 Anne B. Satterthwaite
Anne B. Satterthwaite- 1Department of Internal Medicine, The University of Texas Southwestern Medical Center, Dallas, TX, United States
- 2Department of Immunology, The University of Texas Southwestern Medical Center, Dallas, TX, United States
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by the loss of adaptive immune tolerance to nucleic acid-containing antigens. The resulting autoantibodies form immune complexes that promote inflammation and tissue damage. Defining the signals that drive pathogenic autoantibody production is an important step in the development of more targeted therapeutic approaches for lupus, which is currently treated primarily with non-specific immunosuppression. Here, we review the contribution of Bruton’s tyrosine kinase (Btk), a component of B and myeloid cell signaling pathways, to disease in murine lupus models. Both gain- and loss-of-function genetic studies have revealed that Btk plays multiple roles in the production of autoantibodies. These include promoting the activation, plasma cell differentiation, and class switching of autoreactive B cells. Small molecule inhibitors of Btk are effective at reducing autoantibody levels, B cell activation, and kidney damage in several lupus models. These studies suggest that Btk may promote end-organ damage both by facilitating the production of autoantibodies and by mediating the inflammatory response of myeloid cells to these immune complexes. While Btk has not been associated with SLE in GWAS studies, SLE B cells display signaling defects in components both upstream and downstream of Btk consistent with enhanced activation of Btk signaling pathways. Taken together, these observations indicate that limiting Btk activity is critical for maintaining B cell tolerance and preventing the development of autoimmune disease. Btk inhibitors, generally well-tolerated and approved to treat B cell malignancy, may thus be a useful therapeutic approach for SLE.
Introduction
The development of a B cell repertoire capable of secreting antibodies against a wide range of foreign antigens is crucial for effective immune responses. However, the processes that generate this diversity also result in the production of self-reactive B cells that, if not kept in check, can be pathogenic and lead to autoimmune disease. Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by autoantibodies against nuclear antigens. These autoantibodies promote disease pathogenesis by forming immune complexes that deposit in and damage tissues and synergize with innate immune defects to sustain pro-inflammatory feed-forward loops (1). Autoantibodies arise prior to the development of overt clinical symptoms (2), suggesting that loss of B cell tolerance is an important initiating event in lupus. Understanding the signaling pathways that mediate autoantibody production may reveal new therapeutic targets for SLE, currently treated primarily by non-specific immunosuppression. Here, we review the contribution of Bruton’s tyrosine kinase (Btk) to lupus, with a focus on its role in B cells.
Btk in B Cell Development and Activation
Bruton’s tyrosine kinase is a Tec family tyrosine kinase expressed in B and myeloid cells. It was first identified as the genetic defect in the primary immunodeficiency X-linked agammaglobulinemia (XLA) (3, 4). XLA patients have a block in B cell development at the pre-B stage and a paucity of circulating B cells and immunoglobulin (5). A mutation in the pleckstrin homology (PH) domain of Btk was subsequently found in X-linked immunodeficient (xid) mice (6, 7), which also have a B cell immunodeficiency, although milder than that of XLA patients. Btk-deficient mice phenocopy xid mice, with a block in B cell development at the immature stage, reduced peritoneal B1a cells, and impaired response to T-independent type II antigens (8–10).
Bruton’s tyrosine kinase is an important proximal component of B cell receptor (BCR) signaling pathways. Upon BCR engagement, the Btk PH domain binds to PIP3, a signaling intermediate generated by PI3 kinase (PI3K), thus localizing Btk to the plasma membrane (11–14). This facilitates its phosphorylation and activation by Src kinases (12, 13, 15) and promotes access to its substrates. The most well described of these is PLCγ2, which is activated after phosphorylation by Btk (16, 17) leading to increased Ca++ flux (14, 16, 18) and activation of NF-κB (19–21). BCR-induced proliferation and survival are impaired in the absence of Btk (9, 22–24). Btk is also required for toll-like receptor (TLR)-induced IL-10 expression by B cells (25, 26), and for synergy between the BCR and TLRs in enhancing IL-6 expression (27). Integrin-mediated adhesion of B lineage cells (28) and their response to chemokines, such as SDF-1 (29, 30), are also controlled by Btk.
Btk and Mouse Lupus Models
Btk Is Required for Autoantibody Production and Pathogenesis in Many Lupus Models
The xid mutation has long been known to reduce autoantibody levels in several murine lupus models, including NZB × NZW (31), BXSB (32), MRL.lpr (33), motheaten (34), and Gld (35). Renal disease was also prevented and survival improved by the xid mutation in NZB × NZW (31), BXSB (32), and MRL.lpr (33) mice.
The subsequent finding that Btk is a B cell signaling molecule suggested that enhanced B cell activation through Btk underlies autoantibody production in lupus models. This was tested using mice lacking B cell inhibitory signaling molecules. BCR signaling is normally limited by inhibitory receptors such as FcγRIIb, CD22, SiglecG, PIR-B, and CD72. The ITIMs of these receptors are phosphorylated by the tyrosine kinase Lyn, which results in the recruitment and activation of inhibitory phosphatases, such as SHIP and SHP-1 [reviewed in Ref. (36–38)]. B cell-specific deletion of Lyn, SHIP, or SHP-1 leads to B cell hyper-responsiveness and lupus-like autoimmune disease in mice (39–41). Mutations in inhibitory receptors result in milder autoimmunity, likely due to some degree of redundancy among them (42–47). Several of these inhibitory pathways target activating signals mediated by Btk (14, 16, 48).
Either the xid mutation (49) or Btk-deficiency (50–52) ameliorates the autoimmune phenotype of Lyn−/− mice. Similarly, Btk is required for autoantibodies in FcγRIIb−/−.Yaa mice, which lack the inhibitory receptor FcγRIIb and also have enhanced TLR7 signaling (53). One caveat to these studies is that the reduction in mature B cells in xid and Btk−/− mice is exacerbated in the absence of Lyn. To circumvent this defect, a transgene expressing a low level of Btk in B cells (Btklo) (22) was crossed to Lyn−/−Btk−/− mice (50–52). This normalized mature follicular B cell numbers to that of Lyn−/− mice. However, Lyn−/−Btklo mice failed to produce autoantibodies or develop kidney damage, indicating that Btk signaling in mature B cells, rather than simply effects of Btk on B cell development, is critical for autoimmunity.
The autoimmunity caused by loss of Lyn-dependent inhibitory signaling is likely mediated by heightened Btk responses, as it is mitigated by reducing Btk dosage. This is supported by gain-of-function studies in which either a constitutively active form of Btk [which carries a PH domain mutation that enhances Btk membrane localization (54)] or wild-type Btk were overexpressed in the B lineage (55–57). In both cases, autoimmunity ensued. Limiting Btk signal strength in B cells is thus critical to prevent the loss of B cell tolerance.
Multiple Functions of Btk Contribute to Autoantibody Production
Autoreactive B Cells Are Present in the Periphery in the Absence of Btk
Developing B cells are subjected to a central tolerance checkpoint at the immature B stage in the bone marrow. Those cells that express autoreactive receptors undergo receptor editing, rearranging a new Ig light chain to change their specificity. Cells that remain self-reactive after editing are deleted by apoptosis or rendered anergic. Autoreactive cells that escape are kept in check by peripheral tolerance mechanisms.
Taken together, the following observations suggest that Btk acts primarily in the periphery, rather than the bone marrow, to drive a loss of B cell tolerance. In Btk−/− mice carrying an anti-DNA Ig transgene, anti-DNA B cells are present in the periphery but do not produce antibodies in vivo (26). Single cell repertoire analysis of new emigrant B cells (recently arrived in the periphery from the bone marrow) from XLA patients revealed a higher frequency of autoreactive B cells than in healthy controls (58). This indicates that Btk signaling may actually promote central tolerance, and that Btk-deficiency does not abrogate autoimmunity simply by preventing autoreactive B cells from reaching the periphery. Furthermore, immunoglobulin transgenic mouse models and analysis of XLA patient B cell repertoires have shown that receptor editing is independent of Btk (58–60). A role for Btk in the loss of peripheral B cell tolerance is highlighted by both loss-of-function and overexpression studies. Btk is required for autoimmunity in Lyn−/− mice (49–52), which have intact central tolerance but develop autoantibodies due to a breach of peripheral tolerance (61, 62). Mice overexpressing Btk in mature B cells and myeloid cells, but not at earlier stages of B cell development in the bone marrow, develop autoimmunity (56).
Btk Contributes to Autoantibody Production beyond Its Role in Initial B Cell Activation
How does Btk signaling in the periphery drive autoantibody production? The role of Btk in the initial activation of BCR signals likely contributes, as residual B cells in Lyn−/− xid and Lyn−/−Btk−/− mice proliferate poorly in response to anti-IgM (49, 50). However, Lyn−/−Btklo B cells, like Lyn−/− B cells, have increased proliferative response to BCR engagement (50, 52), suggesting that in the absence of Lyn-mediated inhibitory signaling, low levels of Btk are able to transmit some aspects of BCR signals efficiently. However, Lyn−/−Btklo mice do not develop autoantibodies or autoimmune disease (51, 52). Similarly, although Btk-deficient anti-DNA transgenic mice do not produce autoantibodies (26), Btk is not required for B cells from these mice or from AM14 rheumatoid factor (RF) transgenic mice to proliferate in response to nucleic acid-containing antigens (26, 63). Such autoantigens, common in lupus, activate B cells via both the BCR and nucleic acid-sensing TLRs (1). Thus, Btk has additional functions beyond transmitting proliferative signals from the BCR and TLRs that promote the loss of B cell tolerance.
Btk Drives Plasma Cell (PC) Accumulation
Accumulation of antibody-secreting PCs in the periphery is characteristic of SLE patients (64, 65) and murine lupus models, including Lyn−/− mice (51, 66–74). A subset of inactive SLE patients demonstrate a PC-focused gene expression profile in their B cells, indicating that some patients may have an intrinsic predisposition to inappropriate B cell terminal differentiation (75). Btk is required for PC accumulation, as the increased PC frequency observed in Lyn−/− mice is normalized in Lyn−/−Btklo mice (51). This is likely due to enhanced Btk signaling in B cells, since B cell-specific overexpression of either constitutively active or wild-type Btk also results in elevated splenic PCs (55, 56).
Activating signals by Btk and inhibitory signals by Lyn converge on the transcription factor Ets1 (76). Ets1 is expressed in resting B cells and limits PC differentiation by inhibiting the activity of Blimp1 (77), a master PC transcription factor. Ets1−/− mice accumulate PCs and develop lupus-like autoimmunity, similar to Lyn−/− mice (71). Ets1 levels are significantly reduced in B cells from mice deficient in Lyn or the inhibitory signaling components SHP-1 or CD22 plus SiglecG, but are normalized in Lyn−/−Btklo B cells (76). Restoration of Ets1 expression to Lyn−/− or SHP-1−/− B cells prevents excessive B cell differentiation in vitro (76). These observations indicate that autoreactive PCs accumulate in Lyn−/− mice at least in part because of excessive downregulation of Ets1 by Btk. This is likely an exacerbation of a normal process, as BCR signaling downregulates Ets1 in wild-type B cells in a Btk-dependent manner (76). TLR signaling also downregulates Ets1 in wild-type B cells, and synergizes with BCR signaling to do so (76). In contrast, failure to downregulate Ets1 in response to Btk signals results in decreased steady state PC levels, as demonstrated by the ability of Ets1-deficiency to rescue the reduction in IgM antibody-secreting cells that occurs in Btk−/− mice (78). Thus, a continuum of Btk signaling to Ets1 controls PC frequencies, and can result in autoimmunity, normal responses, or immunodeficiency depending on the signal strength (78).
Btk Promotes Class Switching of Autoreactive B Cells
Class switching to IgG is required for autoantibodies to be pathogenic (79). In Btklo mice carrying the 56R anti-DNA immunoglobulin transgene, anti-DNA IgM, but not IgG, is produced (26). Thus, Btk also promotes class switching of autoreactive B cells separate from its role in their initial activation and terminal differentiation.
Several functions of Btk contribute to this process. Expression of the class switching factors AID and T-bet is reduced in TLR-stimulated Btk−/− and Btklo B cells relative to wild-type cells (26). Btk likely also plays an indirect role in class switching. IL-6 is required for IgG autoantibodies and autoimmune disease in Lyn−/− mice (51, 70, 80). Lyn−/−Btklo mice have decreased serum IL-6 levels and a reduced frequency of myeloid cells expressing IL-6 in response to LPS compared to Lyn−/− mice (51). B cell-derived IL-6 is also increased in Lyn−/− mice (70), and is required in other models for the formation of autoreactive germinal centers, in which class switching occurs (81, 82). Btk is required for the upregulation of IL-6 in B cells in response to synergistic BCR and TLR9 signaling (27), and B cells overexpressing Btk express more IL-6 (57). Btk also promotes expression of IL-21, a Tfh-derived cytokine, in Lyn−/− mice. This likely occurs via IL-6 as splenocytes from both Lyn−/−Btklo and Lyn−/−IL6−/− mice have reduced expression of IL-21 mRNA compared to Lyn−/− mice (80). Furthermore, Btk overexpression in B cells results in increased Tfh cells and IFNγ-producing T cells (57), which are important for autoreactive germinal centers and pathogenic autoantibodies (83–86).
Btk and Innate-Like B Cells
Bruton’s tyrosine kinase is expressed in B1a and marginal zone (MZ) B cells. These innate-like B cells may have both pathogenic and protective roles in autoimmune disease. The relative importance of Btk in these specific roles is not clear.
B1a cells are found predominantly in the peritoneal cavity, have a repertoire enriched in polyreactivity (87, 88), and are increased in several lupus models (41, 42, 88–90). Whether they are elevated in Lyn−/− mice is controversial (52, 72, 91, 92). They secrete protective IgM autoantibodies (93) and the anti-inflammatory cytokine IL-10 (94, 95). In some cases they do not contribute to pathogenic autoantibodies (96), but they can produce IgG autoantibodies and interact with T cells in a pro-inflammatory manner in some lupus models (88, 97, 98). B1a cells are reduced in Lyn−/−Btklo mice (52) and increased in mice expressing constitutively active Btk (55), and Btk is required for their expression of IL-10 (25).
Marginal zone B cells are also enriched in autoreactivity (99–101). Whether they contribute to pathogenic autoantibodies in lupus is model-dependent (71, 101–107), and may be modulated indirectly by alterations in splenic architecture in some strains. Btk is not required for MZ B cell development, but it controls the positive selection of particular B cell specificities into the MZ compartment (108). How this affects autoimmunity is not clear. Skewing of autoreactive B cells to the MZ is promoted by Btk in the 56R anti-DNA immunoglobulin transgenic model (26) and in NOD mice (109), a model of type I diabetes, but RF B cells carrying the xid mutation are enriched in the MZ relative to their wild-type counterparts (63).
Btk Inhibitors Are Effective in Mouse Lupus Models
The genetic evidence described above suggests that small molecule inhibitors of Btk could be an effective therapy for SLE. Preclinical studies with several inhibitors in multiple mouse models suggest that this may indeed be the case (Table 1) (110–117). Kidney damage was ameliorated in all cases and survival increased when measured. Btk inhibitors diminished B cell activity, as measured by reduced CD69 expression, PC frequencies, and/or germinal center B cell frequencies. Autoantibodies were also decreased, although in some cases not all specificities or isotypes were affected. Interestingly, kidney damage was prevented even in the few situations where IgG autoantibodies were not significantly reduced. This indicates that Btk has roles in lupus pathogenesis beyond its contribution to the loss of B cell tolerance. Btk inhibitors were effective in anti-GBM models of kidney disease, which measure only the effector phase of kidney inflammation and damage and do not depend on autoantibody production (113, 117). Btk inhibitors impair pro-inflammatory FcR responses of myeloid cells in vitro (111–113, 116–119), suggesting that Btk-dependent effector functions of myeloid cells may contribute to end-organ damage in vivo. However, Btk-deficiency in myeloid cells can have pro- or anti-inflammatory effects dependent on cell type and stimulus (120–135), and off-target effects of inhibitors cannot be ruled out (136). For instance, the Btk inhibitor ibrutinib also inhibits Itk (137), a related Tec kinase which has important functions in T cells. Further studies of the relative roles of B and myeloid cell-expressed Btk in lupus pathogenesis would be facilitated by the development of cell type-specific Btk knockout mice.
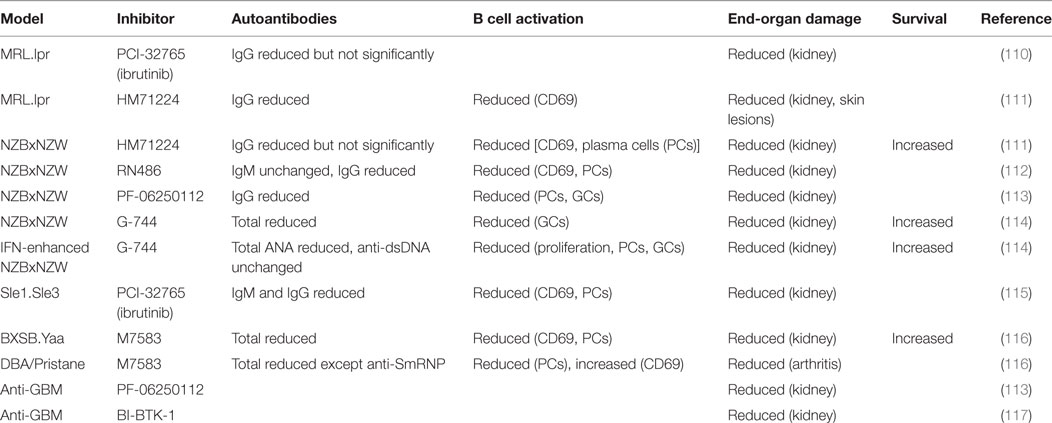
Table 1. Effects of Bruton’s tyrosine kinase (Btk) inhibitors on B cell activation and end-organ damage in murine lupus models.
Btk in Human Autoimmunity
While polymorphisms in Btk have not been identified in GWAS studies of SLE or other autoimmune diseases, several lines of evidence suggest that increased Btk activity may be associated with autoantibody production in humans. Increased Btk expression and phosphorylation was observed in B cells from rheumatoid arthritis patients (138, 139), correlating with RF antibodies among RF-positive patients (139) and enriched in anti-citrullinated protein antibody-positive patients (138). Similarly, increased Btk expression and phosphorylation correlated with RF antibodies in Sjogren’s syndrome patients (138). The frequency of Btk+ cells in the peripheral blood of SLE patients has been reported to correlate with disease activity, anti-dsDNA antibodies, proteinuria, and C3 levels (140), but whether this reflects changes in Btk signaling or cell subset distribution is unclear.
Several SLE-associated signaling defects and polymorphisms likely result in increased activity of Btk signaling pathways in B cells. Reduced expression of PTEN, which counteracts PI3K, has been observed in human lupus B cells (141). Btk activation and function require the binding of its PH domain to the product of PI3K in the plasma membrane (11–14), and PTEN haploinsufficiency enhances the efficiency of Btk signaling in mice (142). The autoimmune phenotype in Lyn−/− mice is mediated by excessive Btk activity in mice (49–52, 76, 80). Polymorphisms in Lyn are associated with SLE (143, 144), and reduced Lyn expression is observed in B cells from a subset of SLE patients (145–147). Expression of CSK, an inhibitor of Lyn, is increased by an SLE-associated polymorphism in the CSK gene. B cells carrying this SNP have reduced Lyn activity and increased responses to BCR signaling (148). Finally, several polymorphisms in Ets1 are associated with SLE, and Ets1 expression is reduced in PBMCs from SLE patients [reviewed in Ref. (149)]. Btk promotes the accumulation of autoreactive PCs in Lyn−/− mice by downregulating Ets1 (76).
Conclusion
Genetic studies demonstrate multiple roles for Btk in the development of murine lupus (Figure 1), including promoting the activation, differentiation, and class switching of autoreactive B cells. Btk inhibitors are effective at reducing autoantibodies and/or autoimmune symptoms in mouse lupus models and may act in both B and myeloid cells to exert these effects. In humans, several components of Btk signaling pathways are altered in B cells from lupus patients, and Btk expression and activation is elevated in B cells from other autoimmune diseases. Btk has dose-dependent effects on B cell activation and autoantibody production as illustrated by the phenotypes of Btklo and Btk-overexpressing mice. Such a rheostat effect of Btk (150) is supported by recent structural analysis indicating that Btk has graded degrees of activity (151), and suggests that partial inhibition of Btk may have significant functional consequences. Btk may thus be a useful therapeutic target for SLE. The Btk inhibitor ibrutinib is well tolerated and approved for treatment of several B cell malignancies (152, 153), and second generation, more specific inhibitors such as acalabrutinib are promising (154, 155). The use of these and other Btk inhibitors in B cell malignancy will be informative with respect to potential off-target and side effects (156, 157) that might be encountered in the context of autoimmune disease.
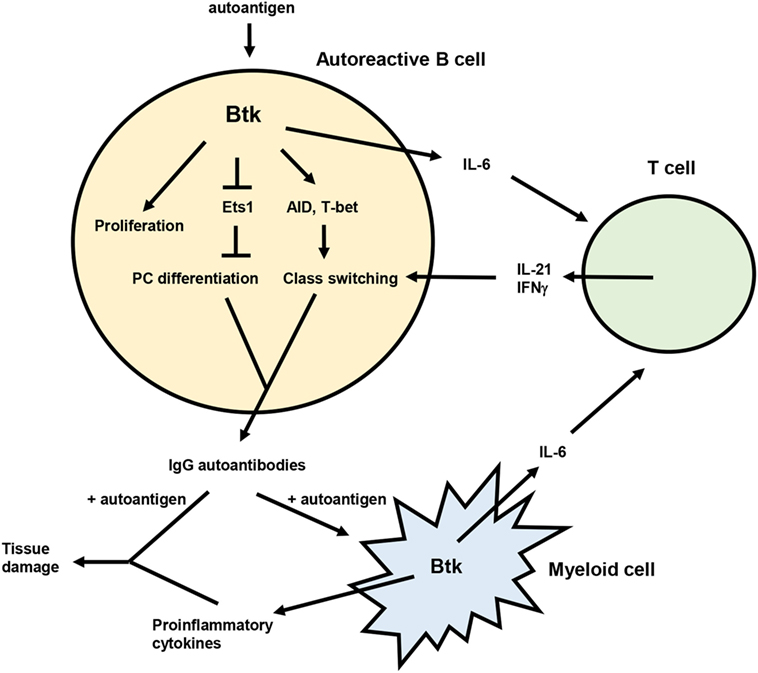
Figure 1. Model for the role of Bruton’s tyrosine kinase (Btk) in lupus pathogenesis. Btk acts in autoreactive B cells to promote proliferation, plasma cell (PC) differentiation, and class switching, resulting in the production of pathogenic IgG autoantibodies. IgG autoantibody production is also facilitated by the ability of Btk to enhance IL-6 expression from both B and myeloid cells. IL-6 then acts on T cells to promote differentiation of Tfh cells and IFNγ producing T cells, which in turn contribute to autoreactive B cell class switching via IL-21 and IFNγ. IgG autoantibodies produced in a Btk-dependent manner can then form immune complexes with autoantigen that deposit in tissues and induce inflammation and damage. These immune complexes can also activate myeloid cells, likely in a Btk-dependent manner, to produce inflammatory mediators that also damage tissues.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ET and handling editor declared their shared affiliation.
Funding
This work was supported by NIH grant AI122720 to AS. AS is a Southwestern Medical Foundation Scholar in Biomedical Research and holds the Peggy Chavellier Professorship in Arthritis Research and Treatment.
References
1. Liu Z, Davidson A. Taming lupus – a new understanding of pathogenesis is leading to clinical advances. Nat Med (2012) 18(6):871–82. doi:10.1038/nm.2752
2. Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med (2003) 349(16):1526–33. doi:10.1056/NEJMoa021933
3. Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the Src family of protein-tyrosine kinases. 1993. Nature (1993) 361(6409):226–33. doi:10.1038/361226a0
4. Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell (1993) 72(2):279–90. doi:10.1016/0092-8674(93)90667-F
5. Ochs HD, Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine (Baltimore) (1996) 75(6):287–99. doi:10.1097/00005792-199611000-00001
6. Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science (1993) 261(5119):358–61. doi:10.1126/science.8332901
7. Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science (1993) 261(5119):355–8. doi:10.1126/science.8332900
8. Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, et al. Impaired expansion of mouse B cell progenitors lacking Btk. Immunity (1995) 3(3):301–12. doi:10.1016/1074-7613(95)90115-9
9. Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, et al. Defective B cell development and function in Btk-deficient mice. Immunity (1995) 3(3):283–99. doi:10.1016/1074-7613(95)90114-0
10. Hendriks RW, de Bruijn MF, Maas A, Dingjan GM, Karis A, Grosveld F. Inactivation of Btk by insertion of lacZ reveals defects in B cell development only past the pre-B cell stage. EMBO J (1996) 15(18):4862–72.
11. Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J (1996) 15(22):6241–50.
12. Li Z, Wahl MI, Eguinoa A, Stephens LR, Hawkins PT, Witte ON. Phosphatidylinositol 3-kinase-gamma activates Bruton’s tyrosine kinase in concert with Src family kinases. Proc Natl Acad Sci U S A (1997) 94(25):13820–5. doi:10.1073/pnas.94.25.13820
13. Saito K, Scharenberg AM, Kinet JP. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates Btk. J Biol Chem (2001) 276(19):16201–6. doi:10.1074/jbc.M100873200
14. Bolland S, Pearse RN, Kurosaki T, Ravetch JV. SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity (1998) 8(4):509–16. doi:10.1016/S1074-7613(00)80555-5
15. Kurosaki T, Kurosaki M. Transphosphorylation of Bruton’s tyrosine kinase on tyrosine 551 is critical for B cell antigen receptor function. J Biol Chem (1997) 272(25):15595–8. doi:10.1074/jbc.272.25.15595
16. Fluckiger AC, Li Z, Kato RM, Wahl MI, Ochs HD, Longnecker R, et al. Btk/Tec kinases regulate sustained increases in intracellular Ca2+ following B-cell receptor activation. EMBO J (1998) 17(7):1973–85. doi:10.1093/emboj/17.7.1973
17. Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med (1996) 184(1):31–40. doi:10.1084/jem.184.1.31
18. Genevier HC, Callard RE. Impaired Ca2+ mobilization by X-linked agammaglobulinaemia (XLA) B cells in response to ligation of the B cell receptor (BCR). Clin Exp Immunol (1997) 110(3):386–91. doi:10.1046/j.1365-2249.1997.4581478.x
19. Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH. Bruton’s tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med (2000) 191(10):1735–44. doi:10.1084/jem.191.10.1735
20. Petro JB, Khan WN. Phospholipase C-gamma 2 couples Bruton’s tyrosine kinase to the NF-kappaB signaling pathway in B lymphocytes. J Biol Chem (2001) 276(3):1715–9. doi:10.1074/jbc.M009137200
21. Petro JB, Rahman SM, Ballard DW, Khan WN. Bruton’s tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med (2000) 191(10):1745–54. doi:10.1084/jem.191.10.1745
22. Satterthwaite AB, Cheroutre H, Khan WN, Sideras P, Witte ON. Btk dosage determines sensitivity to B cell antigen receptor cross-linking. Proc Natl Acad Sci U S A (1997) 94(24):13152–7. doi:10.1073/pnas.94.24.13152
23. Brorson K, Brunswick M, Ezhevsky S, Wei DG, Berg R, Scott D, et al. xid affects events leading to B cell cycle entry. J Immunol (1997) 159(1):135–43.
24. Solvason N, Wu WW, Kabra N, Lund-Johansen F, Roncarolo MG, Behrens TW, et al. Transgene expression of bcl-xL permits anti-immunoglobulin (Ig)-induced proliferation in xid B cells. J Exp Med (1998) 187(7):1081–91. doi:10.1084/jem.187.7.1081
25. Contreras CM, Halcomb KE, Randle L, Hinman RM, Gutierrez T, Clarke SH, et al. Btk regulates multiple stages in the development and survival of B-1 cells. Mol Immunol (2007) 44(10):2719–28. doi:10.1016/j.molimm.2006.11.023
26. Halcomb KE, Musuka S, Gutierrez T, Wright HL, Satterthwaite AB. Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Mol Immunol (2008) 46(2):233–41. doi:10.1016/j.molimm.2008.08.278
27. Kenny EF, Quinn SR, Doyle SL, Vink PM, van Eenennaam H, O’Neill LA. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS One (2013) 8(8):e74103. doi:10.1371/journal.pone.0074103
28. Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J Exp Med (2003) 198(10):1539–50. doi:10.1084/jem.20011866
29. de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity (2007) 26(1):93–104. doi:10.1016/j.immuni.2006.11.012
30. Halcomb KE, Contreras CM, Hinman RM, Coursey TG, Wright HL, Satterthwaite AB. Btk and phospholipase C gamma 2 can function independently during B cell development. Eur J Immunol (2007) 37(4):1033–42. doi:10.1002/eji.200636451
31. Steinberg BJ, Smathers PA, Frederiksen K, Steinberg AD. Ability of the xid gene to prevent autoimmunity in (NZB X NZW)F1 mice during the course of their natural history, after polyclonal stimulation, or following immunization with DNA. J Clin Invest (1982) 70(3):587–97. doi:10.1172/JCI110651
32. Smith HR, Chused TM, Steinberg AD. The effect of the X-linked immune deficiency gene (xid) upon the Y chromosome-related disease of BXSB mice. J Immunol (1983) 131(3):1257–62.
33. Steinberg EB, Santoro TJ, Chused TM, Smathers PA, Steinberg AD. Studies of congenic MRL-Ipr/Ipr.xid mice. J Immunol (1983) 131(6):2789–95.
34. Scribner CL, Hansen CT, Klinman DM, Steinberg AD. The interaction of the xid and me genes. J Immunol (1987) 138(11):3611–7.
35. Seldin MF, Reeves JP, Scribner CL, Roths JB, Davidson WF, Morse HC III, et al. Effect of xid on autoimmune C3H-gld/gld mice. Cell Immunol (1987) 107(1):249–55. doi:10.1016/0008-8749(87)90284-X
36. Nitschke L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr Opin Immunol (2005) 17(3):290–7. doi:10.1016/j.coi.2005.03.005
37. Nitschke L. CD22 and Siglec-G: B-cell inhibitory receptors with distinct functions. Immunol Rev (2009) 230(1):128–43. doi:10.1111/j.1600-065X.2009.00801.x
38. Xu Y, Harder KW, Huntington ND, Hibbs ML, Tarlinton DM. Lyn tyrosine kinase: accentuating the positive and the negative. Immunity (2005) 22(1):9–18. doi:10.1016/j.immuni.2004.12.004
39. Lamagna C, Hu Y, DeFranco AL, Lowell CA. B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol (2014) 192(3):919–28. doi:10.4049/jimmunol.1301979
40. O’Neill SK, Getahun A, Gauld SB, Merrell KT, Tamir I, Smith MJ, et al. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity (2011) 35(5):746–56. doi:10.1016/j.immuni.2011.10.011
41. Pao LI, Lam KP, Henderson JM, Kutok JL, Alimzhanov M, Nitschke L, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity (2007) 27(1):35–48. doi:10.1016/j.immuni.2007.04.016
42. Jellusova J, Wellmann U, Amann K, Winkler TH, Nitschke L. CD22 x Siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol (2010) 184(7):3618–27. doi:10.4049/jimmunol.0902711
43. Muller J, Lunz B, Schwab I, Acs A, Nimmerjahn F, Daniel C, et al. Siglec-G deficiency leads to autoimmunity in aging C57BL/6 mice. J Immunol (2015) 195(1):51–60. doi:10.4049/jimmunol.1403139
44. O’Keefe TL, Williams GT, Batista FD, Neuberger MS. Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med (1999) 189(8):1307–13. doi:10.1084/jem.189.8.1307
45. Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity (2000) 13(2):277–85. doi:10.1016/S1074-7613(00)00027-3
46. Kubo T, Uchida Y, Watanabe Y, Abe M, Nakamura A, Ono M, et al. Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J Exp Med (2009) 206(9):1971–82. doi:10.1084/jem.20082392
47. Li DH, Winslow MM, Cao TM, Chen AH, Davis CR, Mellins ED, et al. Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis Rheum (2008) 58(10):3192–204. doi:10.1002/art.23812
48. Maeda A, Scharenberg AM, Tsukada S, Bolen JB, Kinet JP, Kurosaki T. Paired immunoglobulin-like receptor B (PIR-B) inhibits BCR-induced activation of Syk and Btk by SHP-1. Oncogene (1999) 18(14):2291–7. doi:10.1038/sj.onc.1202552
49. Takeshita H, Taniuchi I, Kato J, Watanabe T. Abrogation of autoimmune disease in Lyn-deficient mice by the mutation of the Btk gene. Int Immunol (1998) 10(4):435–44. doi:10.1093/intimm/10.4.435
50. Satterthwaite AB, Lowell CA, Khan WN, Sideras P, Alt FW, Witte ON. Independent and opposing roles for Btk and lyn in B and myeloid signaling pathways. J Exp Med (1998) 188(5):833–44. doi:10.1084/jem.188.5.833
51. Gutierrez T, Halcomb KE, Coughran AJ, Li QZ, Satterthwaite AB. Separate checkpoints regulate splenic plasma cell accumulation and IgG autoantibody production in Lyn-deficient mice. Eur J Immunol (2010) 40(7):1897–905. doi:10.1002/eji.200940043
52. Whyburn LR, Halcomb KE, Contreras CM, Lowell CA, Witte ON, Satterthwaite AB. Reduced dosage of Bruton’s tyrosine kinase uncouples B cell hyperresponsiveness from autoimmunity in lyn-/- mice. J Immunol (2003) 171(4):1850–8. doi:10.4049/jimmunol.171.4.1850
53. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science (2006) 312(5780):1669–72. doi:10.1126/science.1124978
54. Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K, et al. Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity (1995) 2(5):451–60. doi:10.1016/1074-7613(95)90026-8
55. Kersseboom R, Kil L, Flierman R, van der Zee M, Dingjan GM, Middendorp S, et al. Constitutive activation of Bruton’s tyrosine kinase induces the formation of autoreactive IgM plasma cells. Eur J Immunol (2010) 40(9):2643–54. doi:10.1002/eji.201040521
56. Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood (2012) 119(16):3744–56. doi:10.1182/blood-2011-12-397919
57. Corneth OB, de Bruijn MJ, Rip J, Asmawidjaja PS, Kil LP, Hendriks RW. Enhanced expression of Bruton’s tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J Immunol (2016) 197(1):58–67. doi:10.4049/jimmunol.1600208
58. Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J Exp Med (2004) 200(7):927–34. doi:10.1084/jem.20040920
59. Dingjan GM, Middendorp S, Dahlenborg K, Maas A, Grosveld F, Hendriks RW. Bruton’s tyrosine kinase regulates the activation of gene rearrangements at the lambda light chain locus in precursor B cells in the mouse. J Exp Med (2001) 193(10):1169–78. doi:10.1084/jem.193.10.1169
60. Schram BR, Tze LE, Ramsey LB, Liu J, Najera L, Vegoe AL, et al. B cell receptor basal signaling regulates antigen-induced Ig light chain rearrangements. J Immunol (2008) 180(7):4728–41. doi:10.4049/jimmunol.180.7.4728
61. Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity (1998) 8(4):497–508. doi:10.1016/S1074-7613(00)80554-3
62. Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL. Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol (2009) 182(9):5382–92. doi:10.4049/jimmunol.0803941
63. Nundel K, Busto P, Debatis M, Marshak-Rothstein A. The role of Bruton’s tyrosine kinase in the development and BCR/TLR-dependent activation of AM14 rheumatoid factor B cells. J Leukoc Biol (2013) 94(5):865–75. doi:10.1189/jlb.0313126
64. Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, et al. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J Immunol (2000) 165(10):5970–9. doi:10.4049/jimmunol.165.10.5970
65. Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol (2015) 16(7):755–65. doi:10.1038/ni.3175
66. Bories JC, Willerford DM, Grevin D, Davidson L, Camus A, Martin P, et al. Increased T-cell apoptosis and terminal B-cell differentiation induced by inactivation of the Ets-1 proto-oncogene. Nature (1995) 377(6550):635–8. doi:10.1038/377635a0
67. Erickson LD, Lin LL, Duan B, Morel L, Noelle RJ. A genetic lesion that arrests plasma cell homing to the bone marrow. Proc Natl Acad Sci U S A (2003) 100(22):12905–10. doi:10.1073/pnas.2131686100
68. Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med (2004) 199(11):1577–84. doi:10.1084/jem.20040168
69. Mathian A, Gallegos M, Pascual V, Banchereau J, Koutouzov S. Interferon-alpha induces unabated production of short-lived plasma cells in pre-autoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur J Immunol (2011) 41(3):863–72. doi:10.1002/eji.201040649
70. Tsantikos E, Oracki SA, Quilici C, Anderson GP, Tarlinton DM, Hibbs ML. Autoimmune disease in Lyn-deficient mice is dependent on an inflammatory environment established by IL-6. J Immunol (2010) 184(3):1348–60. doi:10.4049/jimmunol.0901878
71. Wang D, John SA, Clements JL, Percy DH, Barton KP, Garrett-Sinha LA. Ets-1 deficiency leads to altered B cell differentiation, hyperresponsiveness to TLR9 and autoimmune disease. Int Immunol (2005) 17(9):1179–91. doi:10.1093/intimm/dxh295
72. Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, et al. Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell (1995) 83(2):301–11. doi:10.1016/0092-8674(95)90171-X
73. Infantino S, Jones SA, Walker JA, Maxwell MJ, Light A, O’Donnell K, et al. The tyrosine kinase Lyn limits the cytokine responsiveness of plasma cells to restrict their accumulation in mice. Sci Signal (2014) 7(338):ra77. doi:10.1126/scisignal.2005105
74. Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, et al. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity (1995) 3(5):549–60. doi:10.1016/1074-7613(95)90126-4
75. Garaud JC, Schickel JN, Blaison G, Knapp AM, Dembele D, Ruer-Laventie J, et al. B cell signature during inactive systemic lupus is heterogeneous: toward a biological dissection of lupus. PLoS One (2011) 6(8):e23900. doi:10.1371/journal.pone.0023900
76. Luo W, Mayeux J, Gutierrez T, Russell L, Getahun A, Muller J, et al. A balance between B cell receptor and inhibitory receptor signaling controls plasma cell differentiation by maintaining optimal Ets1 levels. J Immunol (2014) 193(2):909–20. doi:10.4049/jimmunol.1400666
77. John SA, Clements JL, Russell LM, Garrett-Sinha LA. Ets-1 regulates plasma cell differentiation by interfering with the activity of the transcription factor Blimp-1. J Biol Chem (2008) 283(2):951–62. doi:10.1074/jbc.M705262200
78. Mayeux J, Skaug B, Luo W, Russell LM, John S, Saelee P, et al. Genetic interaction between Lyn, Ets1, and Btk in the control of antibody levels. J Immunol (2015) 195(5):1955–63. doi:10.4049/jimmunol.1500165
79. Jiang C, Foley J, Clayton N, Kissling G, Jokinen M, Herbert R, et al. Abrogation of lupus nephritis in activation-induced deaminase-deficient MRL/lpr mice. J Immunol (2007) 178(11):7422–31. doi:10.4049/jimmunol.178.11.7422
80. Gutierrez T, Mayeux JM, Ortega SB, Karandikar NJ, Li QZ, Rakheja D, et al. IL-21 promotes the production of anti-DNA IgG but is dispensable for kidney damage in lyn-/- mice. Eur J Immunol (2013) 43(2):382–93. doi:10.1002/eji.201142095
81. Arkatkar T, Du SW, Jacobs HM, Dam EM, Hou B, Buckner JH, et al. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J Exp Med (2017) 214:3207–17. doi:10.1084/jem.20170580
82. de Valle E, Grigoriadis G, O’Reilly LA, Willis SN, Maxwell MJ, Corcoran LM, et al. NFkappaB1 is essential to prevent the development of multiorgan autoimmunity by limiting IL-6 production in follicular B cells. J Exp Med (2016) 213(4):621–41. doi:10.1084/jem.20151182
83. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, Cannons JL, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med (2009) 206(3):561–76. doi:10.1084/jem.20081886
84. Scapini P, Hu Y, Chu CL, Migone TS, Defranco AL, Cassatella MA, et al. Myeloid cells, BAFF, and IFN-gamma establish an inflammatory loop that exacerbates autoimmunity in Lyn-deficient mice. J Exp Med (2010) 207(8):1757–73. doi:10.1084/jem.20100086
85. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med (2016) 213(5):715–32. doi:10.1084/jem.20151722
86. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med (2016) 213(5):733–50. doi:10.1084/jem.20151724
87. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol (2011) 11(1):34–46. doi:10.1038/nri2901
88. Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, et al. A novel subpopulation of B-1 cells is enriched with autoreactivity in normal and lupus-prone mice. Arthritis Rheum (2009) 60(12):3734–43. doi:10.1002/art.25015
89. Mohan C, Morel L, Yang P, Wakeland EK. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum (1998) 41(9):1652–62. doi:10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W
90. Ishida D, Su L, Tamura A, Katayama Y, Kawai Y, Wang SF, et al. Rap1 signal controls B cell receptor repertoire and generation of self-reactive B1a cells. Immunity (2006) 24(4):417–27. doi:10.1016/j.immuni.2006.02.007
91. Chan VW, Meng F, Soriano P, DeFranco AL, Lowell CA. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal initiation and down-regulation. Immunity (1997) 7(1):69–81. doi:10.1016/S1074-7613(00)80511-7
92. Hasegawa M, Fujimoto M, Poe JC, Steeber DA, Lowell CA, Tedder TFA. CD19-dependent signaling pathway regulates autoimmunity in Lyn-deficient mice. J Immunol (2001) 167(5):2469–78. doi:10.4049/jimmunol.167.5.2469
93. Gronwall C, Vas J, Silverman GJ. Protective roles of natural IgM antibodies. Front Immunol (2012) 3:66. doi:10.3389/fimmu.2012.00066
94. Blenman KR, Duan B, Xu Z, Wan S, Atkinson MA, Flotte TR, et al. IL-10 regulation of lupus in the NZM2410 murine model. Lab Invest (2006) 86(11):1136–48. doi:10.1038/labinvest.3700468
95. Baglaenko Y, Manion KP, Chang NH, Loh C, Lajoie G, Wither JE. Suppression of autoimmunity by CD5(+) IL-10-producing B cells in lupus-prone mice. Genes Immun (2015) 16(5):311–20. doi:10.1038/gene.2015.17
96. Reap EA, Sobel ES, Jennette JC, Cohen PL, Eisenberg RA. Conventional B cells, not B1 cells, are the source of autoantibodies in chronic graft-versus-host disease. J Immunol (1993) 151(12):7316–23.
97. Kitabatake M, Soma M, Zhang T, Kuwahara K, Fukushima Y, Nojima T, et al. JNK regulatory molecule G5PR induces IgG autoantibody-producing plasmablasts from peritoneal B1a cells. J Immunol (2015) 194(4):1480–8. doi:10.4049/jimmunol.1401127
98. Enghard P, Humrich JY, Chu VT, Grussie E, Hiepe F, Burmester GR, et al. Class switching and consecutive loss of dsDNA-reactive B1a B cells from the peritoneal cavity during murine lupus development. Eur J Immunol (2010) 40(6):1809–18. doi:10.1002/eji.200940050
99. Chen X, Martin F, Forbush KA, Perlmutter RM, Kearney JF. Evidence for selection of a population of multi-reactive B cells into the splenic marginal zone. Int Immunol (1997) 9(1):27–41. doi:10.1093/intimm/9.1.27
100. Enzler T, Bonizzi G, Silverman GJ, Otero DC, Widhopf GF, Anzelon-Mills A, et al. Alternative and classical NF-kappa B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity (2006) 25(3):403–15. doi:10.1016/j.immuni.2006.07.010
101. Li Y, Li H, Weigert M. Autoreactive B cells in the marginal zone that express dual receptors. J Exp Med (2002) 195(2):181–8. doi:10.1084/jem.20011453
102. Wang JH, Wu Q, Yang P, Li H, Li J, Mountz JD, et al. Type I interferon-dependent CD86(high) marginal zone precursor B cells are potent T cell costimulators in mice. Arthritis Rheum (2011) 63(4):1054–64. doi:10.1002/art.30231
103. Duan B, Niu H, Xu Z, Sharpe AH, Croker BP, Sobel ES, et al. Intrafollicular location of marginal zone/CD1d(hi) B cells is associated with autoimmune pathology in a mouse model of lupus. Lab Invest (2008) 88(9):1008–20. doi:10.1038/labinvest.2008.62
104. Zhou Z, Niu H, Zheng YY, Morel L. Autoreactive marginal zone B cells enter the follicles and interact with CD4+ T cells in lupus-prone mice. BMC Immunol (2011) 12:7. doi:10.1186/1471-2172-12-7
105. Seo S, Buckler J, Erikson J. Novel roles for Lyn in B cell migration and lipopolysaccharide responsiveness revealed using anti-double-stranded DNA Ig transgenic mice. J Immunol (2001) 166(6):3710–6. doi:10.4049/jimmunol.166.6.3710
106. Amano H, Amano E, Moll T, Marinkovic D, Ibnou-Zekri N, Martinez-Soria E, et al. The Yaa mutation promoting murine lupus causes defective development of marginal zone B cells. J Immunol (2003) 170(5):2293–301. doi:10.4049/jimmunol.170.5.2293
107. Kolhatkar NS, Scharping NE, Sullivan JM, Jacobs HM, Schwartz MA, Khim S, et al. B-cell intrinsic TLR7 signals promote depletion of the marginal zone in a murine model of Wiskott-Aldrich syndrome. Eur J Immunol (2015) 45(10):2773–9. doi:10.1002/eji.201545644
108. Martin F, Kearney JF. Positive selection from newly formed to marginal zone B cells depends on the rate of clonal production, CD19, and btk. Immunity (2000) 12(1):39–49. doi:10.1016/S1074-7613(00)80157-0
109. Case JB, Bonami RH, Nyhoff LE, Steinberg HE, Sullivan AM, Kendall PL. Bruton’s tyrosine kinase synergizes with Notch2 To govern marginal zone B cells in nonobese diabetic mice. J Immunol (2015) 195(1):61–70. doi:10.4049/jimmunol.1400803
110. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A (2010) 107(29):13075–80. doi:10.1073/pnas.1004594107
111. Kim YY, Park KT, Jang SY, Lee KH, Byun JY, Suh KH, et al. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther (2017) 19(1):211. doi:10.1186/s13075-017-1402-1
112. Mina-Osorio P, LaStant J, Keirstead N, Whittard T, Ayala J, Stefanova S, et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum (2013) 65(9):2380–91. doi:10.1002/art.38047
113. Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol (2013) 191(9):4540–50. doi:10.4049/jimmunol.1301553
114. Katewa A, Wang Y, Hackney JA, Huang T, Suto E, Ramamoorthi N, et al. Btk-specific inhibition blocks pathogenic plasma cell signatures and myeloid cell-associated damage in IFNalpha-driven lupus nephritis. JCI Insight (2017) 2(7):e90111. doi:10.1172/jci.insight.90111
115. Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther (2012) 14(6):R243. doi:10.1186/ar4086
116. Bender AT, Pereira A, Fu K, Samy E, Wu Y, Liu-Bujalski L, et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin Immunol (2016) 164:65–77. doi:10.1016/j.clim.2016.01.012
117. Chalmers SA, Doerner J, Bosanac T, Khalil S, Smith D, Harcken C, et al. Therapeutic blockade of immune complex-mediated glomerulonephritis by highly selective inhibition of Bruton’s tyrosine kinase. Sci Rep (2016) 6:26164. doi:10.1038/srep26164
118. Hartkamp LM, Fine JS, van Es IE, Tang MW, Smith M, Woods J, et al. Btk inhibition suppresses agonist-induced human macrophage activation and inflammatory gene expression in RA synovial tissue explants. Ann Rheum Dis (2015) 74(8):1603–11. doi:10.1136/annrheumdis-2013-204143
119. Ren L, Campbell A, Fang H, Gautam S, Elavazhagan S, Fatehchand K, et al. Analysis of the effects of the Bruton’s tyrosine kinase (Btk) inhibitor ibrutinib on monocyte Fcgamma receptor (FcgammaR) function. J Biol Chem (2016) 291(6):3043–52. doi:10.1074/jbc.M115.687251
120. Tampella G, Kerns HM, Niu D, Singh S, Khim S, Bosch KA, et al. The Tec kinase-regulated phosphoproteome reveals a mechanism for the regulation of inhibitory signals in murine macrophages. J Immunol (2015) 195(1):246–56. doi:10.4049/jimmunol.1403238
121. Ni Gabhann J, Hams E, Smith S, Wynne C, Byrne JC, Brennan K, et al. Btk regulates macrophage polarization in response to lipopolysaccharide. PLoS One (2014) 9(1):e85834. doi:10.1371/journal.pone.0085834
122. Ni Gabhann J, Spence S, Wynne C, Smith S, Byrne JC, Coffey B, et al. Defects in acute responses to TLR4 in Btk-deficient mice result in impaired dendritic cell-induced IFN-gamma production by natural killer cells. Clin Immunol (2012) 142(3):373–82. doi:10.1016/j.clim.2011.12.009
123. Koprulu AD, Kastner R, Wienerroither S, Lassnig C, Putz EM, Majer O, et al. The tyrosine kinase Btk regulates the macrophage response to Listeria monocytogenes infection. PLoS One (2013) 8(3):e60476. doi:10.1371/journal.pone.0060476
124. Schmidt NW, Thieu VT, Mann BA, Ahyi AN, Kaplan MH. Bruton’s tyrosine kinase is required for TLR-induced IL-10 production. J Immunol (2006) 177(10):7203–10. doi:10.4049/jimmunol.177.10.7203
125. Marron TU, Martinez-Gallo M, Yu JE, Cunningham-Rundles C. Toll-like receptor 4-, 7-, and 8-activated myeloid cells from patients with X-linked agammaglobulinemia produce enhanced inflammatory cytokines. J Allergy Clin Immunol (2012) 129(1):184–90.e1–4. doi:10.1016/j.jaci.2011.10.009
126. Marron TU, Rohr K, Martinez-Gallo M, Yu J, Cunningham-Rundles C. TLR signaling and effector functions are intact in XLA neutrophils. Clin Immunol (2010) 137(1):74–80. doi:10.1016/j.clim.2010.06.011
127. Ormsby T, Schlecker E, Ferdin J, Tessarz AS, Angelisova P, Koprulu AD, et al. Btk is a positive regulator in the TREM-1/DAP12 signaling pathway. Blood (2011) 118(4):936–45. doi:10.1182/blood-2010-11-317016
128. Kawakami Y, Inagaki N, Salek-Ardakani S, Kitaura J, Tanaka H, Nagao K, et al. Regulation of dendritic cell maturation and function by Bruton’s tyrosine kinase via IL-10 and Stat3. Proc Natl Acad Sci U S A (2006) 103(1):153–8. doi:10.1073/pnas.0509784103
129. Sochorova K, Horvath R, Rozkova D, Litzman J, Bartunkova J, Sediva A, et al. Impaired toll-like receptor 8-mediated IL-6 and TNF-alpha production in antigen-presenting cells from patients with X-linked agammaglobulinemia. Blood (2007) 109(6):2553–6. doi:10.1182/blood-2006-07-037960
130. Gagliardi MC, Finocchi A, Orlandi P, Cursi L, Cancrini C, Moschese V, et al. Bruton’s tyrosine kinase defect in dendritic cells from X-linked agammaglobulinaemia patients does not influence their differentiation, maturation and antigen-presenting cell function. Clin Exp Immunol (2003) 133(1):115–22. doi:10.1046/j.1365-2249.2003.t01-1-02178.x
131. Horwood NJ, Mahon T, McDaid JP, Campbell J, Mano H, Brennan FM, et al. Bruton’s tyrosine kinase is required for lipopolysaccharide-induced tumor necrosis factor alpha production. J Exp Med (2003) 197(12):1603–11. doi:10.1084/jem.20021845
132. Horwood NJ, Page TH, McDaid JP, Palmer CD, Campbell J, Mahon T, et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J Immunol (2006) 176(6):3635–41. doi:10.4049/jimmunol.176.6.3635
133. Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, et al. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol (2012) 13(4):369–78. doi:10.1038/ni.2234
134. Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, et al. Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood (2011) 117(4):1329–39. doi:10.1182/blood-2010-04-281170
135. Lopez-Herrera G, Vargas-Hernandez A, Gonzalez-Serrano ME, Berron-Ruiz L, Rodriguez-Alba JC, Espinosa-Rosales F, et al. Bruton’s tyrosine kinase – an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol (2014) 95(2):243–50. doi:10.1189/jlb.0513307
136. Nyhoff LE, Barron BL, Johnson EM, Bonami RH, Maseda D, Fensterheim BA, et al. Bruton’s tyrosine kinase deficiency inhibits autoimmune arthritis in mice but fails to block immune complex-mediated inflammatory arthritis. Arthritis Rheumatol (2016) 68(8):1856–68. doi:10.1002/art.39657
137. Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood (2013) 122(15):2539–49. doi:10.1182/blood-2013-06-507947
138. Corneth OBJ, Verstappen GMP, Paulissen SMJ, de Bruijn MJW, Rip J, Lukkes M, et al. Enhanced Bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol (2017) 69(6):1313–24. doi:10.1002/art.40059
139. Wang SP, Iwata S, Nakayamada S, Niiro H, Jabbarzadeh-Tabrizi S, Kondo M, et al. Amplification of IL-21 signalling pathway through Bruton’s tyrosine kinase in human B cell activation. Rheumatology (Oxford) (2015) 54(8):1488–97. doi:10.1093/rheumatology/keu532
140. Kong W, Deng W, Sun Y, Huang S, Zhang Z, Shi B, et al. Increased expression of Bruton’s tyrosine kinase in peripheral blood is associated with lupus nephritis. Clin Rheumatol (2017) 37(1):43–9. doi:10.1007/s10067-017-3717-3
141. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med (2014) 6(246):246ra99. doi:10.1126/scitranslmed.3009131
142. Satterthwaite AB, Willis F, Kanchanastit P, Fruman D, Cantley LC, Helgason CD, et al. A sensitized genetic system for the analysis of murine B lymphocyte signal transduction pathways dependent on Bruton’s tyrosine kinase. Proc Natl Acad Sci U S A (2000) 97(12):6687–92. doi:10.1073/pnas.110146697
143. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet (2008) 40(2):204–10. doi:10.1038/ng.81
144. Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes Immun (2009) 10(5):397–403. doi:10.1038/gene.2009.19
145. Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP. B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med (2001) 49(2):157–65. doi:10.2310/6650.2001.34042
146. Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum (2005) 52(12):3955–65. doi:10.1002/art.21416
147. Liu Y, Dong J, Mu R, Gao Y, Tan X, Li Y, et al. MicroRNA-30a promotes B cell hyperactivity in patients with systemic lupus erythematosus by direct interaction with Lyn. Arthritis Rheum (2013) 65(6):1603–11. doi:10.1002/art.37912
148. Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, et al. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet (2012) 44(11):1227–30. doi:10.1038/ng.2439
149. Garrett-Sinha LA, Kearly A, Satterthwaite AB. The role of the transcription factor Ets1 in lupus and other autoimmune diseases. Crit Rev Immunol (2016) 36(6):485–510. doi:10.1615/CritRevImmunol.2017020284
150. Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol (2016) 12(7):763–73. doi:10.1586/1744666X.2016.1152888
151. Joseph RE, Wales TE, Fulton DB, Engen JR, Andreotti AH. Achieving a graded immune response: BTK adopts a range of active/inactive conformations dictated by multiple interdomain contacts. Structure (2017) 25(10):1481.e–94.e. doi:10.1016/j.str.2017.07.014
152. Wang Y, Zhang LL, Champlin RE, Wang ML. Targeting Bruton’s tyrosine kinase with ibrutinib in B-cell malignancies. Clin Pharmacol Ther (2015) 97(5):455–68. doi:10.1002/cpt.85
153. Smith MR. Ibrutinib in B lymphoid malignancies. Expert Opin Pharmacother (2015) 16(12):1879–87. doi:10.1517/14656566.2015.1067302
154. Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med (2016) 374(4):323–32. doi:10.1056/NEJMoa1509981
155. Wu J, Zhang M, Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol (2016) 9:21. doi:10.1186/s13045-016-0250-9
156. Aw A, Brown JR. Current status of Bruton’s tyrosine kinase inhibitor development and use in B-cell malignancies. Drugs Aging (2017) 34(7):509–27. doi:10.1007/s40266-017-0468-4
Keywords: Bruton’s tyrosine kinase, lupus, autoantibody, B cell, plasma cell, Lyn
Citation: Satterthwaite AB (2018) Bruton’s Tyrosine Kinase, a Component of B Cell Signaling Pathways, Has Multiple Roles in the Pathogenesis of Lupus. Front. Immunol. 8:1986. doi: 10.3389/fimmu.2017.01986
Received: 29 October 2017; Accepted: 21 December 2017;
Published: 22 January 2018
Edited by:
Margaret Hibbs, Monash University, AustraliaReviewed by:
Rudi W. Hendriks, Erasmus University Rotterdam, NetherlandsPeggy Lyn Kendall, Vanderbilt University School of Medicine, United States
Evelyn Tsantikos, Monash University, Australia
Copyright: © 2018 Satterthwaite. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne B. Satterthwaite, YW5uZS5zYXR0ZXJ0aHdhaXRlQHV0c291dGh3ZXN0ZXJuLmVkdQ==