 Glaucia Souza-Almeida1
Glaucia Souza-Almeida1 Heloisa D’Avila2
Heloisa D’Avila2 Patricia E. Almeida2
Patricia E. Almeida2 Tatiana Luna-Gomes2,3
Tatiana Luna-Gomes2,3 Sally Liechocki1
Sally Liechocki1 Barbara Walzog4
Barbara Walzog4 Ingrid Hepper4
Ingrid Hepper4 Hugo Caire Castro-Faria-Neto1
Hugo Caire Castro-Faria-Neto1 Patricia T. Bozza1
Patricia T. Bozza1 Christianne Bandeira-Melo5
Christianne Bandeira-Melo5 Clarissa M. Maya-Monteiro1*
Clarissa M. Maya-Monteiro1*
- 1Laboratório de Imunofarmacologia, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil
- 2Instituto de Ciências Biológicas, Universidade Federal de Juiz de Fora, Juiz de Fora, Brazil
- 3Departamento de Ciências da Natureza, Instituto de Aplicação Fernando Rodrigues da Silveira, Universidade do Estado do Rio de Janeiro, Rio de Janeiro, Brazil
- 4Walter Brendel Centre of Experimental Medicine, Department of Cardiovascular Physiology and Pathophysiology, Biomedical Center, Ludwig-Maximilians-Universität München, Munich, Germany
- 5Instituto de Biofisica Carlos Chagas Filho, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
Leptin directly activates macrophages and lymphocytes, but the role of leptin in neutrophil activation and migration is still controversial. Here, we investigate the in vivo mechanisms of neutrophil migration induced by leptin. The intraperitoneal injection of leptin (1 mg/kg) induces a time- and concentration-dependent neutrophil influx. We did not observe the enhancement of lipid bodies/droplets in neutrophils, after leptin treatment, as we had observed previously in peritoneal macrophages. The participation of leukotriene B4 (LTB4) in neutrophil recruitment triggered by leptin was investigated using different strategies. Leptin-induced neutrophil recruitment occurs both in the absence of 5-lipoxygenase activity in 5-lipoxygenase (5-LO)−/− mice and after the administration of either 5-LO inhibitor (Zileuton) or the LTB4 receptor antagonist (U-75302). Moreover, no direct induction of LTB4 by leptin could be observed. Neutrophil influx could not be prevented by the mammalian target of rapamycin (mTOR) inhibitor, rapamycin, contrasting with the leptin-induced signaling for lipid body formation in macrophage that is mTOR-dependent. Leptin administration led to tumor necrosis factor-alpha (TNFα) production by the peritoneal cells both in vivo and in vitro. In addition, neutrophil recruitment was inhibited in tumor necrosis factor receptor 1 (TNFR1−/−) mice, indicating a role for TNF in leptin-induced neutrophil recruitment to the peritoneal cavity. Leptin-induced neutrophil influx was PI3Kγ-dependent, as it was absent in PI3Kγ−/− mice. Accordingly, leptin induced the peritoneal cells to produce CXCL1, both in vivo and in vitro, and the neutrophil influx was ablated after using an antibody against CXCL1. Our results establish TNFα/TNFR1- and CXCL1-dependent signaling as important pathways for leptin-induced neutrophil migration in vivo.
Introduction
A growing body of evidence indicates that leptin is a key modulator of the homeostasis of the immune system (1, 2). Although effective leptin therapy for treating obesity and diabetes is not established, mainly due to leptin resistance in obese patients, there are several clinical trials for combined therapies to improve the treatment of obesity-related conditions and lipodystrophy (3–6). In fact, the leptin analog Metreleptin was approved by the U.S. Food and Drug Administration for the long-term treatment of lipodystrophy (3, 7). Leptin is known as an adipokine and modulator of the immune system (8). However, the mechanism of leptin-induced leukocyte activation and recruitment in vivo needs further investigation. Leptin has been shown to act as a direct activator of macrophages, lymphocytes, and other leukocytes (9). Neutrophils, however, do not present the full-length leptin receptor, LepRb. This isoform is responsible for the described intracellular effects of leptin in different cell types. Leptin indirectly induces the in vitro neutrophil expression of CD11b in response to the direct stimulation of macrophages (10). When investigating the in vivo effects of leptin on peritoneal macrophages and lipid droplet formation, we detected the presence of neutrophils in the peritoneal lavage (11). Leptin is known to be important for a proper response to infections and immunological homeostasis, but little is known about the in vivo modulation of neutrophil migration by leptin (12). Leptin is acutely enhanced in infections and chronically enhanced during obesity, and neutrophils play an important role on the innate immune response (13, 14). We decided therefore to investigate how leptin can activate and induce inflammatory mediators indirectly causing neutrophil migration in vivo, with potential implications for a number of inflammatory conditions in different diseases. TNFα was described to act on the priming step of neutrophil activation, while chemokines as well as leukotriene B4 (LTB4) can induce neutrophil adhesion and migration (15). TNFα was also found contributing to neutrophil recruitment stimulated by the chemokine CXCL1/KC, a direct chemoattractant for neutrophils (16). Here, we show that exogenous leptin induces in vivo migration and persistent neutrophil accumulation in the peritoneal cavity, through a mechanism largely dependent on TNFα and CXCL1 but independent of LTB4 production and signaling.
Materials and Methods
Materials
Murine-recombinant leptin, rabbit anti-mouse CXCL1 antibody (catalog # 250-11), and control rabbit IgG (catalog # 500-P00) were purchased from Peprotech, Inc. (Rocky Hill, NJ, USA). Rapamycin was obtained from Sigma-Aldrich, Inc. (Saint Louis, MO, USA). Zileuton was obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). U-75302 was purchased from Cayman Chemical (Ann Arbor, MI, USA). Osmium tetroxide was provided from Ted Pella, Inc. (Redding, CA, USA).
Animals
We used male mice of different strains: C57Bl/6, C3H/HeJ, C3H/He, 5-lipoxygenase (5-LO)-deficient (5-LO−/−), CCL3-deficient (CCL3−/−), tumor necrosis factor receptor 1 (TNFR1-deficient) (TNFR1−/−), and PI3Kγ-deficient (PI3Kγ−/−) mice and respective wild types (WTs) (5-LO+/+, CCL3+/+, TNFR1+/+, and PI3Kγ+/+), obtained as previously described (17–21). Mice were obtained from the FIOCRUZ breeding unit, as well as raised and maintained under the same housing conditions. All animal care and experimental protocols were conducted following the guidelines of the Brazilian Council for Care and Use of Experimentation Animals (CONCEA). The Oswaldo Cruz Institute Animal Welfare Committee (CEUA-IOC license number L-011/2015) approved all protocols used in this study.
In Vivo Leptin Treatments
The in vivo treatments were performed as previously described (11). Briefly, following the intraperitoneal (i.p.) administration of leptin (0.25, 0.5, 1, and 2 mg/kg, depending on the experiment) or vehicle (sterile, apirogenic saline), animals were euthanized at different time points (1, 6, or 24 h, as specified in each experiment). Alternatively, animals received three i.p. injections of rapamycin (12.5 μg/kg), or vehicle, 12 h before, 15 min before, and 12 h after the injection of leptin or saline, and the peritoneal lavage was harvested after 24 h. This treatment was established by us and was proved to be effective for the inhibition of leptin-induced lipid droplets in peritoneal macrophages (11). We also evaluated the effect of i.p. pretreatments, 15 min prior to leptin treatment, with the phospholipase A2 inhibitor, Zileuton (60 μg/cavity), or the LTB4 receptor BLT1-specific inhibitor, U-75302 (5 mg/kg). These drugs were administered according to data from previous works from our group and others (22–28). To block CXCL1, the antibody against CXCL1 (3 μg/animal) or the isotype control (diluted in sterile saline) was injected into the peritoneal cavity, 10 min before the leptin injection. After the time specified in each experiment, the peritoneal cells were harvested as follows. The peritoneal cavity was rinsed with HBSS (Hank’s balanced salted solution, 3 mL/cavity), and a volume of approximately 2.5 mL was recovered. Samples were diluted in Turk fluid (2% acetic acid) for total leukocyte counts using Neubauer chambers. Differential leukocyte counting was performed in cytospin smears stained by May–Grünwald–Giemsa, a classical staining for the differential identification of leukocytes (mononuclear cells, neutrophils, and eosinophils) (29). As a control of vascular integrity, we assured that leptin injection does not modify peritoneal lavage protein concentrations There is no difference in the total protein concentration between the samples at 6 h after injection of saline (0.754 ± 0.032 mg/mL) or leptin (0.729 ± 0.048 mg/mL), or at 24 h after injection of saline (0.776 ± 0.014 mg/mL) or leptin (0.749 ± 0.037 mg/mL).
In Vitro Leptin Incubation
The peritoneal cells including the macrophages were obtained from naïve C57Bl/6 mice, by peritoneal lavage with HBSS (5 mL). Cells were transferred to polypropylene tubes (1 × 106 cells/mL) and then incubated with leptin (20 nM) for 4 h, under 37°C, 5% CO2 atmosphere in RPMI 1604 medium. After incubation, tubes were centrifuged for supernatant collection. Cell viability was always >85% as determined by Trypan blue exclusion. For the in vitro incubation of neutrophils, murine bone marrow neutrophils were obtained as previously described (30). Briefly, the bone marrow from femurs and tibias was washed, and neutrophils were purified in a discontinuous Percoll gradient. Neutrophil viability was greater than 95% as assessed by the Trypan blue exclusion test, and purity was greater than 98% as analyzed by microscopy using Hemacolor staining (Merck, Darmstadt, Germany).
KC, TNF, Leptin, and LTB4 Quantification
Peritoneal lavage or in vitro supernatants were analyzed for TNFα, CXCL1/KC, and LTB4. TNFα and CXCL1 ELISA were performed with Duo Set kit, according to manufacturer’s protocol (R&D Systems, Minneapolis, MN, USA) and LTB4 with enzyme immuno assay (EIA) kit (Cayman Chemical, Ann Arbor, MI, USA). For the investigation of the presence of intracellular LTB4, we used the previously described Eicosacell protocol (31). Briefly, leukocytes were recovered from peritoneal cavities and immediately submitted to fixation and permeabilization with 0.5% 1-ethyl-3-(3-dimethylamino-propyl) carbodiimide (Sigma) in HBSS. After that, a common immunodetection protocol was performed using the following antibodies: the primary antibody anti-LTB4 (Cayman Chemical) or irrelevant IgG and the secondary antibody Alexa 488-labeled anti-rabbit IgG. Images were obtained using an Olympus BX51 fluorescence microscope and equipped with a Plan Apo ×100 objective and a DP72 camera (Olympus Optical, Japan) in conjunction with CellF Imaging Software (Olympus Life Science Europe, Germany).
Statistical Analysis
Data were reported as the mean ± standard error of the mean (SEM). Data were statistically analyzed by the analysis of variance with Newman–Keuls–Student test, or Student’s t-test. Differences were considered to be significant when p ≤ 0.05 (see figure legends).
Results
Leptin-Induced Selective Neutrophil Recruitment In Vivo
Leptin deficiency is associated with impaired cell-mediated immunity and increased susceptibility to infection (1), and neutrophil chemotaxis is a key component of inflammation and host response to infection. There are very few studies though addressing leptin-inflammatory effects in vivo, and the effects of leptin are confounded with the effects of other immunometabolism-modulating factors that are altered in diseases such as infection or obesity (32). To investigate the specific effect of leptin on neutrophil recruitment, here, neutrophil influx was evaluated at different time points after i.p. leptin (1 mg/kg) administration. Leptin-induced neutrophil accumulation in the peritoneal cavity of C57BL/6 mice was significant within 1 h, maximal within 6 h, and sustained for at least until 24 h (Figure 1A). This effect was observed at different leptin concentrations (0.5, 1, and 2 mg/kg) for 24 h (Figure 1B). It is known that after bacterial or LPS stimulation, activated neutrophils present enhanced lipid body numbers, which is considered characteristic of their activation (33–35). In our previous work, the enhancement of lipid droplets in leptin-stimulated macrophages was described (11). However, we did not observe the enhancement of lipid droplets above the basal levels in peritoneal neutrophils after leptin treatment (Figure S1 in Supplementary Material). The i.p. injection of leptin in C3H/HeJ LPS-resistant mice led to significant neutrophil accumulation in the peritoneal cavity within 24 h (Figure 1C). Leptin samples were confirmed negative for LPS contamination by LAL testing (<0.01 UI); therefore, our data indicate that LPS is not involved in the observed leptin response. Moreover, we confirmed the absence of a direct effect of leptin on neutrophils by in vitro neutrophil adhesion experiments (Figure S2 in Supplementary Material). Even with very high leptin concentrations (up to 200 nM), no adhesion was observed. In our preparation, we ensured >98% purity of neutrophils as previously described (30).
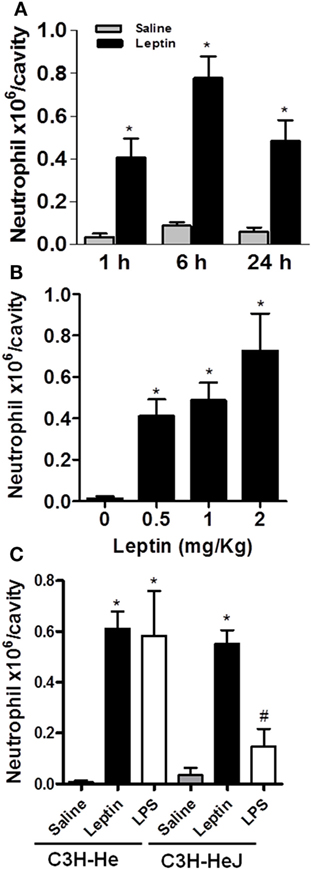
Figure 1. Leptin-induced neutrophil migration. Leptin was injected into the peritoneal cavity of C57Bl/6 mice, and the cells were collected and stained by May–Grünwald–Giemsa for neutrophil recruitment analysis. (A) Neutrophils were counted in the peritoneal cavity at 1, 6, and 24 h after leptin (1 mg/kg) injection. (B) Different concentrations of leptin (0.5, 1, and 2 mg/kg) led to neutrophils presence in the peritoneal cavity in 24 h. (C) Neutrophils accumulated in the peritoneal cavity of C3H-HeJ mice 6 h after leptin injection (1 mg/kg), but not after LPS (80 ng/cavity) or saline injection. Each bar represents the mean ± SEM, n = 5–7 (A,B) and n = 3–6 (C). All experiments were performed at least three times. All data were analyzed by Newman–Keuls–Student test. *Statistically significant (p < 0.05) difference between control and stimulated groups. #Statistically significant (p < 0.05) difference between C3H-Hej and C3H-He LPS-stimulated groups.
Leptin-Induced Neutrophil Migration Is Requisitely Dependent on TNFα
It had been previously demonstrated that leptin indirectly activates human neutrophils (10). This work showed that neutrophil CD11b expression can be enhanced in response to TNFα produced by leptin-stimulated monocytes in vitro. We confirmed that leptin induced TNFα production by the peritoneal cells in vitro (Figure 2A). Moreover, leptin injection into the peritoneal cavity induced in vivo TNFα production (Figure 2B). In order to investigate the role of TNFα receptor on leptin-induced neutrophil recruitment in vivo, TNFR1 knockout mice (TNFR1−/−) or WT (TNFR1+/+) animals (C57Bl/6) were used. As demonstrated in Figure 2C, leptin was unable to induce neutrophil accumulation in TNFR1−/− mice, but interestingly, TNFR1 is not necessary for the effect of leptin in inducing lipid droplets in macrophages (Figure S3 in Supplementary Material). Leptin induced lipid body formation in peritoneal macrophages from TNFR1−/− mice, similar to what was observed in WT (TNFR1+/+ mice). Thus, leptin-induced neutrophil migration is dependent on TNFR1 signaling, but macrophage lipid body formation occurs independently of this TNFα receptor.
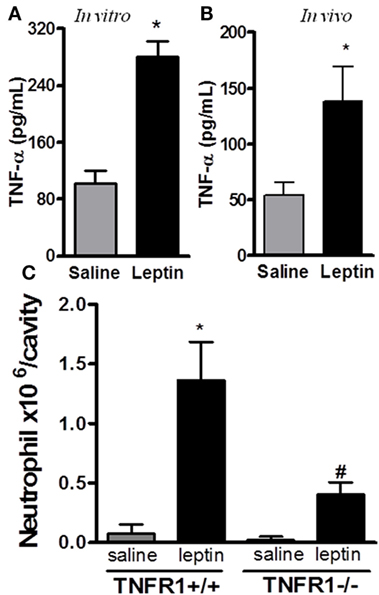
Figure 2. Tumor necrosis factor-alpha (TNFα) participates in leptin-induced neutrophil migration. (A) Peritoneal macrophages were stimulated with 20 nM leptin for 4 h in vitro, before TNFα measurement in the culture supernatant. (B) C57Bl/6 mice were injected intraperitoneally with leptin (1 mg/kg) in vivo and, 6 h afterward, TNFα was measured in the peritoneal washing supernatant. (C) Tumor necrosis factor receptor 1 (TNFR1−/−) or TNFR1+/+ mice received leptin (1 mg/kg) or saline i.p. injection. The analysis of neutrophil recruitment was performed at 24 h after leptin administration. Each bar represents the mean ± SEM, n = 4–5. Experiments A and B were performed at least three times. Data were analyzed by analysis by Student’s t-test (A,B) and by Newman–Keuls–Student test. *Statistically significant difference (p < 0.05) between leptin-stimulated and saline groups. #Statistically significant difference (p < 0.05) between TNFR1−/− and TNFR1+/+ leptin-stimulated groups.
Leptin-Induced Neutrophil Recruitment Is Dependent on the PI3Kγ Pathway
Several studies have demonstrated the importance of PI3K, specifically PI3Kγ, in leukocyte migration and activation in vivo (19, 36–39). To determine the role of PI3Kγ in the acute response induced by leptin, we examined neutrophil in WT mice or mice genetically deficient in PI3Kγ (PI3Kγ−/−), 24 h after leptin i.p. administration. Leptin-induced accumulation of neutrophils in the peritoneal cavity was completely inhibited in PI3Kγ−/− mice in comparison to that in WT control animals (Figure 3A). Thus, the PI3Kγ-signaling pathway is required for leptin-induced neutrophil recruitment.
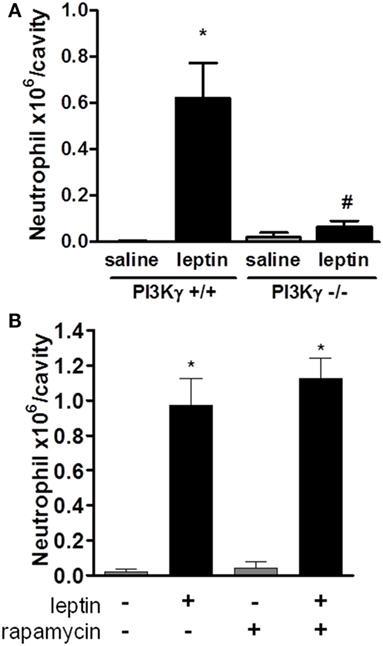
Figure 3. Leptin-induced neutrophil migration is dependent on PI3Kγ but independent of mammalian target of rapamycin (mTOR) signaling. (A) PI3Kγ+/+ or PI3Kγ−/− mice were injected intraperitoneally with leptin (1 mg/kg) or saline. Neutrophils were counted in the peritoneal wash at 24 h after leptin injection. (B) C57Bl/6 mice received rapamycin treatment (12.5 µg/kg per injection) 12 h before, 15 min before, and 12 h after leptin injection. Each bar represents the mean ± SEM, n = 3–6 (A) and 6–8 (B). The experiments were performed at least three times. Data were analyzed by Newman–Keuls–Student test. *Statistically significant difference (p < 0.05) between leptin-stimulated and saline groups. #Statistically significant difference (p < 0.05) between PI3Kγ−/− and PI3Kγ+/+ leptin-stimulated groups.
Leptin-Induced Neutrophil Recruitment Is Independent on mTOR Pathway
We showed previously that mTOR was required for the effects of leptin on lipid droplets formation in macrophages both in vitro and in vivo (11). Others reported that the mTOR pathway was important for GM-CSF-dependent neutrophil activation in vitro (40). We investigated here the functional role of the mTOR pathway for the in vivo leptin-induced neutrophil activation and recruitment. We used the previously described treatment with rapamycin, a specific inhibitor and probe for mTOR activity (11, 41). The treatment of mice with rapamycin failed to modify leptin-induced neutrophil accumulation after 24 h (Figure 3B).
Leptin-Induced Neutrophil Migration Is Independent of LTB4
It had been shown that the lipid mediator LTB4 directly exerts a chemoattractive effect on neutrophils in inflammatory conditions (42, 43). We performed several in vivo experiments in order to evaluate the role of LTB4 in neutrophil migration stimulated by leptin. First, an EIA assay was performed to detect LTB4 presence on peritoneal supernatants obtained after leptin i.p. injection. There was no difference in the concentration of LTB4 between saline and 1-, 6-, or 24-h leptin-stimulated groups (Figure 4A). It could be that LTB4 was produced but not released from the cell; therefore, we performed the Eicosacell assay. We did not observe intracellularly retained LTB4 in the peritoneal cells submitted to the Eicosacell assay as performed before by our group (data not shown) (31). We also investigated the importance of the LTB4 synthesis enzyme, 5-LO for neutrophil migration. Therefore, 5-LO+/+ or 5-LO−/− mice were injected with leptin in the peritoneal cavity (1 mg/kg), and neutrophil migration was investigated. As shown in Figure 4B, leptin induced neutrophil migration (24 h) in 5-LO knockout mice. The pharmacological inhibition of LTB4 synthesis by Zileuton (60 µg/animal i.p.) and signaling on the receptor BTL1 by U-75302 (5 mg/kg i.p.) were tested. Figure 4C shows that peritoneal neutrophils migrate in response to leptin injection, despite pharmacological pretreatments. These results demonstrate that LTB4 does not participate in leptin-induced neutrophil recruitment.
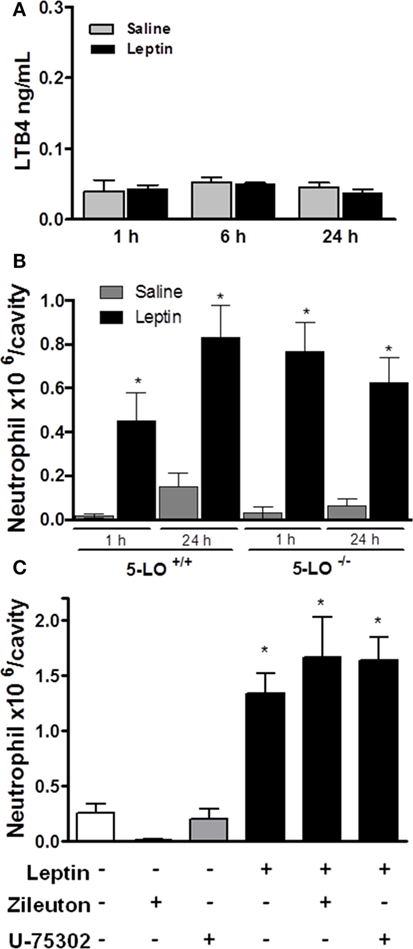
Figure 4. Leptin-induced neutrophil migration is independent of LTB4 signaling. (A) C57Bl/6 mice were injected intraperitoneally with leptin (1 mg/kg), and after 24 h LTB4 EIA was performed in the peritoneal washing supernatant. (B) 5-LO+/+ or 5-LO−/− mice were injected intraperitoneally with leptin (1 mg/kg) or saline. Neutrophils were counted in the peritoneal wash at 24 h after leptin injection. (C) C57Bl/6 mice were pre-treated with U-75302 (5 mg/kg) or Zileuton (60 µg/animal), 10 min before receiving leptin injection (1.5 mg/kg). Neutrophils were counted in peritoneal wash, 3 h after leptin injection. Each bar represents the mean ± SEM, n = 5–8. The experiments were performed at least three times. Data were analyzed by Newman–Keuls–Student test. *Statistically significant difference (p < 0.05) between leptin and saline. There are no statistically significant differences (p > 0.05) between 5-LO−/− and 5-LO+/+ leptin-stimulated groups.
Leptin-Induced Neutrophil Migration Is Dependent on CXCL1
CXCL1 is known to mediate the neutrophil migration through the activation of CXCR2, a PI3Kγ-dependent receptor (44). Neutrophil migration can also be promoted by CCL3/MIP-1α signaling through CCR1, a PI3Kγ-dependent receptor (45). Because our result in Figure 3A showed that leptin-induced neutrophil recruitment is dependent on PI3Kγ, we investigated the chemokines CXCL1 and CCL3 as possible mediators for this effect. We observed that CXCL1 is secreted by the peritoneal cells stimulated by leptin both in vitro and in vivo (Figures 5A,B). Anti-CXCL1-neutralizing antibodies were used in vivo as a pretreatment before leptin injection. A partial but significant inhibition of neutrophil migration to peritoneal cavity was observed (Figure 5C). We investigated the participation of the chemokine CCL3 using the CCL3 knockout mice (CCL3−/−). Mice were injected with leptin i.p. and, as seen in Figure 5D, neutrophil recruitment was not altered when compared to WT mice (CCL3+/+). Taken together, our data show that leptin activates the peritoneal cells to induce neutrophil migration in a specific CXCL1-dependent manner.
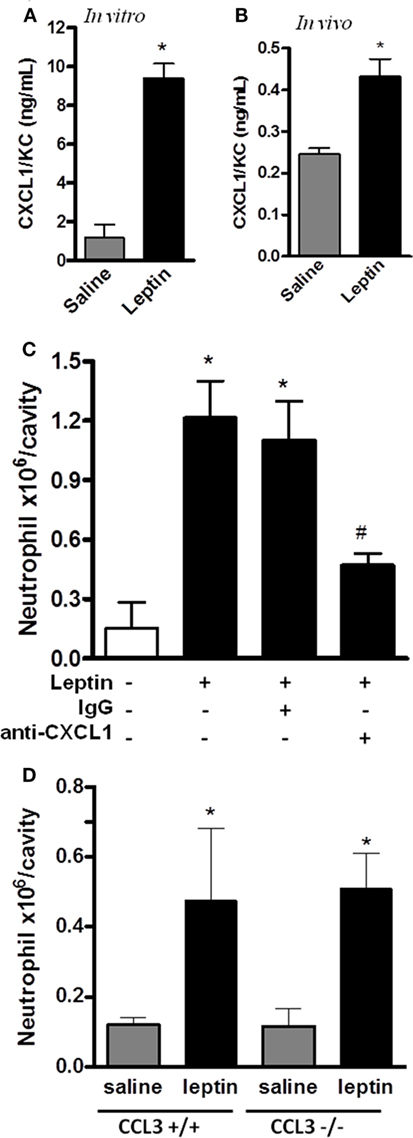
Figure 5. Leptin-induced neutrophil migration is dependent on CXCL1/KC signaling but independent of CCL3 signaling. (A) Peritoneal macrophages were stimulated with 20 nM leptin in vitro, for 4 h, and CXCL1 was measured in the culture supernatant. (B) C57Bl/6 mice were injected intraperitoneally with leptin (1 mg/kg), in vivo, and after 6 h, CXCL1 was measured in the peritoneal washing supernatants. (C) C57BL/6 mice were treated with either non-specific IgG or anti-CXCL1 antibodies (3 microgram/cavity i.p.), 10 min before leptin injection (1 mg/kg; i.p.). Neutrophils were counted in the peritoneal wash at 4 h after leptin injection. (D) CCL3+/+ or CCL3−/− mice were injected intraperitoneally with leptin (1 mg/kg) or saline. Neutrophils were counted in the peritoneal wash at 24 h after leptin injection. Each bar represents the mean ± SEM, n = 4–6 (A,B), n = 7–9 (C), and n = 5–8 (D). The experiments were performed at least three times. Data were analyzed by Student’s t-test (A,B) or by Newman–Keuls–Student test (C,D). *Statistically significant differences (p < 0.05) between leptin-stimulated and saline groups. #Statistically significant differences (p < 0.05) between anti-CXCL1 plus leptin group and leptin-stimulated group. There is no statistically significant difference (p > 0.05) between CCL3−/− and CCL3+/+ leptin-stimulated groups.
Discussion
We demonstrated here that the in vivo administration of leptin significantly induces neutrophil recruitment to the peritoneal cavity. We propose here that this is an indirect effect of leptin, which requires peritoneal resident cell activation with the production of TNFα and CXCL1, mostly by resident monocytes/macrophages. It was shown before that leptin induces TNFα production by macrophages, and under different inflammatory stimuli, peritoneal macrophages produce TNFα and CXCL1 that are important for neutrophil migration (10, 16, 46). Leptin contribution to the modulation of neutrophil activation has been controversial because neutrophils present LepRa but lack LepRb (10, 47). It was suggested that leptin could stimulate chemotaxis and the release of hydrogen peroxide by human neutrophils (48). Others demonstrated that leptin could only indirectly trigger increased CD11b expression on human neutrophils by the induction of TNFα release from monocytes in vitro, and highly purified neutrophils (more than 98%) do not produce TNF under leptin stimulation (10). It was also suggested that leptin could inhibit neutrophil apoptosis in a LepRa-dependent manner (49). More recently, it was demonstrated that only extremely high concentrations of leptin could drive direct changes on neutrophil activation or the inhibition of apoptosis (above 3–25 µg/mL; 200–1,600 nM) (50). Kamp et al. concluded that there is no convincing evidence that physiological leptin concentrations could directly activate neutrophils. Accordingly, we found no significant activation of adhesion, even with 200 nM (Figure S2 in Supplementary Material). These previous studies to evaluate the neutrophil capacity to migrate were performed in vitro. We decided to study the in vivo mechanisms for neutrophil migration under exogenous leptin administration. Our data show in vivo leptin-induced neutrophil recruitment and suggest an indirect activation of other resident cells. Lipid droplet formation is associated with leukocyte activation in different inflammatory models (33, 35, 51, 52). Interestingly, we found that there was no enhanced lipid droplet formation in leptin-dependent-migrating neutrophils (Figure S1 in Supplementary Material). We suggest here that leptin effects are not sufficient for the complete activation of neutrophils in vivo.
The contribution of LTB4 to inflammation progression is well known. This lipid mediator is both an important neutrophil chemotaxis inducer, the activator of phagocytosis, and the main eicosanoid produced by neutrophils under diverse pro-inflammatory stimuli ((24, 53–57)). We had previously shown that leptin-stimulated peritoneal macrophages have a higher capacity to produce LTB4 following calcium ionophore activation (11). The absence of LTB4 in the peritoneal cavity, shown in Figure 4A, provides evidence that the migrating neutrophils, after leptin stimulation, are not fully activated. Moreover, leptin induced neutrophil recruitment despite the genetic 5-LO null background, the pharmacological inhibition of 5-LO, or the pharmacological blockage of BLT1 (Figure 4). These results rule out an involvement of LTB4 in leptin-induced neutrophil activation and recruitment in vivo.
It was known that TNFα induces CD11b expression on neutrophils, an important integrin component that participates in intraluminal adhesion and crawling in the site of inflammation (10, 58). We show here that the leptin effect on neutrophil migration is mediated by TNFα signaling through TNFR1. Indeed, leptin failed to recruit neutrophils to the peritoneal cavity in mice genetically deficient in TNFR1. An indirect action of leptin on neutrophils mediated by TNFα supports the involvement of this pathway in acute inflammatory responses. Chemokines, produced by resident cells under TNFα stimulation, such as the CXCL1/KC, the mouse analog to human IL-8, have been shown to induce the recruitment of neutrophils to inflammatory sites (16). Moreover, leptin induces CXCL1 in epithelial cells (59). Here, we show that resident peritoneal cells secrete CXCL1 after leptin stimulation, both in vivo and in vitro, and CXCL1 neutralization partially inhibits neutrophil migration. We conclude that leptin-induced neutrophil recruitment is dependent on the CXCL1 chemokine produced by the peritoneal cells. It was recently described that adipose stromal cells can be recruited from white adipose tissue to tumor environments through CXCL1 signaling, suggesting that the increased aggressiveness of certain cancers is linked to white adipose tissue overgrowth in obesity (60). Our data offer a possible explanation for the role of leptin in tumor cell recruitment during obesity, when high levels of leptin production by white adipose tissue are observed. The PI3Kγ-signaling pathway was shown to be relevant for neutrophil recruitment induced by CXCL1 in vivo in specific sites of inflammatory stimuli. Neutrophil recruitment to bronchoalveolar space induced by CXCL1 was prevented in PI3Kγ−/− mice (44). However, these authors showed that the administration of CXCL1 in the cremaster muscle of PI3Kγ−/− or WT mice induced similar increases in neutrophil rolling, adhesion, and transmigration. Our data show that leptin-induced neutrophil migration to the peritoneal cavity is prevented in PI3Kγ−/− mice. We suggest that this may be caused by CXCL1 signaling through PI3Kγ, since PI3K-regulatory subunits differentially participate in neutrophil migration either in vivo and in vitro (61). It may also reflect the combination of PI3Kγ activation in endothelium and neutrophils, which is required for leukocyte trafficking to sites of inflammation (62). Based on our findings and previous published data, we conclude that PI3Kγ have both direct and indirect effects on neutrophil recruitment, since PI3Kγ is part of the signaling pathway activated by CXCL1 in neutrophils and is important for the leptin-induced activation of macrophages (11, 44, 63).
Other chemokines were shown to be important for neutrophil recruitment, such as CCL3, and both insulin and TNFα could prime the neutrophils for CCL3/MIP-1α-dependent migration (64, 65). Different from insulin, we observed that the leptin effect was not dependent on CCL3 since neutrophil migration was maintained on the CCL3-deficient mice (Figure 5C).
We previously demonstrated that leptin induced lipid droplet formation in peritoneal macrophages in a PI3K/mTOR-dependent manner (11). Interestingly, the lipid droplet induction by leptin on resident peritoneal macrophages is not dependent on TNFα signaling because this effect was maintained in the TNFR1−/− (Figure S3 in Supplementary Material). Therefore, leptin induces TNFα- and CXCL1-dependent neutrophil migration, by a distinct pathway from lipid droplet formation in the peritoneal cells.
In the immune system, mTOR is involved in signaling downstream from different inflammatory stimuli. Phosphatidic acid and LPS are examples of leukocyte activation inducers that depend on mTOR activation (66–68), and also GM-CSF-induced neutrophil migration was shown to be inhibited by rapamycin (40). The mTOR pathway is important for the PAF-induced activation of neutrophils, inducing the translation of the cytokine IL-6 receptor (69). Nevertheless, we showed here that the indirect effect of leptin-induced migration of neutrophils is independent of the TORC1 pathway (rapamycin-inhibited mTOR complex). Rapamycin is a proapoptotic drug for different cells including neutrophils. Since rapamycin does not alter neutrophil accumulation after 24 h (Figure 3), we suggest that the mTOR-dependent inhibition of apoptosis is not the main effect of leptin on neutrophils in vivo.
It has been shown that LPS-induced systemic inflammation presents neutrophils transmigration into the brain in a leptin-dependent manner (70, 71). Our data are in agreement to these in vivo results, but we show that leptin can promote in vivo neutrophil migration independently of LPS-inflammatory response. In a lung injury model, the intranasal administration of leptin counteracted some LPS effects in neutrophils (72). Such results are in apparent contrast with our hypothesis and data. We think that these results are due to tissue-specific effects. We evaluated the peritoneal cavity, while Landgraf et al. analyzed lung tissue, where leptin can directly activate the alveolar epithelium. We recently studied the direct effects of leptin intestinal epithelial cells and observed an enhancement of TGFβ that could be responsible for inhibitory effects on the tissue macrophages (59). It has also been suggested that there may be different subsets of neutrophils (73). These different neutrophil subsets may then have diverse responses toward direct or indirect leptin stimulation. Therefore, it would be very interesting to further explore the differential effects of leptin on leukocytes migration in distinct tissues, administration sites, and different neutrophil subsets.
Our data reveal the importance of leptin for neutrophil migration to sites of inflammation. We argue that it is crucial to understand these effects in great detail, in particular because of the prescription of recombinant leptin in different human conditions.
Ethics Statement
All animal care and experimental protocols were conducted following the guidelines of the Brazilian Council for Care and Use of Experimentation Animals (CONCEA). The Oswaldo Cruz Institute Animal Welfare Committee (CEUA_IOC, L-011/2015) approved all protocols used in this study.
Author Contributions
All authors had critically revised and approved the final version of the manuscript. GS-A and CM-M performed the conception, designed and performed the experiments, analyzed and interpreted data, and wrote the manuscript draft. HB, PA, TG, SL, IH, and BW participated in the data acquisition, analysis, and interpretation of the data. HC-F-N, PB, and CB-M participated in the conception, design, analysis, and interpretation of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Mauro Teixeira (Universidade Federal de Minas Gerais, Belo Horizonte, Brazil) and Dr. Joseli Lannes Vieira (Instituto Oswaldo Cruz, Rio de Janeiro, Brazil) for the genetically deficient animals and MSc Lohanna Palhinha for the assistance. The authors are in debt with Dr. Jens Rietdorf (CDTS, FIOCRUZ) and Dr. Guy Zimmerman (University of Utah, Salt Lake City, UT, USA) for comments on the work and manuscript.
Funding
This work was supported by the Howard Hughes Medical Institute (HHMI) (to PB), Conselho Nacional de Pesquisa (CNPq, Brazil), Fundação de Amparo à Pesquisa do Rio de Janeiro (FAPERJ, Brazil), CAPES, CAPES-SUS, and PAPES (FIOCRUZ, Brazil) (to CM-M, CB-M, and PB), and Deutsche Forschungsgemeinschaft SFB914/A02 (to BW).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00111/full#supplementary-material.
Abbreviations
5-LO, 5-lipoxygenase; ADRP, adipose differentiation-related protein; EIA, enzyme immuno assay; mTOR, mammalian target of rapamycin; LTB4, leukotriene B4; PLA2, phospholipase A2; TNFR1, tumor necrosis factor receptor 1.
References
1. Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. J Immunol (2005) 174:3137–42. doi:10.4049/jimmunol.174.6.3137
2. Procaccini C, De Rosa V, Galgani M, Carbone F, Cassano S, Greco D, et al. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. J Immunol (2012) 189:2941–53. doi:10.4049/jimmunol.1200935
3. Moon H-S, Matarese G, Brennan AM, Chamberland JP, Liu X, Fiorenza CG, et al. Efficacy of Metreleptin in obese patients with type 2 diabetes: cellular and molecular pathways underlying leptin tolerance. Diabetes (2011) 60:1647–56. doi:10.2337/db10-1791
4. Polyzos SA, Mantzoros CS. Leptin in health and disease: facts and expectations at its twentieth anniversary. Metabolism (2015) 64:5–12. doi:10.1016/j.metabol.2014.10.017
5. Shetty GK, Matarese G, Magkos F, Moon H-S, Liu X, Brennan AM, et al. Leptin administration to overweight and obese subjects for 6 months increases free leptin concentrations but does not alter circulating hormones of the thyroid and IGF axes during weight loss induced by a mild hypocaloric diet. Eur J Endocrinol (2011) 165:249–54. doi:10.1530/EJE-11-0252
6. Wabitsch M, Funcke J-B, von Schnurbein J, Denzer F, Lahr G, Mazen I, et al. Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J Clin Endocrinol Metab (2015) 100:3227–30. doi:10.1210/jc.2015-2263
7. Chou K, Perry CM. Metreleptin: first global approval. Drugs (2013) 73:989–97. doi:10.1007/s40265-013-0074-7
8. La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol (2004) 4:371–9. doi:10.1038/nri1350
9. Abella V, Scotece M, Conde J, Pino J, Gonzalez-Gay MA, Gómez-Reino JJ, et al. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat Rev Rheumatol (2017) 13:100–9. doi:10.1038/nrrheum.2016.209
10. Zarkesh-Esfahani H, Pockley AG, Wu Z, Hellewell PG, Weetman AP, Ross RJM. Leptin indirectly activates human neutrophils via induction of TNF-. J Immunol (2004) 172:1809–14. doi:10.4049/jimmunol.172.3.1809
11. Maya-Monteiro CM, Almeida PE, D’Avila H, Martins AS, Rezende AP, Castro-Faria-Neto H, et al. Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem (2008) 283:2203–10. doi:10.1074/jbc.M706706200
12. Mancuso P, Huffnagle GB, Olszewski M, Phipps J, Peters-Golden M. Leptin corrects host defense defects after acute starvation in murine pneumococcal pneumonia. Am J Respir Crit Care Med (2006) 173:212–8. doi:10.1164/rccm.200506-909OC
13. Behnes M, Brueckmann M, Lang S, Putensen C, Saur J, Borggrefe M, et al. Alterations of leptin in the course of inflammation and severe sepsis. BMC Infect Dis (2012) 12:217. doi:10.1186/1471-2334-12-217
14. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6:173–82. doi:10.1038/nri1785
15. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol (2013) 13:159–75. doi:10.1038/nri3399
16. Vieira SM, Lemos HP, Grespan R, Napimoga MH, Dal-Secco D, Freitas A, et al. A crucial role for TNF-alpha in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br J Pharmacol (2009) 158:779–89. doi:10.1111/j.1476-5381.2009.00367.x
17. Cook D, Beck M, Coffman T, Kirby S, Sheridan J, Pragnell I, et al. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science (1995) 269(5230):1583–5. doi:10.1126/science.7667639
18. Goulet JL, Snouwaert JN, Latour AM, Coffman TM, Koller BH. Altered inflammatory responses in leukotriene-deficient mice. Proc Natl Acad Sci U S A (1994) 91(26):12852–6. doi:10.1073/pnas.91.26.12852
19. Sasaki T. Function of PI3K in thymocyte development, T cell activation, and neutrophil migration. Science (2000) 287:1040–6. doi:10.1126/science.287.5455.1040
20. Watson J, Riblet R. Genetic control of responses to bacterial lipopolysaccharides in mice. I. Evidence for a single gene that influences mitogenic and immunogenic responses to lipopolysaccharides. J Exp Med (1974) 140:1147–61. doi:10.1084/jem.140.5.1147
21. Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature (1995) 377:348–51. doi:10.1038/377348a0
22. Gomides LF, Duarte ID, Ferreira RG, Perez AC, Francischi JN, Klein A. Proteinase-activated receptor-4 plays a major role in the recruitment of neutrophils induced by trypsin or carrageenan during pleurisy in mice. Pharmacology (2012) 89:275–82. doi:10.1159/000337378
23. Li X, Fu H, Yi W, Zhao Y, Wang J, Li J, et al. Dual role of leukotriene B4 receptor type 1 in experimental sepsis. J Surg Res (2015) 193:902–8. doi:10.1016/j.jss.2014.09.013
24. Monteiro APT, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, Porto BN, et al. Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol (2011) 186:6562–7. doi:10.4049/jimmunol.1002400
25. Rao TS, Currie JL, Shaffer AF, Isakson PC. In vivo characterization of zymosan-induced mouse peritoneal inflammation. J Pharmacol Exp Ther (1994) 269:917–25.
26. Rao TS, Yu SS, Djuric SW, Isakson PC. Phorbol ester-induced dermal inflammation in mice: evaluation of inhibitors of 5-lipoxygenase and antagonists of leukotriene B4 receptor. J Lipid Mediat Cell Signal (1994) 10:213–28.
27. Silva AR, de Assis EF, Caiado LFC, Marathe GK, Bozza MT, McIntyre TM, et al. Monocyte chemoattractant protein-1 and 5-lipoxygenase products recruit leukocytes in response to platelet-activating factor-like lipids in oxidized low-density lipoprotein. J Immunol (2002) 168:4112–20. doi:10.4049/jimmunol.168.8.4112
28. Sun M, Wang R, Han Q. Inhibition of leukotriene B4 receptor 1 attenuates lipopolysaccharide-induced cardiac dysfunction: role of AMPK-regulated mitochondrial function. Sci Rep (2017) 7:44352. doi:10.1038/srep44352
29. D’Avila H, Roque NR, Cardoso RM, Castro-Faria-Neto HC, Melo RCN, Bozza PT. Neutrophils recruited to the site of Mycobacterium bovis BCG infection undergo apoptosis and modulate lipid body biogenesis and prostaglandin E production by macrophages. Cell Microbiol (2008) 10:2589–604. doi:10.1111/j.1462-5822.2008.01233.x
30. Schymeinsky J, Sindrilaru A, Frommhold D, Sperandio M, Gerstl R, Then C, et al. The Vav binding site of the non-receptor tyrosine kinase Syk at Tyr 348 is critical for beta2 integrin (CD11/CD18)-mediated neutrophil migration. Blood (2006) 108:3919–27. doi:10.1182/blood-2005-12-030387
31. Bandeira-Melo C, Weller PF, Bozza PT. Eicosacell – an immunofluorescent-based assay to localize newly synthesized eicosanoid lipid mediators at intracellular sites. Methods Mol Biol (2011) 689:163–81. doi:10.1007/978-1-60761-950-5_10
32. Trottier MD, Naaz A, Kacynski K, Yenumula PR, Fraker PJ. Functional capacity of neutrophils from class III obese patients. Obesity (2012) 20:1057–65. doi:10.1038/oby.2011.354
33. Bozza PT, Magalhães KG, Weller PF. Leukocyte lipid bodies – biogenesis and functions in inflammation. Biochim Biophys Acta (2009) 1791:540–51. doi:10.1016/j.bbalip.2009.01.005
34. Melo RCN, Sabban A, Weller PF. Leukocyte lipid bodies: inflammation-related organelles are rapidly detected by wet scanning electron microscopy. J Lipid Res (2006) 47:2589–94. doi:10.1194/jlr.D600028-JLR200
35. Weller PF, Ackerman SJ, Nicholson-Weller A, Dvorak AM. Cytoplasmic lipid bodies of human neutrophilic leukocytes. Am J Pathol (1989) 135:947–59.
36. Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase γ in inflammation. Science (2000) 287:1049–53. doi:10.1126/science.287.5455.1049
37. Rickert P. Leukocytes navigate by compass: roles of PI3Kγ and its lipid products. Trends Cell Biol (2000) 10:466–73. doi:10.1016/S0962-8924(00)01841-9
38. Servant G. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science (2000) 287:1037–40. doi:10.1126/science.287.5455.1037
39. Pinho V, Souza DG, Barsante MM, Hamer FP, De Freitas MS, Rossi AG, et al. Phosphoinositide-3 kinases critically regulate the recruitment and survival of eosinophils in vivo: importance for the resolution of allergic inflammation. J Leukoc Biol (2005) 77:800–10. doi:10.1189/jlb.0704386
40. Gomez-Cambronero J. Rapamycin inhibits GM-CSF-induced neutrophil migration. FEBS Lett (2003) 550:94–100. doi:10.1016/S0014-5793(03)00828-7
41. Shamji AF, Nghiem P, Schreiber SL. Integration of growth factor and nutrient signaling: implications for cancer biology. Mol Cell (2003) 12:271–80. doi:10.1016/j.molcel.2003.08.016
42. Oyoshi MK, He R, Li Y, Mondal S, Yoon J, Afshar R, et al. Leukotriene B4-driven neutrophil recruitment to the skin is essential for allergic skin inflammation. Immunity (2012) 37:747–58. doi:10.1016/j.immuni.2012.06.018
43. Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, Hurley BP. distinct cellular sources of hepoxilin A 3 and leukotriene B 4 are used to coordinate bacterial-induced neutrophil transepithelial migration. J Immunol (2015) 194:1304–15. doi:10.4049/jimmunol.1402489
44. Pinho V, de Castro Russo R, Amaral FA, de Sousa LP, Barsante MM, de Souza DG, et al. Tissue- and stimulus-dependent role of phosphatidylinositol 3-kinase isoforms for neutrophil recruitment induced by chemoattractants in vivo. J Immunol (2007) 179:7891–8. doi:10.4049/jimmunol.179.11.7891
45. Reichel CA, Puhr-Westerheide D, Zuchtriegel G, Uhl B, Berberich N, Zahler S, et al. C-C motif chemokine CCL3 and canonical neutrophil attractants promote neutrophil extravasation through common and distinct mechanisms. Blood (2012) 120:880–90. doi:10.1182/blood-2012-01-402164
46. Lee SM, Choi HJ, Oh CH, Oh JW, Han JS. Leptin increases TNF-α expression and production through phospholipase D1 in Raw 264.7 cells. PLoS One (2014) 9(7):e102373. doi:10.1371/journal.pone.0102373
47. Caldefie-Chezet F, Poulin A, Tridon A, Sion B, Vasson M-P. Leptin: a potential regulator of polymorphonuclear neutrophil bactericidal action? J Leukoc Biol (2001) 69:414–8. doi:10.1189/jlb.69.3.414
48. Caldefie-Chezet F, Poulin A, Vasson M-P. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic Res (2003) 37:809–14. doi:10.1080/1071576031000097526
49. Bruno A, Conus S, Schmid I, Simon H-U. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J Immunol (2005) 174:8090–6. doi:10.4049/jimmunol.174.12.8090
50. Kamp VM, Langereis JD, van Aalst CW, van der Linden JA, Ulfman LH, Koenderman L. Physiological concentrations of leptin do not affect human neutrophils. PLoS One (2013) 8(9):e73170. doi:10.1371/journal.pone.0073170
51. Bozza PT, Melo RCN, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther (2007) 113:30–49. doi:10.1016/j.pharmthera.2006.06.006
52. Weller PF. Leukocyte lipid bodies – structure and functions as “Eicosasomes.”. Trans Am Clin Climatol Assoc (2016) 127:328–40.
53. Palmblad J, Malmsten CL, Udén AM, Rådmark O, Engstedt L, Samuelsson B. Leukotriene B4 is a potent and stereospecific stimulator of neutrophil chemotaxis and adherence. Blood (1981) 58:658–61.
54. Mancuso P, Standiford TJ, Marshall T, Peters-Golden M. 5-Lipoxygenase reaction products modulate alveolar macrophage phagocytosis of Klebsiella pneumoniae. Infect Immun (1998) 66:5140–6.
55. Mancuso P, Nana-Sinkam P, Peters-Golden M. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect Immun (2001) 69:2011–6. doi:10.1128/IAI.69.4.2011-2016.2001
56. Chou RC, Kim ND, Sadik CD, Seung E, Lan Y, Byrne MH, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity (2010) 33:266–78. doi:10.1016/j.immuni.2010.07.018
57. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11:519–31. doi:10.1038/nri3024
58. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med (2006) 203:2569–75. doi:10.1084/jem.20060925
59. Fazolini NPB, Cruz ALS, Werneck MBF, Viola JPB, Maya-Monteiro CM, Bozza PT. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle (2015) 14:2667–76. doi:10.1080/15384101.2015.1041684
60. Zhang T, Tseng C, Zhang Y, Sirin O, Corn PG, Li-Ning-Tapia EM, et al. CXCL1 mediates obesity-associated adipose stromal cell trafficking and function in the tumour microenvironment. Nat Commun (2016) 7:11674. doi:10.1038/ncomms11674
61. Deladeriere A, Gambardella L, Pan D, Anderson KE, Hawkins PT, Stephens LR. The regulatory subunits of PI3K g control distinct neutrophil responses. Sci Signal (2015) 8:1–12. doi:10.1126/scisignal.2005564
62. Puri KD, Doggett TA, Huang C, Douangpanya J, Hayflick JS, Turner M, et al. The role of endothelial PI3Kgamma activity in neutrophil trafficking. Blood (2005) 106:150–7. doi:10.1182/blood-2005-01-0023
63. Camps M, Rückle T, Ji H, Ardissone V, Rintelen F, Shaw J, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med (2005) 11(9):936–43. doi:10.1038/nm1284
64. Montecucco F, Bianchi G, Bertolotto M, Viviani G, Dallegri F, Ottonello L. Insulin primes human neutrophils for CCL3-induced migration: crucial role for JNK 1/2. Ann N Y Acad Sci (2006) 1090:399–407. doi:10.1196/annals.1378.043
65. Montecucco F, Steffens S, Burger F, Da Costa A, Bianchi G, Bertolotto M, et al. Tumor necrosis factor-alpha (TNF-alpha) induces integrin CD11b/CD18 (Mac-1) up-regulation and migration to the CC chemokine CCL3 (MIP-1alpha) on human neutrophils through defined signalling pathways. Cell Signal (2008) 20:557–68. doi:10.1016/j.cellsig.2007.11.008
66. Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Science (2001) 294:1942–5. doi:10.1126/science.1066015
67. Kristof AS, Marks-Konczalik J, Billings E, Moss J. Stimulation of signal transducer and activator of transcription-1 (STAT1)-dependent gene transcription by lipopolysaccharide and interferon-gamma is regulated by mammalian target of rapamycin. J Biol Chem (2003) 278:33637–44. doi:10.1074/jbc.M301053200
68. Lim HK, Choi YA, Park W, Lee T, Ryu SH, Kim SY, et al. Phosphatidic acid regulates systemic inflammatory responses by modulating the Akt-mammalian target of rapamycin-p70 S6 kinase 1 pathway. J Biol Chem (2003) 278:45117–27. doi:10.1074/jbc.M303789200
69. Lindemann SW, Yost CC, Denis MM, McIntyre TM, Weyrich AS, Zimmerman GA. Neutrophils alter the inflammatory milieu by signal-dependent translation of constitutive messenger RNAs. Proc Natl Acad Sci U S A (2004) 101:7076–81. doi:10.1073/pnas.0401901101
70. Rummel C, Inoue W, Poole S, Luheshi GN. Leptin regulates leukocyte recruitment into the brain following systemic LPS-induced inflammation. Mol Psychiatry (2010) 15(5):523–34. doi:10.1038/mp.2009.98
71. Aguliar-Valles A, Kim J, Jung S, Woodside B, Luheshi GN. Role of brain transmigrating neutrophils in depression-like behavior during systemic infection. Mol Psychiatry (2014) 19(5):599–606. doi:10.1038/mp.2013.137
72. Landgraf MA, Silva RC, Corrêa-Costa M, Hiyane MI, Carvalho MHC, Landgraf RG, et al. Leptin downregulates LPS-induced lung injury: role of corticosterone and insulin. Cell Physiol Biochem (2014) 33:835–46. doi:10.1159/000358656
Keywords: leptin, neutrophil, inflammation, tumor necrosis factor-alpha, CXCL1, leukotriene B4
Citation: Souza-Almeida G, D’Avila H, Almeida PE, Luna-Gomes T, Liechocki S, Walzog B, Hepper I, Castro-Faria-Neto HC, Bozza PT, Bandeira-Melo C and Maya-Monteiro CM (2018) Leptin Mediates In Vivo Neutrophil Migration: Involvement of Tumor Necrosis Factor-Alpha and CXCL1. Front. Immunol. 9:111. doi: 10.3389/fimmu.2018.00111
Received: 06 October 2017; Accepted: 15 January 2018;
Published: 06 February 2018
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Alexandre Benani, Centre National de la Recherche Scientifique (CNRS), FranceZhihong Yang, University of Fribourg, Switzerland
Copyright: © 2018 Souza-Almeida, D’Avila, Almeida, Luna-Gomes, Liechocki, Walzog, Hepper, Castro-Faria-Neto, Bozza, Bandeira-Melo and Maya-Monteiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clarissa M. Maya-Monteiro, Y2xhcmlzc2FAaW9jLmZpb2NydXouYnI=, Y2xhcmlzc2FtYXlhbUBnbWFpbC5jb20=