 Simon A. Babayan1*
Simon A. Babayan1* Wei Liu1
Wei Liu1 Graham Hamilton2Elizabeth Kilbride1Evelyn C. Rynkiewicz3Melanie Clerc3
Graham Hamilton2Elizabeth Kilbride1Evelyn C. Rynkiewicz3Melanie Clerc3 Amy B. Pedersen3
Amy B. Pedersen3
- 1Institute of Biodiversity, Animal Health & Comparative Medicine, University of Glasgow, Glasgow, United Kingdom
- 2Glasgow Polyomics, Glasgow, United Kingdom
- 3Institute of Evolutionary Biology and Centre for Immunity, Infection and Evolution, School of Biological Sciences, University of Edinburgh, Edinburgh, United Kingdom
Parasitic helminths are extremely resilient in their ability to maintain chronic infection burdens despite (or maybe because of) their hosts’ immune response. Explaining how parasites maintain these lifelong infections, identifying the protective immune mechanisms that regulate helminth infection burdens, and designing prophylactics and therapeutics that combat helminth infection, while preserving host health requires a far better understanding of how the immune system functions in natural habitats than we have at present. It is, therefore, necessary to complement mechanistic laboratory-based studies with studies on wild populations and their natural parasite communities. Unfortunately, the relative paucity of immunological tools for non-model species has held these types of studies back. Thankfully, recent progress in high-throughput ‘omics platforms provide powerful and increasingly practical means for immunologists to move beyond traditional lab-based model organisms. Yet, assigning both metabolic and immune function to genes, transcripts, and proteins in novel species and assessing how they interact with other physiological and environmental factors requires identifying quantitative relationships between their expression and infection. Here, we used supervised machine learning to identify gene networks robustly associated with burdens of the gastrointestinal nematode Heligmosomoides polygyrus in its natural host, the wild wood mice Apodemus sylvaticus. Across 34 mice spanning two wild populations and across two different seasons, we found 17,639 transcripts that clustered in 131 weighted gene networks. These clusters robustly predicted H. polygyrus burden and included well-known effector and regulatory immune genes, but also revealed a number of genes associated with the maintenance of tissue homeostasis and hematopoiesis that have so far received little attention. We then tested the effect of experimentally reducing helminth burdens through drug treatment on those putatively protective immune factors. Despite the near elimination of H. polygyrus worms, the treatment had surprisingly little effect on gene expression. Taken together, these results suggest that hosts balance tissue homeostasis and protective immunity, resulting in relatively stable immune and, consequently, parasitological profiles. In the future, applying our approach to larger numbers of samples from additional populations will help further increase our ability to detect the immune pathways that determine chronic gastrointestinal helminth burdens in the wild.
Introduction
Chronic helminth infections challenge our understanding of how the immune system functions. In natural populations, while individuals are continually exposed to helminth parasites, there is substantial variability in infection burdens, with some consistently showing no active infection (1, 2). This suggests that individuals can potentially control infection. In addition, while anthelminthics are commonly employed to reduce helminth burdens, after drug clearance, infections typically return to their initial burdens (3–5), suggesting that the immune system does not or cannot easily acquire complete protection against helminths. Such observations have lead to the hypothesis that hosts must balance the costs of helminth infection with the immunopathology and/or protein-energy-related costs associated with eliminating these parasites (6–8). If this trade-off exists, it suggests that the host can regulate the intensity of its immune attack on the parasites, and, alternatively, that these parasites can avoid and suppress host immune responses (9, 10). While these ideas have received substantial theoretical and experimental support, the mechanisms that underlie chronic helminth infection dynamics “in the real world,” and whether or how the balance of resistance and susceptibility might change over the lifetime of an individual, either naturally or in response to vaccination, remain very poorly understood. We suggest that addressing these questions is necessary for the development of more effective and sustainable treatment and immunization against parasitic helminths, which are the leading cause of productivity loss in livestock (11–13) and remain a substantial agent of poverty in the developing world (14, 15). It is, therefore, paramount to study hosts in their natural environment, outside of the controlled laboratory, where the data may be messy, but where relationships between hosts and their natural parasite communities are the result of millions of years of coevolution and more likely to be biomedically relevant.
Identifying protective immune mechanisms in inherently variable natural populations warrants relatively large sample sizes, the possibility to manipulate parasitological, physiological, and environmental parameters, and the ability to monitor individuals repeatedly over time. The wood mouse Apodemus sylvaticus has been extensively studied by disease biologists since the 1960s (16, 17) and is now increasingly used for immunological studies (18, 19). It offers many of the features that originally made small rodents attractive experimental models (i.e., affordable, easy to maintain and handle, prolific breeding, and short time to maturity) and adds features that make it an ecologically and epidemiologically sound model for mammalian, including human, immunology, and parasitology (i.e., high diversity and prevalence of parasites, good trapability, large population sizes, genetic relatedness to Mus musculus). Indeed, A. sylvaticus harbors a diverse and prevalent community of parasite and pathogens that closely resembles those found in larger mammals, including humans (20–24), and domestic animals (25). Importantly, unlike Mus musculus which is naturally infected by very few gastrointestinal helminth parasites (26, 27), A. sylvaticus is the natural host of the nematode Heligmosomoides polygyrus, which is routinely found in >50% of wild wood mice (28) and is closely related to H. bakeri (29), a species extensively studied as a model of human gastrointestinal helminths that is known for its ability to suppress the immune system of its host (30). In this study, we therefore used H. polygyrus as a model for chronic endemic helminthiasis, and aimed to identify immune networks that regulate infection burden in a natural host–helminth system. Due to the preponderance of the laboratory mouse model in immunology, there are few, if any, reagents developed and optimized for non-model organisms (31). We, therefore, utilized a transcriptomics approach. While the genome itself provides invaluable information about immune resistance to infection (32–34), messenger RNA expression profiles provide a time- and context-sensitive picture of the immune system as it responds to antigenic stimuli. Moreover, unlike PCR and other candidate gene-driven approaches, transcriptomics allow discovery of unexpected correlates of immune protection and inclusion of physiological processes in the discovery of determinants of resistance to disease that would otherwise be overlooked in a purely immunology-focused approach.
In this study, we aimed (i) to identify gene networks that were either positively or negatively associated with H. polygyrus infection burden, (ii) to use those genes to identify immune pathways that promoted immunity to H. polygyrus, and (iii) to test whether those protective pathways were affected by sex and anthelminthic drug treatment. We used a newly published A. sylvaticus genome for the assembly and analysis of RNAseq samples generated from the spleens of 34 wild-caught wood mice from two distinct populations showing natural variation in helminth burdens, and then randomly treated a subset with anthelminthics to experimentally reduce their nematode infection burdens. Identifying transcripts predictive of infection burdens in a natural system, and maximizing the chances of these associations to generalize across populations requires detecting potentially weak signals among many variables, while simultaneously avoiding spurious variation (false positives). We addressed this challenge by reducing transcripts into co-expression gene networks, and identifying therein robust predictors of immunity to H. polygyrus using supervised machine learning—a class of statistical models that can integrate multiple variables that each carry weak signal into a stronger predictor by mapping them to a response variable, here H. polygyrus burdens. These types of supervised machine learning approaches hold promise for ecological and immunological studies (35, 36). We then applied a similar supervised approach to a narrower set of transcripts explicitly associated with immunity to assess how the immune system regulates its response to chronic helminth burdens in both male and female mice. To ensure that our models were not specific to a single population, time point, and sequencing platform, and thereby to increase the generalizability of our conclusions, we split samples spanning two wood mouse populations repeatedly at random into training sets on which predictive models were generated, and corresponding test sets on which the predictions of trained models were validated against observed values. Finally, given the widely reported propensity of anthelminthic-treated individuals (humans included) to return to their initial helminth burdens within weeks, we assessed whether the correlates of protection identified by our supervised learning approach were impacted by anthelminthic treatment.
Materials and Methods
Ethics Statement
All procedures on animals were approved by the University of Glasgow ethics committee and the UK Home Office (PPL60/4572) and conducted in accordance with the Animals (Scientific Procedures) Act 1986.
Wood Mouse Field Treatment and Sampling
To minimize false discovery and ensure that our findings could generalize to other wood mouse populations, we included samples from two geographically and temporally distinct A. sylvaticus populations at two woodland sites: Haddon Wood (N 53.16°, W 3.1°, hereafter referred to as HW) in north-west England in 2011, and Callendar Wood (N 55.99°, W 3.78° hereafter referred to as CW) in the Central Lowlands of Scotland in 2015.
Mice were trapped using Sherman live traps (H. B. Sherman 2 × 2.5 × 6.5 inch folding trap, Tallahassee, FL, USA) baited with grain, carrot, and bedding material. Two traps were placed every 10 m in a 70 m × 70 m (total 128 traps per grid). At first capture, each individual was microchipped (AVID microchips, Lewes, UK). In CW only, mice were allocated to one of two sex-balanced groups: anthelminthic drug-treated or control. Age and reproductive status, assessed by the body mass, color of the coat, position of testes, occlusion of the vagina, and visible signs of lactation and pregnancy were also recorded. The drug treatment removed gastrointestinal nematodes, which are typically the most abundant parasites in wood mice (4). Drugs were given as a single oral dose of 2 µl g−1 of body weight of a mixture of 9.4 mg kg−1 Ivermectin and 100 mg kg−1 Pyrantel (IVM + PYR), and controls consisted of oral delivery of 2 µl g−1 of body weight of water (H2O). Mice that were recaptured 14 ± 3 days post first capture were sacrificed on site. After cervical dislocation and exsanguination by cardiac puncture, the spleen of each individual was extracted and immediately transferred to a tube containing 4 ml RNA later, followed by whole removal of the intestine and storage in phosphate-buffered saline for subsequent dissection and parasite identification.
Parasitology
Heligmosomoides polygyrus are ingested at their third larval (L3) stage, penetrate the submucosa of the small intestine within 24 h, and migrate to the muscularis externa, where they develop into L4-stage larvae. 8–10 days after infection, adult worms begin to emerge into the lumen of the intestine and attach to the intestinal villi within ~14 days, where they mate and release eggs. We measured adult H. polygyrus intestinal burdens of all sacrificed mice to assess immune resistance to infection. Worm burdens are preferred over fecal egg counts because they have much lower variability and likely reflect interactions with the host’s immune system during larval development. While it was not possible to measure individual level differences in exposure to parasite infective L3s in the wild, we selected mice of similar ages within both sexes, and assumed lifetime exposure levels were similar across all selected mice. Worm burdens were compared between sites (CW or HW), sexes and treatment groups using negative binomial generalized linear models because of the significant left-skew coupled with high worm prevalence, over a zero-inflated distribution.
RNA Sequencing, Assembly, Annotation, and Quantification
A transversal segment of each spleen was cut under sterile conditions, weighed, and processed following the RNeasy kit (Qiagen). RNA quantity and quality were assessed using a Tapestation (Agilent Technologies), and stored at −80 until RNA sequencing. All RNA samples were evaluated on a Bioanlayzer (Agilent Technologies) immediately prior to Poly-A selection of messenger RNA. Ten wood mice from Haddon Wood were sequenced on an Illumina Solexa 454 with 100 bp paired end reads by Edinburgh Genomics (2011). Twenty-four wood mice from Callendar wood were sequenced on an Illumina NextSeq 500 with 75 pb paired end reads by Glasgow Polyomics (2015). Raw reads generated by the sequencers were quality-checked using FastQC v. 0.11.5 (37) and sequences of low-quality and sequencer adapters trimmed using cutadapt v. 1.14 (38). The resulting trimmed reads were then quality-checked using FastQC and MultiQC (39) as above (see MultiQC report in Supplementary Data Sheet 1), and aligned to a reference transcriptome and quantified using Kallisto v. 0.43.1 (40). An A. sylvaticus transcriptome generated from a recently published genome (assembly ASM130590v1, https://www.ncbi.nlm.nih.gov/nuccore/LIPJ00000000.1) was used as the reference for assembly and read counts.
Transcriptome Analysis
Dimensionality Reduction
To select within the full transcriptome only the genes that had a potential role in resistance or tolerance to H. polygyrus across the full wood mouse transcriptome, we first grouped highly interconnected transcripts that may form distinct biological pathways by applying a weighted correlation network analysis (41) with the R package WGCNA (42). This entailed constructing gene networks (or modules) using a co-expression similarity measure defined as:
where Aij is the pairwise correlation between gene expressions (xi, xj), and β is the soft-threshold weight which is set at 6 in our analysis based on scale-free topology criterion (41). The WGCNA cluster eigengenes, which summarize the expression levels of all transcripts within each cluster, were used for further analysis as combining such clusters into biologically linked “meta-networks” as been shown to help identify biologically meaningful pathways (43–45). In addition to identifying the strongest correlates of protection across the full transcriptome, we sought to describe how genes explicitly associated with immunity might be functionally associated with H. polygyrus infection burdens. We, therefore, selected all genes for which the BLAST annotation contained the following immunological terms: “chemokine,” “cytokine,” “gata3,” “immunoglobulin,” “interferon,” “interleukin,” “platelet,” “relm,” “resistin,” “ror-gamma,” “t-bet,” “TBX21,” “TGF,” “TNF,” or “toll-like” for their reported roles in immunity or regulation of responses to parasitic nematodes (9, 46–49).
Supervised Learning
To identify WGCNA clusters or immune genes (“features”) that may have a role in regulating H. polygyrus burdens, we trained supervised learning models to map them to parasite counts of untreated individuals from both CW and HW populations. All features were scaled to unit variance to ensure homoscedasticity, and worm counts were log10+1-transformed to improve machine learning algorithm performance. To predict H. polygyrus burdens, we used a regression analysis with the task of minimizing mean squared errors (MSE) between parasitic worm burdens observed in a test set and burdens predicted by a model trained on an independent training subset of the full dataset.
To select which algorithm was most likely to generate the best models from our data, we began by comparing the baseline performance of widely used algorithms using their default settings on the full dataset using 10-fold cross-validation: Elastic Net; k-Nearest Neighbors for Regression; Random Forest regressor; and eXtreme Gradient Boosting regressor using either a linear (GLM) booster, or a tree booster.
The full dataset was then split 75/25 into a training set and the corresponding test set 10 times repeatedly at random without replacement to avoid sampling bias affecting our choice of trained model. For each of the random train-test paired subsets, the selected algorithm was tuned on the training set using a wide range of possible parameter settings on the training subset using fivefold cross-validation, and the trained models that achieved the lowest MSE between predicted and observed burdens in the test set were retained. Summary statistics (mean and SEM) of all 50 resulting MSE, and of all 50 weights applied to each cluster within the trained models were used for ranking the importance of each gene transcript or cluster in predicting H. polygyrus burdens.
Statistical Analysis
Immune features (WGCNA clusters and transcripts) that contributed most to the predictive performance of each model were used as response variables in generalized linear models to assess the effects of host sex (two level factor) and anthelminthic treatment (two level factor) on their expression. To reduce type 1 errors due to multiple testing, statistical significant was considered at p ≤ 0.01.
All data processing, machine learning, statistical analyses, and graphs were performed using Python 3.6 packages pandas (50), scikit-learn (51), statsmodels (52), and seaborn (53), respectively.
Results
Infection by H. polygyrus Was Prevalent but Infection Intensity Varied Substantially between Individuals and Populations
As is typical of parasite burdens where a minority of the animals carry most of the infection (54), H. polygyrus infection burdens followed a negative binomial distribution in the 61 individuals sampled in the two field populations (CW and HW), with burdens ranging from 0 to 135 worms (Figure 1A). Of those, 34 adult mice selected at random with H. polygyrus burdens at post mortem ranging 0–86 were retained for further analysis. While worm burdens within each population did not to differ significantly between sexes (Figure 1B), they were markedly higher in CW than in HW (Figure 1C, p < 0.0001, n = 22 untreated wood mice). All males and all females from CW included in subsequent analyses were infected with H. polygyrus, while prevalence was 60% for the females from HW (Figure 1B), amounting to a 91% prevalence of H. polygyrus in our untreated individuals. Half of the males and females caught in CW were treated with anthelminthic drugs to test associations between transcript expression (Illumina RNA-seq counts) and gastrointestinal helminth burdens. Treatment was equally effective at reducing H. polygyrus numbers and prevalence in both sexes but failed to remove the parasites completely (Figure 1C, 60× reduction in burden p < 0.0001; prevalence X2 = 0.0007, n = 24 in CW only). Taken together, these results confirm that this parasite is common in A. sylvaticus, suggest that H. polygyrus varies substantially in intensity between and within populations, and that anthelminthic treatment, while effective, did not completely eliminate gastrointestinal helminths from the treated individuals.
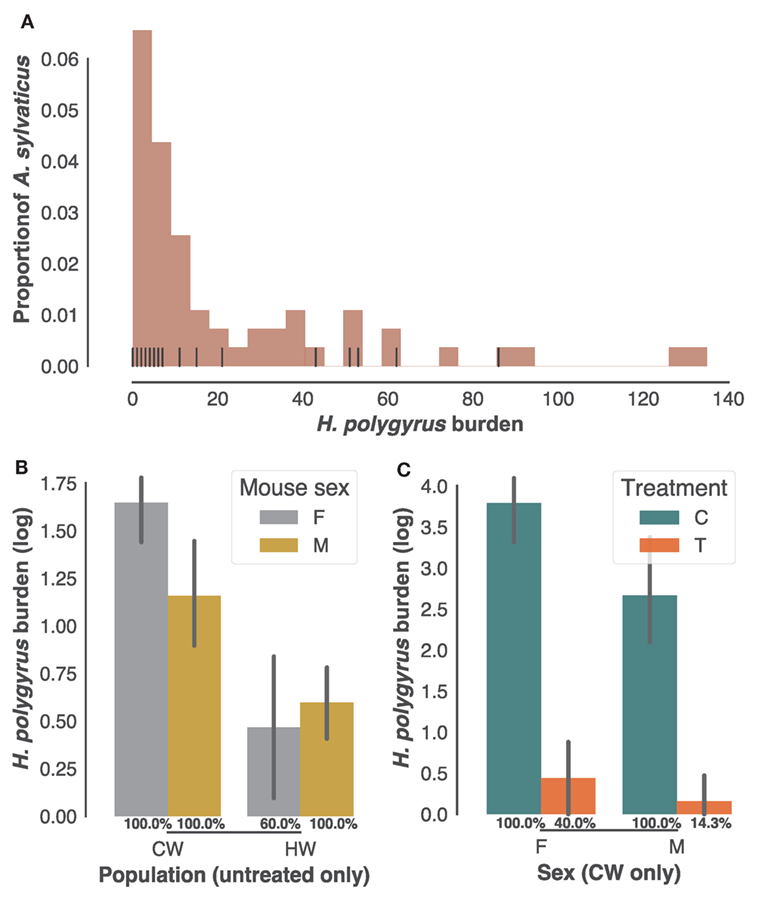
Figure 1. Heligmosomoides polygyrus prevalence and burdens in sampled Apodemus sylvaticus. (A) Distribution of H. polygyrus intestinal burdens in 61 A. sylvaticus sampled in HW and CW (histogram) and among those the 34 individuals randomly selected for the transcriptome analysis (vertical black lines). (B) H. polygyrus burdens of untreated male (M) and female (F) mice from CW and HW. (C) H. polygyrus burdens of treated (T) and untreated controls (C) from CW only. In (B,C) H. polygyrus prevalences in each category are given as percentages, bars represent mean log-transformed worm counts and error bars show the SEM.
Transcriptome-Wide Correlates of Resistance and Susceptibility to H. polygyrus Were Highly Predictive of Worm Burden
To identify transcriptome-wide gene expression profiles that might explain the observed variation in worm burdens and help identify immune mechanisms that regulate H. polygyrus burdens in wild wood mice, we began by clustering transcripts into co-expression networks to reduce the number of variables for further analysis. WGCNA reduced the 17,639 transcripts contained in the full transcriptome to 131 clusters. We used the eigengene (or first principle component) of each cluster to capture the majority of variation of all the genes contained within that cluster. Despite differences in infection burdens reported above, we found no clustering of transcription profiles by sampling origin, sex, or treatment (Figure S2 in Supplementary Material), indicating that our sampling procedure, use of different sequencing platforms, host sex, and drug treatment did not cause transcriptome-wide biases between individuals. All 34 transcriptomes were, thus, treated as belonging to the same statistical population. To identify the clusters associated with chronic H. polygyrus burdens, we used a supervised learning approach with clusters entered as explanatory variables and log10+1-transformed H. polygyrus counts as the response variable. Only mice that were not treated with anthelminthics were considered. A comparison of machine learning algorithms using fivefold cross-validation suggested two classes of algorithms were best suited for generating models mapping gene expression clusters to H. polygyrus burdens, gradient boosting with a linear booster (55) and Elastic Nets (Figure S3 in Supplementary Material). Elastic Nets identified several positively and negatively correlated clusters with robust statistical support. Elastic nets are particularly useful when the number of predictors is larger than the number of observations, as they tend to group together highly correlated features, a desirable behavior when selecting gene expression variables (56). Conversely, the XGBoost algorithm selected few clusters with clear positive of negative associations with worm burdens and appeared too sensitive to outliers, suggesting that it was likely to overfit (Figure S4 in Supplementary Material)—we, therefore, retained only Elastic Nets for further analysis. We then trained Elastic Nets as described in the section “Materials and Methods” on 10 randomly selected training subsets of untreated only mice from both CW and HW. The resulting models predicted log10+1-transformed H. polygyrus burdens in the corresponding test sets with a MSE of 0.24 ± 0.01. To identify clusters that were most informative to the prediction models, we ranked them using the Elastic Net coefficients, of which both positively- and negatively associated transcript expression networks were identified (Figure 2A). Within the 10 top-ranking clusters, we only retained for further analysis the five clusters that correlated significantly with worm burden at p ≤ 0.01 (Figure 2B). The KEGG pathways associated with the gene transcripts that correlated positively with parasite burdens included farnesylated proteins-converting enzyme 1, ubiquitin protein ligase synthesis and terpenoid backbone synthesis, ATP-dependant RNA helicase activity, and the NOD-like receptor signaling pathway (see Table S1 in Supplementary Material for details of all clusters positively associated with worm burdens). Pathways negatively correlated with parasite burdens included apoptosis, phosphatidylinositol 3-kinase regulatory subunit binding, glucosidase activity, C2H2 zinc finger domain binding, disordered domain specific binding, protein tyrosine kinase activity, and ATP-binding cassette transporters (see Table S2 in Supplementary Material for details of all clusters negatively associated with worm burdens). Host sex did not significantly affect the expression of either sets of genes (Figure S5 in Supplementary Material).
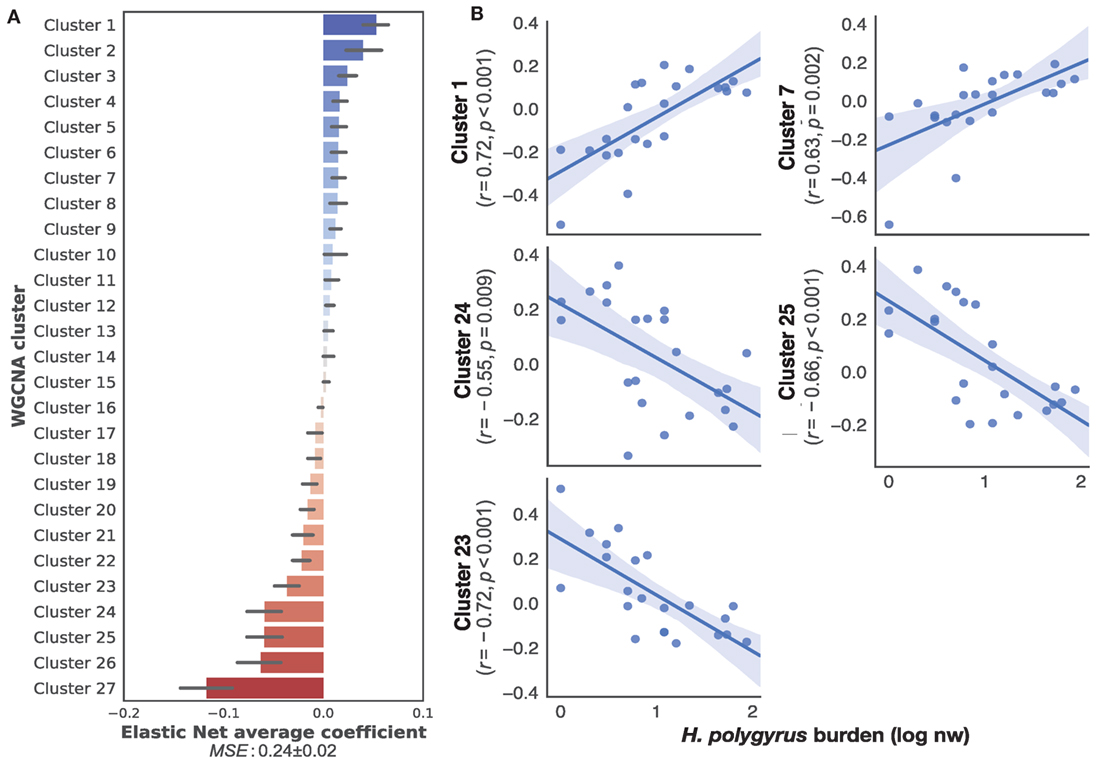
Figure 2. Top gene networks predictive of chronic Heligmosomoides polygyrus worm burdens in wild wood mice. (A) Average ranking of WGCNA clusters based on Elastic Net coefficients of 50 models trained on 22 untreated wood mice from HW and CW (see Materials and Methods for description details). (B) Regression plots of the top five positive and negative clusters based on their coefficients against log-transformed H. polygyrus burdens, for which p ≤ 0.01. Each point represents a wood mouse, the solid line is the regression line and the shaded areas represent the corresponding 95% bootstrapped confidence intervals. KEGG pathways (human and mouse) associated with: Cluster 1: ubiquitin protein ligase synthesis and terpenoid backbone synthesis; Cluster 7: ATP-dependant RNA helicase activity, NOD-like receptor signaling pathway; Cluster 24: phosphatidylinositol 3-kinase regulatory subunit binding, glucosidase activity, apoptosis; Cluster 25: C2H2 zinc finger domain binding, disordered domain specific binding; Cluster 23: protein tyrosine kinase activity, ATP-binding cassette transporters.
Immune Gene Transcription Correlates of Resistance and Susceptibility to H. polygyrus
To specifically identify, within the transcriptome, only immune genes associated with the regulation of H. polygyrus burdens, we applied our supervised learning approach to transcripts selected on the basis of their BLAST annotations containing explicit reference to immune function (see Materials and Methods for details). This retained a list of 222 unique genes out of the 12,437 present in the full transcriptome. Elastic nets mapping those immune transcripts to log10+1-transformed H. polygyrus burdens predicted H. polygyrus burdens of mice in the test datasets with a MSE of 0.33 ± 0.02 (Figure 3A). Among the transcripts that contributed most to the prediction, eight were significantly (p ≤ 0.01) correlated with parasite burden (Figure 3B), 2 positively—which included TGF-β-activated kinase 1 and MAP3K7-binding protein 2 (TAB 2), and dedicator of cytokinesis protein 7 (DOCK7), and 4 negatively—which included interleukin-5 receptor alpha, interleukin-17 receptor alpha, atypical chemokine receptor 3 (ACKR3), and platelet endothelial aggregation receptor 1.
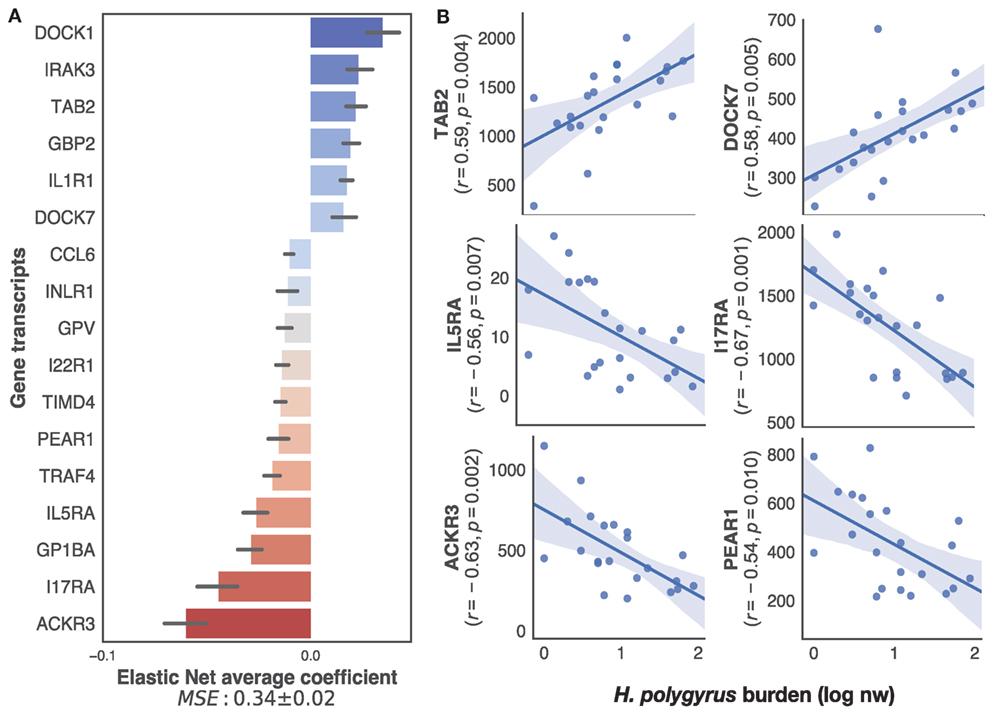
Figure 3. Top immune predictors of chronic Heligmosomoides polygyrus worm burdens in wild wood mice. (A) Average ranking of immune gene transcripts based on Elastic Net coefficients of 50 models trained on 22 untreated wood mice from HW and CW (see Materials and Methods for details on immune gene selection procedure and model training). (B) Regression plots of the top five positive and negative clusters based on their coefficients against log-transformed H. polygyrus burdens, for which p ≤ 0.01. In regression plots, each point represents a wood mouse, the solid line is the regression line and the shaded areas represent the corresponding 95% bootstrapped confidence intervals.
Effect of Sex and Treatment on Immune Pathways Correlated with Parasite Burdens
Sex effects are widely reported to affect gastrointestinal parasite burdens (57–60). Although there was little sex bias in H. polygyrus burdens in our study population (Figure 1B; Figure S1B in Supplementary Material), we did investigate whether the expression of protective pathways identified above differed between sexes. In addition, we predicted that experimental reduction of parasite burdens through anthelminthic treatment, which had resulted in a dramatic reduction in H. polygyrus worm burdens (Figure 1C), would profoundly affect the expression of immune genes associated with responses to these parasites. Contrary to our expectations, neither sex nor drug treatment resulted in major differences in expression of gene networks (Figure S2 in Supplementary Material), nor specifically in the expression of genes that correlated with H. polygyrus burdens in untreated individuals (Figure 4). Because our choice of Elastic Nets favored linear relationships and may, thus, have failed to identify non-linear relationships between gene expression and worm counts, we repeated the analyses above using gradient boosted trees. Consistent with expectations (Figure S3 in Supplementary Material), XGBoost achieved very similar predictive performances to the Elastic Nets (mean MSE [range] = 0.34 [0.11–3.3], Figure S6 in Supplementary Material), and of the top features, two out of the four that were significantly correlated with parasite burden were in agreement with those identified by the Elastic Nets (DOCK7 and IL17RA, Figure S6B in Supplementary Material). The two other genes identified by XGBoost were suppressor of cytokine signaling 2 (SOCS2) and interferon-stimulated 20 kDa exonuclease-like 2 (I20L2), which both showed greater variance in gene expression at intermediate parasite burdens. SOCS2 and I20L2 were expressed at only marginally different levels between treated and untreated animals (p = 0.04 and p = 0.05, respectively), and between sexes (p = 0.15 and p = 0.03, respectively) (Figure S7 in Supplementary Material).
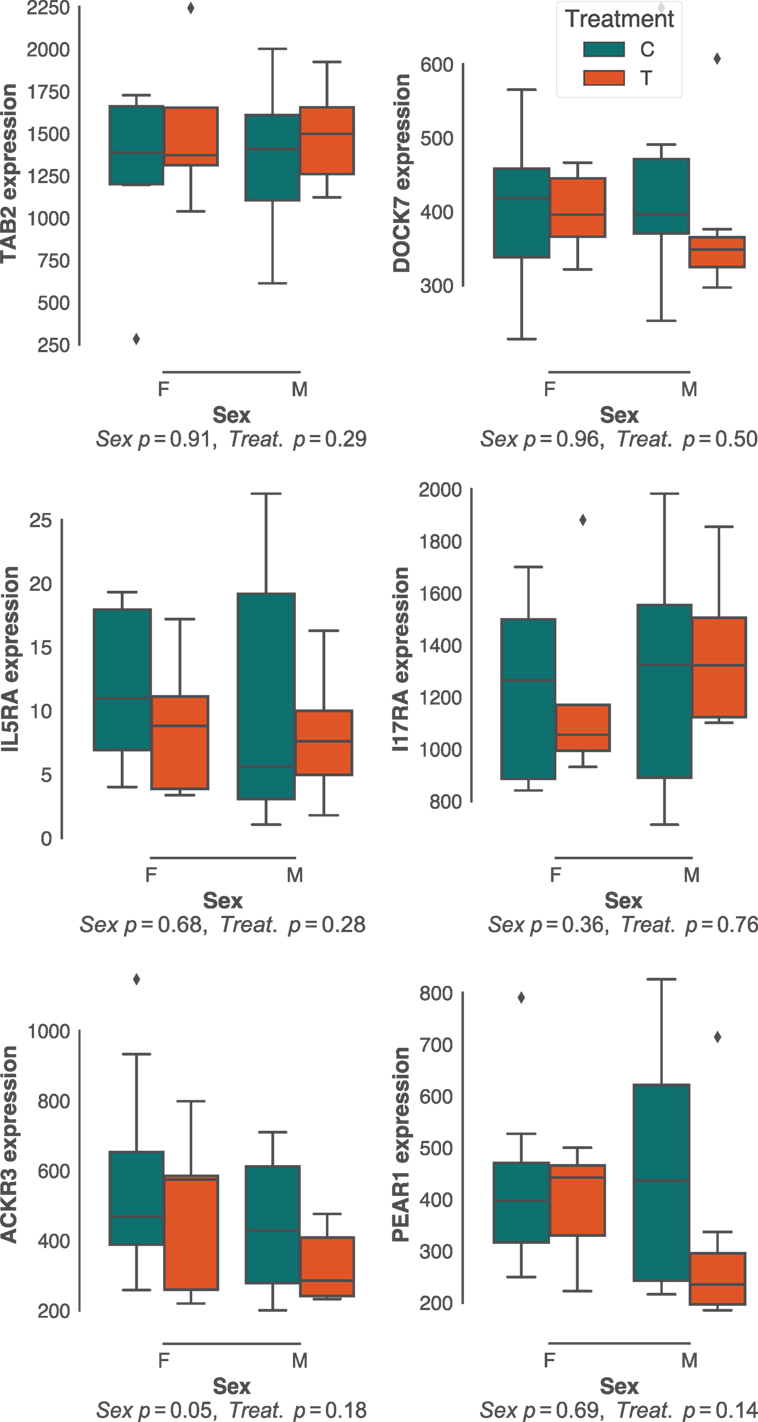
Figure 4. Effects of host sex and drug treatment on protective immune gene expression. No statistically significant effects of host sex and drug treatment were detected among the best predictors of Heligmosomoides polygyrus burdens identified in untreated mice only. Each plot represents transcript counts scaled to unit variance but not centered. Horizontal bars represent the median, boxes represent the interquartile range, whiskers the range, and diamonds transcript counts that lay beyond 1.5× of the interquartile range.
Discussion
The distribution of parasite infection burdens within a population is typically highly dispersed, with a small proportion of the hosts harboring the heaviest infections (61). Here, the prevalence of H. polygyrus was high in both populations studied, and remained detectable even after drug treatment. This suggests, assuming similar exposure to infection at a given age, that most individuals limited their parasite burdens but did not eliminate infection completely, consistent with reports on chronic helminth infections across diverse host species (61–63). Furthermore, drug treatment usually needs to be administered regularly to control helminth infections, as otherwise worms recrudesce within weeks (3, 5, 64). We sought to better understand how the immune system, under natural exposure and chronic helminth infection, regulates its response to infection, what immune traits allow some hosts to maintain low infection burdens, and how anthelminthic treatment and the subsequent reduction in parasite burdens impact those protective immune traits.
Addressing these questions required choosing a species that naturally harbors a diverse community of parasites at high prevalence and densities to maximize our chance of detecting the effects of their removal on the host. This ruled out M. musculus, which despite being a powerful model for immunology, is a relatively poor model for parasitology owing to the uncharacteristically low helminth infections naturally present in that species (26, 27). Wood mice, however, resemble species of greater societal importance in the diversity and prevalence of the parasite communities (21, 65). Here, we chose to focus on the gastrointestinal nematode H. polygyrus due to its prevalence in wood mice, and to its extensive use as a laboratory model (24, 66, 67), thus providing a powerful way to compare lab-based and field-based immune responses to this parasite. In our study populations, H. polygyrus was present in 91% of the wood mice we caught.
The immune system of A. sylvaticus is poorly known, thus we chose to take a transcriptomic approach to discover specific host–parasite interactions. In addition, having the full transcriptome also allowed us to use a candidate gene approach based on BLAST annotations. Moreover, it is likely that a significant amount of variation in chronic helminth burdens could be driven by processes not typically classified as immunological. We, therefore, decided to combine the discovery approach afforded by our sequencing of the full transcriptome with an approach focused on the genes explicitly associated with the immune system. However, RNAseq risks overestimating transcript abundance due to splicing variants, or being dominated by the more abundantly transcribed genes. While a technical solution would be to combine platforms with different read depths and lengths, here we used analytical solutions (e.g., FastQC reports of overrepresented sequences), and supervised learning to focus on transcripts that had statistically significant correlation with a biological read-out, here, parasite burdens. In short, we used parasite burdens to select the genes and co-expression networks for further analysis. By using Elastic Nets to identify correlates of parasite burdens, we ensured that redundant transcripts would feature together in the ranking of coefficients once models were trained. Yet, after removing all exact duplicates from the raw transcript counts, our models generated lists of uniquely represented genes, suggesting that for any that may have had differently expressed isoforms, only one of each correlated significantly with parasite burden.
We found that within the transcriptome, the best predictors of parasite burdens included both immune and non-immune genes. Among non-immune genes that were associated with H. polygyrus, were farnesylated proteins-converting enzyme 1 (FACE1), which is involved in protein hydrolysis (68), dedicator of cytokinesis protein 7 (DOCK7), which is reported to influence lipid regulation (69, 70), and proteases, helicases, and ABC transporters—genes generally involved in protein and energy metabolism, and potentially tissue growth and/or repair. Although nutrition plays an important role in resistance to helminth infections (71), how these specific pathways relate to immunity or exposure to H. polygyrus is currently unclear.
Examining immune genes more specifically confirmed known associations between gastrointestinal helminths and immune factors, and revealed others that merit further study. Among previously reported associations was the negative correlation between parasite burdens and the receptor for IL-5, which is the main cytokine involved in the activation of eosinophils, part of the group-2 innate lymphoid cell-driven responses to helminths in the gut (48). Also consistent with previous reports in laboratory models was the negative relationship between H. polygyrus and IL-17A. Indeed, this parasite has previously been reported to inhibit IL-17 production in the gut mucosa (72), and more recently, that this interaction may be mediated by a subset of gut-resident eosinophils that suppress IL-17 (47, 73–75). Furthermore, interactions between H. polygyrus, Th17, and regulatory T cell responses have been reported to interact with the gut microbiota and be genotype dependent. Indeed, a susceptible mouse strain has elevated IL-17 and increased proportions of Lactobacilli whereas in a resistant mouse strain, H. polygyrus has no effect on Lactobacillus abundance (76). In wild wood mice, these interactions are likely to be driven, in part, by seasonal changes in diet (77), which is consistent with three-way interactions observed between hosts, helminths, and their microbiota in wild seabirds (78). We also detected strong positive associations between H. polygyrus burdens and TAB 2 expression, which is reported to activate IL-1 via JNK and NF-κB (79). This is consistent with their positive correlation with the reactive oxygen species-expressing NOD-like receptor signaling pathway (80) we identified among WGCNA clusters. This suggests either an inflammatory response to H. polygyrus or to the damage it may cause in the gut (81), or that immune systems already skewed toward inflammation are less able to control parasitic helminths. While we did not detect clear signatures of regulatory T cell activation in the spleen, two genes potentially involved in immune regulation correlated strongly with parasite burdens: IL17RA, as mentioned above, and ACKR3, a chemokine scavenger (82) that is widely expressed in the hematopoietic system, heart, vascular endothelial cells, bone, kidney, and brain, and that is reported to be upregulated in many cancers (83) and also mimicked by a herpesvirus agonist (84, 85).
Interestingly, we did not detect significant associations between worm burdens and cytokines or chemokines, but rather with that of their receptors. Likewise, we might expect GATA-3 to correlate with H. polygyrus burdens, since this transcription factor is central to ILC2 initiating Th2 responses to helminths in the gut (48). This suggests that in the spleen associations between infection burdens and RNA expression of cytokine receptor genes are more statistically robust, and thus more functionally interpretable, than those between infection burdens and cytokine messenger RNA.
Having observed no significant difference between male and female mice in their parasite burdens, we also did not detect significant differences in the expression of any of the top predictors of parasite burdens, and host sex only marginally explaining the variation in the expression of ACKR3. More surprising was that a 60-fold reduction in H. polygyrus burdens within an individual mouse would have so little effect on the immune pathways that predict their chronic burdens. This may be due to the target of the anthelminthic drugs we used, ivermectin and pyrantel (86). The combination of immune-regulatory predictors of chronic infection burdens, the mode of action of commonly used anthelminthics, and the ensuing lack of immune response to drug-induced worm death may explain the high reinfection rates post drug-removal (3–5).
Finally, while wood mice infected with high worm burdens may be more tolerant to infection [i.e., the ability of hosts to minimize adverse fitness consequences of increasing parasite burdens (87)], our study could not address this question satisfactorily because we did not collect data to assess fitness-relevant effects of infection nor of drug treatment in the wood mice. While recent studies suggest a complex relationship between protective immunity, resource limitation, and immunopathology (88, 89), how organisms balance the benefits of eliminating helminth infection and the costs of mounting the anthelminthic immune responses to do so is poorly understood. A further limitation of this study is its reliance on the transcriptome to assess immune responses: in the future, it will be important not only to validate our findings in additional wood mouse populations, e.g., using quantitative RT-PCR, but also to integrate protein, cellular, and metabolomic data alongside transcriptomic data (90) to reduce the risk of overlooking important processes that vary in their post-transcriptional regulation.
In conclusion, we have generated the first transcriptome for A. sylvaticus and identified a number of transcriptional predictors of chronic infection chronic infection burdens by H. polygyrus that include previously known immune pathways as well as novel candidates which merit further investigation. Notably, resolving the causal relationships between hosts, parasitic helminths and microbiota in the maintenance of immune homeostasis even in the face of drug-induced parasite removal, merits further attention. By combining two distinct wood mouse populations, integrating two different sequencing platforms, and applying machine-learning-based cross-validation procedures to map transcript expression levels to parasite burdens, we have sought to maximize the generalizability and functional relevance of our analysis of the wood mouse immune system. In the future, longer term studies and the integration of multiple biological levels, from genomes to cells, should help further our understanding of how the immune system maintains the health of the organism in its natural habitat.
Ethics Statement
All procedures on animals were approved by the University of Glasgow ethics committee and the UK Home Office (PPL60/4572) and conducted in accordance with the Animals (Scientific Procedures) Act 1986.
Author Contributions
SB and AP conceived, designed, and secured funding for the study, ran the field sites, participated in the field and lab work, and wrote the manuscript. SB performed all statistics and machine learning analyses. WL performed the digital transcriptomics and helped write the manuscript. GH performed all assembly and annotation of the transcriptomes. EK extracted and quality-checked all RNA samples. MC and ER participated in the field work and analyzed the parasite samples.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work and the next-generation sequencing performed by Glasgow Polyomics were supported by the Wellcome Trust ISSF grant [097821/Z/11/Z] to SB, NBAF grant (#NBAF528) to AP and SB, a targeted Institute of Biodiversity, Animal Health & Comparative Medicine Research Fellowship to SB, a Wellcome Trust Strategic Grant for the Centre for Immunity Infection and Evolution (095831 Advanced Fellowship) to AP, a University of Edinburgh Chancellors Fellowship to AP, and a National Science Foundation Postdoctoral Research Fellowship in Biology (DBI-1306608) to ER.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00056/full#supplementary-material.
References
1. Hoerauf A, Brattig N. Resistance and susceptibility in human onchocerciasis – beyond Th1 vs. Th2. Trends Parasitol (2002) 18:25–31. doi:10.1016/S1471-4922(01)02173-0
2. Nagaraj SH, Harsha HC, Reverter A, Colgrave ML, Sharma R, Andronicos N, et al. Proteomic analysis of the abomasal mucosal response following infection by the nematode, Haemonchus contortus, in genetically resistant and susceptible sheep. J Proteomics (2012) 75:2141–52. doi:10.1016/j.jprot.2012.01.016
3. Nfon CK, Makepeace BL, Njongmeta LM, Tanya VN, Trees AJ. Lack of resistance after re-exposure of cattle cured of Onchocerca ochengi infection with oxytetracycline. Am J Trop Med Hyg (2007) 76:67–72.
4. Knowles SC, Fenton A, Petchey OL, Jones TR, Barber R, Pedersen AB. Stability of within-host-parasite communities in a wild mammal system. Proc Biol Sci (2013) 280:20130598. doi:10.1098/rspb.2013.0598
5. Speich B, Moser W, Ali SM, Ame SM, Albonico M, Hattendorf J, et al. Efficacy and reinfection with soil-transmitted helminths 18-weeks post-treatment with albendazole-ivermectin, albendazole-mebendazole, albendazole-oxantel pamoate and mebendazole. Parasit Vectors (2016) 9:123. doi:10.1186/s13071-016-1406-8
6. Mideo N, Nelson WA, Reece SE, Bell AS, Read AF, Day T. Bridging scales in the evolution of infectious disease life histories: application. Evolution (2011) 65:3298–310. doi:10.1111/j.1558-5646.2011.01382.x
7. Long GH, Graham AL. Consequences of immunopathology for pathogen virulence evolution and public health: malaria as a case study. Evol Appl (2011) 4:278–91. doi:10.1111/j.1752-4571.2010.00178.x
8. Choisy M, de Roode JC. Mixed infections and the evolution of virulence: effects of resource competition, parasite plasticity, and impaired host immunity. Am Nat (2010) 175:E105–18. doi:10.1086/651587
9. Ludwig-Portugall I, Layland LE. TLRs, Treg, and B cells, an interplay of regulation during helminth infection. Front Immunol (2012) 3:8. doi:10.3389/fimmu.2012.00008
10. Guivier E, Bellenger J, Sorci G, Faivre B, Metcalf CJE, Winn AA. Helminth interaction with the host immune system: short-term benefits and costs in relation to the infectious environment. Am Nat (2016) 188:253–63. doi:10.5061/dryad.7017n
11. Charlier J, De Waele V, Ducheyne E, van der Voort M, Vande Velde F, Claerebout E. Decision making on helminths in cattle: diagnostics, economics and human behaviour. Ir Vet J (2015) 69:14. doi:10.1186/s13620-016-0073-6
12. Coop RL, Sykes AR, Angus KW. The effect of three levels of intake of Ostertagia circumcincta larvae on growth rate, food intake and body composition of growing lambs. J Agric Sci (1982) 98:247–55. doi:10.1017/S0021859600041782
13. Coop RL, Kyriazakis I. Influence of host nutrition on the development and consequences of nematode parasitism in ruminants. Trends Parasitol (2001) 17:325–30. doi:10.1016/S1471-4922(01)01900-6
14. Muller O, Krawinkel M. Malnutrition and health in developing countries. CMAJ (2005) 173:279–86. doi:10.1503/cmaj.050342
15. Pullan R, Brooker S. The health impact of polyparasitism in humans: are we under-estimating the burden of parasitic diseases. Parasitology (2008) 135:783–94. doi:10.1017/S0031182008000346
16. Rioux JA, Golvan YJ. [Mange caused by Psorergates musculinus (Michael 1889) (Acari; Myobiidae) in the field mice Apodemus sylvaticus (Linne 1758) of the mass of Caroux (Herault)]. Ann Parasitol Hum Comp (1961) 36:785–7. doi:10.1051/parasite/1961365785
17. Pokorny J, Havlik O. [Experimental infection of Apodemus sylvaticus L. with leptospiras (L. grippotyphosa)]. Cesk Epidemiol Mikrobiol Imunol (1960) 9:88–92.
18. Friberg IM, Little S, Ralli C, Lowe A, Hall A, Jackson JA, et al. Macroparasites at peripheral sites of infection are major and dynamic modifiers of systemic antimicrobial pattern recognition responses. Mol Ecol (2013) 22:2810–26. doi:10.1111/mec.12212
19. Jackson JA, Hall AJ, Friberg IM, Ralli C, Lowe A, Zawadzka M, et al. An immunological marker of tolerance to infection in wild rodents. PLoS Biol (2014) 12(7):e1001901. doi:10.1371/journal.pbio.1001901
20. Goüy de Bellocq J, Charbonnel N, Morand S. Coevolutionary relationship between helminth diversity and MHC class II polymorphism in rodents. J Evol Biol (2008) 21:1144–50. doi:10.1111/j.1420-9101.2008.01538.x
21. Behnke JM, Eira C, Rogan M, Gilbert FS, Torres J, Miquel J, et al. Helminth species richness in wild wood mice, Apodemus sylvaticus, is enhanced by the presence of the intestinal nematode Heligmosomoides polygyrus. Parasitology (2009) 136:793–804. doi:10.1017/S0031182009006039
22. Poulin R. Interactions between species and the structure of helminth communities. Parasitology (2001) 122:S3–11. doi:10.1017/S0031182000016991
23. Montgomery SSJ, Montgomery WI. Structure, stability and species interactions in helminth communities of wood mice, Apodemus sylvaticus. Int J Parasitol (1990) 20:225–42. doi:10.1016/0020-7519(90)90105-V
24. Behnke JM, Menge DM, Noyes H. Heligmosomoides bakeri: a model for exploring the biology and genetics of resistance to chronic gastrointestinal nematode infections. Parasitology (2009) 136:1565–80. doi:10.1017/S0031182009006003
26. Abolins S, King EC, Lazarou L, Weldon L, Hughes L, Drescher P, et al. The comparative immunology of wild and laboratory mice, Mus musculus domesticus. Nat Commun (2017) 8:14811. doi:10.1038/ncomms14811
27. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532(7600):512–6. doi:10.1038/nature17655
28. Behnke JM, Lewis JW, Zain SN, Gilbert FS. Helminth infections in Apodemus sylvaticus in southern England: interactive effects of host age, sex and year on the prevalence and abundance of infections. J Helminthol (1999) 73:31–44.
29. Cable J, Harris PD, Lewis JW, Behnke JM. Molecular evidence that Heligmosomoides polygyrus from laboratory mice and wood mice are separate species. Parasitology (2006) 133:111–22. doi:10.1017/S0031182006000047
30. Maizels RM, Hewitson JP, Murray J, Harcus YM, Dayer B, Filbey KJ, et al. Immune modulation and modulators in Heligmosomoides polygyrus infection. Exp Parasitol (2012) 132:76–89. doi:10.1016/j.exppara.2011.08.011
31. Pedersen AB, Babayan SA. Wild immunology. Mol Ecol (2011) 20:872–80. doi:10.1111/j.1365-294X.2010.04938.x
32. Behnke JM, Menge DM, Nagda S, Noyes H, Iraqi FA, Kemp SJ, et al. Quantitative trait loci for resistance to Heligmosomoides bakeri and associated immunological and pathological traits in mice: comparison of loci on chromosomes 5, 8 and 11 in F2 and F6/7 inter-cross lines of mice. Parasitology (2010) 137:311–20. doi:10.1017/S0031182009991028
33. Beraldi D, McRae AF, Gratten J, Pilkington JG, Slate J, Visscher PM, et al. Quantitative trait loci (QTL) mapping of resistance to strongyles and coccidia in the free-living Soay sheep (Ovis aries). Int J Parasitol (2007) 37:121–9. doi:10.1016/j.ijpara.2006.09.007
34. Venturina VM, Gossner AG, Hopkins J. The immunology and genetics of resistance of sheep to Teladorsagia circumcincta. Vet Res Commun (2013) 37:171–81. doi:10.1007/s11259-013-9559-9
35. Stephens PR, Altizer S, Smith KF, Alonso Aguirre A, Brown JH, Budischak SA, et al. The macroecology of infectious diseases: a new perspective on global-scale drivers of pathogen distributions and impacts. Ecol Lett (2016) 19:1159–71. doi:10.1111/ele.12644
36. Urrutia A, Duffy D, Rouilly V, Posseme C, Djebali R, Illanes G, et al. Standardized whole-blood transcriptional profiling enables the deconvolution of complex induced immune responses. Cell Rep (2016) 16:2777–91. doi:10.1016/j.celrep.2016.08.011
37. Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. (2010). Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
38. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J (2011) 17:10–2. doi:10.14806/ej.17.1.200
39. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics (2016) 32:3047–8. doi:10.1093/bioinformatics/btw354
40. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol (2016) 34:525–7. doi:10.1038/nbt.3519
41. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol (2005) 4:Article17. doi:10.2202/1544-6115.1128
42. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics (2008) 9:559. doi:10.1186/1471-2105-9-559
43. Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol (2007) 1:54. doi:10.1186/1752-0509-1-54
44. Albert R. Scale-free networks in cell biology. J Cell Sci (2005) 118:4947–57. doi:10.1242/jcs.02714
45. Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A (2006) 103:17402–7. doi:10.1073/pnas.0608396103
46. Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol (2015) 15:271–82. doi:10.1038/nri3831
47. Pathak M, Sharma P, Sharma A, Verma M, Srivastava M, Misra-Bhattacharya S. Regulatory T-cell neutralization in mice during filariasis helps in parasite clearance by enhancing T helper type 17-mediated pro-inflammatory response. Immunology (2016) 147:190–203. doi:10.1111/imm.12550
48. Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Rückerl D, Seddon B, et al. IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol (2016) 9(6):1407–17. doi:10.1038/mi.2016.4
49. Anthony RM, Rutitzky LI, Urban JFJ, Stadecker MJ, Gause WC. Protective immune mechanisms in helminth infection. Nat Rev Immunol (2007) 7:975–87. doi:10.1038/nri2199
50. McKinney W. Data structures for statistical computing in python. In: van der Walt S, Millman J, editors. Proceedings of the 9th Python in Science Conference. (Vol. 445), Austin, TX: SciPy (2010). p. 51–6.
51. Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in python. J Mach Learn Res (2011) 12:2825–30.
52. Seabold S, Perktold J. Statsmodels: econometric and statistical modeling with python. In: van der Walt S, Millman J, editors. Proceedings of the 9th Python in Science Conference. Austin, TX: SciPy (2010). p. 57–61.
53. Waskom M, Botvinnik O, O’Kane D, Hobson P, Lukauskas S, Gemperline DC, et al. mwaskom/seaborn: v0.8.1 (September 2017). Zenodo (2017). doi:10.5281/zenodo.883859
54. Loeys T, Moerkerke B, De Smet O, Buysse A. The analysis of zero-inflated count data: beyond zero-inflated Poisson regression. Br J Math Stat Psychol (2012) 65:163–80. doi:10.1111/j.2044-8317.2011.02031.x
55. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. arXiv preprint arXiv:160302754. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. (2016) 785–794.
56. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc Series B Stat Methodol (2005) 67:301–20. doi:10.1111/j.1467-9868.2005.00503.x
57. Hämäläinen A, Raharivololona B, Ravoniarimbinina P, Kraus C. Host sex and age influence endoparasite burdens in the gray mouse lemur. Front Zool (2015) 12:25. doi:10.1186/s12983-015-0118-9
58. Ferrari N, Rosà R, Pugliese A, Hudson PJ. The role of sex in parasite dynamics: model simulations on transmission of Heligmosomoides polygyrus in populations of yellow-necked mice, Apodemus flavicollis. Int J Parasitol (2007) 37:341–9. doi:10.1016/j.ijpara.2006.10.015
59. Hepworth MR, Hardman MJ, Grencis RK. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur J Immunol (2010) 40:406–16. doi:10.1002/eji.200939589
60. Bordes F, Ponlet N, de Bellocq JG, Ribas A, Krasnov BR, Morand S. Is there sex-biased resistance and tolerance in Mediterranean wood mouse (Apodemus sylvaticus) populations facing multiple helminth infections? Oecologia (2012) 170:123–35. doi:10.1007/s00442-012-2300-5
61. Churcher TS, Ferguson NM, Basáñez MG. Density dependence and overdispersion in the transmission of helminth parasites. Parasitology (2005) 131:121–32. doi:10.1017/S0031182005007341
62. Bundy DA, Thompson DE, Golden MH, Cooper ES, Anderson RM, Harland PS. Population distribution of Trichuris trichiura in a community of Jamaican children. Trans R Soc Trop Med Hyg (1985) 79:232–7. doi:10.1016/0035-9203(85)90343-8
63. Barger IA. The statistical distribution of trichostrongylid nematodes in grazing lambs. Int J Parasitol (1985) 15:645–9. doi:10.1016/0020-7519(85)90010-4
64. Jia TW, Melville S, Utzinger J, King CH, Zhou XN. Soil-transmitted helminth reinfection after drug treatment: a systematic review and meta-analysis. PLoS Negl Trop Dis (2012) 6:e1621. doi:10.1371/journal.pntd.0001621
65. Nunn CL, Altizer S, Jones KE, Sechrest W. Comparative tests of parasite species richness in primates. Am Nat (2003) 162:597–614. doi:10.1086/378721
66. Reynolds LA, Filbey KJ, Maizels RM. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol (2012) 34:829–46. doi:10.1007/s00281-012-0347-3
67. Monroy FG, Enriquez FJ. Heligmosomoides polygyrus: a model for chronic gastrointestinal helminthiasis. Parasitol Today (1992) 8:49–54. doi:10.1016/0169-4758(92)90084-F
68. Pendás AM, Zhou Z, Cadiñanos J, Freije JM, Wang J, Hultenby K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet (2002) 31:94–9. doi:10.1038/ng871
69. Varga TV, Kurbasic A, Aine M, Eriksson P, Ali A, Hindy G, et al. Novel genetic loci associated with long-term deterioration in blood lipid concentrations and coronary artery disease in European adults. Int J Epidemiol (2017) 46:1211–22. doi:10.1093/ije/dyw245
70. Guo T, Yin RX, Huang F, Yao LM, Lin WX, Pan SL. Association between the DOCK7, PCSK9 and GALNT2 gene polymorphisms and serum lipid levels. Sci Rep (2016) 6:19079. doi:10.1038/srep19079
71. Ing R, Su Z, Scott ME, Koski KG. Suppressed T helper 2 immunity and prolonged survival of a nematode parasite in protein-malnourished mice. Proc Natl Acad Sci U S A (2000) 97:7078–83. doi:10.1073/pnas.97.13.7078
72. Elliott DE, Metwali A, Leung J, Setiawan T, Blum AM, Ince MN, et al. Colonization with Heligmosomoides polygyrus suppresses mucosal IL-17 production. J Immunol (2008) 181:2414–9. doi:10.4049/jimmunol.181.4.2414
73. Tian BP, Hua W, Xia LX, Jin Y, Lan F, Lee JJ, et al. Exogenous interleukin-17A inhibits eosinophil differentiation and alleviates allergic airway inflammation. Am J Respir Cell Mol Biol (2015) 52:459–70. doi:10.1165/rcmb.2014-0097OC
74. Wiesner DL, Smith KD, Kashem SW, Bohjanen PR, Nielsen K. Different lymphocyte populations direct dichotomous eosinophil or neutrophil responses to pulmonary Cryptococcus infection. J Immunol (2017) 198:1627–37. doi:10.4049/jimmunol.1600821
75. Yang BG, Seoh JY, Jang MH. Regulatory eosinophils in inflammation and metabolic disorders. Immune Netw (2017) 17:41–7. doi:10.4110/in.2017.17.1.41
76. Reynolds L, Smith K, Filbey K, Harcus Y, Hewitson J, Redpath S, et al. Commensal-pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes (2014) 5(4):522–32. doi:10.4161/gmic.32155
77. Maurice CF, Knowles SC, Ladau J, Pollard KS, Fenton A, Pedersen AB, et al. Marked seasonal variation in the wild mouse gut microbiota. ISME J (2015) 9:2423–34. doi:10.1038/ismej.2015.53
78. Newbold LK, Burthe SJ, Oliver AE, Gweon HS, Barnes CJ, Daunt F, et al. Helminth burden and ecological factors associated with alterations in wild host gastrointestinal microbiota. ISME J (2017) 11:663–75. doi:10.1038/ismej.2016.153
79. Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. TAB 2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell (2000) 5:649–58. doi:10.1016/S1097-2765(00)80244-0
80. Tattoli I, Carneiro LA, Jéhanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep (2008) 9:293–300. doi:10.1038/sj.embor.7401161
81. McDermott JR, Bartram RE, Knight PA, Miller HR, Garrod DR, Grencis RK. Mast cells disrupt epithelial barrier function during enteric nematode infection. Proc Natl Acad Sci U S A (2003) 100:7761–6. doi:10.1073/pnas.1231488100
82. Nibbs RJB, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol (2013) 13(11):815–29. doi:10.1038/nri3544
83. Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med (2006) 203:2201–13. doi:10.1084/jem.20052144
84. Freitas C, Desnoyer A, Meuris F, Bachelerie F, Balabanian K, Machelon V. The relevance of the chemokine receptor ACKR3/CXCR7 on CXCL12-mediated effects in cancers with a focus on virus-related cancers. Cytokine Growth Factor Rev (2014) 25:307–16. doi:10.1016/j.cytogfr.2014.04.006
85. Szpakowska M, Dupuis N, Baragli A, Counson M, Hanson J, Piette J, et al. Human herpesvirus 8-encoded chemokine vCCL2/vMIP-II is an agonist of the atypical chemokine receptor ACKR3/CXCR7. Biochem Pharmacol (2016) 114:14–21. doi:10.1016/j.bcp.2016.05.012
86. Beugnet F, Kerboeuf D, Nicolle JC, Soubieux D. Use of free living stages to study the effects of thiabendazole, levamisole, pyrantel and ivermectin on the fine structure of Haemonchus contortus and Heligmosomoides polygyrus. Vet Parasitol (1996) 63:83–94. doi:10.1016/0304-4017(95)00879-9
87. Little TJ, Shuker DM, Colegrave N, Day T, Graham AL. The coevolution of virulence: tolerance in perspective. PLoS Pathog (2010) 6:e1001006. doi:10.1371/journal.ppat.1001006
88. Flies AS, Mansfield LS, Flies EJ, Grant CK, Holekamp KE. Socioecological predictors of immune defences in wild spotted hyenas. Funct Ecol (2016) 30:1549–57. doi:10.1111/1365-2435.12638
89. Graham AL, Hayward AD, Watt KA, Pilkington JG, Pemberton JM, Nussey DH. Fitness correlates of heritable variation in antibody responsiveness in a wild mammal. Science (2010) 330:662–5. doi:10.1126/science.1194878
Keywords: wild immunology, Apodemus sylvaticus, transcriptome, machine learning applied to immunology, Heligmosomoides polygyrus, anthelminthics
Citation: Babayan SA, Liu W, Hamilton G, Kilbride E, Rynkiewicz EC, Clerc M and Pedersen AB (2018) The Immune and Non-Immune Pathways That Drive Chronic Gastrointestinal Helminth Burdens in the Wild. Front. Immunol. 9:56. doi: 10.3389/fimmu.2018.00056
Received: 07 October 2017; Accepted: 09 January 2018;
Published: 05 February 2018
Edited by:
Lluis Tort, Universitat Autònoma de Barcelona, SpainReviewed by:
Katherine Buckley, George Washington University, United StatesJorge Galindo-Villegas, Universidad de Murcia, Spain
Copyright: © 2018 Babayan, Liu, Hamilton, Kilbride, Rynkiewicz, Clerc and Pedersen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon A. Babayan, c2ltb24uYmFiYXlhbkBnbGFzZ293LmFjLnVr