 Jean-Marie Carpier
Jean-Marie Carpier Carrie L. Lucas
Carrie L. Lucas- Immunobiology Department, Yale University School of Medicine, New Haven, CT, United States
Activated PI3Kδ Syndrome (APDS) is an inherited immune disorder caused by heterozygous, gain-of-function mutations in the genes encoding the phosphoinositide 3-kinase delta (PI3Kδ) subunits p110δ or p85δ. This recently described primary immunodeficiency disease (PID) is characterized by recurrent sinopulmonary infections, lymphoproliferation, and susceptibility to herpesviruses, with Epstein–Barr virus (EBV) infection being most notable. A broad range of PIDs having disparate, molecularly defined genetic etiology can cause susceptibility to EBV, lymphoproliferative disease, and lymphoma. Historically, PID patients with loss-of-function mutations causing defective cell-mediated cytotoxicity or antigen receptor signaling were found to be highly susceptible to pathological EBV infection. By contrast, the gain of function in PI3K signaling observed in APDS patients paradoxically renders these patients susceptible to EBV, though the underlying mechanisms are incompletely understood. At a cellular level, APDS patients exhibit deranged B lymphocyte development and defects in class switch recombination, which generally lead to defective immunoglobulin production. Moreover, APDS patients also demonstrate an abnormal skewing of T cells toward terminal effectors with short telomeres and senescence markers. Here, we review APDS with a particular focus on how the altered lymphocyte biology in these patients may confer EBV susceptibility.
Introduction
Epstein–Barr virus (EBV) is a gammaherpesvirus carried by ~95% of the world population. EBV has a tropism for oronasopharyngeal epithelial cells (site of lytic replication) and B lymphocytes (reservoir of latent virus) and is well controlled throughout life in most people. However, immunocompromised patients often show persistent EBV viremia, putting them at risk for B-cell transformation due to viral oncogenes. Indeed, the virus was first identified in a Burkitt’s lymphoma in the 1960s (1) and is also associated with nasopharyngeal (2, 3) and gastric (4–7) cancer. Thus, inherited gene defects causing primary immunodeficiency diseases (PIDs) are often associated with recurrent or persistent EBV infections and related malignancies, and unraveling the genetic and molecular mechanisms underlying PIDs has led to better knowledge of the cellular and molecular components of the immune system that control herpesviruses. Here, we review the features of the recently described PID called Activated PI3Kδ Syndrome (APDS) and discuss the immunological abnormalities that may confer susceptibility to EBV and elucidate the cellular and molecular immune mechanisms normally controlling EBV.
The Class IA phosphoinositide 3-kinase delta (PI3Kδ) complex is recruited to phosphotyrosines and catalyzes the phosphorylation of phosphatidylinositol-4,5-bisphosphate to generate phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) that acts as a second messenger recruiting downstream signaling molecules. As a negative regulator of this signaling, the phosphatase PTEN can reverse this reaction and reduce levels of PIP3. PI3Kδ is a heterodimer of the p110δ catalytic subunit and the p85α, p55α, or p50α regulatory subunit and is known to play a major role in cell survival, cell growth, and cell-cycle entry through downstream mediators including AKT and mTORC1 (8). Loss of PI3Kδ catalytic activity has been described in a single PID patient with severe disease, but EBV susceptibility was not reported (9). Gain-of-function (GoF) mutations in the PIK3CD or PIK3R1 gene encoding p110δ or p85α, respectively, have been identified by us and others in PID patients with a disorder now known as PASLI Disease (PI3Kδ-Activating mutation causing Senescent T cells, Lymphadenopathy, and Immunodeficiency), or APDS for short. In the following sections, we will briefly review the discovery of APDS and its genetic and molecular basis, the clinical and immunological features of APDS, and possible contributors to poor control of EBV in APDS patients.
Genetic and Molecular Basis of APDS
Activated PI3Kδ Syndrome and causative PIK3CD mutations were initially described in two reports with a total of 26 patients in 14 unrelated families (10, 11). Prior to these initial reports, there had been one description of the most frequent mutation in PIK3CD (causing E1021K p110δ) in a single individual being studied for B-cell immunodeficiency, but no causative relationship was established (12). Shortly after discovery of APDS and underlying PIK3CD mutations, two additional reports with eight patients from six unrelated families with similar clinical findings described splice site mutations in PIK3R1 as a second genetic cause for APDS (13, 14). Thus, APDS1 (or PASLI-CD) has been established to denote patients with PIK3CD mutations, and APDS2 (or PASLI-R1) denotes those with PIK3R1 mutations. Another more recent phenocopy of APDS has been called APDS-like (APDS-L) and is caused by loss-of-function PTEN mutations (15, 16). Since the description of APDS in 2013, approximately 214 patients have been described with a spectrum of clinical features described below (10, 11, 13–41).
The PI3Kδ complex forms when p110δ and p85α bind at a 1:1 ratio. This constitutive complex remains stable due to tight binding interactions between the adaptor-binding domain (ABD) of p110δ and the inter-SH2 domain of p85α. To date, all activating APDS mutations affecting p110δ (E81K, G124D, N334K, R405C, C416R, E525K, E525A, R929C, E1021K, E1025G) and p85α (delE11, N564K) have been found or are expected to maintain some level of protein–protein interaction to form a hyperactive PI3Kδ complex, as free p110δ or p85α is unstable and would likely be degraded (Figure 1A). Each evaluated mutant has been found to hyperactivate signaling by disrupting inter- or intra-molecular inhibitory contacts, as observed for tumor-associated GoF mutations in the related PIK3CA (Figure 1A) (42, 43).
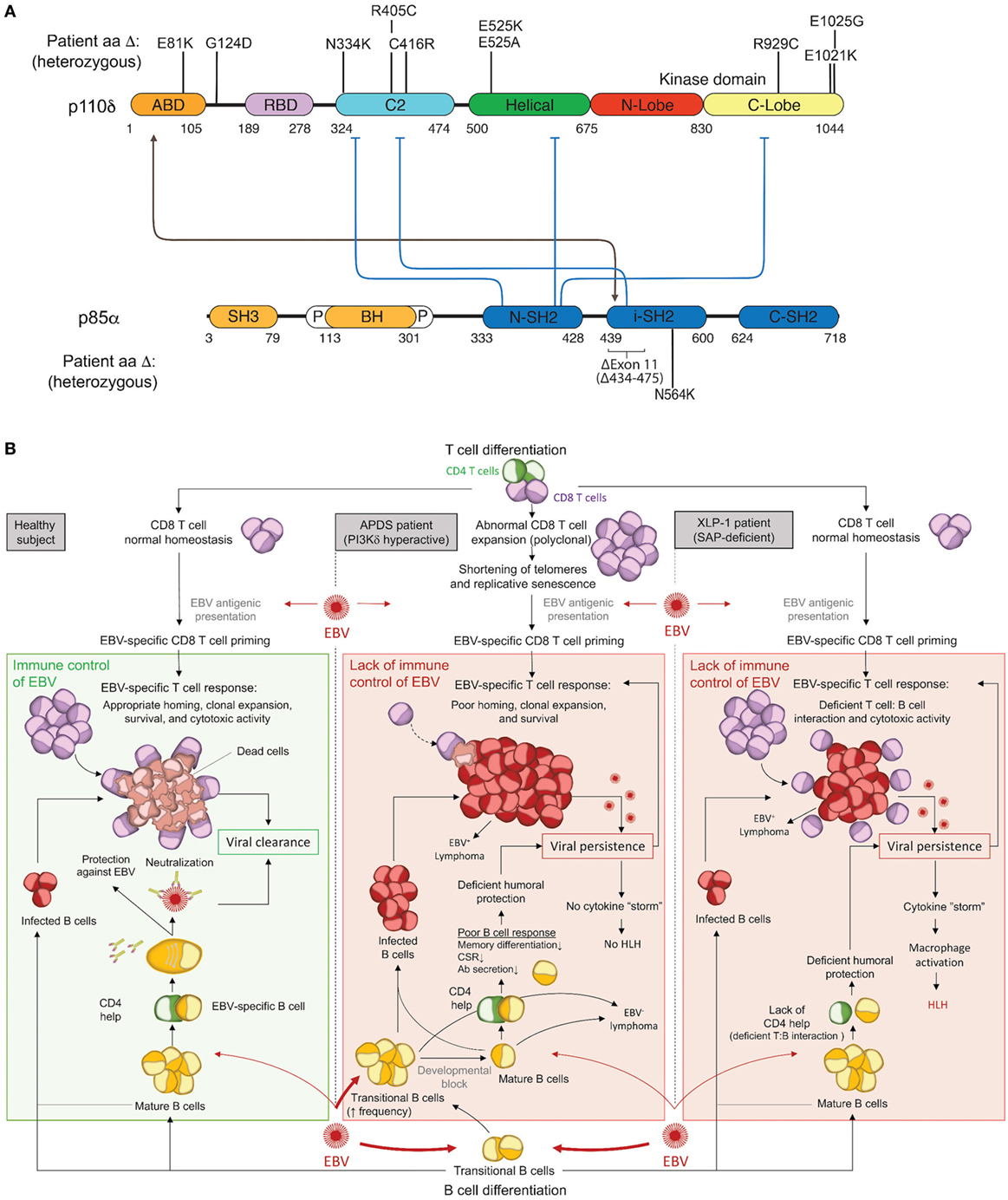
Figure 1. Activated PI3Kδ Syndrome (APDS) GoF mutations in the PI3Kδ complex and associated immune dysfunction responsible for Epstein–Barr virus (EBV) susceptibility. (A) Schematic representation of p110δ and p85α protein domains and APDS mutations reported in patients. The black line depicts the stabilizing interaction, and the blue lines show the inhibitory contacts within the PI3Kδ complex. ABD, adaptor-binding domain; BH, breakpoint-cluster region homology domain; P, proline-rich region; SH, SRC-homology domain; N, amino-terminal; i, inter; C, carboxy-terminal. (B) Schematic representation of the current understanding for the immune control of EBV in healthy subjects (left) and proposed hypothesis for EBV susceptibility in APDS (middle) and XLP1 (right) patients. APDS mutations cause abnormal polyclonal expansion of CD8 T cells that become senescent. Senescent CD8 T cells show an impaired EBV-specific response due to limited homing, expansion, and survival. In conjunction with CD8 T-cell defects, APDS patients exhibit an elevated frequency of transitional B cells, a major cell type for cell entry of EBV, and have defective humoral immunity that may further contribute to EBV susceptibility. In comparison, XLP1 patients, who are susceptible to EBV and develop HLH, are deficient in the SAP adaptor and exhibit defective EBV-specific T cell: B-cell interactions, causing a lack of CD4 help and a failure of CD8 T-cell cytotoxicity. As opposed to APDS, viral persistence in XLP1 patients causes a recurring stimulation/expansion of EBV-specific CD8 T cells and results in a cytokine storm underlying hemophagocytic lymphohistiocytosis (HLH). Antibodies depiction: taken from SMART (Servier Medical Art) licensed under a Creative Commons Attribution 3.0 Unported License.
Clinical and Cellular Features of APDS
The clinical spectrum of APDS1, APDS2, and APDS-L is largely overlapping and consists mostly of immunological abnormalities (Table 1), although growth retardation has also been reported APDS2 and, less frequently, APDS1 (10, 12–14, 17, 21, 24, 26, 27, 29–33, 37). Recurrent upper and lower respiratory tract infections are the most common clinical features affecting 98% of APDS patients and often resulting in progressive airway damage. APDS is associated with lymphoproliferative disease (71%), which commonly presents as lymphoid hyperplasia, splenomegaly, and/or lymphadenopathy. Autoinflammatory disease also occurs in 29% of cases. Importantly, recurrent infection with herpesviruses, such as EBV or cytomegalovirus (CMV), is observed in about 47% of cases but has not been associated with hemophagocytic lymphohistiocytosis (HLH). We hypothesize that HLH does not occur in APDS patients because, as described below, hyperactive PI3K drives polyclonal T-cell senescence, which limits homing, expansion, and survival of EBV-specific T cells and thereby prevents the cytokine storm that causes HLH (Figure 1B). EBV infection is found in 30% of APDS patients and represents an important risk factor for the development of B-cell lymphoma (occurring in 20% of EBV-infected APDS patients). However, the occurrence of EBV-negative lymphomas has overall been reported as higher (19%) than EBV-positive lymphomas (6%), which likely reflects the oncogenic potential of hyperactive PI3K signaling. Thus, intrinsically hyperactive PI3K (rather than EBV infection) appears to be the more dominant driver of B-cell transformation in APDS.
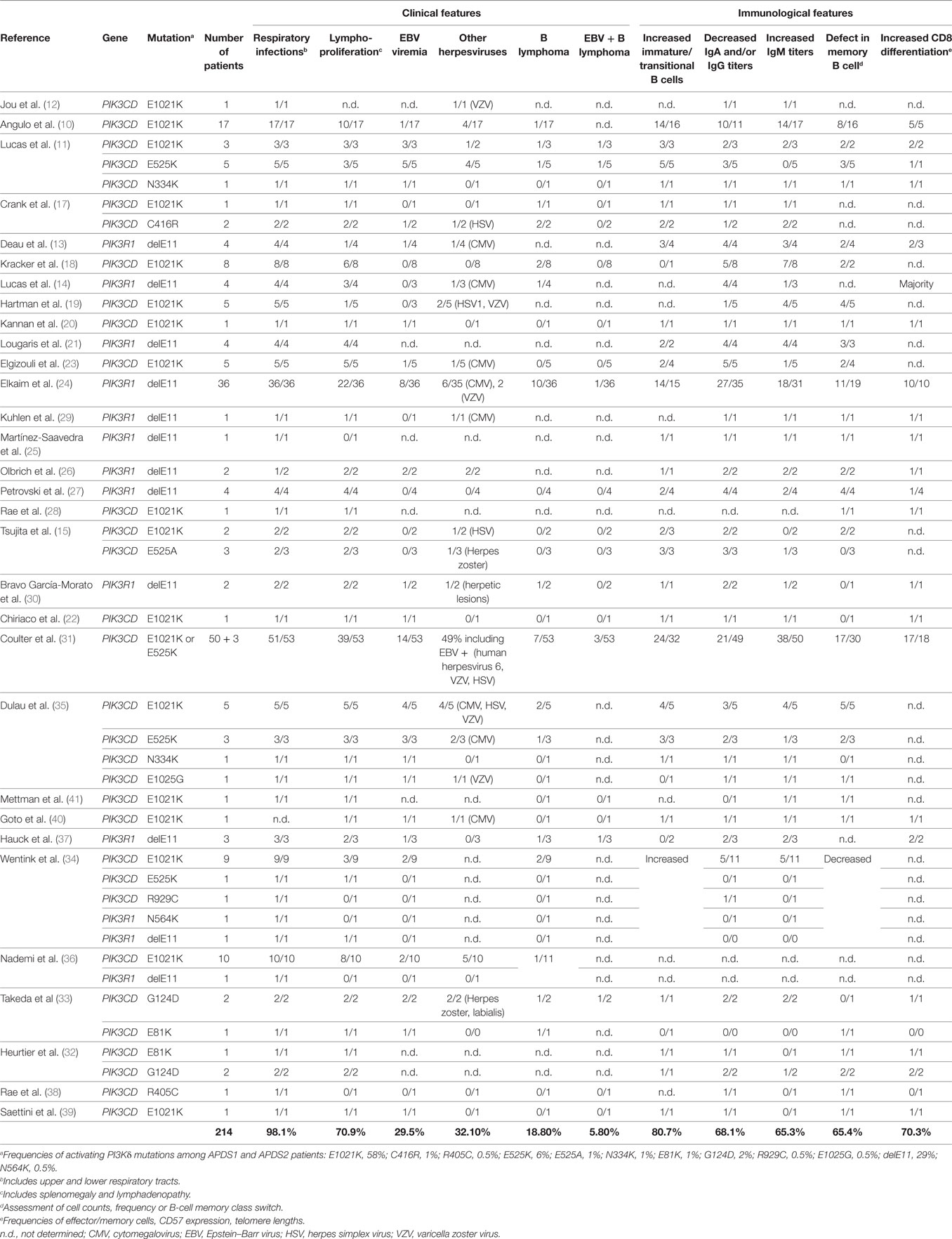
Table 1. Summary of clinical and immunological features of APDS patients.
The susceptibility to infections displayed by APDS patients is associated with deficiencies in both T and B lymphocyte function, a feature that categorizes APDS as a combined immunodeficiency (Table 1). B-cell compartment abnormalities have been universally described in both APDS1 and APDS2. B-cell lymphopenia is found in 74% of patients and may be due to a developmental defect at the transitional stage, as IgD+CD10+ B cells are consistently increased in APDS patient blood (81%). Additionally, humoral defects have been observed in the majority of APDS patients, leading to poor vaccine responses in some patients. Serum concentrations of IgM are increased in 65% of cases, while IgA and at least one IgG isotype are decreased (68%). This phenotype suggests a defect in class-switch recombination (CSR), and in vitro studies have not yet provided a clear conclusion about whether this defect arises predominantly from B-cell-intrinsic or -extrinsic effects of PI3Kδ hyperactivation (11, 17, 22, 44). Although immunodeficiency is a major feature of APDS, expansion of CD8 T cells is commonly observed (70%) and, together with CD4 lymphopenia, explains the inverted CD4:CD8 ratios found in the disease (71%). In addition, the constitutive activation of PI3K is also linked to the progressive differentiation of T cells toward effector memory and terminally differentiated (TEMRA) subtypes. Consistently, CD8 T cells from APDS patients exhibit normal degranulation activity (induced by anti-CD3 stimulation) and TNF/IFNγ production (11) with reduced secretion of IL-2, weak proliferative responses, and enhanced restimulation-induced cell death (RICD) (10, 11, 14, 22).
Thus, APDS is characterized by a complex spectrum of clinical, immunological, and cellular features. Elucidation of the genetic and molecular defects has improved diagnosis and care of APDS patients (45). Because of the recurrent sinopulmonary infections, antibiotics are often given prophylactically, and immunoglobulin replacement is commonly used, although recurrent infections have been reported despite this treatment (15, 20, 26). Chemo- and/or radiotherapy are often used for lymphomas, a major cause of death in APDS patients (about 62% of deaths) (11, 14, 17–19, 24, 30, 31, 37). Beyond the treatment of these specific symptoms, hematopoietic stem-cell transplantation has proven beneficial for restoration of immune function in 67% of APDS patients receiving this therapy, which requires availability of an HLA-compatible donor and is particularly risky in the setting of EBV infection (10, 14, 15, 18, 24, 31, 34, 36). Identification of the genetic and molecular etiology of APDS has also led to more specific treatments, such as the use of the mTORC1 inhibitor (rapamycin) (10, 11, 23, 24, 26, 28, 34, 40) and specific p110δ inhibitors, which are currently being evaluated for APDS treatment in clinical trials.
EBV Susceptibility in APDS Patients
B-Cell Dysfunction
Epstein–Barr virus is usually acquired during childhood and is asymptomatic throughout life, while primary infection in young adulthood can (in ~30–70% of cases) cause infectious mononucleosis (IM) (46). Although control of EBV infection by the immune system has been mainly attributed to CD8 T cells and to a lesser extent to NK cells, a role for humoral immunity in protecting from EBV infection has recently been reevaluated with a focus on IM patients (46–48). Although a neutralizing antibody response against several viral proteins such as gp350, a particularly immunogenic EBV protein, is detectable in these patients (47), the peak of this antibody response occurs after disappearance of IM symptoms and clearance of the virus, and this delay has been attributed to B-cell dysfunction in acutely infected patients (46). Several vaccination strategies have focused on the gp350 protein (49–51) since it acts as a major mediator for entry of EBV into B cells through its interaction with CD21 (52). Interestingly, vaccination using recombinant gp350 in phase-I and-II trials correlated with a gp350-specific antibody response and showed a protective effect in IM development but not in asymptomatic EBV infections (50, 51). Thus, the role of neutralizing antibodies in protecting B cells from infection and lowering the extent of infection during primary exposure can be considered in asymptomatic individuals and especially in children who might carry maternal EBV-specific antibodies. This protection might also be crucial to prevent disease upon reexposure to EBV. As such, the defects in B-cell development and function observed in APDS patients might help explain their increased susceptibility to EBV.
Changes in B-cell differentiation and intrinsic B-cell dysregulation may also be relevant contributors to EBV susceptibility in APDS. The nature of the B-cell compartment primarily infected by EBV has been a matter of debate, and it was first proposed that IgD−CD27+ memory B cells are the major entry point (53). However, in vitro observations as well as data from IM patients suggested that primary infection of B cells occurs in naïve IgD+CD27 cells, which then undergo differentiation in germinal center reactions, resulting in the emergence of class-switched memory B cells carrying EBV (54, 55). The observation that APDS patients exhibit an increased frequency of immature transitional CD10+ B cells and have a low frequency of memory CD27+ B cells (11) while remaining highly susceptible to EBV may support the possibility that EBV can also infect developing B cells. Indeed, several studies performed in mice have reported the ability of developing B cells to be infected by EBV (56) or the homologous γ-herpesvirus MHV68 (57, 58). The idea that transitional B cells might be a critical entry point and reservoir for EBV has been proposed before and fits with a model in which recurrent seeding of the developing B-cell compartment with EBV virions promotes establishment of long-term B-cell infection (57). In agreement with this hypothesis, depletion of transitional B cells in mice reduces EBV in the mature B-cell compartment (58). Therefore, it is possible that persistent EBV infection is facilitated in APDS patients by the predominant transitional B-cell compartment that would provide a pathologically increased reservoir of EBV, although additional studies are required to evaluate this hypothesis.
The EBV latency proteins LMP2a and LMP1 are thought to be key players in hijacking B-cell maturation by EBV since they mimic B-cell receptor and CD40 signaling, respectively (59, 60). LMP1 in particular is sufficient to transform several cell types, activates PI3K signaling, and promotes B-cell survival, growth, and proliferation programs (59–61). As p110δ is the main Class IA PI3K isoform expressed in EBV-positive B-cell lymphomas, this isoform might be a major target for LMP1 (62), and EBV-driven lymphomas in APDS may thus be facilitated in B cells expressing hyperactive forms of PI3Kδ. Moreover, several studies have demonstrated that PI3K inhibition reduces EBV reactivation (59, 63, 64), suggesting that the increased PI3Kδ activity displayed by APDS patients would favor a constitutive lytic program and may contribute to persistent viremia.
Thus, APDS patients harbor abnormal B cells that likely promote EBV susceptibility through several mechanisms. These may include, among others, poor anti-EBV antibody responses, increased transitional B cells serving as an EBV reservoir, and heightened cell-intrinsic PI3K signaling that may promote EBV-driven B-cell transformation and/or EBV reactivation.
T-Cell Dysfunction
T lymphocytes are a crucial immune cell type for control of EBV infection (65, 66). Substantial expansion of EBV-specific CD8 T cells has been observed in IM patients (67), and EBV control in healthy carriers has been correlated with the presence of functional EBV-specific CD8 T cells (68). However, the major arguments supporting a functional role for CD8 T cells in controlling EBV in vivo come from immunocompromised patients. Indeed, post-transplant lymphoproliferative disease (PTLD) is an important clinical concern in immunosuppressed transplant patients. In these patients, PTLD is caused by EBV-driven B-cell expansion and can be overcome by infusing EBV-specific cytotoxic T cells (69–72). Moreover, immunodeficiency syndromes, particularly HLH and X-linked lymphoproliferative diseases, have also provided valuable lessons and advanced our understanding of the role for CD8 T cells in EBV immunity (73, 74).
Monogenic causes of EBV-associated HLH have demonstrated that defective cytotoxicity machinery most commonly underlies disease (66, 75). However, these more general defects are not present in APDS patients, highlighting a more nuanced mechanism conferring EBV susceptibility when PI3K signaling is hyperactive. XLP1 patients deficient in the signaling lymphocytic activation molecule-associated protein (SAP) adaptor exhibit a very specific vulnerability to EBV viremia, and uncovering the genetic mutations responsible for disease contributed to defining crucial and non-redundant molecular pathways for EBV control by cytotoxic cells (76–79). Indeed, mutations in SH2D1A encoding SAP result in failure of T cell: B-cell interactions and inability to propagate 2B4- and NTBA-mediated signals promoting cytotoxicity and instead favor an inflammatory cytokine storm that drives HLH (77, 80–84). Although XLP1 and APDS patients fail to control EBV infection, both patient cohorts harbor EBV-specific T cells and their CD8 T cells show normal in vitro effector functions in response to SAP-independent stimuli (82, 85). Interestingly, positive signaling for cytotoxicity induced by receptors of the SLAM family (e.g., 2B4 and NTBA) that utilize the SAP adaptor involves PI3K/AKT activity (86, 87). Thus, both APDS and XLP1 share the feature of EBV susceptibility; however, unlike XLP1 patients, APDS patients are not susceptible to HLH. We hypothesize that hyperactive PI3K T-cell intrinsically drives polyclonal senescence and prevents a cytokine storm and HLH by limiting homing, expansion, and survival of EBV-specific T cells, as described further below (Figure 1B). Indeed, T cells from APDS patients exhibit enhanced stimulation-induced apoptosis (10), which is a feature shared with patients deficient in the anti-apoptotic factor XIAP who are susceptible to EBV and HLH (88, 89). Poor survival of EBV-reactive T cells may be a common underlying feature of EBV susceptibility in both XIAP deficiency and APDS, although the HLH phenotype in XIAP deficiency is poorly understood (90, 91).
The PI3K-driven expansion of effector CD8 T cells in APDS (11, 14) raises the question of why they cannot control EBV infection. The answer might come from the differentiation state of CD8 T cells since peripheral blood T cells in APDS patients are terminally differentiated with characteristics of senescence (92) (Table 1), including low IL-2 secretion, shortened telomeres, and poor proliferative capacity. Studies in mouse tumor models have similarly shown that senescent T cells exhibit in vivo defects including reduced survival, proliferation, IL-2 production, lymphoid homing, and tumor rejection (Figure 1B) (93, 94). Replicative senescence occurs when telomere erosion that occurs with each cell division reaches a critical point, leading to irreversible cell-cycle arrest through activation of the DNA damage response that is thought to protect from cellular transformation by preventing genomic instability and infinite proliferation (95). CD8 and CD4 T-cell immunosenescence has been observed in elderly individuals (96), and numerous studies demonstrate a high correlation between T-cell aging and persistent infections (e.g., CMV, EBV and HIV) (97–99) or the development of tumors (100, 101). A closer look at CMV-specific T cells has revealed a link between aging and increased frequency of CMV-specific CD8 T cells with a senescent phenotype (102, 103), suggesting that chronic antigen stimulation might drive T-cell senescence. Consistent with this hypothesis, the expression of the telomerase reverse transcriptase (TERT) that regulates the length of telomeres drastically declines in CD8 T cells after repeated antigen stimulation and acquisition of a senescent phenotype (104). Interestingly, overexpression of TERT increases the proliferative capacity of stimulated T cells (105), and using a pharmacological activator of TERT enhances CD8 T-cell-mediated control HIV infection in vitro (106).
Thus, immunosenescence represents a plausible contributor to defective EBV control in APDS patients, as CD8 T cells might not be able to clonally expand and mount a robust and specific response against EBV despite their prominent effector phenotype (11). While repeated EBV antigen stimulation seems to be an attractive hypothesis for driving T-cell immunosenescence in APDS, patients without active herpesviruses still have a high frequency of senescent T cells (Table 1), indicating that immunosenescence is likely not restricted to antigen-specific T cells. Instead, the hyperactivation of PI3K, a signaling pathway known to play multiple roles in survival, metabolism, cell growth, and cell-cycle progression (107–109), likely drives senescence by promoting exuberant in vivo CD8 T-cell proliferation (and resulting in clinical features of lymphoproliferation). Moreover, several studies have linked increased PI3K/AKT/mTORC1 activity with senescence in immortalized and primary cells (110–115). Interestingly, studies in cells with hyperactive PI3K signaling or mTORC1 inhibition with rapamycin have led to a model in which PI3K/AKT/mTORC1 signaling plays an early role in cell senescence induction without hyperproliferation as a prerequisite (110). While this latter set of data suggests that DNA damage is not a driving factor for PI3K-dependent senescence, other studies further proposed that PI3K/AKT contributes to reactive oxygen species production to cause irreparable chromosomal damage and irreversible cell-cycle arrest (111, 116). Although it is clear that the PI3K pathway plays an important role in senescence, further investigation is required to fully understand senescence of CD8 T cells in APDS patients. As such, APDS provides an invaluable opportunity to study immunosenescence and roles for PI3K in its regulation in humans.
Thus, hyperactive PI3Kδ may drive CD8 T-cell growth, terminal differentiation, and immunosenescence, although the detailed molecular basis of T-cell senescence in APDS patients remains to be fully elucidated. This state is associated with altered CD8 T-cell functions, including decreased proliferation and increased TCR restimulation-induced cell death, that might contribute to failure of APDS patients to adequately control EBV.
Conclusion
Genomics has greatly advanced studies of PIDs (117, 118), shedding light on genes critical for human immunity. The recently solved PID called APDS highlights important roles for regulated PI3Kδ signaling in control of EBV through effects on B- and T-cell development and function.
Author Contributions
JMC and CL prepared and wrote the minireview manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the patients and their families as well as referring clinicians and their teams. The authors also thank Mr. Andrew Takeda for discussions and feedback on the manuscript and Servier Medical Art for providing visual elements in Figure 1 (https://smart.servier.com/smart_image/antibody-5/).
Funding
This work was supported by Yale University (grant number NHLBI R00HL125668) and the Anderson Postdoctoral Fellowship (Yale).
References
1. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet (1964) 1:702–3. doi:10.1016/S0140-6736(64)91524-7
2. zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, et al. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature (1970) 228:1056–8. doi:10.1038/2281056a0
3. Desgranges C, Wolf H, De-The G, Shanmugaratnam K, Cammoun N, Ellouz R, et al. Nasopharyngeal carcinoma. X. Presence of Epstein-Barr genomes in separated epithelial cells of tumours in patients from Singapore, Tunisia and Kenya. Int J Cancer (1975) 16:7–15. doi:10.1002/ijc.2910160103
4. Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod Pathol (1990) 3:377–80.
5. Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol (1992) 140:769–74.
6. Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E. Epstein-Barr virus in gastric carcinoma. Am J Pathol (1993) 143:1250–4.
7. Takada K. Epstein-Barr virus and gastric carcinoma. Mol Pathol (2000) 53:255–61. doi:10.1136/mp.53.5.255
8. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kdelta and primary immunodeficiencies. Nat Rev Immunol (2016) 16:702–14. doi:10.1038/nri.2016.93
9. Zhang KJ, Husami A, Marsh R, Jordan MB. Identification of a phosphoinositide 3-kinase (PI-3K) p110δ (PIK3CD) deficient individual. J Clin Immunol (2013) 33:671–709. doi:10.1007/s10875-013-9869-2
10. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi:10.1126/science.1243292
11. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi:10.1038/ni.2771
12. Jou ST, Chien YH, Yang YH, Wang TC, Shyur SD, Chou CC, et al. Identification of variations in the human phosphoinositide 3-kinase p110delta gene in children with primary B-cell immunodeficiency of unknown aetiology. Int J Immunogenet (2006) 33:361–9. doi:10.1111/j.1744-313X.2006.00627.x
13. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124:3923–8. doi:10.1172/jci75746
14. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211:2537–47. doi:10.1084/jem.20141759
15. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase delta syndrome-like immunodeficiency. J Allergy Clin Immunol (2016) 138:1672–80.e1610. doi:10.1016/j.jaci.2016.03.055
16. Driessen GJ, IJspeert H, Wentink M, Yntema HG, van Hagen PM, van Strien A, et al. Increased PI3K/Akt activity and deregulated humoral immune response in human PTEN deficiency. J Allergy Clin Immunol (2016) 138:1744–7.e1745. doi:10.1016/j.jaci.2016.07.010
17. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34:272–6. doi:10.1007/s10875-014-0012-9
18. Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. J Allergy Clin Immunol (2014) 134:233–6. doi:10.1016/j.jaci.2014.02.020
19. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol (2015) 35:11–4. doi:10.1007/s10875-014-0109-1
20. Kannan JA, Davila-Saldana BJ, Zhang K, Filipovich AH, Kucuk ZY. Activated phosphoinositide 3-kinase delta syndrome in a patient with a former diagnosis of common variable immune deficiency, bronchiectasis, and lymphoproliferative disease. Ann Allergy Asthma Immunol (2015) 115:452–4. doi:10.1016/j.anai.2015.08.009
21. Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol (2015) 159:33–6. doi:10.1016/j.clim.2015.04.014
22. Chiriaco M, Brigida I, Ariganello P, Di Cesare S, Di Matteo G, Taus F, et al. The case of an APDS patient: defects in maturation and function and decreased in vitro anti-mycobacterial activity in the myeloid compartment. Clin Immunol (2017) 178:20–8. doi:10.1016/j.clim.2015.12.008
23. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hulsdunker J, Eskandarian Z, et al. Activating PI3Kdelta mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2016) 183:221–9. doi:10.1111/cei.12706
24. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138:210–8.e219. doi:10.1016/j.jaci.2016.03.022
25. Martínez-Saavedra MT, García-Gomez S, Domínguez Acosta A, Mendoza Quintana JJ, Páez JP, García-Reino EJ, et al. Gain-of-function mutation in PIK3R1 in a patient with a narrow clinical phenotype of respiratory infections. Clin Immunol (2016) 173:117–20. doi:10.1016/j.clim.2016.09.011
26. Olbrich P, Lorenz M, Cura Daball P, Lucena JM, Rensing-Ehl A, Sanchez B, et al. Activated PI3Kdelta syndrome type 2: two patients, a novel mutation, and review of the literature. Pediatr Allergy Immunol (2016) 27:640–4. doi:10.1111/pai.12585
27. Petrovski S, Parrott RE, Roberts JL, Huang H, Yang J, Gorentla B, et al. Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. J Clin Immunol (2016) 36:462–71. doi:10.1007/s10875-016-0281-6
28. Rae W, Ramakrishnan KA, Gao Y, Ashton-Key M, Pengelly RJ, Patel SV, et al. Precision treatment with sirolimus in a case of activated phosphoinositide 3-kinase delta syndrome. Clin Immunol (2016) 171:38–40. doi:10.1016/j.clim.2016.07.017
29. Kuhlen M, Hönscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P, et al. De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol (2016) 162:27–30. doi:10.1016/j.clim.2015.10.008
30. Bravo García-Morato M, García-Miñaúr S, Molina Garicano J, Santos Simarro F, Del Pino Molina L, López-Granados E, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol (2017) 179:77–80. doi:10.1016/j.clim.2017.03.004
31. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol (2017) 139:597–606.e594. doi:10.1016/j.jaci.2016.06.021
32. Heurtier L, Lamrini H, Chentout L, Deau MC, Bouafia A, Rosain J, et al. Mutations in the adaptor-binding domain and associated linker region of p110delta cause activated PI3K-delta syndrome 1 (APDS1). Haematologica (2017) 102:e278–81. doi:10.3324/haematol.2017.167601
33. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110delta cause activated PI3Kdelta syndrome (APDS) in humans. J Allergy Clin Immunol (2017). doi:10.1016/j.jaci.2017.03.026
34. Wentink M, Dalm V, Lankester AC, van Schouwenburg PA, Scholvinck L, Kalina T, et al. Genetic defects in PI3Kdelta affect B-cell differentiation and maturation leading to hypogammaglobulineamia and recurrent infections. Clin Immunol (2017) 176:77–86. doi:10.1016/j.clim.2017.01.004
35. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Daub J, Goyal RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol (2017) 139:1032–5.e1036. doi:10.1016/j.jaci.2016.08.028
36. Nademi Z, Slatter MA, Dvorak CC, Neven B, Fischer A, Suarez F, et al. Hematopoietic stem cell transplant in patients with activated PI3K delta syndrome. J Allergy Clin Immunol (2017) 139:1046–9. doi:10.1016/j.jaci.2016.09.040
37. Hauck F, Magg T, Krolo A, Bilic I, Hirschmugl T, Laass M, et al. Variant PIK3R1 hypermorphic mutation and clinical phenotypes in a family with short statures, mild immunodeficiency and lymphoma. Klin Padiatr (2017) 229:113–7. doi:10.1055/s-0043-104218
38. Rae W, Gao Y, Ward D, Mattocks CJ, Eren E, Williams AP. A novel germline gain-of-function variant in PIK3CD. Clin Immunol (2017) 181:29–31. doi:10.1016/j.clim.2017.05.020
39. Saettini F, Pelagatti MA, Sala D, Moratto D, Giliani S, Badolato R, et al. Early diagnosis of PI3Kdelta syndrome in a 2 years old girl with recurrent otitis and enlarged spleen. Immunol Lett (2017) 190:279–81. doi:10.1016/j.imlet.2017.08.021
40. Goto F, Uchiyama T, Nakazawa Y, Imai K, Kawai T, Onodera M. Persistent impairment of T-cell regeneration in a patient with activated PI3K delta syndrome. J Clin Immunol (2017) 37:347–50. doi:10.1007/s10875-017-0393-7
41. Mettman D, Thiffault I, Dinakar C, Saunders C. Immunodeficiency-associated lymphoid hyperplasia as a cause of intussusception in a case of activated PI3K-delta syndrome. Front Pediatr (2017) 5:71. doi:10.3389/fped.2017.00071
42. Dornan GL, Siempelkamp BD, Jenkins ML, Vadas O, Lucas CL, Burke JE. Conformational disruption of PI3Kdelta regulation by immunodeficiency mutations in PIK3CD and PIK3R1. Proc Natl Acad Sci U S A (2017) 114:1982–7. doi:10.1073/pnas.1617244114
43. Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci (2015) 40:88–100. doi:10.1016/j.tibs.2014.12.003
44. Compagno M, Wang Q, Pighi C, Cheong TC, Meng FL, Poggio T, et al. Phosphatidylinositol 3-kinase delta blockade increases genomic instability in B cells. Nature (2017) 542:489–93. doi:10.1038/nature21406
45. Garcelon N, Neuraz A, Benoit V, Salomon R, Kracker S, Suarez F, et al. Finding patients using similarity measures in a rare diseases-oriented clinical data warehouse: Dr. Warehouse and the needle in the needle stack. J Biomed Inform (2017) 73:51–61. doi:10.1016/j.jbi.2017.07.016
46. Panikkar A, Smith C, Hislop A, Tellam N, Dasari V, Hogquist KA, et al. Impaired Epstein-Barr virus-specific neutralizing antibody response during acute infectious mononucleosis is coincident with global B-cell dysfunction. J Virol (2015) 89:9137–41. doi:10.1128/jvi.01293-15
47. Bu W, Hayes GM, Liu H, Gemmell L, Schmeling DO, Radecki P, et al. Kinetics of Epstein-Barr virus (EBV) neutralizing and virus-specific antibodies after primary infection with EBV. Clin Vaccine Immunol (2016) 23:363–9. doi:10.1128/cvi.00674-15
48. Jayasooriya S, de Silva TI, Njie-jobe J, Sanyang C, Leese AM, Bell AI, et al. Early virological and immunological events in asymptomatic Epstein-Barr virus infection in African children. PLoS Pathog (2015) 11:e1004746. doi:10.1371/journal.ppat.1004746
49. Kanekiyo M, Bu W, Joyce MG, Meng G, Whittle JR, Baxa U, et al. Rational design of an Epstein-Barr virus vaccine targeting the receptor-binding site. Cell (2015) 162:1090–100. doi:10.1016/j.cell.2015.07.043
50. Moutschen M, Léonard P, Sokal EM, Smets F, Haumont M, Mazzu P, et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine (2007) 25:4697–705. doi:10.1016/j.vaccine.2007.04.008
51. Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Léonard P, Moreels A, et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis (2007) 196:1749–53. doi:10.1086/523813
52. Martin DR, Marlowe RL, Ahearn JM. Determination of the role for CD21 during Epstein-Barr virus infection of B-lymphoblastoid cells. J Virol (1994) 68:4716–26.
53. Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, et al. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity (2000) 13:485–95. doi:10.1016/S1074-7613(00)00048-0
54. Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol (2001) 1:75–82. doi:10.1038/35095584
55. Chaganti S, Heath EM, Bergler W, Kuo M, Buettner M, Niedobitek G, et al. Epstein-Barr virus colonization of tonsillar and peripheral blood B-cell subsets in primary infection and persistence. Blood (2009) 113:6372–81. doi:10.1182/blood-2008-08-175828
56. Lee EK, Joo EH, Song KA, Choi B, Kim M, Kim SH, et al. Effects of lymphocyte profile on development of EBV-induced lymphoma subtypes in humanized mice. Proc Natl Acad Sci U S A (2015) 112:13081–6. doi:10.1073/pnas.1407075112
57. Coleman CB, Nealy MS, Tibbetts SA. Immature and transitional B cells are latency reservoirs for a gammaherpesvirus. J Virol (2010) 84:13045–52. doi:10.1128/jvi.01455-10
58. Coleman CB, McGraw JE, Feldman ER, Roth AN, Keyes LR, Grau KR, et al. A gammaherpesvirus Bcl-2 ortholog blocks B cell receptor-mediated apoptosis and promotes the survival of developing B cells in vivo. PLoS Pathog (2014) 10:e1003916. doi:10.1371/journal.ppat.1003916
59. Liu X, Cohen JI. The role of PI3K/Akt in human herpesvirus infection: from the bench to the bedside. Virology (2015) 479-480:568–77. doi:10.1016/j.virol.2015.02.040
60. Hatton OL, Harris-Arnold A, Schaffert S, Krams SM, Martinez OM. The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol Res (2014) 58:268–76. doi:10.1007/s12026-014-8496-1
61. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem (2003) 278:3694–704. doi:10.1074/jbc.M209840200
62. Furukawa S, Wei L, Krams SM, Esquivel CO, Martinez OM. PI3Kdelta inhibition augments the efficacy of rapamycin in suppressing proliferation of Epstein-Barr virus (EBV)+ B cell lymphomas. Am J Transplant (2013) 13:2035–43. doi:10.1111/ajt.12328
63. Goswami R, Gershburg S, Satorius A, Gershburg E. Protein kinase inhibitors that inhibit induction of lytic program and replication of Epstein-Barr virus. Antiviral Res (2012) 96:296–304. doi:10.1016/j.antiviral.2012.09.021
64. Oussaief L, Hippocrate A, Ramirez V, Rampanou A, Zhang W, Meyers D, et al. Phosphatidylinositol 3-kinase/Akt pathway targets acetylation of Smad3 through Smad3/CREB-binding protein interaction: contribution to transforming growth factor beta1-induced Epstein-Barr virus reactivation. J Biol Chem (2009) 284:23912–24. doi:10.1074/jbc.M109.036483
65. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi:10.1146/annurev-immunol-032414-112326
66. Cohen JI. Primary immunodeficiencies associated with EBV disease. Curr Top Microbiol Immunol (2015) 390:241–65. doi:10.1007/978-3-319-22822-8_10
67. Callan MF, Fazou C, Yang H, Rostron T, Poon K, Hatton C, et al. CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J Clin Invest (2000) 106:1251–61. doi:10.1172/jci10590
68. Ning RJ, Xu XQ, Chan KH, Chiang AK. Long-term carriers generate Epstein-Barr virus (EBV)-specific CD4(+) and CD8(+) polyfunctional T-cell responses which show immunodominance hierarchies of EBV proteins. Immunology (2011) 134:161–71. doi:10.1111/j.1365-2567.2011.03476.x
69. Naik S, Nicholas SK, Martinez CA, Leen AM, Hanley PJ, Gottschalk SM, et al. Adoptive immunotherapy for primary immunodeficiency disorders with virus-specific T lymphocytes. J Allergy Clin Immunol (2016) 137:1498–505.e1491. doi:10.1016/j.jaci.2015.12.1311
70. Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet (1995) 345:9–13. doi:10.1016/S0140-6736(95)91150-2
71. Rooney CM, Smith CA, Ng CY, Loftin SK, Sixbey JW, Gan Y, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood (1998) 92:1549–55.
72. Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood (2012) 119:2644–56. doi:10.1182/blood-2011-08-371971
73. Palendira U, Low C, Bell AI, Ma CS, Abbott RJ, Phan TG, et al. Expansion of somatically reverted memory CD8+ T cells in patients with X-linked lymphoproliferative disease caused by selective pressure from Epstein-Barr virus. J Exp Med (2012) 209:913–24. doi:10.1084/jem.20112391
74. Palendira U, Rickinson AB. Primary immunodeficiencies and the control of Epstein-Barr virus infection. Ann N Y Acad Sci (2015) 1356:22–44. doi:10.1111/nyas.12937
75. Tangye SG, Palendira U, Edwards ES. Human immunity against EBV-lessons from the clinic. J Exp Med (2017) 214:269–83. doi:10.1084/jem.20161846
76. Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol (2014) 34:772–9. doi:10.1007/s10875-014-0083-7
77. Sayos J, Wu C, Morra M, Wang N, Zhang X, Allen D, et al. The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature (1998) 395:462–9. doi:10.1038/26683
78. Coffey AJ, Brooksbank RA, Brandau O, Oohashi T, Howell GR, Bye JM, et al. Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nat Genet (1998) 20:129–35. doi:10.1038/2424
79. Nichols KE, Harkin DP, Levitz S, Krainer M, Kolquist KA, Genovese C, et al. Inactivating mutations in an SH2 domain-encoding gene in X-linked lymphoproliferative syndrome. Proc Natl Acad Sci U S A (1998) 95:13765–70. doi:10.1073/pnas.95.23.13765
80. Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med (2001) 194:235–46. doi:10.1084/jem.194.3.235
81. Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest (2009) 119:2976–89. doi:10.1172/jci39518
82. Hislop AD, Palendira U, Leese AM, Arkwright PD, Rohrlich PS, Tangye SG, et al. Impaired Epstein-Barr virus-specific CD8+ T-cell function in X-linked lymphoproliferative disease is restricted to SLAM family-positive B-cell targets. Blood (2010) 116:3249–57. doi:10.1182/blood-2009-09-238832
83. Nakajima H, Cella M, Bouchon A, Grierson HL, Lewis J, Duckett CS, et al. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. Eur J Immunol (2000) 30:3309–18. doi:10.1002/1521-4141(200011)30:11<3309::AID-IMMU3309>3.0.CO;2-3
84. Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature (2008) 455:764–9. doi:10.1038/nature07345
85. Dupré L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Aricò M, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood (2005) 105:4383–9. doi:10.1182/blood-2004-08-3269
86. Aoukaty A, Tan R. Association of the X-linked lymphoproliferative disease gene product SAP/SH2D1A with 2B4, a natural killer cell-activating molecule, is dependent on phosphoinositide 3-kinase. J Biol Chem (2002) 277:13331–7. doi:10.1074/jbc.M112029200
87. Mikhalap SV, Shlapatska LM, Berdova AG, Law CL, Clark EA, Sidorenko SP. CDw150 associates with src-homology 2-containing inositol phosphatase and modulates CD95-mediated apoptosis. J Immunol (1999) 162:5719–27.
88. Rigaud S, Fondanèche MC, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature (2006) 444:110–4. doi:10.1038/nature05257
89. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. J Clin Immunol (2015) 35:331–8. doi:10.1007/s10875-015-0141-9
90. Hatton O, Phillips LK, Vaysberg M, Hurwich J, Krams SM, Martinez OM. Syk activation of phosphatidylinositol 3-kinase/Akt prevents HtrA2-dependent loss of X-linked inhibitor of apoptosis protein (XIAP) to promote survival of Epstein-Barr virus+ (EBV+) B cell lymphomas. J Biol Chem (2011) 286:37368–78. doi:10.1074/jbc.M111.255125
91. Carter BZ, Milella M, Tsao T, McQueen T, Schober WD, Hu W, et al. Regulation and targeting of antiapoptotic XIAP in acute myeloid leukemia. Leukemia (2003) 17:2081–9. doi:10.1038/sj.leu.2403113
92. Chou JP, Effros RB. T cell replicative senescence in human aging. Curr Pharm Des (2013) 19:1680–98. doi:10.2174/138161213805219711
93. Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest (2005) 115:1616–26. doi:10.1172/jci24480
94. Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol (2006) 6:383–93. doi:10.1038/nri1842
95. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol (2013) 75:685–705. doi:10.1146/annurev-physiol-030212-183653
96. Tarazona R, DelaRosa O, Alonso C, Ostos B, Espejo J, Peña J, et al. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech Ageing Dev (2000) 121:77–88. doi:10.1016/S0047-6374(00)00199-8
97. Ouyang Q, Wagner WM, Walter S, Müller CA, Wikby A, Aubert G, et al. An age-related increase in the number of CD8+ T cells carrying receptors for an immunodominant Epstein-Barr virus (EBV) epitope is counteracted by a decreased frequency of their antigen-specific responsiveness. Mech Ageing Dev (2003) 124:477–85. doi:10.1016/S0047-6374(03)00026-5
98. Sansoni P, Vescovini R, Fagnoni FF, Akbar A, Arens R, Chiu YL, et al. New advances in CMV and immunosenescence. Exp Gerontol (2014) 55:54–62. doi:10.1016/j.exger.2014.03.020
99. Dock JN, Effros RB. Role of CD8 T cell replicative senescence in human aging and in HIV-mediated immunosenescence. Aging Dis (2011) 2:382–97.
100. Poschke I, De Boniface J, Mao Y, Kiessling R. Tumor-induced changes in the phenotype of blood-derived and tumor-associated T cells of early stage breast cancer patients. Int J Cancer (2012) 131:1611–20. doi:10.1002/ijc.27410
101. Ye J, Peng G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology (2015) 4:e994398. doi:10.4161/2162402x.2014.994398
102. Pita-Lopez ML, Gayoso I, DelaRosa O, Casado JG, Alonso C, Muñoz-Gomariz E, et al. Effect of ageing on CMV-specific CD8 T cells from CMV seropositive healthy donors. Immun Ageing (2009) 6:11. doi:10.1186/1742-4933-6-11
103. Tu W, Rao S. Mechanisms underlying T cell immunosenescence: aging and cytomegalovirus infection. Front Microbiol (2016) 7:2111. doi:10.3389/fmicb.2016.02111
104. Valenzuela HF, Effros RB. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol (2002) 105:117–25. doi:10.1006/clim.2002.5271
105. Dagarag M, Evazyan T, Rao N, Effros RB. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J Immunol (2004) 173:6303–11. doi:10.4049/jimmunol.173.10.6303
106. Fauce SR, Jamieson BD, Chin AC, Mitsuyasu RT, Parish ST, Ng HL, et al. Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J Immunol (2008) 181:7400–6. doi:10.4049/jimmunol.181.10.7400
107. Appleman LJ, van Puijenbroek AA, Shu KM, Nadler LM, Boussiotis VA. CD28 costimulation mediates down-regulation of p27kip1 and cell cycle progression by activation of the PI3K/PKB signaling pathway in primary human T cells. J Immunol (2002) 168:2729–36. doi:10.4049/jimmunol.168.6.2729
108. Cappellini A, Tabellini G, Zweyer M, Bortul R, Tazzari PL, Billi AM, et al. The phosphoinositide 3-kinase/Akt pathway regulates cell cycle progression of HL60 human leukemia cells through cytoplasmic relocalization of the cyclin-dependent kinase inhibitor p27(Kip1) and control of cyclin D1 expression. Leukemia (2003) 17:2157–67. doi:10.1038/sj.leu.2403111
109. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov (2014) 13:140–56. doi:10.1038/nrd4204
110. Astle MV, Hannan KM, Ng PY, Lee RS, George AJ, Hsu AK, et al. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: implications for targeting mTOR during malignancy. Oncogene (2012) 31:1949–62. doi:10.1038/onc.2011.394
111. Imai Y, Takahashi A, Hanyu A, Hori S, Sato S, Naka K, et al. Crosstalk between the Rb pathway and AKT signaling forms a quiescence-senescence switch. Cell Rep (2014) 7:194–207. doi:10.1016/j.celrep.2014.03.006
112. Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature (2005) 436:725–30. doi:10.1038/nature03918
113. Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J (2004) 23:212–20. doi:10.1038/sj.emboj.7600045
114. Mavrakis KJ, Zhu H, Silva RL, Mills JR, Teruya-Feldstein J, Lowe SW, et al. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev (2008) 22:2178–88. doi:10.1101/gad.1690808
115. Moral M, Segrelles C, Lara MF, Martínez-Cruz AB, Lorz C, Santos M, et al. Akt activation synergizes with Trp53 loss in oral epithelium to produce a novel mouse model for head and neck squamous cell carcinoma. Cancer Res (2009) 69:1099–108. doi:10.1158/0008-5472.can-08-3240
116. Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, et al. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell (2008) 14:458–70. doi:10.1016/j.ccr.2008.11.003
117. Lucas CL, Lenardo MJ. Identifying genetic determinants of autoimmunity and immune dysregulation. Curr Opin Immunol (2015) 37:28–33. doi:10.1016/j.coi.2015.09.001
Keywords: Activated PI3Kδ Syndrome, PASLI, PI3K/AKT/mTOR, Epstein–Barr virus, immunodeficiency, B cell, T cell
Citation: Carpier JM and Lucas CL (2018) Epstein–Barr Virus Susceptibility in Activated PI3Kδ Syndrome (APDS) Immunodeficiency. Front. Immunol. 8:2005. doi: 10.3389/fimmu.2017.02005
Received: 14 October 2017; Accepted: 26 December 2017;
Published: 16 January 2018
Edited by:
Jeffrey I. Cohen, National Institutes of Health (NIH), United StatesReviewed by:
Shigeaki Nonoyama, National Defense Medical College, JapanKohsuke Imai, Tokyo Medical and Dental University, Japan
Copyright: © 2018 Carpier and Lucas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carrie L. Lucas, Y2FycmllLmx1Y2FzQHlhbGUuZWR1