Abstract
Fungal biofilms are communities of adherent cells surrounded by an extracellular matrix. These biofilms are commonly found during infection caused by a variety of fungal pathogens. Clinically, biofilm infections can be extremely difficult to eradicate due to their resistance to antifungals and host defenses. Biofilm formation can protect fungal pathogens from many aspects of the innate immune system, including killing by neutrophils and monocytes. Altered immune recognition during this phase of growth is also evident by changes in the cytokine profiles of monocytes and macrophages exposed to biofilm. In this manuscript, we review the host response to fungal biofilms, focusing on how these structures are recognized by the innate immune system. Biofilms formed by Candida, Aspergillus, and Cryptococcus have received the most attention and are highlighted. We describe common themes involved in the resilience of fungal biofilms to host immunity and give examples of biofilm defenses that are pathogen-specific.
Introduction
Fungi frequently flourish as biofilms, which are aggregated communities encased in a protective extracellular matrix (1, 2). Many clinically relevant fungi have been shown to form biofilms, such as Candida spp., Aspergillus spp., Cryptococcus neoformans, Fusarium spp., Blastoschizomyces capitatus, Malassezia pachydermatis, Pneumocystis spp., Trichosporon asahii, Rhizopus spp., and Rhizomucor spp (3–13). In the clinical setting, fungal biofilms can propagate on artificial medical devices, such as catheters, as well as on epithelial and endothelial surfaces (3, 14–19) (Figure 1A). In addition, during invasive infection, fungal pathogens can proliferate as non-surface associated microcolonies embedded in extracellular matrix (18) (Figure 1B). Biofilms resist antifungal therapies and host defenses, making them notoriously difficult to eradicate (4, 20–36). One defining trait of biofilm formation is the production of a microbial-produced extracellular matrix, or the “glue” necessary for adhesion, which also serves as a shield that creates protected reservoirs of infection (4, 5, 20, 33, 37). As investigations reveal the complex composition of the extracellular matrix for several fungal pathogens, it has become increasing clear that this material is distinct from the cell wall (4, 5, 18, 38–41). Therefore, biofilm growth provides a means to present unique moieties and conceal cell wall ligands. Studies are just beginning to shed light on how biofilm formation and matrix production influence host recognition (5, 15, 27, 29–32, 42–50). In this review, we examine the innate immune response to fungal biofilms, highlighting how production of the extracellular matrix alters immunity. While a variety of fungal pathogens have been shown to produce biofilms, investigations examining host interactions have primarily utilized model pathogens Candida albicans, Aspergillus fumigatus, and C. neoformans, which will be the focus of our discussion.
Figure 1
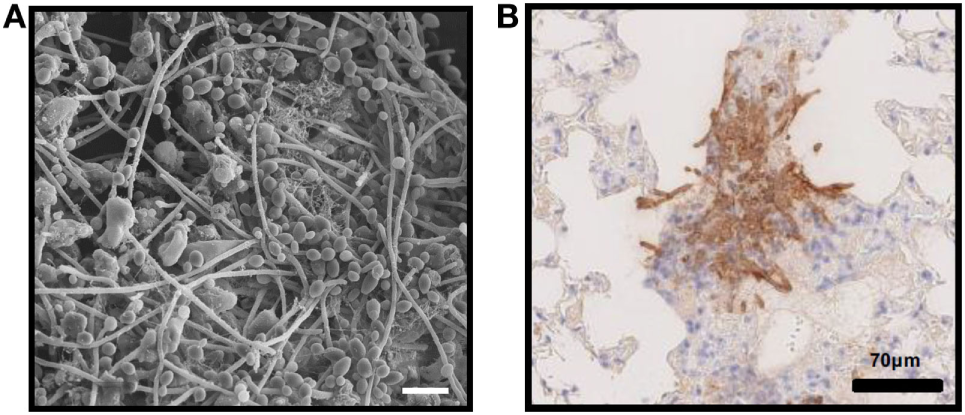
In vivo fungal biofilms. (A)Candida albicans biofilm growing on the luminal surface of a rat venous catheter for 24 h. Scanning electron microscopy reveals adherent organisms growing within in an extracellular matrix. (B) Immunohistochemistry of pulmonary tissue from an immunocompromised mouse infected with Aspergillus fumigatus and stained with an anti-galactosaminogalactan antibody. Brown indicates accumulation of galactosaminogalactan-containing biofilm matrix surrounding hyphae growing within pulmonary tissues.
Candida Biofilms
Biofilm Formation
Candida spp., commensal fungi of the gastrointestinal tract, can cause severe, disseminated disease with high mortality, particularly in patients with implanted medical devices or compromised immune systems (17, 51–57). The vast majority of these infections involve biofilm formation on either an artificial or a biotic surface (13, 58, 59) (Figure 2). Clinical Candida biofilms grow on diverse medical devices, including central venous catheters, urinary catheters, prosthetic valves, left ventricular assist devices, and oral devices, such as dentures (3, 60). Both vaginal and oral mucosal surfaces promote biofilm formation as well (16, 61). The majority of the in vitro and in vivo biofilm studies have utilized C. albicans, the most widespread species. However, non-albicans species, including Candida tropicalis, Candida parapsilosis, and Candida glabrata, similarly produce clinically relevant biofilms (3, 62–68). In addition, this virulence trait has been described for the emerging pathogen Candida auris (69). Candida biofilm formation involves the adherence of yeast to a substrate, the proliferation of cells to form a fungal community, and the production of an extracellular matrix (37, 70–72). C. albicans biofilm development often involves the production of hyphae, although the degree of filamentation varies among strains and niches. Non-albicans strains lacking the ability to filament produce biofilms composed of layers of yeast embedded in an extracellular matrix (73).
Figure 2
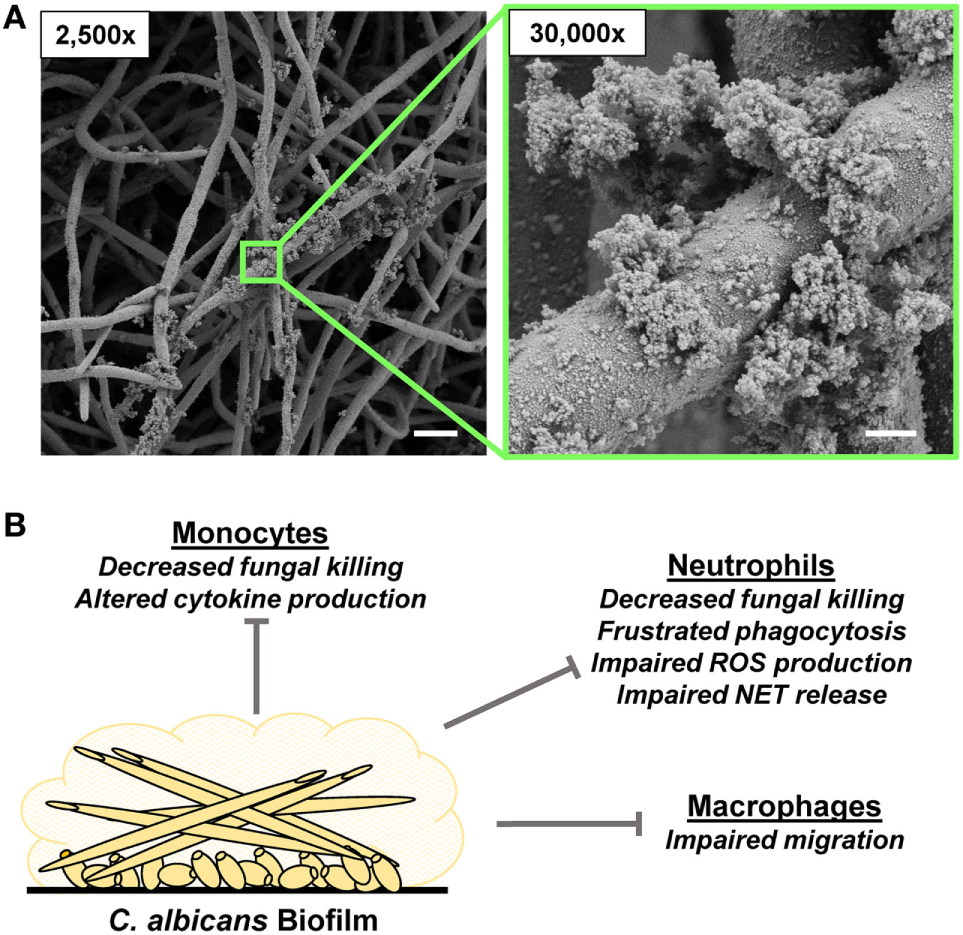
Candida albicans biofilm formation and innate immune response. (A) Scanning electron microscopy images reveal C. albicans biofilms grown on coverslips. Biofilms were grown for 48 h. Measurement bars represent 10 and 1 µm for 2,500× and 30,000×, respectively. (B) Summary of innate immune responses impaired by C. albicans biofilms.
Matrix Composition
Upon encounter with biofilm, immune cells are first confronted with the extracellular matrix covering the fungal cells. A combination of both in vitro and in vivo models has been integral for the dissection of Candida biofilm matrix assembly and composition (20, 22, 39, 40, 58, 67, 70, 74–79). For C. albicans, in vitro studies have revealed that the mature biofilm matrix consists of a variety of macromolecules, including protein (55%), carbohydrate (25%), lipid (15%), and DNA (5%) (23, 38, 40, 80). Interestingly, many of the matrix components vastly differ from the cell wall components that would be initially recognized by immune cells in the absence of biofilm (40). For example, the main polysaccharide of C. albicans biofilm matrix is α-1,2-branched α-1,6 mannan, which is found in high molecular weight structures of approximately 12,000 residues. In contrast, mannans of the outer cell wall layer are 5- to 10-fold smaller. Furthermore, the matrix mannans associate with β-1,6 glucans, forming a mannan–glucan complex that assembles extracellularly, and this structure has not been identified in the cell wall of C. albicans (40, 75). In addition, matrix β-1,6 glucan exists in a linear conformation, while the β-1,6 glucan of the cell wall is highly branched (81). Proteomic analysis of in vitro biofilms shows some similarities between matrix-associated proteins and those released into the media during planktonic growth (79). However, the extracellular matrix lacks many of the proteins associated with the cell wall (40, 82). In vivo, host proteins contribute to the construction of biofilm, with an astonishing majority (>95%) of the matrix-associated proteins of host origin (76). This finding demonstrates variation in the content of fungal biofilm matrix in vivo and highlights the importance of including animal models for investigation of Candida biofilms.
Neutrophil–Candida Biofilm Interactions
Neutrophils are primary responders to C. albicans infection, with neutropenic patients prone to severe, lethal candidiasis (83–86). Neutrophils respond to chemokines and other signals during recruitment to the site of infection. Upon pathogen encounter, neutrophils elicit various responses important for control of infection, including phagocytosis, degranulation, reactive oxygen species (ROS) production, and neutrophil extracellular trap (NET) release (87). In the context of C. albicans biofilms, neutrophils are the primary leukocyte recruited to the site of infection (15, 76, 88–90). In fact, neutrophil recruitment has been observed in animal models mimicking diverse clinical biofilms, including catheter-related infections (vascular and urinary), denture stomatitis, oral candidiasis, and vaginal candidiasis (15, 76, 91). Despite their presence at the site of infection, neutrophils fail to eradicate Candida biofilms. Pioneering studies with human neutrophils revealed that C. albicans biofilms resist neutrophil attack, in comparison to their planktonic counterparts (29, 30). Intact biofilm structure is required for this resistance, as resuspension of the biofilm cells reverses the phenotype (29). Furthermore, biofilm impairment of neutrophils is robust, persisting despite neutrophil priming by IFN-γ or G-CSF (92).
The ability of Candida biofilms to withstand immune attack appears to vary by strain and species. For C. albicans, biofilms are approximately twofold to fivefold more resistant to killing when compared to planktonic cells (29–31, 92). Investigation of C. parapsilosis found a similar trend, but did not identify a significant difference in susceptibility to neutrophils between biofilms and planktonic organisms (43, 93). The lack of statistical significance was attributed to the heterogeneity of biofilm formation, resulting in high assay variability. A recent investigation found C. glabrata biofilms also resist neutrophil killing, exhibiting a threefold higher resistance for biofilm over planktonic organisms (45).
For killing and containment of C. albicans, neutrophils release NETs, which are structures of DNA, histones, and antimicrobial proteins (94, 95). These structures are particularly well-suited to combat large organisms, such as hyphae, which are unable to be ingested by phagocytosis (95). As C. albicans biofilms consist of aggregated cells and hyphal elements, NETosis would seemingly be an efficient method of attack. However, a recent study has revealed that neutrophils fail to release NETs in the presence of C. albicans biofilms (31). This phenomenon is conserved across a variety of C. albicans strains exhibiting differing degrees of filamentation and biofilm architecture (44). This inhibitory pathway appears to be closely linked to the production of an extracellular matrix, as physical or genetic disruption of this process restores NET release (31). Remarkably, when neutrophils are induced to generate NETs prior to biofilms exposure, biofilm inhibition is observed (31). This suggests that inhibition of NETosis is an adaptation by C. albicans biofilms to prevent killing by neutrophils. Other species appear to employ this mechanism as well. For example, C. glabrata biofilms also impair NET release, although the inhibition is not as pronounced (45).
Recent studies have begun to shed light on the planktonic C. albicans cell surface components that induce NET release. The process appears to be multifactorial, as β-glucan, mannan, and secreted aspartic proteases all variably trigger NETosis (93, 96, 97). NET inhibition by biofilm likely involves concealment of cell surface ligands by the extracellular matrix, as disruption of this process permits NET release (31). In particular, disruption of the matrix mannan–glucan complex in a pmr1Δ/Δ mutant strain reverses the NET inhibition phenotype, suggesting a role for this unique polysaccharide complex (31, 75). In addition, studies by Zawrotniak et al. show that NET induction by cell wall mannan is concentration-dependent, with higher concentrations failing to trigger NETosis in vitro (97). Further investigation would be of interest to explore a similar pattern for polysaccharides of the biofilm matrix in NET inhibition.
Upon encounter with C. albicans biofilms, the generation of ROS by neutrophils is dampened compared to the response observed for planktonic organisms (30, 31, 44). Multiple pathways govern NET release and a subset of these depend on ROS production (96, 98–103). In response to planktonic C. albicans, both ROS-dependent and ROS-independent pathways trigger NETosis, which may not be surprising given the numerous cell surface ligands expected to be involved (96, 97, 103). While further investigation is needed to dissect pathways impairing neutrophil function by biofilms, inhibition of ROS production is likely involved. A study of C. glabrata–neutrophil interactions demonstrates a similar neutrophil response, with reduced ROS production upon encounter with biofilm (45). Taken together, these studies show that C. albicans biofilms inhibit the release of NETs and resist killing by neutrophils. The pathway appears to involve the production of an extracellular matrix and dampening of neutrophil ROS production. Based on studies with C. glabrata, it may be conserved, in part, among Candida spp.
Monocytes and Macrophages Interactions with Candida Biofilms
Chandra et al. first demonstrated that Candida biofilms resist attack by monocytes and can alter their cytokine profile (42). Peripheral blood mononuclear cells (PBMCs) fail to phagocytose biofilm-associated C. albicans, in contrast to planktonic organisms (42). However, these cells remain viable, migrating within the biofilm, even providing a stimulus for biofilm proliferation through an unknown mechanism (29, 42). Compared to planktonic organisms, C. albicans biofilms are twofold to threefold more resistant to killing by monocytes (29).
Encounter with Candida biofilms influences cytokine release by mononuclear cells. One of the more intriguing alterations is the downregulation of TNF-α, a cytokine which facilitates phagocyte activation. Compared to planktonic organisms, exposure to C. albicans biofilms significantly diminishes the production of TNF-α by monocytic cell line THP-1 (29). Not only is this predicted to impact phagocyte function in the host but the alteration in production of TNF-α may also have a direct impact on the biofilm. Application of exogenous TNF-α has been shown to prevent C. albicans biofilm formation, through a TNF receptor-independent pathway (104). Furthermore, this activity is blocked by preincubation of TNF-α with N,N′-diacetylchitobiose, a major carbohydrate component of C. albicans cell wall (104). Therefore, inhibition of TNF-α by biofilms may represent an evolutionary adaption and mechanism of immune evasion. However, much remains a mystery about how the cytokine response influences the host response to biofilm infection. For example, when compared to planktonic cells, PBMCs exposed to C. albicans biofilms produce elevated levels of IL-1β, IL-10, and MCP-1 and reduced levels of IL-6 and MIP1β (42). How these combinations of both pro- and anti-inflammatory cytokines are triggered and their influence on host response to biofilm infection is unknown.
Recent work by Alonso et al. revealed that formation of C. albicans biofilm impairs the migratory capacity of macrophages (105). Upon exposure to biofilm, the migration of murine macrophages (J774.1 cell line) is reduced approximately twofold when compared to encounter with planktonic organisms. A pmr1Δ/Δ mutant similarly impaired macrophage migration during biofilm growth. As this biofilm is deficient in matrix mannan production, the macrophage inhibition is likely related to another factor, such as physical structure (75, 105). Therefore, Candida biofilms may elicit distinct inhibitory pathways for neutrophils and macrophages (31, 105).
Aspergillus Biofilms
Biofilm Formation
Aspergillus spp. grow ubiquitously in the environment and individuals are constantly exposed to their spores, which are released into the air (106). Immunocompetent individuals clear these spores after inhalation, but those with impaired immunity are at risk for development of severe disease. Aspergillus spp. can cause a variety of clinical diseases, including invasive, chronic, and allergic forms (106). The chronic form of disease typically involves formation of an aspergilloma, or fungal ball, in the sinus or lung cavity. These dense structures consist of agglutinated hyphae with occasional conidial heads growing as a biofilm encased in an adhesive extracellular matrix (18) (Figure 3). As the community matures, the inner cells loose viability, likely due to starvation. A. fumigatus also produces extracellular matrix material during invasive pulmonary aspergillosis (18). However, during this mode of growth, the hyphae remain separated without an inner core of decaying fungal mass. In vitro, A. fumigatus forms biofilms on agar medium in aerial, static conditions that mimic the host niches for aspergilloma formation (4).
Figure 3

Aspergillus fumigatus biofilm formation and innate immune response. (A) Scanning electron microscopy images reveal A. fumigatus biofilms grown on coverslips. Biofilms were grown for 24 h. Measurement bars represent 10 and 1 µm for 2,500× and 30,000×, respectively. (B) Summary of innate immune responses impaired by A. fumigatus biofilms.
Matrix Production
Aspergillus fumigatus produces a unique extracellular matrix during biofilm growth in vitro and in vivo (4, 18, 107). By biochemical analysis, this material consists of 40% protein, 43% carbohydrates, 14% lipids, and 3% aromatic-containing compounds, as well as DNA (108–110). The polysaccharides of the extracellular matrix exhibit cohesion properties and provide immune protection. The main matrix polysaccharides include galactomannan and galactosaminogalactan (GAG), of which, GAG has received the most attention (18). A. fumigatus strains deficient in GAG production lack the capacity to form biofilms or produce extracellular matrix (47). GAG is an α-1,4-linked linear heteroglycan composed of variable combinations of galactose and N-acetyl-galactosamine (GalNAc) (111, 112). GalNAc residues within the GAG polymer are partially deacetyated by the secreted enzyme Agd3, rendering mature GAG polycationic (113). Deacetylation is required for GAG to mediate adhesion between hyphae and other anionic surfaces such as host cells, plastic, and glass (113). GAG production has also been reported for other Aspergillus spp., including Aspergillus parasiticus, Aspergillus niger, and Aspergillus nidulans, although the relative proportion of galactose and GalNAc varies between strains and likely influences matrix function as detailed below (114–117). While galactomannan and GAG are universally present in A. fumigatus matrix, key differences exist between the clinical niche biofilms. For example, aspergilloma biofilms produce a thicker extracellular matrix, which contains α-glucan, a polysaccharide absent in biofilms formed during invasive aspergillosis (18). Aspergilloma biofilms also produce melanin, an immune modulator (4, 18, 33, 118). However, its specific role during biofilm formation is unknown.
Innate Immunity to Aspergillus Biofilms
Neutrophils are key players in the innate immune defense against Aspergillus. Neutropenia, often in the face of chemotherapy or hematologic malignancy, places patients at high risk factor for invasive pulmonary aspergillosis, which can progress to disseminated lethal disease (119, 120). Neutrophils are recruited to Aspergillus spores in vivo and are critical for their engulfment through phagocytosis (121–123). Neutrophils also release NETs in response to hyphal elements (46, 124–126). While NETs lack significant activity against conidia, they exhibit modest inhibitory activity against the larger hyphal forms of A. fumigatus (46, 126). Production of an extracellular matrix shields A. fumigatus from neutrophil attack and much of this protection is attributed to GAG (32). A. fumigatus mutants deficient in GAG synthesis or deacetylation exhibit attenuated virulence (47, 113), and treatment with recombinant glycoside hydrolases that degrade GAG reduces fungal growth in a murine model of aspergillosis (127).
Studies of differences in GAG composition between A. fumigatus and A. nidulans have suggested that protection against neutrophil attack is mediated by hyphal-associated GalNAc-rich GAG (32). Unlike A. fumigatus, A. nidulans produces GalNAc-poor GAG, which contains over fivefold higher levels of galactose and produces poorly adherent biofilms containing minimal extracellular matrix. A. nidulans is also less virulent in a murine model of pulmonary aspergillosis and more than twofold more susceptible to killing by human neutrophils. Heterologous expression of the A. fumigatus uge3 gene encoding a GalNAc-epimerase in A. nidulans results in the production of A. fumigatus-like GalNAc-rich GAG (32). Unlike wild-type A. nidulans, the GalNAc-rich GAG-producing strain of A. nidulans forms biofilm, produces extracellular matrix, and resists killing by neutrophils. The protective effects of GAG are dependent on NADPH oxidase and likely involve defense against NETs released through activation of this pathway. It has been hypothesized that GAG-mediated protection against NETs is mediated by electrostatic repulsion between this partially deacetylated cationic polysaccharide and cationic antimicrobial peptides or histones contained within NETs (32).
The immunomodulatory effects of GAG during biofilm formation are likely multifactorial. Hyphal-associated GAG masks β-glucans on the cell wall of hyphae (47, 128) and alters recognition by murine bone marrow-derived dendritic cells in vitro (47). Genetic disruption of GAG synthesis leads to increased pro-inflammatory cytokine release through Dectin-1 signaling (47). In a non-neutropenic murine model of pulmonary aspergillosis, genetic disruption of GAG synthesis results in production of a non-protective, hyper-inflammatory response marked by increased neutrophil recruitment (47). This observation suggests that GAG impairs neutrophil recruitment during biofilm growth and is consistent with the reports that soluble GAG can modulate immunity through induction of apoptosis in neutrophils and stimulation of anti-inflammatory IL-1Ra production by macrophages in vitro (49, 129). Further, in vivo studies are needed to evaluate the relative role of these functions of GAG in invasive and chronic Aspergillus infections.
Recent studies have begun to shed light on the mechanisms involved in NET release in response to fungi (32, 46, 93, 97, 124–126, 130). Specific ligands triggering this response to Aspergillus remain largely unknown, and how the extracellular matrix may influence these pathways is of great interest. For example, NET production is reduced in response to resting conidia when compared to hyphae (46). This inhibition is linked to RodA, a hydrophobin on the surface of conidia that masks pathogen-associated molecular patterns, including β-glucan (46, 48). As transcriptional analysis shows abundance of RodA during A. fumigatus biofilm growth when compared to planktonic conditions, it is interesting to postulate a role for RodA production in immune evasion during biofilm growth (131). Also, as melanin production has been described for some Aspergillus biofilms, investigation of a role for this immune modulator is also intriguing (4, 18, 33, 118).
Cryptococcus Biofilms
Biofilm Formation
Cryptococcus spp. are opportunistic environmental fungal pathogens that cause life-threatening meningoencephalitis, particularly in patients with suppressed immunity in the setting of HIV or organ transplantation (132). Following inhalation of spores from the environment, C. neoformans disseminates from the lungs, with a propensity for the central nervous system. C. neoformans also exhibits a predilection for artificial surfaces and forms biofilms on medical devices, such as cerebrospinal fluid shunts, vascular catheters, and prosthetic dialysis fistulae (133–136). These adherent communities are composed of yeast encased in an extracellular matrix (5, 41, 137). In vitro, C. neoformans biofilms mature in 24–48 h and display a multiple-drug-resistance phenotype (5).
Matrix Production
Cryptococcus neoformans produces a protective polysaccharide capsule composed of glucuronoxylomannan (GXM), galactoxylomannan, and mannoprotein (137, 138). During biofilm growth, these capsular polysaccharides are shed into the surrounding milieu, ultimately providing extracellular matrix material for surface adhesion and cell–cell cohesion (5). Acapsular C. neoformans mutants are unable to form biofilms (5). Martinez and Casadevall identified GXM as the principle polysaccharide of the C. neoformans biofilm matrix (41). This polysaccharide has received the most attention due to its immunomodulatory properties and high abundance in the biofilm matrix (5, 138–141). However, biochemical analysis also shows the presence of sugars not found in GXM, suggesting that the biofilm matrix contains additional polysaccharides (41). Little is known about the structure of these polysaccharides and how they may influence immunity to Cryptococcus biofilms.
Innate Immunity to Cryptococcus Biofilms
While studies have begun to dissect the impact of biofilm formation on immunity to Cryptococcus, much of this host–fungal interaction remains a mystery. Production of a GXM-rich extracellular matrix appears to be the key defense against host immunity. Genetic or antibody-mediated disruption of GXM impairs biofilm formation and diminishes virulence (5, 141). As a capsule polysaccharide, GXM is responsible for a multifaceted inhibition of neutrophil function, impeding chemotaxis, phagocytosis, NET production, and antifungal activity (138, 139, 142, 143). Similar mechanisms of diminished neutrophil function are anticipated in response to C. neoformans biofilms and may even be augmented given the high GXM content of biofilm matrix (41). Furthermore, capsular GXM can impair phagocytosis by monocytes and macrophages (138). However, it is unknown if phagocytosis would be an effective response against Cryptococcus biofilms, given the large structure of cohesive, aggregated yeast (41).
In addition to the immunomodulatory activity of the extracellular matrix, C. neoformans biofilms also resist antimicrobial host defenses. Compared to planktonic C. neoformans, biofilms tolerate higher concentrations of defensins, including PG-1, β-defensin-1, and β-defensin-3 (27). This resistance is even further augmented when biofilms are induced to produce melanin through l-Dopa supplementation. Biofilm formation also protects C. neoformans from oxidative stress induced by a variety of stimuli (27). Taken together, these studies show that Cryptococcus biofilms withstand innate immunity through both immune modulation and resistance to immune attack.
Conclusion
Adoption of a biofilm lifestyle during fungal infection is increasingly recognized as a mechanism to avoid host immune attack and provide a protective niche. In this environment, the extracellular matrix can shield the fungal cell wall from host cellular recognition, modulating the immune response. In addition, the extracellular matrix can provide protection from antimicrobial defenses, such as defensins, oxidative stress, and NETs. Furthermore, biofilm formation produces an aggregated community that may resist engulfment by phagocytosis.
While it is clear that biofilm formation significantly influences immunity, studies are just beginning to shed light on the many mechanisms underlying this modulation of host response. As biofilms are heterogeneous structures with variations in architecture and composition based on their environmental niche, mechanisms impairing immunity likely vary among clinical biofilms. Therefore, inclusion of conditions closely mimicking the host and animal models of biofilm infection remains critical for future studies. While recent studies have revealed the influence of biofilm formation on the innate immune response, still little is known about how these structures may modulate adaptive immunity.
Fungal biofilms are among the most difficult infections to treat due to their high tolerance of antifungals and immune evasion strategies. The incidence of fungal biofilm infections is likely to rise given the growing number of patients with artificial medical devices and immunocompromising conditions. Anti-biofilm therapies are urgently needed. Understanding the dynamics of biofilm formation, matrix production, and how these processes induce resistance to multiple facets of the innate immune system may lead to biofilm-specific antifungal strategies.
Statements
Author contributions
JK, DS, and JN wrote the manuscript. JK and BS constructed the figures.
Funding
JN is supported by the National Institutes of Health (K08 AI108727), the Burroughs Wellcome Fund (1012299), and the Doris Duke Charitable Foundation (112580130). BS has been supported by graduate scholarships from CFC and CIHR. DS is supported by a Research Chair from the Fonds de Recherche Quebec Santé.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
DonlanRM. Biofilm formation: a clinically relevant microbiological process. Clin Infect Dis (2001) 33(8):1387–92.10.1086/322972
2
HoyleBDJassJCostertonJW. The biofilm glycocalyx as a resistance factor. J Antimicrob Chemother (1990) 26(1):1–5.10.1093/jac/26.1.1
3
KojicEMDarouicheRO. Candida infections of medical devices. Clin Microbiol Rev (2004) 17(2):255–67.10.1128/CMR.17.2.255-267.2004
4
BeauvaisASchmidtCGuadagniniSRouxPPerretEHenryCet alAn extracellular matrix glues together the aerial-grown hyphae of Aspergillus fumigatus. Cell Microbiol (2007) 9(6):1588–600.10.1111/j.1462-5822.2007.00895.x
5
MartinezLRCasadevallA. Specific antibody can prevent fungal biofilm formation and this effect correlates with protective efficacy. Infect Immun (2005) 73(10):6350–62.10.1128/iai.73.10.6350-6362.2005
6
ImamuraYChandraJMukherjeePKLattifAASzczotka-FlynnLBPearlmanEet alFusarium and Candida albicans biofilms on soft contact lenses: model development, influence of lens type, and susceptibility to lens care solutions. Antimicrob Agents Chemother (2008) 52(1):171–82.10.1128/aac.00387-07
7
D’AntonioDParrutiGPontieriEDi BonaventuraGManzoliLSferraRet alSlime production by clinical isolates of Blastoschizomyces capitatus from patients with hematological malignancies and catheter-related fungemia. Eur J Clin Microbiol Infect Dis (2004) 23(10):787–9.10.1007/s10096-004-1207-4
8
CannizzoFTErasoEEzkurraPAVillar-VidalMBolloECastellaGet alBiofilm development by clinical isolates of Malassezia pachydermatis. Med Mycol (2007) 45(4):357–61.10.1080/13693780701225767
9
CushionMTCollinsMSLinkeMJ. Biofilm formation by Pneumocystis spp. Eukaryot Cell (2009) 8(2):197–206.10.1128/ec.00202-08
10
Di BonaventuraGPompilioAPiccianiCIezziMD’AntonioDPiccolominiR. Biofilm formation by the emerging fungal pathogen Trichosporon asahii: development, architecture, and antifungal resistance. Antimicrob Agents Chemother (2006) 50(10):3269–76.10.1128/aac.00556-06
11
SinghRShivaprakashMRChakrabartiA. Biofilm formation by zygomycetes: quantification, structure and matrix composition. Microbiology (2011) 157(Pt 9):2611–8.10.1099/mic.0.048504-0
12
SunYChandraJMukherjeePSzczotka-FlynnLGhannoumMAPearlmanE. A murine model of contact lens-associated Fusarium keratitis. Invest Ophthalmol Vis Sci (2010) 51(3):1511–6.10.1167/iovs.09-4237
13
NobileCJJohnsonAD. Candida albicans biofilms and human disease. Annu Rev Microbiol (2015) 69:71–92.10.1146/annurev-micro-091014-104330
14
EscandeWFayadGModineTVerbruggeEKoussaMSennevilleEet alCulture of a prosthetic valve excised for streptococcal endocarditis positive for Aspergillus fumigatus 20 years after previous A. fumigatus endocarditis. Ann Thorac Surg (2011) 91(6):e92–3.10.1016/j.athoracsur.2011.01.102
15
Dongari-BagtzoglouAKashlevaHDwivediPDiazPVasilakosJ. Characterization of mucosal Candida albicans biofilms. PLoS One (2009) 4(11):e7967.10.1371/journal.pone.0007967
16
HarriottMMLillyEARodriguezTEFidelPLJrNoverrMC. Candida albicans forms biofilms on the vaginal mucosa. Microbiology (2010) 156(Pt 12):3635–44.10.1099/mic.0.039354-0
17
GangulySMitchellAP. Mucosal biofilms of Candida albicans. Curr Opin Microbiol (2011) 14(4):380–5.10.1016/j.mib.2011.06.001
18
LoussertCSchmittCPrevostMCBalloyVFadelEPhilippeBet alIn vivo biofilm composition of Aspergillus fumigatus. Cell Microbiol (2010) 12(3):405–10.10.1111/j.1462-5822.2009.01409.x
19
SeidlerMJSalvenmoserSMullerFM. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob Agents Chemother (2008) 52(11):4130–6.10.1128/aac.00234-08
20
Al-FattaniMADouglasLJ. Biofilm matrix of Candida albicans and Candida tropicalis: chemical composition and role in drug resistance. J Med Microbiol (2006) 55(Pt 8):999–1008.10.1099/jmm.0.46569-0
21
MukherjeePKChandraJKuhnDMGhannoumMA. Mechanism of fluconazole resistance in Candida albicans biofilms: phase-specific role of efflux pumps and membrane sterols. Infect Immun (2003) 71(8):4333–40.10.1128/IAI.71.8.4333-4340.2003
22
NettJLincolnLMarchilloKMasseyRHoloydaKHoffBet alPutative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrob Agents Chemother (2007) 51(2):510–20.10.1128/aac.01056-06
23
RamageGVandewalleKWickesBLLopez-RibotJL. Characteristics of biofilm formation by Candida albicans. Rev Iberoam Micol (2001) 18(4):163–70.
24
MowatEButcherJLangSWilliamsCRamageG. Development of a simple model for studying the effects of antifungal agents on multicellular communities of Aspergillus fumigatus. J Med Microbiol (2007) 56(Pt 9):1205–12.10.1099/jmm.0.47247-0
25
SeidlerMJSalvenmoserSMüllerFMC. Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob Agents Chemother (2008) 52(11):4130–6.10.1128/aac.00234-08
26
MowatELangSWilliamsCMcCullochEJonesBRamageG. Phase-dependent antifungal activity against Aspergillus fumigatus developing multicellular filamentous biofilms. J Antimicrob Chemother (2008) 62(6):1281–4.10.1093/jac/dkn402
27
MartinezLRCasadevallA. Cryptococcus neoformans cells in biofilms are less susceptible than planktonic cells to antimicrobial molecules produced by the innate immune system. Infect Immun (2006) 74(11):6118–23.10.1128/iai.00995-06
28
RamageGRajendranRSherryLWilliamsC. Fungal biofilm resistance. Int J Microbiol (2012) 2012:528521.10.1155/2012/528521
29
KatragkouAKruhlakMJSimitsopoulouMChatzimoschouATaparkouACottenCJet alInteractions between human phagocytes and Candida albicans biofilms alone and in combination with antifungal agents. J Infect Dis (2010) 201(12):1941–9.10.1086/652783
30
XieZThompsonASobueTKashlevaHXuHVasilakosJet alCandida albicans biofilms do not trigger reactive oxygen species and evade neutrophil killing. J Infect Dis (2012) 206(12):1936–45.10.1093/infdis/jis607
31
JohnsonCJCabezas-OlcozJKernienJFWangSXBeebeDJHuttenlocherAet alThe extracellular matrix of Candida albicans biofilms impairs formation of neutrophil extracellular traps. PLoS Pathog (2016) 12(9):1–23.10.1371/journal.ppat.1005884
32
LeeMJLiuHBarkerBMSnarrBDGravelatFNAl AbdallahQet alThe fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog (2015) 11(10):e1005187.10.1371/journal.ppat.1005187
33
BeauvaisALatgeJP. Aspergillus biofilm in vitro and in vivo. Microbiol Spectr (2015) 3(4):1–10.10.1128/microbiolspec.MB-0017-2015
34
ChandraJMukherjeePK. Candida biofilms: development, architecture, and resistance. Microbiol Spectr (2015) 3(4):1–14.10.1128/microbiolspec.MB-0020-2015
35
DesaiJVMitchellAPAndesDR. Fungal biofilms, drug resistance, and recurrent infection. Cold Spring Harb Perspect Med (2014) 4(10):1–18.10.1101/cshperspect.a019729
36
NettJE. Future directions for anti-biofilm therapeutics targeting Candida. Expert Rev Anti Infect Ther (2014) 12(3):375–82.10.1586/14787210.2014.885838
37
FoxEPNobileCJ. A sticky situation: untangling the transcriptional network controlling biofilm development in Candida albicans. Transcription (2012) 3(6):315–22.10.4161/trns.22281
38
BaillieGSDouglasLJ. Matrix polymers of Candida biofilms and their possible role in biofilm resistance to antifungal agents. J Antimicrob Chemother (2000) 46(3):397–403.10.1093/jac/46.3.397
39
HawserSPBaillieGSDouglasLJ. Production of extracellular matrix by Candida albicans biofilms. J Med Microbiol (1998) 47(3):253–6.10.1099/00222615-47-3-253
40
ZarnowskiRWestlerWMLacmbouhGAMaritaJMBotheJRBernhardtJet alNovel entries in a fungal biofilm matrix encyclopedia. MBio (2014) 5(4):e1333–1314.10.1128/mBio.01333-14
41
MartinezLRCasadevallA. Cryptococcus neoformans biofilm formation depends on surface support and carbon source and reduces fungal cell susceptibility to heat, cold, and UV light. Appl Environ Microbiol (2007) 73(14):4592–601.10.1128/aem.02506-06
42
ChandraJMcCormickTSImamuraYMukherjeePKGhannoumMA. Interaction of Candida albicans with adherent human peripheral blood mononuclear cells increases C. albicans biofilm formation and results in differential expression of pro- and anti-inflammatory cytokines. Infect Immun (2007) 75(5):2612–20.10.1128/iai.01841-06
43
KatragkouAChatzimoschouASimitsopoulouMGeorgiadouERoilidesE. Additive antifungal activity of anidulafungin and human neutrophils against Candida parapsilosis biofilms. J Antimicrob Chemother (2011) 66(3):588–91.10.1093/jac/dkq466
44
KernienJJohnsonCNettJ. Conserved inhibition of neutrophil extracellular trap release by clinical Candida albicans biofilms. J Fungi (Basel) (2017) 3(3):49.10.3390/jof3030049
45
JohnsonCJKernienJFHoyerARNettJE. Mechanisms involved in the triggering of neutrophil extracellular traps (NETs) by Candida glabrata during planktonic and biofilm growth. Sci Rep (2017) 7(1):13065.10.1038/s41598-017-13588-6
46
BrunsSKniemeyerOHasenbergMAimaniandaVNietzscheSThywissenAet alProduction of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog (2010) 6(4):e1000873.10.1371/journal.ppat.1000873
47
GravelatFNBeauvaisALiuHLeeMJSnarrBDChenDet alAspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog (2013) 9(8):e1003575.10.1371/journal.ppat.1003575
48
de Jesus CarrionSLealSMGhannoumMAPearlmanE. The RodA hydrophobin on Aspergillus fumigatus spores masks Dectin-1 and Dectin-2 dependent responses and enhances fungal survival in vivo. J Immunol (2013) 191(5):2581–8.10.4049/jimmunol.1300748
49
GresnigtMSBozzaSBeckerKLJoostenLAAbdollahi-RoodsazSvan der BergWBet alA polysaccharide virulence factor from Aspergillus fumigatus elicits anti-inflammatory effects through induction of interleukin-1 receptor antagonist. PLoS Pathog (2014) 10(3):e1003936.10.1371/journal.ppat.1003936
50
ChaiLYNeteaMGSuguiJVonkAGVan De SandeWWWarrisAet alAspergillus fumigatus conidial melanin modulates host cytokine response. Immunobiology (2010) 215(11):915–20.10.1016/j.imbio.2009.10.002
51
AchkarJMFriesBC. Candida infections of the genitourinary tract. Clin Microbiol Rev (2010) 23(2):253–73.10.1128/cmr.00076-09
52
KennedyMJVolzPA. Ecology of Candida albicans gut colonization: inhibition of Candida adhesion, colonization, and dissemination from the gastrointestinal tract by bacterial antagonism. Infect Immun (1985) 49(3):654–63.
53
KumamotoCA. Candida biofilms. Curr Opin Microbiol (2002) 5(6):608–11.10.1016/S1369-5274(02)00371-5
54
KumamotoCA. Inflammation and gastrointestinal Candida colonization. Curr Opin Microbiol (2011) 14(4):386–91.10.1016/j.mib.2011.07.015
55
WebbBCThomasCJWillcoxMDHartyDWKnoxKW. Candida-associated denture stomatitis. Aetiology and management: a review. Part 2. Oral diseases caused by Candida species. Aust Dent J (1998) 43(3):160–6.10.1111/j.1834-7819.1998.tb00172.x
56
VazquezJA. Options for the management of mucosal candidiasis in patients with AIDS and HIV infection. Pharmacother J Hum Pharmacol Drug Ther (1999) 19(1):76–87.10.1592/phco.19.1.76.30509
57
LaunayOLortholaryOBouges-MichelCJarrousseBBentataMGuillevinL. Candidemia: a nosocomial complication in adults with late-stage AIDS. Clin Infect Dis (1998) 26(5):1134–41.10.1086/520291
58
DouglasLJ. Candida biofilms and their role in infection. Trends Microbiol (2003) 11(1):30–6.10.1016/S0966-842X(02)00002-1
59
KumamotoCAVincesMD. Alternative Candida albicans lifestyles: growth on surfaces. Annu Rev Microbiol (2005) 59:113–33.10.1146/annurev.micro.59.030804.121034
60
UppuluriPPierceCGLopez-RibotJL. Candida albicans biofilm formation and its clinical consequences. Future Microbiol (2009) 4(10):1235–7.10.2217/fmb.09.85
61
Dongari-BagtzoglouA. Mucosal biofilms: challenges and future directions. Expert Rev Anti Infect Ther (2008) 6(2):141–4.10.1586/14787210.6.2.141
62
ShinJHKeeSJShinMGKimSHShinDHLeeSKet alBiofilm production by isolates of Candida species recovered from nonneutropenic patients: comparison of bloodstream isolates with isolates from other sources. J Clin Microbiol (2002) 40(4):1244–8.10.1128/jcm.40.4.1244-1248.2002
63
BizerraFCNakamuraCVde PoerschCEstivalet SvidzinskiTIBorsato QuesadaRMGoldenbergSet alCharacteristics of biofilm formation by Candida tropicalis and antifungal resistance. FEMS Yeast Res (2008) 8(3):442–50.10.1111/j.1567-1364.2007.00347.x
64
KuhnDMChandraJMukherjeePKGhannoumMA. Comparison of biofilms formed by Candida albicans and Candida parapsilosis on bioprosthetic surfaces. Infect Immun (2002) 70(2):878–88.10.1128/IAI.70.2.878-888.2002
65
JainNKohliRCookEGialanellaPChangTFriesBC. Biofilm formation by and antifungal susceptibility of Candida isolates from urine. Appl Environ Microbiol (2007) 73(6):1697–703.10.1128/aem.02439-06
66
LewisREKontoyiannisDPDarouicheRORaadIIPrinceRA. Antifungal activity of amphotericin B, fluconazole, and voriconazole in an in vitro model of Candida catheter-related bloodstream infection. Antimicrob Agents Chemother (2002) 46(11):3499–505.10.1128/AAC.46.11.3499-3505.2002
67
TheinZMSamaranayakeYHSamaranayakeLP. In vitro biofilm formation of Candida albicans and non-albicans Candida species under dynamic and anaerobic conditions. Arch Oral Biol (2007) 52(8):761–7.10.1016/j.archoralbio.2007.01.009
68
TumbarelloMPosteraroBTrecarichiEMFioriBRossiMPortaRet alBiofilm production by Candida species and inadequate antifungal therapy as predictors of mortality for patients with candidemia. J Clin Microbiol (2007) 45(6):1843–50.10.1128/jcm.00131-07
69
SherryLRamageGKeanRBormanAJohnsonEMRichardsonMDet alBiofilm-forming capability of highly virulent, multidrug-resistant Candida auris. Emerg Infect Dis (2017) 23(2):328–31.10.3201/eid2302.161320
70
ChandraJKuhnDMMukherjeePKHoyerLLMcCormickTGhannoumMA. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J Bacteriol (2001) 183(18):5385–94.10.1128/JB.183.18.5385-5394.2001
71
RamageGMowatEJonesBWilliamsCLopez-RibotJ. Our current understanding of fungal biofilms. Crit Rev Microbiol (2009) 35(4):340–55.10.3109/10408410903241436
72
RamageGSavilleSPThomasDPLópez-RibotJL. Candida biofilms: an update. Eukaryot Cell (2005) 4(4):633–8.10.1128/ec.4.4.633-638.2005
73
SilvaSHenriquesMMartinsAOliveiraRWilliamsDAzeredoJ. Biofilms of non-Candida albicans Candida species: quantification, structure and matrix composition. Med Mycol (2009) 47(7):681–9.10.3109/13693780802549594
74
MitchellKFTaffHTCuevasMAReinickeELSanchezHAndesDR. Role of matrix β-1,3 glucan in antifungal resistance of non-albicans Candida biofilms. Antimicrob Agents Chemother (2013) 57(4):1918–20.10.1128/aac.02378-12
75
MitchellKFZarnowskiRSanchezHEdwardJAReinickeELNettJEet alCommunity participation in biofilm matrix assembly and function. Proc Natl Acad Sci U S A (2015) 112(13):4092–7.10.1073/pnas.1421437112
76
NettJEZarnowskiRCabezas-OlcozJBrooksEGBernhardtJMarchilloKet alHost contributions to construction of three device-associated Candida albicans biofilms. Infect Immun (2015) 83(12):4630–8.10.1128/iai.00931-15
77
MartinsMHenriquesMLopez-RibotJLOliveiraR. Addition of DNase improves the in vitro activity of antifungal drugs against Candida albicans biofilms. Mycoses (2012) 55(1):80–5.10.1111/j.1439-0507.2011.02047.x
78
NettJESanchezHCainMTRossKMAndesDR. Interface of Candida albicans biofilm matrix-associated drug resistance and cell wall integrity regulation. Eukaryot Cell (2011) 10(12):1660–9.10.1128/EC.05126-11
79
ThomasDPBachmannSPLopez-RibotJL. Proteomics for the analysis of the Candida albicans biofilm lifestyle. Proteomics (2006) 6(21):5795–804.10.1002/pmic.200600332
80
MartinsMUppuluriPThomasDPClearyIAHenriquesMLopez-RibotJLet alPresence of extracellular DNA in the Candida albicans biofilm matrix and its contribution to biofilms. Mycopathologia (2010) 169(5):323–31.10.1007/s11046-009-9264-y
81
KlisFMde GrootPHellingwerfK. Molecular organization of the cell wall of Candida albicans. Med Mycol (2001) 39(Suppl 1):1–8.10.1080/mmy.39.1.1.8-0
82
ChaffinWLLópez-RibotJLCasanovaMGozalboDMartínezJP. Cell wall and secreted proteins of Candida albicans: identification, function, and expression. Microbiol Mol Biol Rev (1998) 62(1):130–80.
83
EdwardsJEJrLehrerRIStiehmERFischerTJYoungLS. Severe candidal infections: clinical perspective, immune defense mechanisms, and current concepts of therapy. Ann Intern Med (1978) 89(1):91–106.10.7326/0003-4819-89-1-91
84
FidelPLJr. Immunity to Candida. Oral Dis (2002) 8(Suppl 2):69–75.10.1034/j.1601-0825.2002.00015.x
85
ErwigLPGowNA. Interactions of fungal pathogens with phagocytes. Nat Rev Microbiol (2016) 14(3):163–76.10.1038/nrmicro.2015.21
86
PappasPGKauffmanCAAndesDRClancyCJMarrKAOstrosky-ZeichnerLet alClinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis (2016) 62(4):e1–50.10.1093/cid/civ933
87
KolaczkowskaEKubesP. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13(3):159–75.10.1038/nri3399
88
NettJE. The host’s reply to Candida biofilm. Pathogens (2016) 5(1):1–10.10.3390/pathogens5010033
89
YanoJNoverrMCFidelPLJr. Cytokines in the host response to Candida vaginitis: identifying a role for non-classical immune mediators, S100 alarmins. Cytokine (2012) 58(1):118–28.10.1016/j.cyto.2011.11.021
90
NieminenMTHernandezMNovak-FrazerLKuulaHRamageGBowyerPet alDL-2-hydroxyisocaproic acid attenuates inflammatory responses in a murine Candida albicans biofilm model. Clin Vaccine Immunol (2014) 21(9):1240–5.10.1128/CVI.00339-14
91
FidelPLBarousseMEspinosaTFicarraMSturtevantJMartinDHet alAn intravaginal live Candida challenge in humans leads to new hypotheses for the immunopathogenesis of vulvovaginal candidiasis. Infect Immun (2004) 72(5):2939–46.10.1128/iai.72.5.2939-2946.2004
92
KatragkouASimitsopoulouMChatzimoschouAGeorgiadouEWalshTJRoilidesE. Effects of interferon-gamma and granulocyte colony-stimulating factor on antifungal activity of human polymorphonuclear neutrophils against Candida albicans grown as biofilms or planktonic cells. Cytokine (2011) 55(3):330–4.10.1016/j.cyto.2011.05.007
93
BranzkNLubojemskaAHardisonSEWangQGutierrezMGBrownGDet alNeutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol (2014) 15(11):1017–25.10.1038/ni.2987
94
BrinkmannVReichardUGoosmannCFaulerBUhlemannYWeissDSet alNeutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5.10.1126/science.1092385
95
UrbanCFReichardUBrinkmannVZychlinskyA. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol (2006) 8(4):668–76.10.1111/j.1462-5822.2005.00659.x
96
ByrdASO’BrienXMJohnsonCMLavigneLMReichnerJS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J Immunol (2013) 190(8):4136–48.10.4049/jimmunol.1202671
97
ZawrotniakMBochenskaOKarkowska-KuletaJSeweryn-OzogKAokiWUedaMet alAspartic proteases and major cell wall components in Candida albicans trigger the release of neutrophil extracellular traps. Front Cell Infect Microbiol (2017) 7:414.10.3389/fcimb.2017.00414
98
KennyEFHerzigAKrugerRMuthAMondalSThompsonPRet alDiverse stimuli engage different neutrophil extracellular trap pathways. Elife (2017) 6:e24437.10.7554/eLife.24437
99
BjornsdottirHWelinAMichaelssonEOslaVBergSChristensonKet alNeutrophil NET formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med (2015) 89:1024–35.10.1016/j.freeradbiomed.2015.10.398
100
StoiberWObermayerASteinbacherPKrautgartnerWD. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules (2015) 5(2):702–23.10.3390/biom5020702
101
GrayRDLucasCDMackellarALiFHiersemenzelKHaslettCet alActivation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J Inflamm (Lond) (2013) 10(1):12.10.1186/1476-9255-10-12
102
HoganDWheelerRT. The complex roles of NADPH oxidases in fungal infection. Cell Microbiol (2014) 16(8):1156–67.10.1111/cmi.12320
103
MetzlerKDFuchsTANauseefWMReumauxDRoeslerJSchulzeIet alMyeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood (2011) 117(3):953–9.10.1182/blood-2010-06-290171
104
RochaFACAlvesARochaMFGCordeiroRABrilhanteRSNPintoAet alTumor necrosis factor prevents Candida albicans biofilm formation. Sci Rep (2017) 7(1):1206.10.1038/s41598-017-01400-4
105
AlonsoMGowNErwigLBainJ. Macrophage migration is impaired within Candida albicans biofilms. J Fungi (2017) 3(3):31.10.3390/jof3030031
106
PattersonTFThompsonGRIIIDenningDWFishmanJAHadleySHerbrechtRet alPractice guidelines for the diagnosis and management of aspergillosis: 2016 update by the infectious diseases society of America. Clin Infect Dis (2016) 63(4):e1–60.10.1093/cid/ciw326
107
SheppardDCHowellPL. Biofilm exopolysaccharides of pathogenic fungi: lessons from bacteria. J Biol Chem (2016) 291(24):12529–37.10.1074/jbc.R116.720995
108
ReichhardtCFerreiraJAJoubertLMClemonsKVStevensDACegelskiL. Analysis of the Aspergillus fumigatus biofilm extracellular matrix by solid-state nuclear magnetic resonance spectroscopy. Eukaryot Cell (2015) 14(11):1064–72.10.1128/ec.00050-15
109
RajendranRWilliamsCLappinDFMillingtonOMartinsMRamageG. Extracellular DNA release acts as an antifungal resistance mechanism in mature Aspergillus fumigatus biofilms. Eukaryot Cell (2013) 12(3):420–9.10.1128/EC.00287-12
110
ShopovaIBrunsSThywissenAKniemeyerOBrakhageAAHillmannF. Extrinsic extracellular DNA leads to biofilm formation and colocalizes with matrix polysaccharides in the human pathogenic fungus Aspergillus fumigatus. Front Microbiol (2013) 4:141.10.3389/fmicb.2013.00141
111
FontaineTDelangleASimenelCCoddevilleBvan VlietSJvan KooykYet alGalactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog (2011) 7(11):e1002372.10.1371/journal.ppat.1002372
112
LeeMJGravelatFNCeroneRPBaptistaSDCampoliPVChoeSIet alOverlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J Biol Chem (2014) 289(3):1243–56.10.1074/jbc.M113.522516
113
LeeMJGellerAMBamfordNCLiuHGravelatFNSnarrBDet alDeacetylation of fungal exopolysaccharide mediates adhesion and biofilm formation. MBio (2016) 7(2):1–14.10.1128/mBio.00252-16
114
RuperezPLealJA. Extracellular galactosaminogalactan from Aspergillus parasiticus. Trans Br Mycol Soc (1981) 77(3):621–5.10.1016/S0007-1536(81)80111-8
115
BardalayePCNordinJH. Galactosaminogalactan from cell walls of Aspergillus niger. J Bacteriol (1976) 125(2):655–69.
116
GorinPAEveleighDE. Extracellular 2-acetamido-2-deoxy-d-galacto-d-galactan from Aspergillus nidulans. Biochemistry (1970) 9(25):5023–7.10.1021/bi00827a029
117
LealJARupérezP. Extracellular polysaccharide production by Aspergillus nidulans. Trans Br Mycol Soc (1978) 70(1):115–20.10.1016/S0007-1536(78)80180-6
118
AkoumianakiTKyrmiziIValsecchiIGresnigtMSSamonisGDrakosEet alAspergillus cell wall melanin blocks LC3-associated phagocytosis to promote pathogenicity. Cell Host Microbe (2016) 19(1):79–90.10.1016/j.chom.2015.12.002
119
FeldmesserM. Role of neutrophils in invasive aspergillosis. Infect Immun (2006) 74(12):6514–6.10.1128/iai.01551-06
120
GersonSLTalbotGHHurwitzSStromBLLuskEJCassilethPA. Prolonged granulocytopenia: the major risk factor for invasive pulmonary aspergillosis in patients with acute leukemia. Ann Intern Med (1984) 100(3):345–51.10.7326/0003-4819-100-3-345
121
BonnettCRCornishEJHarmsenAGBurrittJB. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infect Immun (2006) 74(12):6528–39.10.1128/iai.00909-06
122
SturtevantJLatgeJP. Participation of complement in the phagocytosis of the conidia of Aspergillus fumigatus by human polymorphonuclear cells. J Infect Dis (1992) 166(3):580–6.10.1093/infdis/166.3.580
123
BehnsenJNarangPHasenbergMGunzerFBilitewskiUKlippelNet alEnvironmental dimensionality controls the interaction of phagocytes with the pathogenic fungi Aspergillus fumigatus and Candida albicans. PLoS Pathog (2007) 3(2):e13.10.1371/journal.ppat.0030013
124
JaillonSPeriGDelnesteYFremauxIDoniAMoalliFet alThe humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med (2007) 204(4):793–804.10.1084/jem.20061301
125
BianchiMHakkimABrinkmannVSilerUSegerRAZychlinskyAet alRestoration of NET formation by gene therapy in CGD controls aspergillosis. Blood (2009) 114(13):2619–22.10.1182/blood-2009-05-221606
126
McCormickAHeesemannLWagenerJMarcosVHartlDLoefflerJet alNETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect (2010) 12(12–13):928–36.10.1016/j.micinf.2010.06.009
127
SnarrBDBakerPBamfordNCSatoYLiuHLehouxMet alMicrobial glycoside hydrolases as antibiofilm agents with cross-kingdom activity. Proc Natl Acad Sci U S A (2017) 114(27):7124–9.10.1073/pnas.1702798114
128
GravelatFNEjzykowiczDEChiangLYChabotJCUrbMMacdonaldKDet alAspergillus fumigatus MedA governs adherence, host cell interactions and virulence. Cell Microbiol (2010) 12(4):473–88.10.1111/j.1462-5822.2009.01408.x
129
RobinetPBaychelierFFontaineTPicardCDebrePVieillardVet alA polysaccharide virulence factor of a human fungal pathogen induces neutrophil apoptosis via NK cells. J Immunol (2014) 192(11):5332–42.10.4049/jimmunol.1303180
130
RohmMGrimmMJD’AuriaACAlmyroudisNGSegalBHUrbanCF. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect Immun (2014) 82(5):1766–77.10.1128/IAI.00096-14
131
GibbonsJGBeauvaisABeauRMcGaryKLLatgeJPRokasA. Global transcriptome changes underlying colony growth in the opportunistic human pathogen Aspergillus fumigatus. Eukaryot Cell (2012) 11(1):68–78.10.1128/ec.05102-11
132
PerfectJRDismukesWEDromerFGoldmanDLGraybillJRHamillRJet alClinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of America. Clin Infect Dis (2010) 50(3):291–322.10.1086/649858
133
WalshTJSchlegelRMoodyMMCostertonJWSalcmanM. Ventriculoatrial shunt infection due to Cryptococcus neoformans: an ultrastructural and quantitative microbiological study. Neurosurgery (1986) 18(3):373–5.10.1227/00006123-198603000-00025
134
BachMCTallyPWGodofskyEW. Use of cerebrospinal fluid shunts in patients having acquired immunodeficiency syndrome with cryptococcal meningitis and uncontrollable intracranial hypertension. Neurosurgery (1997) 41(6):1280–2; discussion 2–3.10.1097/00006123-199712000-00008
135
BraunDKJanssenDAMarcusJRKauffmanCA. Cryptococcal infection of a prosthetic dialysis fistula. Am J Kidney Dis (1994) 24(5):864–7.10.1016/S0272-6386(12)80683-4
136
BanerjeeUGuptaKVenugopalP. A case of prosthetic valve endocarditis caused by Cryptococcus neoformans var. neoformans. J Med Vet Mycol (1997) 35(2):139–41.10.1080/02681219780001031
137
MartinezLRCasadevallA. Biofilm formation by Cryptococcus neoformans. Microbiol Spectr (2015) 3(3):1–11.10.1128/microbiolspec.MB-0006-2014
138
VecchiarelliA. Immunoregulation by capsular components of Cryptococcus neoformans. Med Mycol (2000) 38(6):407–17.10.1080/714030973
139
DongZMMurphyJW. Intravascular cryptococcal culture filtrate (CneF) and its major component, glucuronoxylomannan, are potent inhibitors of leukocyte accumulation. Infect Immun (1995) 63(3):770–8.
140
Baena-MonroyTMoreno-MaldonadoVFranco-MartinezFAldape-BarriosBQuindosGSanchez-VargasLO. Candida albicans, Staphylococcus aureus and Streptococcus mutans colonization in patients wearing dental prosthesis. Med Oral Patol Oral Cir Bucal (2005) 10(Suppl 1):E27–39.
141
MartinezLRChristakiECasadevallA. Specific antibody to Cryptococcus neoformans glucurunoxylomannan antagonizes antifungal drug action against cryptococcal biofilms in vitro. J Infect Dis (2006) 194(2):261–6.10.1086/504722
142
RochaJDNascimentoMTDecote-RicardoDCorte-RealSMorrotAHeiseNet alCapsular polysaccharides from Cryptococcus neoformans modulate production of neutrophil extracellular traps (NETs) by human neutrophils. Sci Rep (2015) 5:8008.10.1038/srep08008
143
MonariCCasadevallARetiniCBaldelliFBistoniFVecchiarelliA. Antibody to capsular polysaccharide enhances the function of neutrophils from patients with AIDS against Cryptococcus neoformans. AIDS (1999) 13(6):653–60.10.1097/00002030-199904160-00005
Summary
Keywords
biofilm, matrix, fungi, neutrophil extracellular trap, innate immunity, neutrophil, Aspergillus, Candida
Citation
Kernien JF, Snarr BD, Sheppard DC and Nett JE (2018) The Interface between Fungal Biofilms and Innate Immunity. Front. Immunol. 8:1968. doi: 10.3389/fimmu.2017.01968
Received
07 November 2017
Accepted
19 December 2017
Published
10 January 2018
Volume
8 - 2017
Edited by
Steven Templeton, Indiana University School of Medicine – Terre Haute, United States
Reviewed by
Teresa Zelante, University of Perugia, Italy; Luis R. Martinez, University of Texas at El Paso, United States
Updates
Copyright
© 2018 Kernien, Snarr, Sheppard and Nett.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeniel E. Nett, jenett@medicince.wisc.edu
Specialty section: This article was submitted to Microbial Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.