in the bioinformatics section.
Conflict of Interest Statement
All authors declare they have no competing interests. All studies were undertaken with the appropriate consent and institutional ethics approvals.
Acknowledgments
We are extremely grateful to this family for allowing us to undertake these studies for the benefit of others. We hope this discovery will benefit them and any future families with digenic disorders. We thank AMRF, A+ Trust, IDFNZ, ASCIA, and the Australian National Health and Medical Research Council (NHMRC, Program Grant 1054925) for grant support. We also receive support from Bloody Long Way (BLW). CS is supported by NHMRC postgraduate scholarship 1075666. We thank Dr. Kitty Croxson, ADHB, and LabPlus management for ongoing support. Bioinformatic analysis was supported by the New Zealand eScience Infrastructure. All studies were approved by Auckland Hospital (3435), NZ Ministry of Health (MEC/06/10/134), and the Walter and Eliza Hall Institute (WEHI) Human Research Ethics Committee (HREC 10/02).
References
1. Bateson W. Discussion on the influence of heredity on disease, with special reference to tuberculosis, cancer, and diseases of the nervous system: introductory address. Proc R Soc Med (1909) 2:22–30.
3. Tanaka Y, Brugliera F. Flower colour and cytochromes P450. Philos Trans R Soc Lond B Biol Sci (2013) 368(1612):20120432. doi:10.1098/rstb.2012.0432
4. Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet (2002) 11:2463–8. doi:10.1093/hmg/11.20.2463
6. Wong A. Epistasis and the evolution of antimicrobial resistance. Front Microbiol (2017) 8:246. doi:10.3389/fmicb.2017.00246
7. Boone C, Bussey H, Andrews BJ. Exploring genetic interactions and networks with yeast. Nat Rev Genet (2007) 8:437–49. doi:10.1038/nrg2085
8. Moffat J, Sabatini DM. Building mammalian signalling pathways with RNAi screens. Nat Rev Mol Cell Biol (2006) 7:177–87. doi:10.1038/nrm1860
9. Ehrenreich IM. Epistasis: searching for interacting genetic variants using crosses. G3 (Bethesda, Md) (2017) 7:1619–22. doi:10.1534/g3.117.042770
10. Bacchelli C, Buckland KF, Buckridge S, Salzer U, Schneider P, Thrasher AJ, et al. The C76R transmembrane activator and calcium modulator cyclophilin ligand interactor mutation disrupts antibody production and B-cell homeostasis in heterozygous and homozygous mice. J Allergy Clin Immunol (2011) 127(1253–9):e13. doi:10.1016/j.jaci.2011.02.037
11. Ameratunga R, Bartlett A, McCall J, Steele R, Woon ST, Katelaris CH. Hereditary angioedema as a metabolic liver disorder: novel therapeutic options and prospects for cure. Front Immunol (2016) 7:547. doi:10.3389/fimmu.2016.00547
12. Massaad MJ, Zhou J, Tsuchimoto D, Chou J, Jabara H, Janssen E, et al. Deficiency of base excision repair enzyme NEIL3 drives increased predisposition to autoimmunity. J Clin Invest (2016) 126:4219–36. doi:10.1172/JCI85647
13. Ameratunga R, Woon ST, Brewerton M, Koopmans W, Jordan A, Brothers S, et al. Primary immune deficiency disorders in the South Pacific: the clinical utility of a customized genetic testing program in New Zealand. Ann N Y Acad Sci (2011) 1238:53–64. doi:10.1111/j.1749-6632.2011.06238.x
14. Ameratunga R, Woon ST, Neas K, Love DR. The clinical utility of molecular diagnostic testing for primary immune deficiency disorders: a case based review. Allergy Asthma Clin Immunol (2010) 6:12. doi:10.1186/1710-1492-6-12
15. Gazzo AM, Daneels D, Cilia E, Bonduelle M, Abramowicz M, Van Dooren S, et al. DIDA: a curated and annotated digenic diseases database. Nucleic Acids Res (2016) 44:D900–7. doi:10.1093/nar/gkv1068
16. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol (2014) 5:162. doi:10.3389/fimmu.2014.00162
17. Fliegauf M, Bryant VL, Frede N, Slade C, Woon ST, Lehnert K, et al. Haploinsufficiency of the NF-κB1 subunit p50 in common variable immunodeficiency. Am J Hum Genet (2015) 97:389–403. doi:10.1016/j.ajhg.2015.07.008
18. Grimbacher B, Hutloff A, Schlesier M, Glocker E, Warnatz K, Dräger R, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol (2003) 4:261–8. doi:10.1038/ni902
19. van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJ, van Tol MJ, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med (2006) 354:1901–12. doi:10.1056/NEJMoa051568
20. van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest (2010) 120:1265–74. doi:10.1172/JCI39748
21. Ameratunga R, Koopmans W, Woon ST, Leung E, Lehnert K, Slade CA, et al. Epistatic interactions between mutations of TACI (TNFRS13B) and TCF3 result in a severe primary immunodeficiency disorder and systemic lupus erythematosus. Clin Trans Immunol (2017) 6(10):e159. doi:10.1038/cti.2017.41
22. Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol (2014) 5:415. doi:10.3389/fimmu.2014.00415
23. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol (2013) 174:203–11. doi:10.1111/cei.12178
24. Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions on immunoglobulin replacement. J Allergy Clin Immunol (2013) 131:1699–701. doi:10.1016/j.jaci.2013.01.036
25. Hoyos-Bachiloglu R, Chou J, Sodroski CN, Beano A, Bainter W, Angelova M, et al. A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J Clin Invest (2017). doi:10.1172/JCI93486
26. Garibyan L, Lobito AA, Siegel RM, Call ME, Wucherpfennig KW, Geha RS. Dominant-negative effect of the heterozygous C104R TACI mutation in common variable immunodeficiency (CVID). J Clin Invest (2007) 117:1550–7. doi:10.1172/JCI31023
27. Martinez-Gallo M, Radigan L, Almejun MB, Martinez-Pomar N, Matamoros N, Cunningham-Rundles C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J Allergy Clin Immunol (2013) 131:468–76. doi:10.1016/j.jaci.2012.10.029
28. Quong MW, Harris DP, Swain SL, Murre C. E2A activity is induced during B-cell activation to promote immunoglobulin class switch recombination. EMBO J (1999) 18:6307–18. doi:10.1093/emboj/18.22.6307
29. Pan-Hammarstrom Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet (2007) 39:429–30. doi:10.1038/ng0407-429
30. Koopmans W, Woon ST, Brooks AE, Dunbar PR, Browett P, Ameratunga R. Clinical variability of family members with the C104R mutation in transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI). J Clin Immunol (2013) 33:68–73. doi:10.1007/s10875-012-9793-x
31. Ameratunga R, Lehnert K, Woon ST, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol (2017). doi:10.1007/s12016-017-8645-0
32. Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med (2017) 376:21–31. doi:10.1056/NEJMoa1516767
33. Woon ST, Ameratunga R. Comprehensive genetic testing for primary immunodeficiency disorders in a tertiary hospital: 10-year experience in Auckland, New Zealand. Allergy Asthma Clin Immunol (2016) 12:65. doi:10.1186/s13223-016-0169-2
34. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature (2016) 536:285–91. doi:10.1038/nature19057
35. Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet (2016) 48:1107–11. doi:10.1038/ng.3638
Keywords: common variable immunodeficiency disorders, epistasis, digenic mutation, primary immunodeficiency disorder, monogenic syndromes
Citation: Ameratunga R, Woon S-T, Bryant VL, Steele R, Slade C, Leung EY and Lehnert K (2018) Clinical Implications of Digenic Inheritance and Epistasis in Primary Immunodeficiency Disorders. Front. Immunol. 8:1965. doi: 10.3389/fimmu.2017.01965
Received: 03 November 2017; Accepted: 19 December 2017;
Published: 26 January 2018
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Amos Etzioni, University of Haifa, IsraelMichail Lionakis, National Institute of Allergy and Infectious Diseases (NIH), United States
Copyright: © 2018 Ameratunga, Woon, Bryant, Steele, Slade, Leung and Lehnert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rohan Ameratunga, cm9oYW5hQGFkaGIuZ292dC5ueg==