 Maddalena Migliavacca1
Maddalena Migliavacca1 Andrea Assanelli2
Andrea Assanelli2 Maurilio Ponzoni3,4
Maurilio Ponzoni3,4 Roberta Pajno1Federica Barzaghi1Fabio Giglio2Francesca Ferrua1,4
Roberta Pajno1Federica Barzaghi1Fabio Giglio2Francesca Ferrua1,4 Marta Frittoli1
Marta Frittoli1 Immacolata Brigida5
Immacolata Brigida5 Francesca Dionisio5Roberto Nicoletti6Miriam Casiraghi1
Francesca Dionisio5Roberto Nicoletti6Miriam Casiraghi1 Maria Grazia Roncarolo1,7
Maria Grazia Roncarolo1,7 Claudio Doglioni3,4
Claudio Doglioni3,4 Jacopo Peccatori2
Jacopo Peccatori2 Fabio Ciceri2,4
Fabio Ciceri2,4 Maria Pia Cicalese1
Maria Pia Cicalese1 Alessandro Aiuti1,4*
Alessandro Aiuti1,4*
- 1San Raffaele Telethon Institute for Gene Therapy (SR-TIGET), Pediatric Immunohematology and Bone Marrow Transplantation Unit, Scientific Institute San Raffaele (IRCCS), Milan, Italy
- 2Hematology and Bone Marrow Transplantation Unit, Scientific Institute San Raffaele (IRCCS), Milan, Italy
- 3Pathology Unit, Scientific Institute San Raffaele (IRCCS), Milan, Italy
- 4Vita-Salute San Raffaele University, Milan, Italy
- 5San Raffaele Telethon Institute for Gene Therapy (SR-TIGET), Scientific Institute San Raffaele (IRCCS), Milan, Italy
- 6Department of Radiology, Scientific Institute San Raffaele (IRCCS), Milan, Italy
- 7Division of Stem Cell Transplantation and Regenerative Medicine, Department of Pediatrics, Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, United States
Adenosine deaminase-deficient severe combined immunodeficiency disease (ADA-SCID) is a primary immune deficiency characterized by mutations in the ADA gene resulting in accumulation of toxic compounds affecting multiple districts. Hematopoietic stem cell transplantation (HSCT) from a matched donor and hematopoietic stem cell gene therapy are the preferred options for definitive treatment. Enzyme replacement therapy (ERT) is used to manage the disease in the short term, while a decreased efficacy is reported in the medium-long term. To date, eight cases of lymphomas have been described in ADA-SCID patients. Here we report the first case of plasmablastic lymphoma occurring in a young adult with ADA-SCID on long-term ERT, which turned out to be Epstein–Barr virus associated. The patient previously received infusions of genetically modified T cells. A cumulative analysis of the eight published cases of lymphoma from 1992 to date, and the case here described, reveals a high mortality (89%). The most common form is diffuse large B-cell lymphoma, which predominantly occurs in extra nodal sites. Seven cases occurred in patients on ERT and two after haploidentical HSCT. The significant incidence of immunodeficiency-associated lymphoproliferative disorders and poor survival of patients developing this complication highlight the priority in finding a prompt curative treatment for ADA-SCID.
Introduction
The development of immunodeficiency-associated lymphoproliferative disorders (IALDs), especially related to Epstein–Barr virus (EBV), is a life-threatening phenomenon. The incidence of IALD in primary immune deficiencies due to various gene defects ranges from 0.7 to 18% of patients (1). Adenosine deaminase-deficient severe combined immunodeficiency disease (ADA-SCID) is an inherited defect that results in the accumulation of enzyme substrates, such as adenosine, 2′-deoxyadenosine and deoxyribonucleotides (dAXP) leading to severe lymphopenia with absence of cellular and humoral immunity and recurrent, severe infections. High adenosine levels block the differentiation of thymocytes, induce thymic hypoplasia, and lead to apoptosis resulting in T, B, and NK depletion. Delayed and late onset forms have a wide spectrum of manifestations mostly involving immunological and autoimmune system (i.e., hemolytic anemia and immune thrombocytopenia) as the most severe manifestation (2). Since ADA gene is ubiquitously expressed, non-immunological alterations have been described frequently in ADA-deficient patients involving liver, kidney, bone, and central nervous system (3–6).
Background
Eight cases of lymphoma have been reported to date in ADA-SCID patients, and half of them are EBV associated (7–13).
Herein, we present the first case of a fatal EBV-associated plasmablastic lymphoma, which developed in an 18-year-old ADA-SCID girl, after enzyme replacement therapy (ERT) and multiple infusions of genetically modified T cells. The patient was diagnosed with ADA deficiency at 3 months of age due to pneumonia and failure to thrive (14). PEG-ADA was started with partial benefit on immune functions and clinical conditions but the lymphocyte count remained low (450–650/μL with 200/μL T cells). No matched bone marrow (BM) donor was available. Between 1 and 5 years of age, she was treated with repeated infusions of autologous peripheral blood (PB) T lymphocytes transduced ex vivo with a gammaretroviral vector encoding ADA (14) (NCT00599781, GIADAl). The patient also received a small amount of autologous BM cells transduced with a similar gammaretroviral vector (differing only at a restriction enzyme site) without any conditioning, after which there was no evidence of engraftment. PEG-ADA was reduced until complete discontinuation to favor the expansion of gene corrected cells (14). Despite the improvement of T cell count and functions with evidence of antibody responses to neoantigens, ERT was restarted at a low dose due to the suboptimal systemic detoxification, maintained at 11.2–12.5 U/kg/week i.m. and continued for further 16 years. The patient experienced mainly self-limited upper respiratory tract infections and autoimmune hemolytic anemia at the age of 6 years, which was treated with high-dose methylprednisolone (15 mg/kg i.v.) followed by prednisone 2 mg/kg/die p.o. which was slowly tapered and eventually stopped 1 year after onset. A mild pulmonary restrictive syndrome with bronchiectasis and arterial hypertension were controlled with specific therapies. At the age of 17, lymphocyte and T cell counts were 800 and 600/μL, respectively. Proliferative responses to mitogens were normal, while responses to antigens (candida and alloantigens) were reduced. The TCR Vbeta repertoire by flow cytometry was polyclonal with two Vbeta families reduced. dAXP toxic metabolites were 6 nmol/mL (0.46%) on PB (normal range: <1%) (10) EBV serology and search for EBV sequence (EBV PCR) were negative.
One year later the patient was hospitalized in a local hospital due to persistent fever, multiple lymphadenopathies and bilateral periorbital edema with intense conjunctival hyperemia, unresponsive to i.v. antibiotics. At admission to our Unit, total body positron emission tomography–computed tomography showed diffuse lymphadenopathies (cervical, spinal-accessorial, supraclavicular, axillary, retropectoral, tonsillar, upper mediastinal, and right paratracheal) and splenomegaly (Figure 1). Interstitial lung involvement accompanied by pleural effusion resulted in respiratory impairment, requiring oxygen supplementation. Brain MRI detected severe edema of periocular tissue with exophthalmos and lymphoid hyperplasia. Blood tests revealed high plasma EBV DNA load (10 million copies/mL) and elevated anti-VCA EBV IgG with absence of anti-VCA IgM, anti-EBV early antigen and anti-EBV nuclear antigen antibodies. An Ig kappa monoclonal component (10 g/L) was present. Cervical lymphadenectomy and BM examination were performed, and methylprednisolone (1 mg/kg/daily) and anti-CD20 (rituximab 375 mg/m2 in two doses) were started before pathological diagnosis was confirmed. Histopathological examination showed an EBV-related plasmablastic B-cell lymphoma (Figure 2). Lymphoma cells displayed a high proliferation index (>90%), were monoclonal for kappa light chain and expressed plasma cell-associated markers such as CD138, CD30, and MUM-1. In addition, these elements were positive for PAX-5 and BCL2 as well as partially positive (40% of cells) for CD20. The EBV marker LMP-1 was present. On the other hand, neoplastic elements scored negative for CD10, CD3, CD4, CD8, BCL6, and CD56. HHV8 was also negative. A minor component of mature-looking plasma cells sharing the same light chain restriction with large neoplastic cells was present at the edge of the lesion, as reported (15). BM evaluation showed reactive dyshematopoiesis with slight increase of plasma cells, which were monoclonal for kappa light chain.
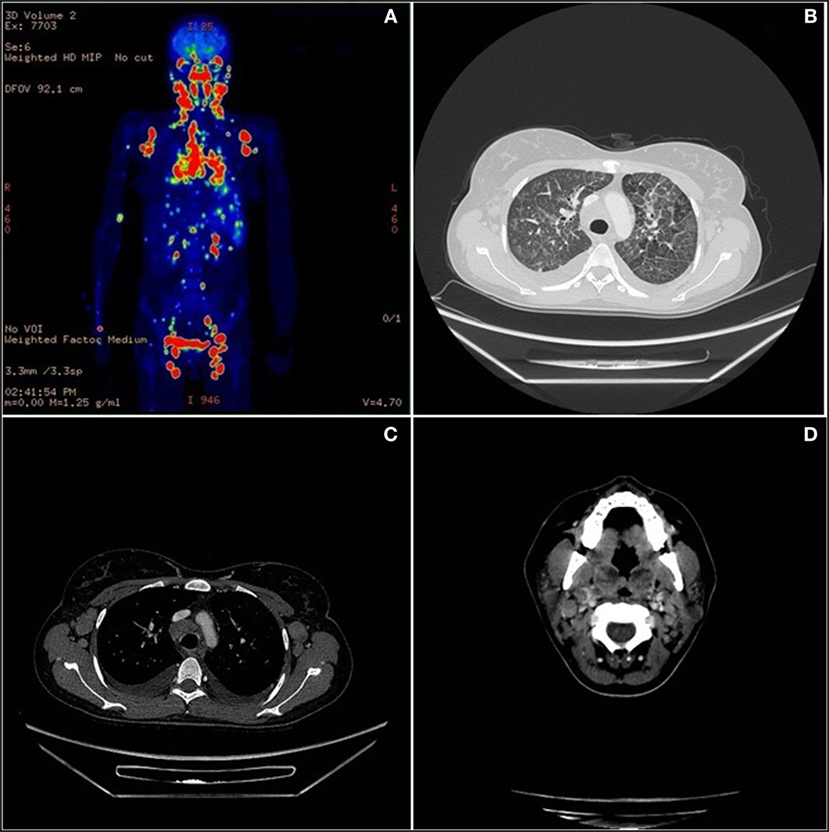
Figure 1. Total body positron emission tomography (PET)–computed tomography (CT). (A) Diffuse lymph nodes involvement in PET. (B) Lymphoma lung interstitial reticular thickening in CT scan. (C) Axillary and mediastinum enlargement lymph nodes in CT scan. (D) Neoplastic involvement of neck nodes and palatine tonsils in CT scan.
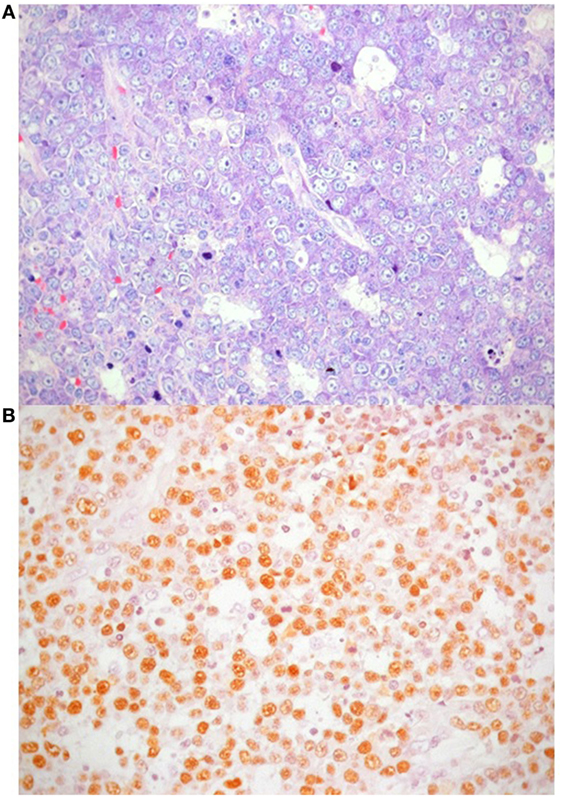
Figure 2. Plasmablastic lymphoma accompanied by areas of necrosis. (A) Giemsa stain highlights nuclear features of neoplastic cells, with particular reference to central, single prominent nucleolus (40×); (B) in situ hybridization for Epstein–Barr virus shows positive nuclear signal in neoplastic lymphocytes.
The disease was classified as stage IV B Es. Therapy consisted of continued rituximab weekly infusions and a cycle of APO (adriamycin, vincristine, and prednisone) with no response. Accordingly, cyclophosphamide (300 mg/m2 BID for 3 days) and dexamethasone were started. Twelve days after the initiation of chemotherapy, the patient died for a hemorrhagic alveolitis eventually causing worsening respiratory failure, refractory to continuous positive airway pressure and biphasic positive airway pressure mechanical ventilation.
To investigate the potential relationship with PB gene therapy, T cells and B cells were purified from BM, PB, and lymph node, and vector transduction measured by quantitative PCR (14). T cells were highly positive for the vector (BM: >100%; PB: >100%; lymph node: 96.1%) in line with previous follow-up analyses. By contrast, B cells showed low vector signal (BM: 0.43%; PB: 3.1%; lymph node: 6.1%), a level compatible with contamination from T cells, as sample purity was <90% after purification. These data rule out the possibility that the lymphoproliferative event originated from the infused transduced cells.
Discussion
The most frequently reported histotype in primary immune deficiencies is diffuse large B-cell lymphoma, which mostly occurs in extra nodal sites (16). Nine cases of lymphoma have been described in ADA-SCID patients, including the present case (Table 1). Of these, five were related to EBV infection (7, 10, 11) with two arising in the lung (10) and the brain (11), respectively, while another was described as a Hodgkin lymphoma (10). An EBV-negative Burkitt’s lymphoma has also been described (12). Overall, six of nine patients developed a malignant disease more than 3 years after the start of PEG-ADA (range 3–17 years) (Table 1). In three patients, alternative treatment was attempted before lymphoma onset, including haploidentical hematopoietic stem cell transplantation (HSCT) (n = 2) and PB T-cell gene therapy. Unfortunately, all patients died despite rescue attempts with several lines of treatments, with the exception of a Burkitt’s hip lymphoma that persisted in complete remission after 20 months from the onset of the malignant disease (12). According to data available in literature seven of nine patients received chemotherapy to cure lymphoma, mostly based on high dose of steroids (prednisone and dexamethasone), anti-mitotic vinca alkaloids (i.e., vincristine), anthracyclines (i.e., doxorubicin), alkylating agents (i.e., cyclophosphamide). Three out of ten patients underwent HSCT after lymphoma, a patient two consecutive haplo-HSCT, and the last two from cord blood and matched unrelated donor, respectively. Unfortunately, all of them (3/3) had a fatal exitus.
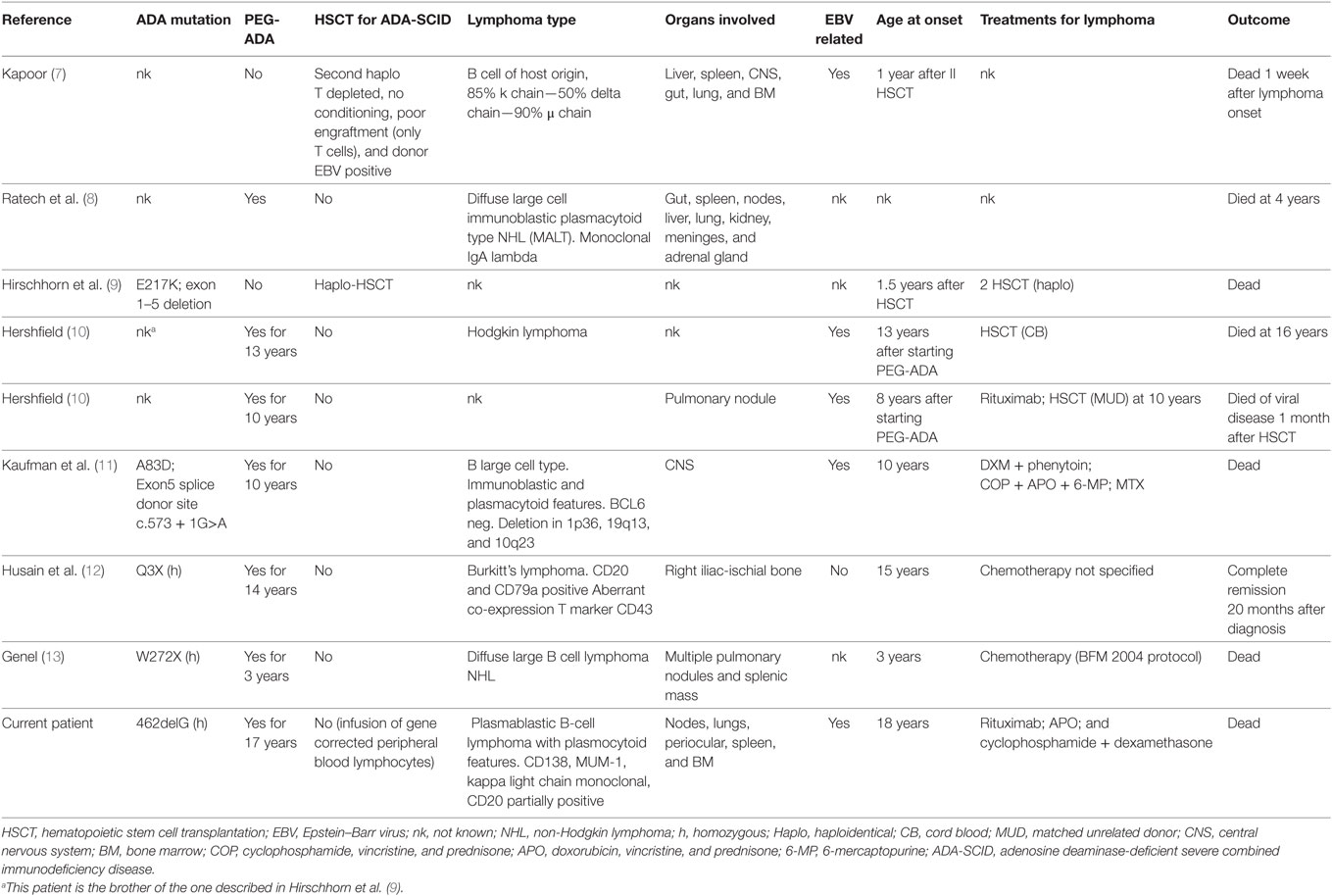
Table 1. Clinical, genetics, and biological characteristics of ADA-SCID patients who developed lymphoma (review of the published cases).
To the best of our knowledge, this is the first report of a plasmablastic lymphoma in an ADA-SCID patient. Lymphoma in patients with primary immune deficiency is frequently EBV associated (16). EBV infection indeed seems to play an important role on lymphocyte proliferation thorough suppression of T-cell surveillance and modulation on apoptosis and cytokine balance in B-cells (8, 17).
In particular, low T-cell count and/or abnormal T-cell function seem to impact on lymphoproliferative disorder prognosis more than histology or gene rearrangements (16). Moreover, dysregulated secretion of IL-6 has a pivotal role in lymphoproliferative disorders in an autocrine and paracrine cytokine stimulated manner, indeed, it contributes to the development of the auto-maintenance and neoplastic growth of EBV-immortalized B cells (17).
The pathogenesis of lymphomas in ADA-SCID patients remains still unclear. The lack of ADA would be expected to decrease the oncogenic risk since ADA inhibitors have been used for the treatment of lymphomas (18) and the accumulation of adenosine and purine metabolites in ADA-deficient patients leads to increased apoptosis in T and B cells (19). Since all patients described here were either under PEG-ADA treatment or received HSCT, one can speculate that in these patients adenosine metabolites were adequately detoxified. It would be also interesting to investigate whether lymphoma cells spontaneously reverted the ADA mutation, as observed in selected cases of ADA-SCID patients (20). At present, the most likely explanation remains a lack of appropriate immune surveillance similarly to other primary immune deficiencies.
The B-cell clonality identified in our patient by PCR, both on lymph node and on BM, has been associated with poor prognosis, confirming the negative predictive role of this feature previously suggested in a small cohort of patients with lymphoproliferative disorders (16).
Our patient continued long-term ERT since an HLA-identical donor was not available and data of overall survival after matched unrelated or alternative donors (i.e., haploidentical) were not encouraging at that time (66 and 43%, respectively) (21). ERT often leads to a transient improvement of the immune function and clinical parameters, including growth (22, 23), but a gradual immunological decline is actually observed within a few years of treatment (23–26). Our patient also received multiple infusions of genetically corrected T cells and presented a T-cell compartment mainly represented by transduced cells. It is likely that, despite being polyclonal, the T-cell repertoire of the transduced T cells, which were ex vivo cultured, was not sufficiently broad to survey and control the B cell transformation induced by the EBV infection. It is also possible that defective functions of other untransduced cell types, such as NK cells, could have contributed to the impaired immune response to EBV. It will be important to continue the strict monitoring of ADA-SCID patients on long-term ERT, including those who received infusions of gene corrected T cells (27–29).
On the other hand, retroviral vector GT with autologous CD34+ cells transduced with ADA has emerged as a safe and efficacious therapeutic strategy for patients lacking HLA-identical sibling donor (26, 30–33). Noteworthy, in comparison with clinical trials conducted on other primary immunodeficiencies (SCIDX1, CGD, and WAS), no lymphoproliferative disorders or aberrant clonal expansion have been observed in more than 40 patients treated worldwide with an available follow-up of more than one decade in some of them (34).
Concluding Remarks
These observations underline the importance of defining a potential lifelong effective treatment for these patients as early as possible. Attending physicians must be aware that the delay of a definitive treatment could expose these fragile patients to multiple severe complications, including aggressive lymphoproliferative disorders.
Ethics Statement
The study was performed in accordance with the Declaration of Helsinki. The patient was enrolled in a clinical trial approved by the San Raffaele Ethical Committee (NCT00599781). Written informed consent was obtained from parents for treatment, biological sample collection, and data publication.
Author Contributions
M.M., AndreaA., M.P., R.P, F.B., F.G., F.F., M.F., I.B., F.D., R.N., M.C., M.G.R., C.D., J.P., F.C., M.P.C, AlessandroA. participated to clinical care of the patient and related clinical data. M.M. and AlessandroA. designed the study, analyzed results and wrote the paper.
Conflict of Interest Statement
AlessandroA is the Principal investigator of gene therapy studies sponsored by GlaxoSmithKline (GSK), which holds the licence for the medicinal product based on autologous CD34+ cell gene therapy for ADA-SCID. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported by grants of the Italian Ministero della Salute (Programma di rete, NET-2011-02350069), the European Commission (ERARE-3-JTC 2015 EUROCID), and Fondazione Telethon. IB is supported by the Italian Ministero della Salute (GR-2011-02346985). The authors are grateful all medical and nursing staff of the TIGET Pediatric Clinical Research Unit and the Pediatric Immunohematology and Bone Marrow Transplant Unit; L. Castagnaro, G. Tomaselli, and all TCTO personnel for clinical trial management and support.
References
1. Oertel SH, Riess H. Immunosurveillance, immunodeficiency and lymphoproliferations. Recent Results Cancer Res (2002) 159:1–8. doi:10.1007/978-3-642-56352-2_1
2. Sauer AV, Brigida I, Carriglio N, Aiuti A. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front Immunol (2012) 3:265. doi:10.3389/fimmu.2012.00265
3. Hirschhorn R, Candotti F. Immunodeficiency due to defects of purine metabolism. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases. Oxford: Oxford University Press (2006). p. 169–96.
4. Aiuti A, Ficara F, Cattaneo F, Bordignon C, Roncarolo MG. Gene therapy for adenosine deaminase deficiency. Curr Opin Allergy Clin Immunol (2003) 3:461–6. doi:10.1097/00130832-200312000-00007
5. Honig M, Albert MH, Schulz A, Sparber-Sauer M, Schutz C, Belohradsky B, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood (2007) 109:3595–602. doi:10.1182/blood-2006-07-034678
6. Sauer AV, Hernandez RJ, Fumagalli F, Bianchi V, Poliani PL, Dallatomasina C, et al. Alterations in the brain adenosine metabolism cause behavioral and neurological impairment in ADA-deficient mice and patients. Sci Rep (2017) 7:40136. doi:10.1038/srep40136
7. Kapoor N. Lymphoma in a patient with severe combined immunodeficiency with adenosine deaminase deficiency, following unsustained engraftment of histoincompatible T cell-depleted bone marrow. J Pediatr (1986) 108(3):435–8. doi:10.1016/S0022-3476(86)80892-7
8. Ratech H, Hirschhorn R, Greco MA. Pathologic findings in adenosine deaminase deficient-severe combined immunodeficiency. II. Thymus, spleen, lymph node, and gastrointestinal tract lymphoid tissue alterations. Am J Pathol (1989) 135(6):1145–56.
9. Hirschhorn R, Nicknam MN, Eng F, Yang DR, Borkowsky W. Novel deletion and a new missense mutation (Glu 217 Lys) at the catalytic site in two adenosine deaminase alleles of a patient with neonatal onset adenosine deaminase-severe combined immunodeficiency. J Immunol (1992) 149(9):3107–12.
10. Hershfield MS. Combined Immune Deficiencies due to Purine Enzyme DefectsIn: Stiehm ER, Ochs HD, Winkelstein JA, editors. Immunologic Disorders in Infants and Children. Philadelphia, PA: WB Saunders (2004). p. 480–504.
11. Kaufman DA, Hershfield MS, Bocchini JA, Moissidis IJ, Jeroudi M, Bahna SL. Cerebral lymphoma in an adenosine deaminase deficient patient with severe combined immunodeficiency receiving polyethylene glycol conjugated adenosine deaminase. Pediatrics (2005) 116(6):e876–9. doi:10.1542/peds.2005-1068
12. Husain Met al. Burkitt’s lymphoma in a patient with adenosine deaminase deficiency-severe combined immunodeficiency treated with polyethylene glycol-adenosine deaminase. J Pediatr (2007) 151:93–5. doi:10.1016/j.jpeds.2007.03.059
13. Genel F. Adenosine deaminase-deficient severe combined immunodeficiency and diffuse large B-cell lymphoma. Pediatr Allergy Immunolo Pulmonol (2015) 28(2):138–42. doi:10.1089/ped.2014.0478
14. Aiuti A. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med (2002) 8(5):423–5. doi:10.1038/nm0502-423
15. Stein H, Harris NL, Campo E, Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissue. Lyon: IARC (2008).
16. Canioni D, Jabado N, MacIntyre E, Patey N, Emile JF, Brousse N. Lymphoproliferative disorders in children with primary immunodeficiencies: immunological status may be more predictive of the outcome than other criteria. Histopathology (2001) 38(2):146–59. doi:10.1046/j.1365-2559.2001.01039.x
17. Ohga S, Nomura A, Takada H, Hara T. Immunological aspects of Epstein-Barr virus infection. Crit Rev Oncol Hematol (2002) 44(3):203–15. doi:10.1016/S1040-8428(02)00112-9
18. Argnani L, Broccoli A, Zinzani PL. Cutaneous T-cell lymphomas: focusing on novel agents in relapsed and refractory disease. Cancer Treat Rev (2017) 61:61–9. doi:10.1016/j.ctrv.2017.10.007
19. Sauer AV, Di Lorenzo B, Carriglio N, Aiuti A. Progress in gene therapy for primary immunodeficiencies using lentiviral vectors. Curr Opin Allergy Clin Immunol (2014) 14(6):527–34. doi:10.1097/ACI.0000000000000114
20. Arredondo-Vega FXet al. Adenosine deaminase deficiency with mosaicism for a “second-site suppressor” of a splicing mutation: decline in revertant T lymphocytes during enzyme replacement therapy. Blood (2002) 99(3):1005–13. doi:10.1182/blood.V99.3.1005
21. Hassan Aet al. Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood (2012) 120(17):3615–24. doi:10.1182/blood-2011-12-396879
22. Weinberg Ket al. T lymphocyte ontogeny in adenosine deaminase-deficient severe combined immune deficiency after treatment with polyethylene glycol-modified adenosine deaminase. J Clin Invest (1993) 92(2):596–602. doi:10.1172/JCI116626
23. Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biologics (2009) 3:349–58. doi:10.214TT.S3103
24. Kohn DB. Gene therapy for childhood immunological diseases. Bone Marrow Transplant (2008) 41(2):199–205. doi:10.1038/sj.bmt.1705895
25. Serana Fet al. The different extent of B and T cell immune reconstitution after hematopoietic stem cell transplantation and enzyme replacement therapies in SCID patients with adenosine deaminase deficiency. J Immunol (2010) 185(12):7713–22. doi:10.4049/jimmunol.1001770
26. Cicalese MP, Ferrua F, Castagnaro L, Pajno R, Barzaghi F, Giannelli S, et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood (2016) 128(1):45–54. doi:10.1182/blood-2016-01-688226
27. Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science (1995) 270(5235):475–80. doi:10.1126/science.270.5235.475
28. Bordignon C, Notarangelo LD, Nobili N, Ferrari G, Casorati G, Panina P, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA-immunodeficient patients. Science (1995) 270(5235):470–5. doi:10.1126/science.270.5235.470
29. Onodera M, Ariga T, Kawamura N, Kobayashi I, Ohtsu M, Yamada M, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood (1998) 91(1):30–6.
30. Gaspar HB, Qasim W, Davies EG, Rao K, Amrolia PJ, Veys P. How i treat severe combined immunodeficiency. Blood (2013) 122(23):3749–58. doi:10.1182/blood-2013-02-380105
31. Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med (2011) 3(97):97–80. doi:10.1126/scitranslmed.3002716
32. Candotti Fet al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood (2012) 120(18):3635–46. doi:10.1182/blood-2012-02-400937
33. Scott O, Kim VH, Reid B, Pham-Huy A, Atkinson AR, Aiuti A, et al. Long-term outcome of adenosine deaminase-deficient patients-a single-center experience. J Clin Immunol (2017) 37(6):582–91. doi:10.1007/s10875-017-0421-7
Keywords: adenosine deaminase-deficient severe combined immunodeficiency disease, lymphoma, plasmablastic lymphoma, primary immunodeficiency, gene therapy, gene therapy for rare diseases, review of literature
Citation: Migliavacca M, Assanelli A, Ponzoni M, Pajno R, Barzaghi F, Giglio F, Ferrua F, Frittoli M, Brigida I, Dionisio F, Nicoletti R, Casiraghi M, Roncarolo MG, Doglioni C, Peccatori J, Ciceri F, Cicalese MP and Aiuti A (2018) First Occurrence of Plasmablastic Lymphoma in Adenosine Deaminase-Deficient Severe Combined Immunodeficiency Disease Patient and Review of the Literature. Front. Immunol. 9:113. doi: 10.3389/fimmu.2018.00113
Received: 19 November 2017; Accepted: 15 January 2018;
Published: 02 February 2018
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Claire Booth, UCL Great Ormond Street Institute of Child Health (ICH), United KingdomNizar MAHLAOUI, Necker-Enfants Malades Hospital, France
Copyright: © 2018 Migliavacca, Assanelli, Ponzoni, Pajno, Barzaghi, Giglio, Ferrua, Frittoli, Brigida, Dionisio, Nicoletti, Casiraghi, Roncarolo, Doglioni, Peccatori, Ciceri, Cicalese and Aiuti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Aiuti, YWxlc3NhbmRyby5haXV0aUBoc3IuaXQ=