 Katie L. Reagin
Katie L. Reagin Kimberly D. Klonowski
Kimberly D. Klonowski- Department of Cellular Biology, University of Georgia, Athens, GA, United States
The yearly, cyclic impact of viruses like influenza on human health and the economy is due to the high rates of mutation of traditional antibody targets, which negate any preexisting humoral immunity. However, the seasonality of influenza infections can equally be attributed to an absent or defective memory CD8 T cell response since the epitopes recognized by these cells are derived from essential virus proteins that mutate infrequently. Experiments in mouse models show that protection from heterologous influenza infection is temporally limited and conferred by a population of tissue-resident memory (TRM) cells residing in the lung and lung airways. TRM are elicited by a diverse set of pathogens penetrating mucosal barriers and broadly identified by extravascular staining and expression of the activation and adhesion molecules CD69 and CD103. Interestingly, lung TRM fail to express these molecules, which could limit tissue retention, resulting in airway expulsion or death with concomitant loss of heterologous protection. Here, we make the case that respiratory infections uniquely evoke a form of natural immunosuppression whereby specific cytokines and cell–cell interactions negatively impact memory cell programming and differentiation. Respiratory memory is not only short-lived but most of the memory cells in the lung parenchyma may not be bona fide TRM. Given the quantity of microbes humans inhale over a lifetime, limiting cellular residence could be a mechanism employed by the respiratory tract to preserve organismal vitality. Therefore, successful efforts to improve respiratory immunity must carefully and selectively breach these inherent tissue barriers.
Introduction
Respiratory infections continue to be one of the leading causes of morbidity and mortality worldwide (1). Approximately four million annual outpatient visits are associated with viral respiratory infections, including influenza and respiratory syncytial virus (RSV) (2, 3). While a RSV vaccine remains elusive, available influenza vaccines induce specific antiviral neutralizing antibodies that recognize the external antigens hemagglutinin and neuraminidase and are protective against a homologous infection. However, host immune pressure promotes mutations of these antigens between seasons rendering the elicited antibodies and those derived from a natural infection ineffective at providing long-term cross-protection against mismatched or heterologous viral strains (3).
Activated CD8 T cells lyse infected lung epithelial cells and produce antiviral cytokines, ultimately eliminating viral reservoirs (4). In the case of influenza infection, CD8 T cells recognize epitopes derived from internal viral proteins that are conserved across 80–100% of circulating influenza strains (4–8), indicating that elicitation of CD8 T cell immunity could offer a broad range of protection against heterologous influenza infection. This protection would rely on the development of memory CD8 T cells (Tmem) capable of responding rapidly upon challenge (9). However, evidence from murine (6, 10–12) and human (13, 14) studies suggest that long-lived protective Tmem does not form in response to influenza infection. While human studies are lacking, murine models indicate that respiratory anti-influenza Tmem numbers wane coordinate with loss of heterosubtypic immunity to influenza infection (10). This observation, paired with the knowledge that humans are susceptible to seasonal infections following both natural infection and vaccination with the live, attenuated vaccine (3) shows that respiratory Tmem are not stable which we believe is partly due to the incomplete generation of a specific population of Tmem in the lung.
TRM: The Other Memory Cell
Infection with various pathogens elicits a heterogeneous Tmem pool that was previously thought to consist of predominately two distinct populations: central memory cells (TCM) located primarily in lymph nodes and effector memory cells (TEM) which circulate through lymphoid and non-lymphoid tissues (15). The preferential localization of TCM is due to expression of CD62L and CCR7 (15), whereas TEM express low levels of these molecules. Tmem develop under a transcriptional program regulated by Eomes (16) and require IL-7 signaling for their survival through T cell contraction (17). However, IL-15 and IL-2 signaling bias Tmem toward a TCM or TEM lineage, respectively (18). In many cases, TEM provide initial pathogen control at portals of entry, while TCM are positioned to broadly patrol lymph nodes (19). Indeed, TCM provide protection against systemic lymphocytic choriomeningitis virus infection (20), while TEM protect against respiratory Sendai virus challenge (21). However, often this is not a true division of labor and, even in the case of non-lymphoid infections, reactivated TCM will also contribute to the generation of new effector cells, albeit with delayed kinetics.
Subsequent studies using parabiotic mice demonstrated the existence of stationary, non-migratory populations of Tmem within the brain and small intestine, and to a lesser extent, other tissues like the lung and liver (22). These cells are now commonly referred to as tissue-resident memory cells (TRM). TRM have a core transcriptional profile that distinguishes them from their TCM and TEM counterparts (23), including expression of transcription factor Hobit (24). How TRM cells developmentally diverge from other Tmem is unclear; however, it is likely to involve early programming followed by acquisition of tissue-specific factors that promote survival and tissue retention (23, 25). In most cases, CD8+ TRM have been identified by expression CD69 and CD103 (αE integrin) which are upregulated on TRM in both humans (26, 27) and mice (28, 29). The ligand of CD103, E-cadherin, is expressed exclusively by epithelial cells and CD69 expression limits tissue egress (30, 31), suggesting these markers are responsible for locking TRM within tissues. In fact, TRM fail to develop in the intestines of CD103−/− mice, and absence of CD69 and CD103 limits TRM formation in the skin (23), indicating that upregulation of CD103 and CD69 are crucial steps for the establishment of TRM. Expression of CD103 and CD69 is regulated by TGF-β (32), which is highly expressed in mucosal sites such as the gut (33) where stable populations of TRM cells have been observed (34). In most cases, TRM are maintained through IL-7- and IL-15-mediated homeostatic proliferation (35, 36). TRM are confirmed to exist in the skin (28, 37), brain (38), liver (39), and female reproductive tract (40, 41) where they are stably maintained. TRM can persist for up to 120 days in the brain following vesicular stomatitis virus (VSV) infection (38), and skin-resident TRM are the most durable, up to a lifetime in mice following cutaneous herpes simplex virus infection (42).
While a secondary, recall response can be delayed by several days for the activation of Tmem and recruitment of new effectors to the infection site, TRM respond immediately to pathogen re-exposure (12). Upon antigen re-encounter, TRM produce IFN-γ (9) to recruit circulating TEM and other immune cells from the blood (43). In addition, TRM can directly kill target cells ex vivo (44), suggesting a cytotoxic potential. TRM have been shown to mediate long-term protection in vivo to infections in the intestine (34), female reproductive tract (40, 41), brain (45), and skin (28, 37). Regarding the latter, the smallpox vaccine, administered by skin scarification, generated Tmem which survived for decades (46). While the specific role of TRM in the success of this vaccine is unclear, mice vaccinated via scarification of recombinant vaccinia virus (VacV) generate skin-resident TRM that mediate protection against subsequent VacV infection (47). However, not every infection generates stable Tmem pools. While TRM cells populate the lung and lung airways after influenza infection (12), protection between influenza seasons following natural infection or vaccination with the live-attenuated vaccine is lost (3), suggesting TRM responses may be uniquely regulated in the lung.
TRM in the Lung
TRM cells exist within the lung in two distinct compartments: the lung airways and the lung parenchyma. Influenza-specific airway-resident TRM are CD11aloCXCR3hi (48, 49) and can be isolated by bronchoalveolar lavage. It is estimated that anti-influenza TRM in the lung airways have a half-life of only 14 days, and for some period of time are continually replenished from the circulating TEM pool (48). Interestingly, airway TRM have a low cytolytic capacity and fail to proliferate upon antigen re-encounter but rapidly produce antiviral cytokines such as IFN-γ (44). TRM embedded in the lung parenchyma are CD11ahiCXCR3lo, highly cytolytic and undergo rapid proliferation after antigen re-exposure (44). We have known for some time that regional Tmem are responsible for limited heterologous immunity after respiratory infection (10). A careful study of the kinetics of Tmem decay after Sendai and influenza virus infections demonstrated a rapid decline in Tmem numbers in the lung and lung airways by 90 days postinfection. Importantly, this loss of influenza-specific Tmem in the lung coincided with loss of heterosubtypic immunity to influenza infection (10). The attrition of influenza-specific cells is restricted to the lung, as splenic memory cell numbers do not decline, indicating this is likely loss of the TEM or TRM pools. Subsequent experiments demonstrated that airway CD103+ cells are responsible for protection against a secondary, heterologous virus challenge. However, this pool declines rapidly after infection and is undetectable within 7 months postinfection (12), in part due to the inhospitable environment of the lung airways.
TRM in the airways reside at the frontline, adjacent to influenza-susceptible epithelial cells. However, lung parenchymal TRM and circulating TEM are also available within the lung tissue and can serve as a secondary line of defense. Recent evidence indicates that over time, TRM cells in the lung airways wane and are replaced by circulating TEM cells; however, these TEM also decline and lose the ability to convert to TRM (50). This, coupled with a loss of TRM in the lung parenchyma, results in a gradual decline in the overall TRM population in the lung. Decline in the lung parenchymal TRM pool could be due to increased cell death, limited proliferation, or emigration. Unlike TRM in other sites (28, 34, 38), most lung Tmem do not undergo homeostatic proliferation (50, 51). However, a small pool is replenished from proliferating Tmem that have recently emigrated from secondary lymphoid tissues (50). In addition, there is no evidence that TRM cells in the airways egress from the lung or re-enter circulation (48). Therefore, we propose that Tmem embedded in the lung tissue are either eventually lost to the airways or do not represent a bona fide, protective TRM pool. Our opinion that lung parenchymal TRM do not exist is based on two observations. The first is that few Tmem truly penetrate into the tissue and the second is that those Tmem that do, are not CD103+CD69+.
Many techniques can identify TRM (Table 1) and each has pros and cons. We believe that the most effective methodology is the combination of two of these approaches: intravascular staining and CD103/69 phenotyping. Intravascular staining distinguishes between cells circulating through the blood and those embedded within a tissue (52). Approximately 99% of the TRM within the epithelial layer of the small intestine are protected from the intravascular staining (Figure 1) (52, 53), validating similar results observed in parabiotic mice (22). In contrast, the majority of the memory cells within the lung parenchyma 35 days after respiratory infection with either influenza, VSV, or Listeria monocytogenes are part of the circulating TEM pool, with only 10–20% of the cells in the lung parenchyma truly within the tissue (52) (Figure 1). These data do contrast with other respiratory infections that are skewed toward the upper respiratory tract (54) or are chronic (55), both cases generating CD103+CD69+ TRM. With regard to the latter study, it is possible that persistent antigen and inflammation is required for the successful development of TRM within this site. In addition to antigen access, antigen competition can regulate TRM populations at the clonal level (56). Moreover, many studies identify lung TRM via CD103 and CD69 expression on isolated lymphocytes (57, 58), independent of intravascular staining. However, expression of these markers does not always correlate with tissue residency. For example, some TRM cells in the lamina propria of the gut (59), the liver (39), and the brain (60) are CD103−, and human splenic Tmem can be CD69+ (26). In fact, less than 30% of the IV protected TRM cells isolated from the lung parenchyma express CD69 and CD103 (Figure 1) compared to TRM isolated from other mucosal sites, where expression ranges from approximately 50–99% (59). Therefore, Tmem located in the lung parenchyma after respiratory infection lack one of the key attributes associated with bona fide TRM, expression of CD69 and CD103. CD103− TRM in the brain are maintained for a few months (60) which may be due to modified tissue localization and enhanced access to IL-15. However, lung parenchymal TRM are maintained independent of IL-15 (61), at least in the short-term, so gained proximity to IL-15 may not matter. However, acquisition of other survival signals dependent on CD103 positioning may be altered, leading to either cell death or assimilation into the TEM pool. Coupled with loss of airway-associated TRM, this situation leaves circulating TEM as the only viable responders. Whether the TEM temporally supplementing the TRM pool are CX3CR1hi and classified as the recently described “peripheral” memory cells (TpM) (62, 63) is unknown. Nonetheless, as TEM induced from respiratory infection decline over time (64), hosts will then be susceptible to infection. Therefore, an inferior CD69+CD103+ TRM response underpins loss of heterosubtypic immunity in the lung and raises the question of why long-lived, stable TRM does not form in the lung following respiratory infection.

Table 1. Common methods used for the identification of TRM cells in peripheral sites.
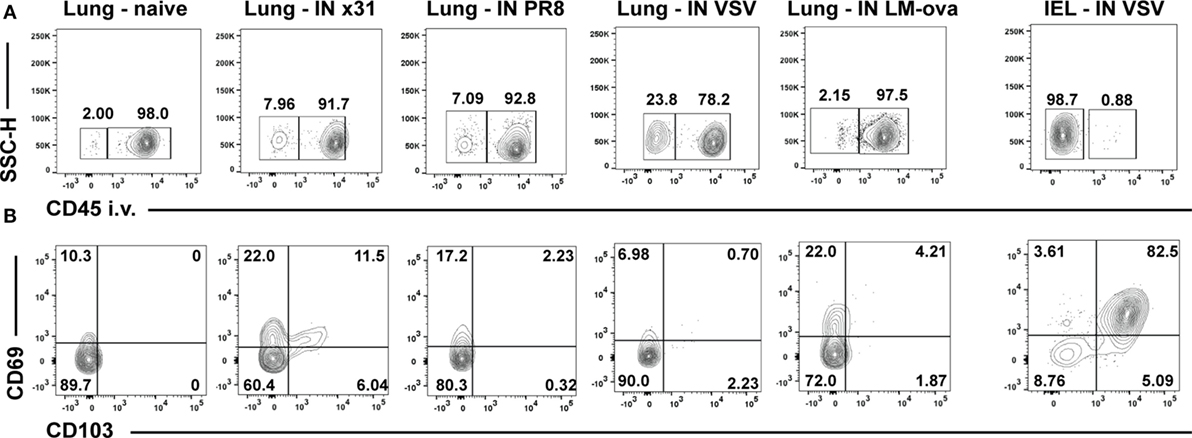
Figure 1. Lung TRM cells express low levels of CD69 and CD103 after respiratory infection with various pathogens. Age- and sex-matched C57/BL6 mice were infected intranasally with a 50-μl inoculum of PBS alone (naïve) or containing sublethal doses of either influenza (103 pfu of strain HKx31 and 10 pfu of PR8), VSV (104 pfu, Indiana strain), or Listeria monocytogenes expressing the recombinant ovalbumin (ova) (LM-ova) (104 cfu). One group of mice was additionally intravenously (i.v.) infected with 104 pfu VSV. Animals were sacrificed 35 days later and TRM assessed by intravascular staining. Briefly, mice were injected i.v. with 3 μg FITC labeled αCD45 antibody 3 min before sacrifice, lungs or small intestine were harvested, and lymphocytes isolated as previously described (22). (A) Representative i.v. staining of lymphocytes isolated from the lungs or intraepithelial lymphocytes (IEL) of naïve mice or following the indicated infections. All samples were first gated on CD8+CD44+ memory phenotype cells and gates in (A) were set by FMO controls within each experiment. For the influenza and VSV-infected animals, an additional MHC-class I tetramer gate was applied to identify antigen-specific CD8 T cells [as in Ref. (61)]. Numbers in the right box represent the frequency of the gated cells that stained with the i.v. injected antibody (αCD45-FITC+) and are in the vasculature (IV+). (B) Representative CD103 and CD69 staining of IV− [resident cells, left box in (A)] cells from the various infections.
The Respiratory Environment Subverts the Development of TRM
As the lung is exposed to both infectious agents and innocuous environmental antigens, immune responses must be tightly controlled to prevent immunopathology (25). Similar regulation is also required in the liver and brain, additional tolerogenic sites. In part, this regulation is accomplished via tissue segregation. Indeed, liver TRM are exclusively segregated from tissue stroma, retained within the sinusoids (74), whereas brain TRM are preferentially localized in the meninges and perivascular areas (60), sequestered from the parenchyma. The lung is no different, with the development of BAL TRM and parenchymal TRM. However, unlike TRM in the brain and liver, BAL TRM are directly exposed to the external environment and easily lost, whereas the lung parenchymal TRM are imbedded in the parenchyma and require an additional level of regulation to prevent immunopathology.
One potential mechanism is through altered mammalian target of rapamycin (mTOR) signaling within the respiratory tract. mTOR is responsible for regulating cellular metabolism, proliferation, and differentiation (75), including memory cell development (76). High levels of mTOR activation reduces the total number of antigen-specific cells expressing CD127, required for the development of memory precursor cells (77), and the subsequent TCM pool (76). While reducing mTOR signaling with rapamycin reverses the effects on TCM (76), TRM formation and retention within the intestinal mucosa was also increased via enhanced expression of gut-specific homing molecules (78). To date, no study has linked reduced mTOR signaling to enhance lung homing and/or respiratory TRM formation. However, evidence from viral respiratory infection models support a role for mTOR in TRM formation. Rapamycin treatment during influenza infection increases the total number of antigen-specific CD8 Tmem circulating in the blood (79) similar to studies in the gut (78). In addition, activated CD8 T cells isolated from infants infected with RSV and treated with rapamycin during in vitro re-stimulation express higher levels of CD127 compared to those cells stimulated without rapamycin. Rapamycin treatment also enhanced the effector response of RSV-specific cells by increasing their proliferation and production of granzyme B (80). While increased infiltration of RSV-specific effector cells into the lung may be important for viral clearance, this can also result in damaging pathology within the lung tissue itself. This indicates that perhaps careful regulation of mTOR signaling during respiratory infection is important for limiting potential immunopathology (80) and Tmem development; however, further studies are needed to directly implicate mTOR as a player in lung TRM formation.
The lung environment is inherently immunosuppressive. In the steady state, a large reservoir of Tregs populate this tissue and contribute to significant IL-10 post-influenza infection (81). Moreover, bronchial and alveolar epithelial cells are known to express moderate levels of the programmed death-1 (PD-1) ligands PD-L1 and PD-L2, both of which are significantly upregulated upon RSV (82) and influenza infection (83). In addition, antigen-specific CD8 T cells infiltrating the lung following RSV and influenza infection have an increased expression of PD-1 (83, 84). Both IL-10 and PD-1 signaling can modulate CD8 T cell activation both individually (85, 86) and cooperatively (87) by tuning TCR signaling. IL-10 suppresses IL-12 signaling which, like PD-1 signaling, activates mTOR. However, PD-1 signaling is not exclusively through mTOR and can affect transcriptional networks and other cell cycle regulators which can impact the fate and function of CD8+ T cells (86). Memory phenotype cells isolated from PD-1−/− versus wild-type mice are preferentially TEM (88). Reciprocal adoptive transfer experiments demonstrated this bias was inherent to the T cell. As PD-1 blockade during RSV infection results in enhanced inflammation and lung injury, PD-1/PD-L1 expression in the respiratory tract may serve to limit the expanding CD8+ T cell pool, thereby restricting developing TRM. Thus, while enhanced PD-1 expression within the respiratory tract may be important for regulating inflammation, this may create an environment that is inhospitable to the formation of TRM.
It is also possible that respiratory infections alter TRM programming via inhibition of CD103 and CD69 expression, which negatively affects the formation and/or retention of TRM cells in the respiratory tract. Constitutive expression of TGF-β in mucosal sites such as the gut (33) is crucial for the development of long-lived TRM through induction of CD103 expression (89). Epithelial cells also provide survival signals such as IL-15 (90), thus high CD103 expression may not only facilitate TRM retention but aid in their development or survival via tissue positioning. However, high levels of TGF-β in the respiratory tract can be detrimental, leading to the development of cystic fibrosis within the lung (91). Although TGF-β expression is induced by influenza infection (92, 93), it may only be transiently expressed to limit immunopathology, albeit at the expense of TRM formation. In fact, the TRM in peripheral sites can cause semi-permanent scarring in tissues that worsens after TRM re-activation and production of IFN-γ in situ (94). Since high levels of IFN-γ production (95), in addition to scarring and fibrosis in the lung, can cause respiratory failure (96), the retention of TRM long term may be inherently limited to maintain host fitness. If this is the case, promoting TRM formation within the respiratory tract could have severe consequences for host respiratory health. Therefore, by reducing TGF-β, and coordinately CD103 expression, lung memory precursor cells would perhaps be ill positioned to receive homeostatic signals responsible for the development, survival, and/or retention of TRM and could be either be lost or assimilated into the TEM pool.
While airway-resident TRM cells confer protection against secondary influenza infection, they rapidly wane, leaving only parenchyma resident TRM and circulating TEM to maintain protection against subsequent infection. However, TEM also wane over time (64) and the formation of bona fide TRM in the lung parenchyma is limited (Figure 1). These incomplete memories leave the host susceptible to recurring influenza infection. We believe the lung evokes a form of natural immunosuppression whereby inhibitory signals in the site protect the host from debilitating tissue damage while simultaneously suppressing the formation of bona fide TRM within the lung tissue. While the exact mechanisms that underlie altered TRM formation within the respiratory tract are still not fully understood, future efforts to improve the maintenance and stability of this population must bear caution due to potentially negative, long-term effects on the host. Moreover, in developing vaccines against respiratory pathogens, it will be important to identify strategies that will prevent re-infection with respiratory viruses without compromising host respiratory health.
Ethics Statement
All animal studies were conducted under guidelines approved by the Institutional Animal Care and Use Committee of the University of Georgia.
Author Contributions
Both KK and KR conceived and wrote the perspective.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Special thanks to Dr. Hillary Shane for critical reading of the manuscript.
Funding
This work was sponsored by NIH grant AI131093 to KDK.
References
2. Haynes AK, Prill MM, Iwane MK, Gerber SI. Respiratory syncytial virus – United States, July 2012–June 2014, National Center for Immunization and Respiratory Diseases, CDC. MMWR Morb Mortal Wkly Rep (2014) 63:1133–6.
3. Grohskopf LA, Sokolow LZ, Broder KR, Olsen SJ, Karron RA, Jernigan DB, et al. Prevention and control of seasonal influenza with vaccines. MMWR Recomm Rep (2016) 65:1–54. doi:10.15585/mmwr.rr6505a1
4. Yewdell JW, Bennink JR, Smith GL, Moss B. Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc Natl Acad Sci U S A (1985) 82:1785–9. doi:10.1073/pnas.82.6.1785
5. La Gruta NL, Turner SJ. T cell mediated immunity to influenza: mechanisms of viral control. Trends Immunol (2014) 35:396–402. doi:10.1016/j.it.2014.06.004
6. Liang S, Mozdzanowska K, Palladino G, Gerhard W. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J Immunol (1994) 152:1653–61.
7. Bush RM, Bender CA, Subbarao K, Cox JJ, Fitch WM. Predicting the evolution of human influenza A. Science (1999) 286:183–5. doi:10.1126/science.286.5446.1921
8. Heiny AT, Miotto O, Srinivasan KN, Khan AM, Zhang GL, Brusic V, et al. Evolutionarily conserved protein sequences of influenza a viruses, avian and human, as vaccine targets. PLoS One (2007) 2:e1190. doi:10.1371/journal.pone.0001190
9. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41:886–97. doi:10.1016/j.immuni.2014.12.007
10. Hogan RJ, Usherwood EJ, Zhong W, Roberts AA, Dutton RW, Harmsen AG, et al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol (2001) 166:1813–22. doi:10.4049/jimmunol.166.3.1813
11. Chang J, Braciale TJ. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat Med (2002) 8:54–60. doi:10.1038/nm0102-54
12. Wu T, Hu Y, Lee Y-TY-T, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol (2014) 95:215–24. doi:10.1189/jlb.0313180
13. Wagar LE, Rosella L, Crowcroft N, Lowcock B, Drohomyrecky PC, Foisy J, et al. Humoral and cell-mediated immunity to pandemic H1N1 influenza in a Canadian cohort one year post-pandemic: implications for vaccination. PLoS One (2011) 6:e28063. doi:10.1371/journal.pone.0028063
14. Hillaire MLB, van Trierum SE, Bodewes R, van Baalen CA, van Binnendijk RS, Koopmans MP, et al. Characterization of the human CD8+ T cell response following infection with 2009 pandemic influenza H1N1 virus. J Virol (2011) 85:12057–61. doi:10.1128/JVI.05204-11
15. Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi:10.1038/44385
16. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, et al. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J Immunol (2010) 185:4988–92. doi:10.4049/jimmunol.1002042
17. Schluns KS, Lefrançois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol (2003) 3:269–79. doi:10.1038/nri1052
18. Obar JJ, Lefrançois L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol (2010) 185:263–72. doi:10.4049/jimmunol.1000492
19. Roberts AD, Ely KH, Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med (2005) 202:123–33. doi:10.1084/jem.20050137
20. Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, et al. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol (2003) 4:225–34. doi:10.1038/ni889
21. Roberts AD, Woodland DL. Cutting edge: effector memory CD8+ T cells play a prominent role in recall responses to secondary viral infection in the lung. J Immunol (2004) 172:6533–7. doi:10.4049/jimmunol.172.11.6533
22. Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrançois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20:551–62. doi:10.1016/S1074-7613(04)00103-7
23. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol (2013) 14:1294–301. doi:10.1038/ni.2744
24. Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352:459–63. doi:10.1126/science.aad2035
25. Shane HL, Klonowski KD. Every breath you take: the impact of environment on resident memory CD8 T cells in the lung. Front Immunol (2014) 5:320. doi:10.3389/fimmu.2014.00320
26. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJC, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38:187–97. doi:10.1016/j.immuni.2012.09.020
27. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (TRM) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6:e16245. doi:10.1371/journal.pone.0016245
28. Jiang X, Clark RA, Luzheng L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature (2012) 483:227–31. doi:10.1038/nature10851
29. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol (2006) 176:2079–83. doi:10.4049/jimmunol.176.4.2079
30. Schön MP, Arya A, Murphy EA, Adams CM, Strauch UG, Agace WW, et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J Immunol (1999) 162:6641–9.
31. Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14:1285–93. doi:10.1038/ni.2745
32. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188:4866–75. doi:10.4049/jimmunol.1200402
33. Koyama SY, Podolsky DK. Differential expression of transforming growth factors alpha and beta in rat intestinal epithelial cells. J Clin Invest (1989) 83:1768–73. doi:10.1172/JCI114080
34. Masopust D, Choo D, Vezys V, Wherry JE, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med (2010) 207:553–64. doi:10.1084/jem.20090858
35. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol (2003) 4:1191–8. doi:10.1038/ni1009
36. Schluns KS, Williams K, Ma A, Zheng XX, Lefrançois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol (2002) 168:4827–31. doi:10.4049/jimmunol.168.10.4827
37. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol (2009) 10:524–30. doi:10.1038/ni.1718
38. Wakim LM, Woodward-davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107:17872–9. doi:10.1073/pnas.1010201107
39. Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-resident memory CD8+ T cells form a front-line defense against malaria liver-stage infection. Immunity (2016) 45:889–902. doi:10.1016/j.immuni.2016.08.011
40. Shin H, Iwasaki A. A vaccine strategy protects against genital herpes by establishing local memory T cells. Nature (2012) 491:463–7. doi:10.1038/nature11522
41. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A (2012) 109:7037–42. doi:10.1073/pnas.1202288109
42. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A (2014) 111:5307–12. doi:10.1073/pnas.1322292111
43. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8+ T cells. Nat Immunol (2013) 14:509–13. doi:10.1038/ni0813-876c
44. McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-γ production. J Immunol (2015) 195:203–9. doi:10.4049/jimmunol.1402975
45. Wakim LM, Woodward-davis A, Liu R, Hu Y, Smyth G, Bevan MJ. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189:1–21. doi:10.4049/jimmunol.1201305
46. Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity (2008) 28:710–22. doi:10.1016/j.immuni.2008.02.020
47. Hobbs SJ, Osborn JF, Nolz JC. Activation and trafficking of CD8+ T cells during viral skin infection: immunological lessons learned from vaccinia virus. Curr Opin Virol (2017) 28:12–9. doi:10.1016/j.coviro.2017.10.001
48. Ely KH, Cookenham T, Roberts AD, Woodland DL. Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol (2006) 176:537–43. doi:10.4049/jimmunol.176.1.537
49. Slutter B, Pewe LL, Kaech SM, Harty JT. Lung airway-surveilling CXCR3hi memory CD8+ T cells are critical for protection against influenza A virus. Immunity (2013) 39:319–35. doi:10.1016/j.immuni.2013.09.013
50. Hogan RJ, Cauley LS, Ely KH, Cookenham T, Roberts AD, Brennan JW, et al. Long-term maintenance of virus-specific effector memory CD8+ T cells in the lung airways depends on proliferation. J Immunol (2002) 169:4976–81. doi:10.4049/jimmunol.169.9.4976
51. Ely KH, Roberts AD, Woodland DL. Cutting edge: effector memory CD8+ T cells in the lung airways retain the potential to mediate recall responses. J Immunol (2003) 171:3338–42. doi:10.4049/jimmunol.171.7.3338
52. Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol (2012) 189:2702–6. doi:10.4049/jimmunol.1201682
53. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161:737–41. doi:10.1016/j.cell.2015.03.031
54. Pizzolla A, Nguyen THO, Smith JM, Brooks AG, Kedzieska K, Heath WR, et al. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci Immunol (2017) 2:1–14. doi:10.1126/sciimmunol.aam6970
55. Perdomo C, Zedler U, Kühl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. MBio (2016) 7:1–11. doi:10.1128/mBio.01686-16
56. Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, König P-A, Tao S, et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med (2016) 213:3075–86. doi:10.1084/jem.20160888
57. Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Hu Y, Cauley LS, et al. CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity (2014) 41:633–45. doi:10.1016/j.immuni.2014.09.007
58. Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol (2014) 7:501–10. doi:10.1038/mi.2013.67
59. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol (2015) 16:406–14. doi:10.1038/ni.3108
60. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med (2016) 213:1571–87. doi:10.1084/jem.20151916
61. Verbist KC, Field MB, Klonowski KD. IL-15 independent maintenance of mucosally generated memory CD8 T cells. J Immunol (2011) 186:6667–71. doi:10.4049/jimmunol.1004022
62. Böttcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Höchst B, et al. Functional classification of memory CD8+ T cells by CX3CR1 expression. Nat Commun (2015) 6:8306. doi:10.1038/ncomms9306
63. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity (2016) 45:1270–84. doi:10.1016/j.immuni.2016.10.018
64. Slütter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol (2017) 2:eaag2031. doi:10.1126/sciimmunol.aag2031
65. Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc (2014) 9:209–22. doi:10.1038/nprot.2014.005
66. Ono S, Egawa G, Kabashima K. Regulation of blood vascular permeability in the skin. Inflamm Regen (2017) 37:11. doi:10.1186/s41232-017-0042-9
67. Tabrizi M, Bornstein GG, Suria H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J (2010) 12:33–43. doi:10.1208/s12248-009-9157-5
68. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med (2016) 213:951–66. doi:10.1084/jem.20151855
69. Bingaman AW, Patke DS, Mane VR, Ahmadzadeh M, Ndejembi M, Bartlett ST, et al. Novel phenotypes and migratory properties distinguish memory CD4 T cell subsets in lymphoid and lung tissue. Eur J Immunol (2005) 35:3173–86. doi:10.1002/eji.200526004
70. Benechet AP, Menon M, Khanna KM. Visualizing T cell migration in situ. Front Immunol (2014) 5:1–12. doi:10.3389/fimmu.2014.00363
71. Kamran P, Sereti K-I, Zhao P, Ali SR, Weissman IL, Ardehali R. Parabiosis in mice: a detailed protocol. J Vis Exp (2013) 80:e50556. doi:10.3791/50556
72. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med (2016) 213(13):3057–73. doi:10.1084/jem.20160938
73. Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology (2008) 71:1261–7. doi:10.1212/01.wnl.0000327609.57688.ea
74. McNamara HA, Cai Y, Wagle MV, Sontani Y, Roots CM, Miosge LA, et al. Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol (2017) 2:eaaj1996. doi:10.1126/sciimmunol.aaj1996
75. Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol (2013) 4:1–11. doi:10.3389/fimmu.2013.00020
76. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T cell differentiation. Nature (2009) 460:108–12. doi:10.1038/nature08155
77. Huster KM, Busch V, Schiemann M, Linkemann K, Kerksiek KM, Wagner H, et al. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc Natl Acad Sci U S A (2004) 101:5610–5. doi:10.1073/pnas.0308054101
78. Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol (2014) 193:2067–71. doi:10.4049/jimmunol.1400074
79. Keating R, Hertz T, Wehenkel M, Harris TL, Edwards BA, McClaren JL, et al. The kinase mTOR modulates the antibody response to provide cross-protective immunity to lethal infection with influenza virus. Nat Immunol (2013) 14:1266–76. doi:10.1038/ni.2741
80. de Souza A, de Freitas DN, de Freitas KE, Antuntes Fernandes M, D’Avila da Cunha JL, Antunes Fernandes R, et al. Respiratory syncytial virus induces phosphorylation of mTOR at ser2448 in CD8 T cells from nasal washes of infected infants. Clin Exp Immunol (2016) 183:248–57. doi:10.1111/cei.12720
81. Bedoya F, Cheng G-S, Leibow A, Zakhary N, Weissler K, Garcia V, et al. Viral antigen induces differentiation of Foxp3+ natural regulatory T cells in influenza virus-infected mice. J Immunol (2013) 190:6115–25. doi:10.4049/jimmunol.1203302
82. Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A, Johnston SL. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis (2006) 193:404–12. doi:10.1086/499275
83. McNally B, Ye F, Willette M, Flaño E. Local blockade of epithelial PDL-1 in the airways enhances T cell function and viral clearance during influenza virus infection. J Virol (2013) 87:12916–24. doi:10.1128/JVI.02423-13
84. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol (1996) 8:765–72. doi:10.1093/intimm/8.5.765
85. Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief Funct Genomics (2013) 12:489–98. doi:10.1093/bfgp/elt028
86. Riley JL. PD-1 signaling in primary T cells. Immunol Rev (2009) 229:114–25. doi:10.1111/j.1600-065X.2009.00767.x
87. Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med (2010) 16:452–9. doi:10.1038/nm.2106
88. Charlton JJ, Chatzidakis I, Tsoukatou D, Boumpas DT, Garinis GA, Mamalaki C. Programmed death-1 shapes memory phenotype CD8 T cell subsets in a cell-intrinsic manner. J Immunol (2013) 190:6104–14. doi:10.4049/jimmunol.1201617
89. Zhang N, Bevan M. Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi:10.1016/j.immuni.2013.08.019
90. Iijima N, Iwasaki A. Tissue instruction for migration and retention of TRM cells. Trends Immunol (2015) 36:556–64. doi:10.1016/j.it.2015.07.002
91. Wilson M, Wynn T. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol (2009) 2:103–21. doi:10.1038/mi.2008.85
92. Schultz-Cherry S, Hinshaw VS. Influenza virus neuraminidase activates latent transforming growth factor beta. J Virol (1996) 70:8624–9.
93. Carlson CM, Turpin EA, Moser LA, O’Brien KB, Cline TD, Jones JC, et al. Transforming growth factor-β: Activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog (2010) 6:e1001136. doi:10.1371/journal.ppat.1001136
94. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med (2015) 7:269rv1. doi:10.1126/scitranslmed.3010641
95. Yao S, Jiang L, Moser EK, Jewett LB, Wright J, Du J, et al. Control of pathogenic effector T-cell activities in situ by PD-L1 expression on respiratory inflammatory dendritic cells during respiratory syncytial virus infection. Mucosal Immunol (2014) 8:746–59. doi:10.1038/mi.2014.106
Keywords: respiratory immunity, influenza infection, CD8+ T cells, CD8 memory, heterologous immunity, tissue-resident memory cells
Citation: Reagin KL and Klonowski KD (2018) Incomplete Memories: The Natural Suppression of Tissue-Resident Memory CD8 T Cells in the Lung. Front. Immunol. 9:17. doi: 10.3389/fimmu.2018.00017
Received: 16 November 2017; Accepted: 04 January 2018;
Published: 22 January 2018
Edited by:
Steven Varga, University of Iowa, United StatesReviewed by:
Sara Hamilton, University of Minnesota, United StatesWolfgang Kastenmüller, University of Würzburg, Germany
Copyright: © 2018 Reagin and Klonowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kimberly D. Klonowski, a2xvbm93c2tAdWdhLmVkdQ==