 Margot Zöller
Margot Zöller- Tumor Cell Biology, University Hospital of Surgery, University of Heidelberg, Heidelberg, Germany
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells originally described to hamper immune responses in chronic infections. Meanwhile, they are known to be a major obstacle in cancer immunotherapy. On the other hand, MDSC can interfere with allogeneic transplant rejection and may dampen autoreactive T cell activity. Whether MDSC-Exosomes (Exo) can cope with the dangerous and potentially therapeutic activities of MDSC is not yet fully explored. After introducing MDSC and Exo, it will be discussed, whether a blockade of MDSC-Exo could foster the efficacy of immunotherapy in cancer and mitigate tumor progression supporting activities of MDSC. It also will be outlined, whether application of native or tailored MDSC-Exo might prohibit autoimmune disease progression. These considerations are based on the steadily increasing knowledge on Exo composition, their capacity to distribute throughout the organism combined with selectivity of targeting, and the ease to tailor Exo and includes open questions that answers will facilitate optimizing protocols for a MDSC-Exo blockade in cancer as well as for strengthening their therapeutic efficacy in autoimmune disease.
Introduction
Myeloid-Derived Suppressor Cells (MDSCs) and Cancer
Cancer is one of the most frequent causes of death (1), which in part is due to the resistance of tumor cells to chemo-, radio-, and immunotherapy (2–4). This implies that after tumor spread, which might prohibit surgical excision, the likeliness of curative therapy steeply declines. Disappointing efficacy of adjuvant cancer therapies accounts particularly for immunotherapy, where frequently no or only weak responses are noted despite the presence of immunogenic tumor-associated antigens (5). In several tumor entities, MDSCs were found to account for resistance toward cancer immunotherapy (6) and additionally for poor responses to chemotherapy. Therefore, drugs were designed that, besides directly attacking the tumor cells, should hamper MDSC development or activation or drive MDSC into apoptosis (7). So far therapeutic trials with a focus on MDSC elimination to improve chemotherapy or immunotherapy are rare, which also accounts for combinations of adjuvant therapeutics (8, 9). The current options and possible modes of improvement attacking MDSC/MDSC-exosomes (MDSC-Exo) to support cancer chemo- and/or immunotherapy will be discussed.
MDSC in Autoimmune Disease and Allograft Transplantation
Autoimmune disease incidence is steadily increasing (10). Autoimmune diseases frequently exacerbate in young adults and progress in waves, which get more severe during time and become life threatening (11). Corticosteroid therapy, frequently used in progressed disease stages (12), is burdened by severe side effects, including dampening immune responses against bacteria and viruses (13). The option to booster autoimmune disease therapy with MDSC (14) gained in weight, when it was realized that MDSC are a strong stimulus for regulatory T cell (Treg) activation, a deficit in Treg contributing to autoreactive T cell expansion (15). There are several trials to integrate MDSC in autoimmune disease therapy, such as myasthenia gravis, arthritis, inflammatory bowel disease, and others, where good response rates were reported (14, 16–21).
Myeloid-derived suppressor cell-promoted downregulation of immune reactivity also is advantageous in allograft transplantation. This accounts for organ as well as hematopoietic stem cell (HSC) transplantation (22–26). Accordingly, drugs promoting MDSC expansion and/or activation and the transfer of MDSC were reported to support long-term allograft survival (27–30).
Having introduced the two faces of MDSC, this review will focus on MDSC and MDSC-Exo in cancer and autoimmune disease. After introducing MDSC and Exo, their mode of action in disease will be outlined. Knowledge on the crosstalk between MDSC/MDSC-Exo and their targets provides the fundament for established and forthcoming therapeutic interference.
MDSC: Phenotypic and Functional Characterization
Myeloid-derived suppressor cells are a heterogeneous group of cells, characterized by myeloid origin, immature state, and mostly functional activity. In humans, MDSC are still difficult to isolate due to an inconclusive surface marker expression profile. However, there is consent on the differentiation between two subgroups defined as monocytic (M) and granulocytic MDSC (G-MDSC), which are differentiated on the basis of Ly6C high [monocytic MDSC (M-MDSC)] or Ly6G high (G-MDSC), M-MDSC exerting stronger suppressive activity (31–33). MDSC account for T cell exhaustion in chronic infections (34, 35), play a crucial role in cancer progression (31, 36), and are a major hindrance in cancer immunotherapy, hampering T cell recruitment and activation, while promoting M1 and Treg expansion (14, 37). On the other hand, MDSC are beneficial in overshooting immune reactions such as autoimmune diseases (24, 33) and allogeneic transplantation (18, 33, 38). Finally, though the activity of MDSC may vary with the pathophysiological conditions promoting their expansion, there is consent that T cells are major targets and that the response of the adaptive immune system is most severely affected (39).
Myeloid-derived suppressor cell expansion is promoted by lipopolysaccharide, macrophage colony stimulating factor, GM-CSF, SCF, IL6, interferon (IFN)γ, IL1β, vascular endothelial growth factor (VEGF), heat shock protein (HSP)72, IL13, C5a, PGE2, and S100A8/A9 (32, 40). Downstream activation of the JAK– signal transducer and activator of transcription (STAT)3/STAT5 pathway with stimulation of cyclinD1, Bcl2-like (BclXl), survivin, c-myc, and S100A8/A9 contribute to inhibition of differentiation into mature myeloid cells. MDSC recruitment is supported by provision of CCL2, C-X-C motif chemokine ligand (CXCL)12, and CXCL15, the corresponding ligands being expressed by MDSC. Intracellular molecules involved in MDSC function are STAT3, COX2, HIF1α, C/EBPβ, inducible nitric oxide synthase (iNOS), arginase (Arg), HO-1, and indoleamine 2,3-dioxygenase (IDO) (32, 40, 41). Downstream effector molecules are Arg-1 and iNOS. Arg-1 and iNOS account for depletion of l-arginine in T cells, which contributes to ζ-chain downregulation. iNOS induces NO, NO and ROS inhibiting T cell proliferation and inducing apoptosis. HO-1 inhibits T cell proliferation via CO production. Membrane-bound TGFβ1 promotes natural killer cell (NK cell) anergy. IL10 and TGFβ foster Treg expansion (40), which become recruited by CXCL10. TGFβ, and IL10 also account for downregulation of IFNγ (40). IL10 promotes TH2 deviation and type 2 macrophage (Mϕ) polarization. Finally, ADAM17 leads to CD62L cleavage, which prohibits T cell homing (42, 43).
In cancer, drugs have been developed and are further improved to selectively attack MDSC maturation and/or activation. These include, besides others, all-transretinoic acid (ATRA) driving progenitors into differentiation, the tyrosine kinase inhibitor sunitinib, gemcitabine, COX-2 inhibitors, and the phosphodiesterase-5 inhibitor sildanefil (44–48). In autoimmune diseases and allogeneic bone marrow (BM) transplantation, the transfer of MDCS was demonstrated as a therapeutic option (18, 33, 38, 42, 49).
Taken together, MDSC are immature myeloid cells that hamper mostly T cell, but also B cell and NK activity, at least in part by supporting Treg expansion and activation. They are a severe hindrance in cancer immunotherapy and in chronic infections. Mostly in cancer immunotherapy drugs and drug combinations to prevent MDSC induction, activation and targeting as well as drugs to drive MDSC into apoptosis are experimentally and clinically explored to improve the efficacy of immunotherapy. Based on the same principle MDSC activity is suited to control undesired immunoreactivity in transplantation and autoimmune disease, the transfer of MDSC being a therapeutic option.
Exosomes (Exo)
Exosomes are small 40–100 nm vesicles delivered by most cells of an organism (50). They distribute throughout the body and are recovered in all body fluids (51). Exo express donor cell-derived components. This finding stimulated Exo research as a non-invasive/minimally invasive tool for diagnosis, prognosis and therapy control (51, 52). Of particular importance was the notion that Exo components are function competent and deliver their messages into target cells (53, 54) such that Exo binding and uptake can severely modulate target structures and suffices for reprogramming target cells (54–57). Furthermore, Exo easily can be modulated in vitro (58). Thus, Exo are a most powerful intercellular communication system and are supposed to become a highly effective therapeutic tool in the near future (59, 60).
Exo Biogenesis
Exosome biogenesis starts with the formation of early endosomes (EE), which can derive from the trans-Golgi network or from different internalized membrane microdomains, such as clathrin-coated pits, tetraspanin and glycolipid-enriched membrane domains (GEM), or proteolipids in cholesterol- and ceramide-rich compartments (61). EE move toward multivesicular bodies (MVB), the transport machinery varying for the different types of EE (62). During inward budding of EE into MVB, called intraluminal vesicles (ILV), vesicles receive their cargo. Loading of the small plasma that could contain ~100 proteins and 10,000 nucleotides (63) with proteins, coding and non-coding RNA and DNA are non-random processes (61). Sorting of proteins is facilitated by mono-ubiquitination, acylation or myristoylation (64, 65). For GEM-derived Exo, higher order oligomerization is important (66), where protein complexes and attached cytoplasmic components are retained (67). In raft-derived ILV, sphingolipids forming ceramide also contribute to vesicle loading (68). miRNA recruitment is guided by a zip code in the 3′-UTR and by coupling of RNA-induced silencing complex to components of the sorting complex. A specific EXOmotif (GGAG) controls miRNAs loading by binding to the heterogeneous ribonucleoprotein A2B1 (hnRNPA2B1), which binds to an RNA transport signal (A2RE) (69). Annexin-II plays a role in RNA sorting into ILV by binding specific RNAs (70). lncRNA also are selectively recruited by so far unknown mechanisms (71). Ras-related proteins regulate MVB movement toward the cell membrane (72). MVB fuse with the plasma membrane, ILV are released and are then called Exo (61).
Though there remain open questions on the precise biogenesis pathways, it is important to remember that due to differences in biogenesis, single cells can deliver different Exo (73, 74). For judging on potential diagnostic and therapeutic validity, information on the Exo composition is a prerequisite.
Exo Composition
Exosomes are composed of a lipid bilayer, which contains transmembrane proteins. The intravesicular content is composed of proteins, coding and non-coding RNA and DNA.
The lipid envelop of Exo contains phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, prostaglandins, and lysobisphosphatidic acid and is enriched in sphingomyelin, cholesterol, GM3, and phosphatidylserine (75). The high phosphatidylserine content allows differentiating Exo from microvesicles (76) and tumor-derived Exo (TEX) lipid composition may be suited for diagnosis (77, 78). Progress in lipidomics will provide further informations.
Improvement in mass spectrometry (79) has greatly facilitated the characterization of Exo proteins, where >7,000 were identified so far (80). Constitutive Exo proteins are structural vesicle component or are involved in vesicle biogenesis and vesicle trafficking. Most abundant are tetraspanins (81), enriched 7- to 124-fold in Exo compared to the parental cells (82). Adhesion molecules, proteases, MHC molecules, HSPs, TSG101, Alix, annexins, cytoskeleton proteins, metabolic enzymes, cytosolic signal transduction molecules, and ribosomal proteins, some of which are recruited via their association with proteins engaged in biogenesis, are also abundantly recovered (83, 84). Cell type-specific Exo proteins are so far most comprehensively explored for cancer and cancer stem cells (CSC), such as MART1, EGFRVIII, multidrug resistance gene 1, EpCAM, MET, mutant KRAS, and tissue factor (73, 85–88). Notably, due to their location in internalization prone microdomains, all known CSC markers are recovered in TEX (89, 90), which implies recovery of CSC marker expressing Exo in body fluids as most reliable for diagnosis.
Next generation sequencing allowed for rapid progress in Exo DNA, coding and non-coding RNA identification (91). Previous studies were mostly concerned about miRNA, which constitutes only 1–3% of the human genome, but due to multiple targets, controls about 30% of the coding genes. miRNA cleaves mRNA via argonaute (AGO) (perfect base pairing) or represses translation (imperfect binding) (92). Knowledge on miRNA greatly fostered progress in oncology, where miRNA could be linked to prognosis, disease progression, local recurrence, and metastasis (93), particularly the miR-200 family playing an important role in epithelial–mesenchymal transition (EMT) (94). miRNA also accounts for CSC maintenance (95), angiogenesis (96), and chemoresistance (97). Other miRNA, such as miR-34, -34a, -340, act as tumor and metastasis suppressors (98, 99).
miRNA also regulates tolerance induction and inflammation (100–102). A knockout of Dicer and Ago2 in HSC results in increased apoptosis and loss of hematopoietic cell reconstitution; Ago2 deletion is accompanied by deficient B and erythroid cell differentiation (103). A knockdown of Dicer results in T cell reduction (104, 105) and a shortened survival rate and reduced antibody repertoire in B cells (106). Dicer and Drosha also are required for Treg regulation (107, 108), a ko promoting a lethal inflammatory disease. MiR-155 regulates NK maturation and activation by suppressing suppressor of cytokine signaling 1 and phorbol-12-myristate-13-acetate-induced protein 1 (109). In MDSC, upregulated miR-494 and -21 target phosphatase and tensin homolog (Pten), miR-155 targets Ship-1 and miR-210 Arg1, whereas downregulated miR-17-5p and -20a target STAT3 (110). In asthma, miR-20b promotes G-MDSC accumulation associated with a decrease in IL-3 and IL-13 (111) and miR-223 suppresses Arg1 and STAT3 in multiple sclerosis and autoimmune encephalitis (112). Thus, miRNA, besides being important in oncogenesis and tumor progression, regulates T cells, B cells, and components of the innate immune system including MDSC, which has severe bearing on inflammation and autoimmune diseases.
Long non-coding (lnc)RNA makes up ~3% of total exosomal RNA and is transferred into host cells (113). Several exosomal lncRNAs, such as MALAT-1, linc-POU3F3, ZFAS1, promote tumor growth and migration, prevent tumor cell apoptosis, or induce angiogenesis (114–117). A most comprehensive study on colorectal cancer revealed 1,028 lncRNAs selectively enriched in Exo, where the co-existence of U1 and U2 rib nucleoproteins and their cognate shrines in the Exo suggests a possible link to recipient cell splicing events (118). Exosomal lncRNA (GAS5) also regulates apoptosis of Mϕ (119), hematopoietic, innate, and adaptive immune responses (120, 121). The recovery of exosomal lncRNA and novel splicing/fusion genes will be important in developing Exo-based therapeutics.
Taken together the ongoing analysis of Exo composition provided a plethora of informations, which strongly sustain the initial hypothesis of Exo as important intercellular communicators allowing sessile cells a systemic communication, which is equally important in physiology and pathology. For optimal therapeutic translation, further analyses with a focus on donor-dependent differences in Exo profiles are desirable.
Exo Targeting and Uptake
Answering the questions how Exo find their targets and are there options to guide targeting is urgent for therapeutic considerations (122). Exo can bind to the extracellular matrix (ECM) or cells via specific receptor–ligand pairs, where binding accounts for matrix and cell modulation (123, 124). Exo uptake also depending on target cell ligands, may require different target structures then binding and can have distinct consequences for the target cell (125, 126).
Exosomes bind to and are taken up by selected target cells. Exo binding frequently involves (tetraspanin-associated) integrants (124, 127), where ICAM1 will be one potential partner (128, 129). Notably, different integrants bind distinct target cells. Thus, the α6β4 integrant binds cells in the premetastatic niche of the lung, whereas integrin αvβ5 binds cells in the premetastatic niche of the liver (127). A Tspan8–α4β1 complex binds to endothelial cells (EC) and EC progenitors, but a Tspan8–α6β4 complex hampers Exo uptake by EC (130). Other known binding partners are proteoglycans prevalently binding to galectins, selectins, and sialic acid binding lectins (131–134). According to our experience, Exo binding is greatly facilitated by clusters of adhesion molecules in both Exo and target cells (84).
Exosome binding mostly is followed by uptake. There are two modalities for Exo uptake, fusion with the cell membrane (135, 136) and, dominating, endocytosis, an active process that requires modulation of the actin cytoskeleton (135, 137–139). There are at least four modes of uptake, phagocytosis, macropinocytosis, clathrin-dependent endocytosis, and uptake by lipid rafts and caveolae. Phagocytosis proceeds via the formation of cup-like extensions, where the tips fuse and become internalized. Phagocytic markers like lysosomal-associated membrane protein 1 on Exo (140) and T-cell immunoglobulin and mucin domain containing (TIM)4 that recognizes phosphatidylserine on Exo facilitate the process (136, 137, 141). Exo uptake by macropinocytosis occurs, when lamellipodia fold back and fuse with the plasma membrane (142, 143). Most frequently, Exo endocytosis proceeds via clathrin-coated pits, where dynamin contributes to the scission of clathrin-coated endocytosed pits (84, 139, 140). Finally, Exo can be internalized by rafts, cholesterol-, and glycolipid-enriched membrane microdomains, such as tetraspanin webs (84, 139, 144) or caveolae (145).
In brief, Exo uptake is an active process with a contribution of the cytoskeleton as well as fission and scission machineries to detach from the plasma membrane. Intracellular processing of the uptaken Exo varies between cells and requires further exploration (146). Though Exo may itinerate (147), they mostly are digested, their content modulating the target cell both directly or by stimulating signaling cascades, transcription, and silencing processes via the target cell’s equipment (148–151).
Exo and Target Cell Reprogramming
Whether Exo-induced changes in target cells are due to the transferred content of Exo or to transfer-induced target cell reactions is still disputed. When Exo bind to the ECM, the Exo membrane-coat accounts for changes observed in matrix proteins and matrix structure. Instead, when Exo bind to, but are not taken up by the target cell, target cell modulation is promoted by Exo-initiated signal transduction and/or cleavage of proteins on the target cell membrane. When Exo are taken up by the target cell, an unequivocal answer is more critical. There are examples, demonstrating that changes in the target cell are directly due to the transferred Exo content. Thus, in prostate cancer cells, αvβ6 is transferred via Exo into an αvβ6-negative recipient cell and localizes to the cell surface, de novo αvβ6 expression by the recipient cell being excluded (152). Also, after dendritic cell (DC) loading with TEX, tumor antigens are processed and loaded into newly synthesized MHC molecules (137, 153). The same principle will be valid for therapeutically tailored Exo loaded with large amounts of therapeutics drugs or miRNA or signaling checkpoint inhibitors (154–156). However, whether the naturally available amount of one type of Exo contains sufficient load to directly modulate targets is questionable. First, the small Exo plasma homes a limited amount of proteins and nucleotides; second, a TEX preparation from a cloned tumor line distinctly affects tumor cells, fibroblasts, EC, and hematopoietic cells. Thus, an impact on signal transduction and/or transcription/translation likely represents the dominating mode of uptaken Exo activity. The strong impact of DC-Exo uptake by the immune synapse also supports an initiator role of the transferred Exo content (157, 158). The hypothesis is backed by activation or inhibition of B cells, NK, and neutrophils initiated by DC-, Mϕ-, stem cell (SC)-, or tumor cell-derived Exo (159–163). The important role of Exo for anterograde and retrograde information transfer via neurological synapses also argues for Exo-initiated activation of signal transduction, where Exo-promoted activation of signaling cascades was described to maintain plasticity under physiological conditions as well as to account for the spread of pathological proteins (164–167).
Thus, without excluding target modulation by uptaken Exo content, in most instances an incentive push by Exo superiorly covers the wide range of Exo activities.
Exo Transfer and the Life Span of Exo
Information on the natural life span and that of transferred Exo is an additional prerequisite for therapeutic trials.
“Therapeutic” Exo recovery in serum after intravenous application declines toward ~50% within 2 min–1 h (168, 169). Instead, uptaken Exo are recovered for several days (170–172). Thus, gold-labeled Exo could be tracked for over 24 h after intranasal or i.v. application (173) and were recovered in liver, lung, BM, peripheral blood leukocytes, and spleen cells for up to 48 h, with particularly high recovery in monocytic cells, including MDSC (168, 169).
Where required, therapeutic rescuing can be further improved by tailoring with docking molecules (174, 175). Tetraspanins and RGD peptides were described to target tumor cells or EC (176, 177). Targeting oncogenic receptors or SC receptors offers an alternative strategy (178). Bacterial-derived extracellular mimetics additionally could facilitate generating large quantities of homogeneous Exo for vaccination and drug delivery (179).
Taken together, available data strongly support the feasibility of therapeutic Exo application to interfere with cancer progression, to balance angiogenesis, blood coagulation, and to regulate native or adaptive immune system responses. MDSC-Exo are engaged in all these processes.
MDSC-Exo Characterization
Myeloid-derived suppressor cells are well characterized and there is a wealth of information on the impact of TEX on MDSC. Information on MDSC-Exo is limited and was mostly collected using MDSC-Exo derived from tumor-induced, immunosuppressive MDSC, which resemble the inflammatory MDSC in chronic infections. Thus, these data are valid for the differentiation between resting versus inflammatory MDSC-Exo in general.
Myeloid-derived suppressor cell-exosomes contain common Exo components such as annexins, tetraspanins, glycosylphosphatidylinositol-anchored CD177, cytoskeletal proteins, proteins engaged in vesicle biogenesis, and HSP. There is an abundance of proteasome subunits, histone variants and elongation factors, and metabolic enzymes that recovery in MDSC-Exo mostly corresponds to the recovery in MDSC. Comparing inflammatory with conventional MDSC-Exo showed a decrease of 33 proteins, some of which being involved in innate immune responses, such as complement components and chemotactic proteins. In addition, some cytoskeletal proteins like spectrin, ankyrin, and tubulin were reduced in inflammatory MDSC-Exo. Thirty proteins increased in inflammatory MDSC-Exo included GTP and ATP-binding proteins and proteins engaged in Exo-biogenesis facilitating budding or sorting (180).
The same group also reported on the abundance of ubiquitinated proteins in MDSC-Exo, a posttranslational modification that contributes to internalization of membrane proteins and the sorting of endosomal proteins (181), which also accounts for five newly recovered ubiquitinated proteins [sortin nexin 13, two keratins (krts), leucine zipper, and EF-hand containing transmembrane protein 1 < LETM1 > and endoplasmin] (182). Furthermore, MDSC-Exo abundantly carry ubiquitinated histones, the non-histone nuclear protein high mobility group box (HMBG)1 as well as all the enzymes required to catalyze ubiquitination (183). A proteome analysis of low mass proteins in MDSC-Exo confirmed, besides the abundance of proinflammatory S100 proteins the abundance of histones, which made up 56% of the MDSC-Exo protein cargo (184). Of special interest also is the analysis of MDSC-Exo surface glycoproteins, which include fibronectin (FN), olfactomedin4, galectin-3-binding protein, myeloperoxidase, thrombospondin1 (Tsp1), a cytoskeletal krt (Krt77), fibrinogen (Fbg), mast cell expressed protein 1, transmembrane 9 superfamily member 3, endothelial lipase, and the CD molecules CD44, CD157, CD11b, CD97, CD39, CD18, CD321, CD41 (185). Searching for potential ligands revealed several shared ligand receptor pairs, like CD41: Tsp, FN, Fbg; CD11b: haptoglobin (Hp), FN, Fbg; CD18: Hp, Fbg; CD47: Tsp, CD172a (SIRPa); CD44: Tsp, CD29, CD47, indicating that MDSC-Exo are well equipped for binding. However, there were no hints toward a pronounced selectivity of binding. Instead, several of these MDSC-Exo membrane proteins could be of functional interest. The do not-eat me CD47, whose dominating ligand is Tsp1, promotes MDSC migration (186). Furthermore, MDSC express the advanced glycosylation end-product specific receptor ligand S100A8/9, which could contribute to the activation of inflammatory/immunosuppressive genes (185–187).
Information on the RNA and DNA load of MDSC-Exo is largely missing, but is well explored in MDSC (188, 189). To give a few examples, TGFβ promotes G- and M-MDSC induction and expansion via upregulation of miR-155 and miR-21, which target inositol phosphate-5-phosphatase D and Pten, leading to activation of STAT3 (190). During sepsis, miR-21 and miR-18b become strikingly upregulated, which is accompanied by pronounced immunosuppressive activity of MDSC prohibiting bacterial clearance (191). Mir-9 overexpression enhances MDSC functional activity. This is due to miR-9 targeting Runx1, an essential transcription factor in promoting MDSC differentiation (192). Doxorubicin treatment promotes miR-126a induction in MDSC. miR-126a + MDSC-Exo induce IL13+Th2 cells and rescue MDSC death in a S100A8/A9-dependent manner (193).
Taken together, the inflammatory MDSC-Exo membrane protein profile provides hints toward receptor–ligand pairs. Unfortunately, so far no selective ligands, e.g., for binding Treg or activated T cells were recovered. The reduced recovery of some inflammatory proteins in inflammatory MDSC-Exo suggests a possible contribution to the inefficacy of immune-response induction in cancer and chronic infections. The abundance of proteasome subunits as well as of histones and HMBG1, which is inflammation independent, is of great interest and should be further elaborated, some functional consequences being already defined. The finding that Doxorubicin treatment affects the MDSC-Exo miRNA profile with severe functional consequences also should spur research on this topic.
Coping with MDSC-Exo in Cancer
MDSC-Exo Activities in Cancer
As Exo are supposed to be most important intercellular communicators, can be easily modulated in vitro and are simple to store for therapeutic application, a detailed knowledge on MDSC-Exo activities will open a wide range of new and promising therapeutic applications. However, gaining insight is a demanding task. This relates to the heterogeneity of MDSC, the delivery of distinct Exo subpopulations by individual cells, the differences in Exo delivered by MDSC during maturation in the BM versus “inflammatory” MDSC. In addition, Exo have more than one target, which becomes aggravated by the distribution of Exo throughout the body and the cooperativity of different cells/subpopulations particularly in the immune system. This implies that a whole range of potential targets needs to be analyzed for MDSC-Exo promoted alterations.
Though not directly approaching MDSC-Exo, there is compelling evidence that TEX induce and affect MDSC. TEX are taken up by myeloid cells in the BM and switch their differentiation toward MDSC. Also, tumor growth-promoting activity of MDSC depends on PGE2 and TEX-provided TGFβ that induce upregulation of Cox2, IL6, VEGF, and Arg1 in MDSC (194). Furthermore, TEX-associated Hsp72 triggers toll-like receptor (TLR)2/myeloid differentiation primary response gene 88 (MyD88)-dependent Stat3 activation in MDSC, which exert pronounced immunosuppressive activity (195). The finding was confirmed using MyD88-ko mice, which additionally revealed a reduction in CCL2 (196). TEX also promotes MDSC expansion in the BM through activation of STAT3, upregulated iNOS, which strengthens the immunosuppressive capacity of MDSC (197). Breast cancer-TEX distribute to the lung are taken up bone-marrow-derived cells and promote accumulation of MDSC in lung and liver. In addition, TEX inhibit through activation of M-MDSC T cell activation and TH1 cytokine production (198). BM stroma cell Exo, which are crucial in multiple myeloma development, are taken up by MDSC, induce their expansion, and survival through STAT3 and STAT1 pathway activation and induction of anti-apoptotic BclXl and Bcl2 family apoptosis regulator and promote NO release by MDSC increasing their suppressive activity on T cells (199).
Functional analysis of freshly ex vivo harvested MDSC-Exo was mostly restricted to the impact on myeloid cells. The authors report that the proinflammatory S100A8/9 heterodimer is chemotactic for MDSC (180). Furthermore, MDSC and, less prominently, MDSC-Exo convert tumoricidal M1–Mϕ to tumor growth-promoting M2-Mϕ by switching off IL12 production (180). Of special functional interest is the recovery of ubiquitinated histones and HMGB1 (182, 183), which exert proinflammatory activity, contribute to systemic inflammation and organ failure, and drive autoimmune diseases (200–202). HMBG1, a chaperone for many inflammatory molecules in MDSC, promotes the development of MDSC from BM progenitors, increases IL10 production by MDSC and contributes to downregulation of the T cell homing receptor CD62L (203, 204). The conversion of monocytes into MDSC-like cells and the differentiation of bone marrow cell into M-MDSC proceeds via the p38/NFκB/Erk1/2 pathway (205). In the context of chemoresistance, which in part relies on MDSC, MDSC-Exo miR-126a induces expansion of TH2, inhibits TH1 proliferation, and IFNγ secretion and supports angiogenesis. In a feedback loop, chemoresistance is transferred into the donor MDSC (193).
Therapeutic Interference with MDSC-Exo in Cancer
There are excellent reviews on the therapeutic use of Exo (206, 207) as well as on attacking MDSC in cancer (208, 209) including approaches with a focus on improving the efficacy of immunotherapy (6, 210). So far, only a limited number of reports was concerned about directly attacking MDSC-Exo in cancer as a therapeutic option.
In brief, attacking tumor-infiltrating MDSC can be achieved by cytotoxic drugs, where ATRA blocks MDSC maturation (211, 212), which was explored in cancer immunotherapy (9, 20, 213) as well as in chronic infections (214). Gemcitabine particularly drives MDSC into apoptosis by a not yet fully explained mechanism (44), its efficacy in improving immunotherapy in cancer being repeatedly described (9, 47, 215). Sunitinib, a checkpoint signaling inhibitor, preferentially attacks MDSC and was reported in several tumor models to support immunotherapy (9, 216–218). Ongoing trials aim to increase vaccination efficacy by using combinations of drugs or radiotherapy (9, 219, 220).
Alternatively, an antibody blockade may be envisaged that prevents MDSC-Exo docking on target cells. According to the enrichment of tetraspanins, anti-CD9 was shown to prohibit breast cancer cell metastasis (221). We used anti-Tspan8 to block pancreatic cancer TEX, Tspan8 being abundantly expressed on pancreatic CSC-TEX (222). The antibody blockade sufficed to hamper angiogenesis and premetastatic niche establishment, but had a minor impact on MDSC (169). A MDSC-Exo selective antibody blockade remains to be explored.
An interesting approach is the use of proton pump inhibitors, toxic byproducts generated by the altered metabolism of cancer cells being expelled by proton transporters (223). Proton pump inhibitors concomitantly contributing to hamper the release of Exo by affecting the acid milieu in the tumor surrounding (224), the release of MDSC-Exo may be inhibited concomitantly, which could contribute to facilitate recruitment of effector immune cells. Similar considerations account for a blockade of the S100A8/9 marker on MDSC-Exo (180). Alternatively, a blockade of premetastatic niche formation was achieved by a blockade of CCL2 that prevented MDSC-Exo recruitment (225). A blockade of CD47 and its ligand Tsp and, less efficiently the signal regulator protein α, highly expressed on MDSC-Exo, also hampered MDSC-Exo chemotaxis and migration (185).
As an alternative approach, extracorporal hemofiltration is used for Exo elimination. Originally established as an affinity plasmapheresis for the elimination of TEX, it is being adapted to remove hepatitis C virions and is being explored to remove immunosuppressive Exo (226, 227). Progress in MDSC-Exo proteomics may provide means for a selective removal. There remains the problem of MDSC-Exo in cancer being mostly located within the tumor tissue or recruited to potential target, e.g., EC and premetastatic organ tissue rather than in the serum. Coping with Exo regeneration may also become demanding.
Last not least, Exo or Exo surrogates can be loaded with drugs, toxins, non-coding RNA to be delivered toward MDSC or MDSC-Exo to directly prohibit their immunosuppressive, angiogenesis and cancer-spread promoting activities (175, 178, 228, 229). The field is rapidly expanding, tailoring Exo, or surrogates also for repair, e.g., in artherosclerosis or thrombosis (230–232). There remains the demand for selective binding as, e.g., miRNA interfering with MDSC activities may promote tumor growth (189, 193, 233). Finally, great efforts are taken to replace Exo by nanoparticles that could allow for easier and homogeneous production (179). First trials attacking MDSC to improve cancer immunotherapy revealed encouraging results (234–237).
Thus, there are several promising options to interfere with the immunosuppressive and tumor growth-promoting activity of “inflammatory” MDSC and MDSC-Exo. There is need improving target selectivity. However, as the tumor milieu/TEX contribute to the recruitment and expansion of “inflammatory” MDSC/MDSC-Exo, targeting TEX may be considered under selected conditions as an alternative. Targeting TEX would be less demanding, as TEX mostly are equipped with oncogenes or CSC markers (222, 238, 239) that are not as widespread as inflammatory MDSC-Exo markers.
Recruiting MDSC and MDSC-Exo in Autoimmune Disease
MDSC and MDSC-Exo in Autoimmune Disease
While the abundance of MDCS/MDSC-Exo in cancer creates a milieu of therapy resistance, autoimmune disease progression is favored by the inefficacy of immune response regulation by immunosuppressive cells and factors (240). This accounts for the paucity of MDSC and Treg (240, 241), where the latter may be linked or be due to the former (242) and frequently is accompanied by an increase in TH17 (243). However, opposing findings were also reported.
Myeloid-derived suppressor cells only recently achieved attention in autoimmune diseases, initially in animal models such as experimental autoimmune encephalomyelitis (EAE), where a deficit in CCR2, which is required for MDSC recruitment, was accompanied by milder EAE (244). However, depending on the model and the readout system, opposing findings were also reported (245). This diversity of findings accounts for a wide range of studies on the recovery of MDSC and their suppressive activity in autoimmune diseases. There are, at least, two reasons for this confusion. First, the disease state is important. With progressive tissue destruction concomitantly to the dysregulated autoimmune effector cells, an inflammatory milieu is generated, which, in fact, supports MDSC activation. Second, a failure to detect a decrease in MDSC and/or Treg in the peripheral blood or peripheral lymphoid organs in autoimmune disease may be irrelevant (246), as the frequency in the autoimmune disease-affected organ can differ significantly. To give an example, while Treg are rare in the peripheral blood, in non-lymphoid tissues, the frequency of Treg ranges from 30–60% of the total CD4+ population (247).
Besides being concerned about the numeric MDCS/MDSC-Exo deficits, several studies elaborated a contribution of miRNA in their regulating. Thus, miR-181a is engaged in the maturation of myeloid progenitor cells, miR-17-5p, miR-20a, miR-106a, and miR-155 play a role in the differentiation of myeloid progenitor cells toward monocytes and miR-146a/b and miR-155 in the maturation toward Mϕ, where miR-155 and miR-181 additionally contribute to T cell differentiation (248). Monocytes in Sjoegren Syndrome show upregulated miR-34b-3p, -300, -609 expression; in psoriasis miR-223 was high and miR-193b was low in Th17 (249). In the antiphospholipid syndrome, which promotes trophoblast inflammation, changes in miR 146a-3p, -155, and -210 affected TLR signaling. These changes, also observed in MDSC-Exo, suggest a contribution to disease progression (250).
At the present state of knowledge, there is an urgent need for additional information on MDSC/MDSC-Exo presence and activity in autoimmune disease-affected organs. Instead, there is consent that chronic infections rely on an abundance of “inflammatory” MDSC/MDSC-Exo, which prevent appropriate activation of the adaptive and the innate immune system (251, 252). This knowledge, in fact, could provide a helpful guide toward MDSC/MDSC-Exo as a therapeutic option in autoimmune disease.
MDSC and MDSC-Exo as a Therapeutic Option in Autoimmune Disease
There are excellent reviews on the link between chronic infections, immune regulation, and the associated hindrance of autoimmune disease development and progression (14, 39). MDSC/MDSC-Exo playing an important role, these inflammatory MDSC/MDSC-Exo may well provide a guide toward correcting overshooting reactions in autoimmune disease.
Thus, several reports demonstrating parasite infections being associated with a significant decrease in incidence or severity of immune diseases in animal models, the protective effect being due to Treg, alternatively activated Mϕ and changes in the cytokine profile (253–255). In chronic hepatitis C virus infection, a striking increase in M-MDSC was noted that expressed high level pSTAT3 and IL-10 and induced Treg expansion, where depletion of MDSC increased IFNγ production by CD4+ effector T cells (256). In human immunodeficiency virus-1 infections, too, MDSC-promoted Treg expansion and inhibited T cell function, a hallmark of chronic infections (257). In tuberculosis, the accumulation of MDSC prevented immune effector cell-mediated bacteria evasion (258). The interference of inflammatory MDSC/MDSC-Exo in cancer with immunotherapy was already outlined in detail.
As bacteria, parasites and viruses that cause chronic inflammation would rather provide a danger than a therapeutic option, chemical compounds that provoke delayed type hypersensitivity may be better suited to induce “inflammatory” MDSC. This option is well explored in alopecia areata (AA), most efficiently treated by the contact sensitizer squaric dibutylester (SADBE) (259, 260). SADBE treatment provokes a strong expansion of MDSC that inhibit autoreactive T cell activation and support Treg expansion. The effect is abolished by ATRA treatment (261). Notably, SADBE treatment can be replaced by the transfer of MDSC (262). In EAE, it was demonstrated that helminth products stimulate the production of TH2 cytokines and suppress TH1 and TH17 responses, the therapeutic efficacy exceeding that of corticosteroid treatment (263). Another option are statins, which are cholesterol lowering drugs, also described to induce immunosuppression. This was confirmed in acute and chronic dextran sodium sulfate (DSS)-induced colitis in mice, where statin-induced attenuation of colitis was due to expansion of MDSC (264).
Thus, the exploration of inflammatory MDSC has opened a path toward their therapeutic use in autoimmune disease. These studies clearly demonstrated therapeutic efficacy of MDSC in experimental autoimmune disease models (14, 265). In addition, good progress already was achieved replacing the infectious agents by synthetic compounds.
Autoimmune disease corrections by Exo, mostly by mesenchymal stem cell (MSC)–Exo, but also by DC-Exo were repeatedly described. To give a few examples, in diabetes susceptible mice, islet MSC release Exo that express endogenous retroviral antigens, which induce potent T and B cell responses (266). Application of MSC–Exo in experimental autoimmune uveitis exerted a therapeutic effect that was due to inhibiting the chemoattractive effects of CCL2 and CCL21 on inflammatory cells (267). Exo from miR-146a overexpressing DC suppress experimental myasthenia gravis by inducing an antigen-specific shift from TH1/TH17 to TH2/Treg (268). However, Exo from different donor cells or at different stages of disease may exert opposing activities. Thus, at early stages in chronic HBV infection, hepatic NK produce IFNγ in response to hepatic Mϕ. Hepatic Mϕ are stimulated by infected hepatocyte-Exo, which contain viral nucleic acids, via MyD88, toll-like receptor adaptor molecule (TICAM) and mitochondrial antiviral signaling protein to express NKG2D ligand. On the other hand, immunoregulatory miR-21 becomes upregulated in infected hepatocytes and is transferred via Exo in Mϕ suppressing IL12p35 expression, which counteracts the host innate immune response (269). For more comprehensive information, excellent reviews are recommended that outline the interplay between Exo from different donor cells and the activity of MDSC in autoimmune disease (270–272).
There is, to my knowledge, only one report explicitly describing the role of MDSC-Exo in autoimmune disease. Mice with DSS-induced colitis were treated with G-MDSC-Exo. G-MDSC-Exo sufficed for a significant decrease in disease severity and a reduction in the inflammatory cell infiltrate. TH1 cells were reduced and Tregs were augmented in the draining lymph node; in the serum IFNγ and TNFα were reduced. Inhibition studies pointed toward the impact of G-MDSC-Exo largely depending on Arg-1 (273).
Having described that the therapeutic efficacy of a chronic contact eczema in AA largely depends on the expansion of MDSC and that SADBE treatment can be replaced by MDSC application (261, 274), we proceeded controlling for the activity of MDSC-Exo in AA-affected mice. MDSC-Exo preferentially target in vitro and in vivo activated T cells, NK and most avidly Treg. Furthermore, an mRNA analysis of spleen cells of MDSC-Exo treated AA-affected mice showed a most striking increase in FoxP3 and Arg-1. These findings suggest MDSC-Exo strongly promoting Treg expansion and hampering innate immune reactions as well as T cell activation directly and via Treg.
Taking the knowledge collected in cancer and chronic infections on the power of inflammatory MDSC-Exo opened a path for a new wave of autoimmune disease treatment. Modalities to circumvent the potential danger of naturally arising inflammatory MDSC/MDSC-Exo have been suggested and are further explored in ongoing studies.
Conclusion, Open Questions, and Outlook
I. The discovery of Exo and other extracellular vesicles has revolutionized cell biology offering sessile cells to communicate over long distance (275). Though difficult to catch due to their heterogeneity (Figure 1A), where even a single cell delivers distinct Exo, great efforts are taken to answer open questions
– We still poorly understand the process of loading the Exo plasma during biogenesis, including the enrichment of nuclear proteins, proteasome subunits, and components of the splicing machinery (Figure 1B).
– The question of target molecules/complexes of target molecules is not comprehensively answered.
– The availability of free versus bound/uptaken Exo requires further exploration (Figure 1C).
II. MDSC, a heterogeneous population of immature myeloid cells, are important immune response regulators targeting T cells, B cells, Treg, NK, DC, and other cells of the innate immune system. This also accounts for MDSC-Exo, where target cells may become distinctly affected by MDSC binding versus MDSC-Exo uptake. MDSC/MDSC-Exo induce depletion of essential amino acids through ARG1, iNOS and IDO, NO, and ROS generation through iNOS and NOX2, anti-inflammatory cytokine production and Treg induction and activation (276). However, it is not well defined, whether
– the equipment of MDSC/MDSC-Exo or of the target cell directs the mode of reprogramming.
– MDSC and MDSC-Exo act via alike or different mechanisms.
III. MDSC/MDSC-Exo are a major hindrance in tumor immunotherapy and chronic infections (39, 277, 278). Instead, autoimmune diseases may progress due to insufficient MDSC/MDSC-Exo (14, 19, 245). The inefficacy of MDSC/MDSC-Exo including the induction of Treg in autoimmune diseases is still debated. But, on site studies strongly support paucity of MDSC and Treg. Nonetheless,
– the poor activation state of MDSC in the diseased tissue and
– the mode whereby MDSC/MDSC-Exo provoke Treg expansion and activation requires further elaboration.
IV. The therapeutic efficacy of eliminating MDSC in cancer and of providing MDSC in autoimmune disease is well documented (44, 209, 245, 279). So far, only few reports were concerned about replacement by MDSC-Exo. However, MDSC-Exo are not only easier to handle, but also can be expected to act systemically and via their uptake to more severely affect their targets. The lack of information on selective markers of “inflammatory” MDSC-Exo provides a handicap. In cancer, MDSC recruitment and expansion are driven by TEX, which express cancer-related markers. Therefore, depletion of TEX instead of MDSC-Exo could provide an alternative. In concern of therapeutic MDSC-Exo substitution, “inflammatory” MDSC-Exo preferentially should be generated from synthetic compound stimulated MDSC, which avoids unwanted support of immunosuppression in response to naturally inflammatory stimuli. Irrespective of these alternatives,
– the high prevalence of MDSC-Exo uptake by Treg and activated T cells suggests selective targets, which should be defined.
V. This review focuses on MDSC-Exo and their activities in cancer and autoimmune disease. Nonetheless, the widespread activity particularly of SC-Exo (280) in physiology, including developmental patterning and the embryonic-maternal crosstalk (281, 282), in rejuvenation, regeneration, and repair (283) should, at least, be mentioned. SC-Exo act via signal transduction and the transfer of non-coding RNA (284, 285) and are suggested being a most potent therapeutics by maintaining stemness and inducing reparative programs (286, 287). There is justified hope on their therapeutic efficacy in SC transplantation, repair, and transplant acceptance (24, 288, 289).
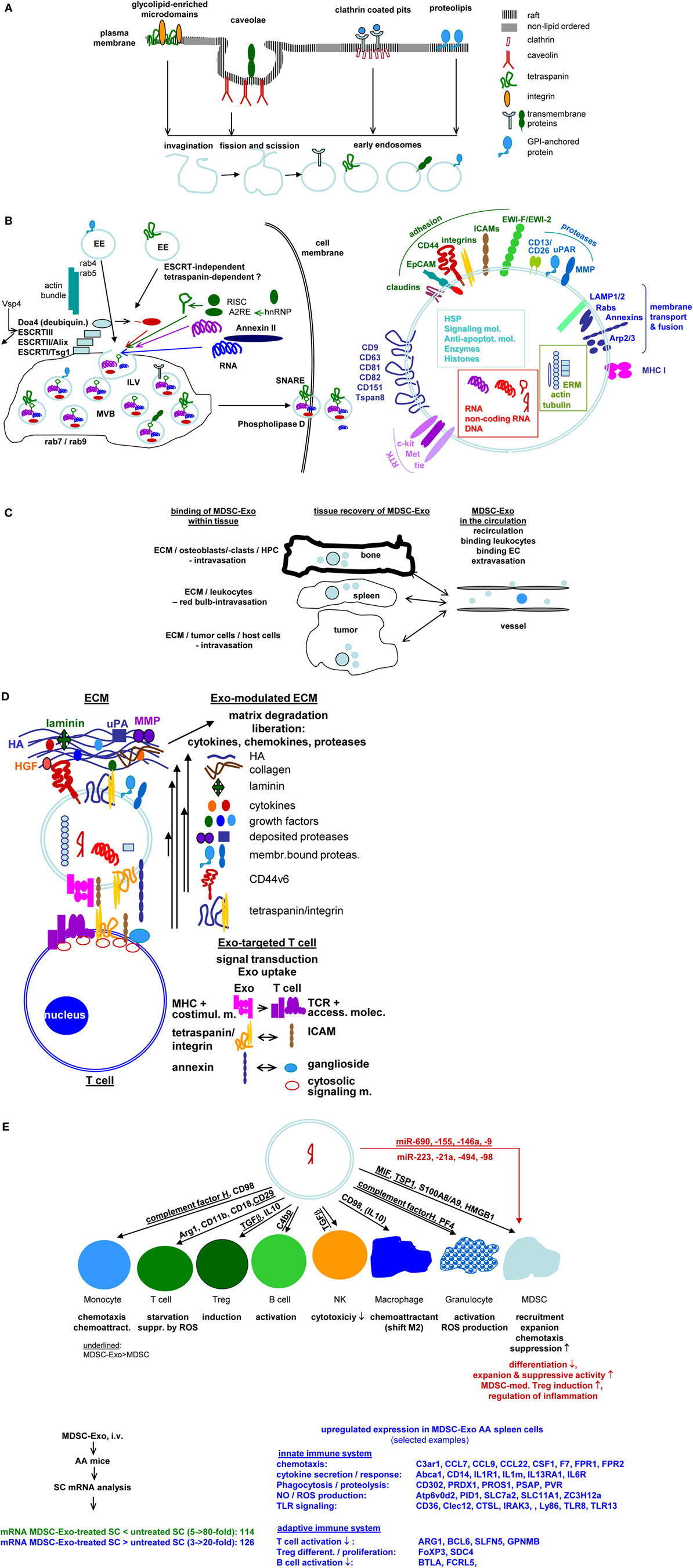
Figure 1. Exosomes (Exo) and therapy: open questions. (A) Exo derive from different membrane raft compartments, which are plane or invaginated, but are all prone for internalization due to enrichment for cholesterol and sphingolipids. Distinct lipid rafts harbor selective membrane-attached and transmembrane proteins, which are retained during invagination. This implies Exo derived from a single cell to be equipped by a different membrane coat. This poses the question on how to select for the appropriate myeloid-derived suppressor cells (MDSC)-Exo subpopulation. (B) After fission and scission, EE use different transporters toward MV, where ILV are loaded with proteins, coding and non-coding RNA and DNA during invagination into MVB. This is a selective process and includes components of different intracellular compartments. The abundance of selected molecules recruitment is only partly understood. Information is urgently required to judge on potential Exo activities. The released Exo are composed of the lipid membrane, the membrane-integrated and membrane-attached molecules, the components transferred into ILV and the majority of molecules engaged in vesicle transport, loading and Exo delivery. An arbitrarily selection is shown. (Alix: ALG-2 interacting protein X, Doad4: deubiquitinase, EE: early endosome, ESCRT: endosomal sortin complex, ILV, intraluminal vesicle; MVB, multivesicular body; SNARE, soluble-N-ethylmaleimide-sensitive fusion protein-attachment protein receptors; Vsp4, ATPase vacuolar protein sorting 4). (C) Exo distribute throughout the body and bind to matrix proteins and cells. Information on the availability of “free” Exo is limited. Yet, it is essential to judge on diagnostic and prognostic validity of Exo, including MSCD-Exo. (D) Exo binding to matrix proteins and cells are selective processes, where cells may use different ligands for binding and uptake. It is suggested that uptake depends on clustered ligands, possibly in invagination prone membrane domains. Binding, too, may be facilitated by clustered ligand. This is important for tailoring “therapeutic” Exo/Exo mimetics to facilitate binding/uptake or to prevent uptake. Selective examples are shown for ECM and T cell binding (ECM, extracellular matrix; HA, hyaluronic acid; HFG, hepatocyte growth factor; MHC, major histocompatibility complex; MMP, matrixmetallo proteinase; TCR, T cell receptor complex). (E) Exo binding and uptake modulates the target. Uptake initiated target cell modulation could proceed directly via incorporation of the Exo content, which recently was evaluated including miRNA (363) or by the target cell equipment after an initial hit by the Exo content. Both modalities were described. In view of the small Exo plasma and unpublished findings on changes in spleen cell mRNA after in vivo treatment of AA mice with MDSC-Exo, an initiating trigger may be more likely (AA, alopecia areata; Abca1, ATP-binding cassette sub-family A member 1; ARG1, Arginase-1; Atp6v0d2, V-type proton ATPase subunit d2; BCL, B cell leukemia; BTLA, B and T lymphocyte associated; CCL, chemokine ligand; C3ar1, C3a anaphylatoxin chemotactic receptor, Clec: C-type lectin domain family; MCSF1R, macrophage colony-stimulating factor 1 receptor; CTSL, Cathepsin L1; F7, coagulation factor VII; FCRL5, Fc receptor like 5; FOXP3, Forkhead box protein P3; FPR, fMet-Leu-Phe receptor; GPNMB, Transmembrane glycoprotein nMB; IL1R1, interleukin-1 receptor type 1; IL13RA1, IL13 receptor subunit alpha 1; IL6R, Interleukin6 receptor; IRAK, Interleukin-1 receptor-associated kinase, Ly, lymphocyte antigen; PID1, PTB-containing, cubilin and LRP1-interacting protein; SC, spleen cells; SDC, syndecan; SLC, Solute carrier family; SLFN5, Schlafen family member 5; TLR, toll-like receptor; ZC3H12a, zinc finger CCCH-type containing 12A). The nature of initiating triggers, target structures, and molecular pathways of progression remain to be defined. Clarification would greatly assist “therapeutic” Exo/Exo mimetic furnishing. Personal view: recovery of selected membrane markers of MDSC-Exo would be highly desirable. Should there be no selective markers, a binding unit for the target cell could be introduced. In concern about vesicle loading during biogenesis, the abundant recovery of proteasome subunits, histones, and splicing complex components requires special attention. It is conceivable that integration of these components rather than the small amount of transferred proteins, coding/non-coding RNA and DNA initiates target cell reprogramming by modulating transcription, translation, and metabolism. These activities will be well supported by MDSC-Exo binding to the T-cell and B-cell synapses, the receptor complexes and the adjacent accessory molecules being targeted by their counterparts on MDSC-Exo and being prone for internalization and initiation of signaling cascades. FcR and FcR-like molecules may cope with similar tasks in NK, granulocytes and Mϕ. Further progress in MDSC-Exo content elaboration and recovery in target cells will provide the answer, whether it appears more suitable loading MDSC-Exo with effector or initiator molecules.
Patrolling through the body to control for burglar and killers and to start the alarm clock was the privilege of cells of the innate immune system. For a long time, it was missed that they also control via Exo the response of the adaptive immune system, they had initiated. Taking into account that Exo are still newcomers in cell biology and all the excellent work collected during a short period, for which I apologize having cited only few, I am confident that open questions are quickly answered. This will provide a means to correct for overshooting and vanishing responses evolving in long-lasting diseases, such as cancer, chronic infections, and autoimmune diseases. The ease of tailoring Exo (290) will fortify therapeutic efficacy. Last, not least, provided open questions on Exo targeting and function-relevant components are answered (Figures 1D,E), Exo mimetics are expected to provide a homogeneous and reliably reproducible therapeutic agent (179, 291).
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (MZ, Zo40/14-3) and the funding programme Open Access Publishing, by the Baden-Württemberg Ministry of Science, Research and the Arts and by Ruprecht-Karls-Universität Heidelberg.
Abbreviations
AA, alopecia areata; AGO, argonaute; Arg, arginase; ATRA, all-transretinoic acid; BclXl, Bcl2-like; BMC, bone marrow cells; BMSC, BM stroma cells; C, complement; CCL, C-C motif chemokine ligand; CEBP, CCAAT/enhancer binding protein; COX, cytochrome C oxidase; CSC, cancer stem cell; CXCL, C-X-C motif chemokine ligand; DC, dendritic cells; DSS, dextran sodium sulfate; EAE, experimental autoimmune encephalomyelitis; EC, endothelial cell; ECM, extracellular matrix; EMT, epithelial–mesenchymal transition; Dicer, ribonuclease; ESCRT, endosomal sorting complex required for transport; Exo, exosomes; Fbg, fibrinogen; FN, fibronectin; FOXO, forkhead box O; G-MDSC, granulocytic-MDSC; GM-CSF, granulocyte-macrophage-colony stimulating factor; GEM, glycolipid-enriched membrane domain; GPI, glycosylphosphatidylinositol; HCV, hepatitis C virus; HIF, hypoxia inducible factor; HIV, human immunodeficiency virus; HMBG, high motility group box; hnRNP, heterogeneous ribonucleoprotein; HO-1, heme oxygenase-1; Hp, haptoglobin; HSC, hematopoietic stem cells; HSP, heat shock protein; IDO, indoleamine 2,3-dioxygenase; ILV, intraluminal vesicles; iNOS, inducible nitric oxide synthase; IL, interleukin; IFN, interferon; JAK, Janus kinase; krt, keratin; LAMP, lysosomal-associated membrane protein; LETM1, leucine zipper and EF-hand containing transmembrane protein 1; LIPG, endothelial lipase; LN, laminin; lnc, long non-coding; LPS, lipopolysaccharide; MAVS, mitochondrial antiviral signaling protein; MCEMP1, mast cell expressed protein 1; Mcl1, Bcl2 family apoptosis regulator; M-CSF, macrophage colony-stimulating factor; Mϕ, macrophage; MDR, multidrug resistance gene; MDSC, myeloid-derived suppressor cells; M-MDSC, monocytic MDSC; MMP, metalloproteinase; MPO, myeloperoxidase; mTOR, mechanistic target of rapamycin; MSC, mesenchymal stem cells; MVB, multivesicular bodies; Myd88, myeloid differentiation primary response gene 88; NFκB, nuclear factor kappa B; NK, natural killer cell; NOXA, phorbol-12-myristate-13-acetate-induced protein 1; PBL, peripheral blood leukocytes; PG, prostaglandin; Pten, phosphatase and tensin homolog; Rab, ras-related proteins; RAGE, advanced glycosylation end-product specific receptor; RISC, RNA-induced silencing complex; ROS, reactive oxygen species; SADBE, squaric dibutylester; SC, stem cell; SCF, stem cell factor; Ship1, inositol phosphate-5-phosphatase D; SNARE, soluble-N-ethylmaleimide-sensitive fusion protein-attachment protein receptor; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TEX, tumor-derived Exo; TGF, transforming growth factor; TH, helper T cell; TLR, Toll-like receptor; TIM, T-cell immunoglobulin and mucin domain containing; TM9SF3, transmembrane 9 superfamily member 3; Treg, regulatory T cells; Tsp; thrombospondin; Tsg, tumor susceptibility gene; VEGF, vascular endothelial growth factor; VsP, ATPase vacuolar protein sorting.
References
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends – an update. Cancer Epidemiol Biomarkers Prev (2016) 25:16–27. doi:10.1158/1055-9965.EPI-15-0578
2. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug resistance in cancer: an overview. Cancers (Basel) (2014) 6:1769–92. doi:10.3390/cancers6031769
3. Chevalier F, Hamdi DH, Saintigny Y, Lefaix JL. Proteomic overview and perspectives of the radiation-induced bystander effects. Mutat Res Rev Mutat Res (2015) 763:280–93. doi:10.1016/j.mrrev.2014.11.008
4. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol (2014) 11:509–24. doi:10.1038/nrclinonc.2014.111
5. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther (2016) 38:1551–66. doi:10.1016/j.clinthera.2016.03.026
6. Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol (2017) 102:727–40. doi:10.1189/jlb.5VMR1116-458RRR
7. Huang A, Zhang B, Wang B, Zhang F, Fan KX, Guo YJ. Increased CD14(+)HLA-DR (-/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother (2013) 62:1439–51. doi:10.1007/s00262-013-1450-6
8. Zhu X, Lang J. Programmed death-1 pathway blockade produces a synergistic antitumor effect: combined application in ovarian cancer. J Gynecol Oncol (2017) 28:e64. doi:10.3802/jgo.2017.28.e64
9. Xiao L, Erb U, Zhao K, Hackert T, Zöller M. Efficacy of vaccination with tumor-exosome loaded dendritic cells combined with cytotoxic drug treatment in pancreatic cancer. Oncoimmunology (2017) 6:e1319044. doi:10.1080/2162402X.2017.1319044
10. Osnes LT, Nakken B, Bodolay E, Szodoray P. Assessment of intracellular cytokines and regulatory cells in patients with autoimmune diseases and primary immunodeficiencies – novel tool for diagnostics and patient follow-up. Autoimmun Rev (2013) 12:967–71. doi:10.1016/j.autrev.2013.02.003
11. Jabri B, Terhorst C. Editorial overview: autoimmunity. Curr Opin Immunol (2014) 31:v–vii. doi:10.1016/j.coi.2014.10.010
12. Flachenecker P. Autoimmune diseases and rehabilitation. Autoimmun Rev (2012) 11:219–25. doi:10.1016/j.autrev.2011.05.016
13. Petta I, Dejager L, Ballegeer M, Lievens S, Tavernier J, De Bosscher K, et al. The interactome of the glucocorticoid receptor and its influence on the actions of glucocorticoids in combatting inflammatory and infectious diseases. Microbiol Mol Biol Rev (2016) 80:495–522. doi:10.1128/MMBR.00064-15
14. Boros P, Ochando J, Zeher M. Myeloid derived suppressor cells and autoimmunity. Hum Immunol (2016) 77:631–6. doi:10.1016/j.humimm.2016.05.024
15. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
16. Saleem SJ, Conrad DH. Hematopoietic cytokine-induced transcriptional regulation and notch signaling as modulators of MDSC expansion. Int Immunopharmacol (2011) 11:808–15. doi:10.1016/j.intimp.2011.03.010
17. Natarajan S, Thomson AW. Tolerogenic dendritic cells and myeloid-derived suppressor cells: potential for regulation and therapy of liver auto- and alloimmunity. Immunobiology (2010) 215:698–703. doi:10.1016/j.imbio.2010.05.024
18. Wang Y, Tian J, Wang S. The potential therapeutic role of myeloid-derived suppressor cells in autoimmune arthritis. Semin Arthritis Rheum (2016) 45:490–5. doi:10.1016/j.semarthrit.2015.07.003
19. Yang H, Bi Y, Han F, Lu Y, Wang J, Zhang Z, et al. Myeloid-derived suppressor cells in immunity and autoimmunity. Expert Rev Clin Immunol (2015) 11:911–9. doi:10.1586/1744666X.2015.1052794
20. Li Y, Tu Z, Qian S, Fung JJ, Markowitz SD, Kusner LL, et al. Myeloid-derived suppressor cells as a potential therapy for experimental autoimmune myasthenia gravis. J Immunol (2014) 193:2127–34. doi:10.4049/jimmunol.1400857
21. Ostanin DV, Bhattacharya D. Myeloid-derived suppressor cells in the inflammatory bowel diseases. Inflamm Bowel Dis (2013) 19:2468–77. doi:10.1097/MIB.0b013e3182902b11
22. Dilek N, van Rompaey N, Le Moine A, Vanhove B. Myeloid-derived suppressor cells in transplantation. Curr Opin Organ Transplant (2010) 15:765–8. doi:10.1097/MOT.0b013e3283401742
23. Lee YK, Kang M, Choi EY. TLR/MyD88-mediated innate immunity in intestinal graft-versus-host disease. Immune Netw (2017) 17:144–51. doi:10.4110/in.2017.17.3.144
24. Koehn BH, Blazar BR. Role of myeloid-derived suppressor cells in allogeneic hematopoietic cell transplantation. J Leukoc Biol (2017) 102:335–41. doi:10.1189/jlb.5MR1116-464R
25. Wang D, Yu Y, Haarberg K, Fu J, Kaosaard K, Nagaraj S, et al. Dynamic change and impact of myeloid-derived suppressor cells in allogeneic bone marrow transplantation in mice. Biol Blood Marrow Transplant (2013) 19:692–702. doi:10.1016/j.bbmt.2013.01.008
26. Lv M, Zhao XS, Hu Y, Chang YJ, Zhao XY, Kong Y, et al. Monocytic and promyelocytic myeloid-derived suppressor cells may contribute to G-CSF-induced immune tolerance in haplo-identical allogeneic hematopoietic stem cell transplantation. Am J Hematol (2015) 90:E9–16. doi:10.1002/ajh.23865
27. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116:5738–47. doi:10.1182/blood-2010-06-287839
28. Wang Y, Gu X, Xiang J, Chen Z. Myeloid-derived suppressor cells participate in preventing graft rejection. Clin Dev Immunol (2012) 2012:731486. doi:10.1155/2012/731486
29. Drujont L, Carretero-Iglesia L, Bouchet-Delbos L, Beriou G, Merieau E, Hill M, et al. Evaluation of the therapeutic potential of bone marrow-derived myeloid suppressor cell (MDSC) adoptive transfer in mouse models of autoimmunity and allograft rejection. PLoS One (2014) 9:e100013. doi:10.1371/journal.pone.0100013
30. Imberti B, Monti M, Casiraghi F. Pluripotent stem cells and tolerance induction in organ transplantation. Curr Opin Organ Transplant (2015) 20:86–93. doi:10.1097/MOT.0000000000000144
31. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi:10.1038/nrc3581
32. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology (2015) 5:e1004983. doi:10.1080/2162402X.2015.1004983
33. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
34. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
35. Dorhoi A, Kaufmann SH. Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. Eur J Immunol (2015) 45:2191–202. doi:10.1002/eji.201545493
36. Diaz-Montero CM, Finke J, Montero AJ. Myeloid-derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol (2014) 41:174–84. doi:10.1053/j.seminoncol.2014.02.003
37. Chen W, Jiang J, Xia W, Huang J. Tumor-related exosomes contribute to tumor-promoting microenvironment: an immunological perspective. J Immunol Res (2017) 2017:1073947. doi:10.1155/2017/1073947
38. Nakamura T, Nakao T, Ashihara E, Yoshimura N. Myeloid-derived suppressor cells recruit CD4(+)/Foxp3(+) regulatory T cells in a murine cardiac allograft. Transplant Proc (2016) 48(4):1275–8. doi:10.1016/j.transproceed.2015.10.060
39. Goh C, Narayanan S, Hahn YS. Myeloid-derived suppressor cells: the dark knight or the joker in viral infections? Immunol Rev (2013) 255:210–21. doi:10.1111/imr.12084
40. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol (2011) 32:19–25. doi:10.1016/j.it.2010.10.002
41. Trikha P, Carson WE 3rd. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta (2014) 1846:55–65. doi:10.1016/j.bbcan.2014.04.003
42. Jayashankar L, Hafner R. Adjunct strategies for tuberculosis vaccines: modulating key immune cell regulatory mechanisms to potentiate vaccination. Front Immunol (2016) 7:577. doi:10.3389/fimmu.2016.00577
43. Camisaschi C, Vallacchi V, Vergani E, Tazzari M, Ferro S, Tuccitto A, et al. Targeting immune regulatory networks to counteract immune suppression in cancer. Vaccines (Basel) (2016) 4:4. doi:10.3390/vaccines4040038
44. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells – an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4:e954829. doi:10.4161/21624011.2014.954829
45. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res (2006) 66:9299–307. doi:10.1158/0008-5472.CAN-06-1690
46. Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res (2009) 69:2506–13. doi:10.1158/0008-5472.CAN-08-4323
47. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11:6713–21. doi:10.1158/1078-0432.CCR-05-0883
48. Talmadge JE, Hood KC, Zobel LC, Shafer LR, Coles M, Toth B. Chemoprevention by cyclooxygenase-2 inhibition reduces immature myeloid suppressor cell expansion. Int Immunopharmacol (2007) 7:140–51. doi:10.1016/j.intimp.2006.09.021
49. Wu T, Zhao Y, Zhao Y. The roles of myeloid-derived suppressor cells in transplantation. Expert Rev Clin Immunol (2014) 10:1385–94. doi:10.1586/1744666X.2014.948424
50. Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta (2012) 1820:940–8. doi:10.1016/j.bbagen.2012.03.017
51. Boukouris S, Mathivanan S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteomics Clin Appl (2015) 9:358–67. doi:10.1002/prca.201400114
52. Jia S, Zocco D, Samuels ML, Chou MF, Chammas R, Skog J, et al. Emerging technologies in extracellular vesicle-based molecular diagnostics. Expert Rev Mol Diagn (2014) 14:307–21. doi:10.1586/14737159.2014.893828
53. Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, Lötvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol (2007) 9:654–9. doi:10.1038/ncb1596
54. Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol (2015) 35:69–77. doi:10.1016/j.ceb.2015.04.013
55. Abd Elmageed ZY, Yang Y, Thomas R, Ranjan M, Mondal D, Moroz K, et al. Neoplastic reprogramming of patient-derived adipose stem cells by prostate cancer cell-associated exosomes. Stem Cells (2014) 32:983–97. doi:10.1002/stem.1619
56. Hao S, Ye Z, Li F, Meng Q, Qureshi M, Yang J, et al. Epigenetic transfer of metastatic activity by uptake of highly metastatic B16 melanoma cell-released exosomes. Exp Oncol (2006) 28:126–31.
57. Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res (2011) 71:5346–56. doi:10.1158/0008-5472.CAN-11-0241
58. Conlan RS, Pisano S, Oliveira MI, Ferrari M, Mendes Pinto I. Exosomes as reconfigurable therapeutic systems. Trends Mol Med (2017) 23:636–50. doi:10.1016/j.molmed.2017.05.003
59. Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta (2014) 1846:75–87. doi:10.1007/s10616-016-9952-7
60. Kotmakçı M, Bozok Çetintaş V. Extracellular vesicles as natural nanosized delivery systems for small-molecule drugs and genetic material: steps towards the future nanomedicines. J Pharm Pharm Sci (2015) 18:396–413. doi:10.18433/J36W3X
61. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi:10.1146/annurev-cellbio-101512-122326
62. vanNiel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, et al. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell (2011) 21:708–21. doi:10.1016/j.devcel.2011.08.019
63. Abels ER, Breakefield XO. Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol (2016) 36:301–12. doi:10.1007/s10571-016-0366-z
64. Kajimoto T, Okada T, Miya S, Zhang L, Nakamura S. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multi-vesicular endosomes. Nat Commun (2013) 4:2712. doi:10.1038/ncomms3712
65. Shen B, Wu N, Yang JM, Gould SJ. Protein targeting to exosomes/microvesicles by plasma membrane anchors. J Biol Chem (2011) 286:14383–95. doi:10.1074/jbc.M110.208660
66. Shen B, Fang Y, Wu N, Gould SJ. Biogenesis of the posterior pole is mediated by the exosome/microvesicle protein-sorting pathway. J Biol Chem (2011) 286:44162–76. doi:10.1074/jbc.M111.274803
67. Rana S, Claas C, Kretz CC, Nazarenko I, Zoeller M. Activation-induced internalization differs for the tetraspanins CD9 and Tspan8: impact on tumor cell motility. Int J Biochem Cell Biol (2011) 43:106–19. doi:10.1016/j.biocel.2010.10.002
68. Guo BB, Bellingham SA, Hill AF. The neutral sphingomyelinase pathway regulates packaging of the prion protein into exosomes. J Biol Chem (2015) 290:3455–67. doi:10.1074/jbc.M115.684258
69. Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun (2013) 4:2980. doi:10.1038/ncomms3980
70. Vedeler A, Hollås H, Grindheim AK, Raddum AM. Multiple roles of annexin A2 in post-transcriptional regulation of gene expression. Curr Protein Pept Sci (2012) 13:401–12. doi:10.2174/138920312801619402
71. Kogure T, Yan IK, Lin WL, Patel T. Extracellular vesicle-mediated transfer of a novel long noncoding RNA TUC339: a mechanism of intercellular signaling in human hepatocellular cancer. Genes Cancer (2013) 4:261–72. doi:10.1177/1947601913499020
72. Kelly EE, Horgan CP, Goud B, McCaffrey MW. The Rab family of proteins: 25 years on. Biochem Soc Trans (2012) 40:1337–47. doi:10.1042/BST20120203
73. Ji H, Greening DW, Barnes TW, Lim JW, Tauro BJ, Rai A, et al. Proteome profiling of exosomes derived from human primary and metastatic colorectal cancer cells reveal differential expression of key metastatic factors and signal transduction components. Proteomics (2013) 13:1672–86. doi:10.1002/pmic.201200562
74. Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A (2016) 113:E968–77. doi:10.1073/pnas.1521230113
75. Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res (2010) 51:2105–20. doi:10.1194/jlr.M003657
76. Record M, Carayon K, Poirot M, Stella A, Lambeau G, Paillasse M, et al. Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim Biophys Acta (2014) 1841:108–20. doi:10.1016/j.bbalip.2013.10.004
77. Llorente A, Skotland T, Sylvänne T, Kauhanen D, Róg T, Orłowski A, et al. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim Biophys Acta (2013) 1831:1302–9. doi:10.1016/j.bbalip.2013.04.011
78. Sharma R, Huang X, Brekken RA, Schroit AJ. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br J Cancer (2017) 117:545–52. doi:10.1038/bjc.2017.183
79. Lesur A, Domon B. Advances in high-resolution accurate mass spectrometry application to targeted proteomics. Proteomics (2015) 15:880–90. doi:10.1002/pmic.201400450
80. Schey KL, Luther JM, Rose KL. Proteomics characterization of exosome cargo. Methods (2015) 87:75–82. doi:10.1016/j.ymeth.2015.03.018
81. Zöller M. Tetraspanins: push and pull in suppressing and promoting metastasis. Nat Rev Cancer (2009) 9:40–55. doi:10.1038/nrc2543
82. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics (2010) 73:1907–20. doi:10.1016/j.jprot.2010.06.006
83. Choi DS, Yang JS, Choi EJ, Jang SC, Park S, Kim OY, et al. The protein interaction network of extracellular vesicles derived from human colorectal cancer cells. J Proteome Res (2012) 11:1144–51. doi:10.1021/pr200842h
84. Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol (2012) 44:1574–84. doi:10.1016/j.biocel.2012.06.018
85. Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol (2008) 10:619–24. doi:10.1038/ncb1725
86. Corcoran C, Rani S, O’Brien K, O’Neill A, Prencipe M, Sheikh R, et al. Docetaxel-resistance in prostate cancer: evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS One (2012) 7:e50999. doi:10.1371/journal.pone.0050999
87. Park JA, Sharif AS, Tschumperlin DJ, Lau L, Limbrey R, Howarth P, et al. Tissue factor-bearing exosome secretion from human mechanically stimulated bronchial epithelial cells in vitro and in vivo. J Allergy Clin Immunol (2012) 130:1375–80. doi:10.1016/j.jaci.2012.05.031
88. Demory Beckler M, Higginbotham JN, Franklin JL, Ham AJ, Halvey PJ, Imasuen IE, et al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol Cell Proteomics (2013) 12:343–55. doi:10.1074/mcp.M112.022806
89. Kumar D, Gupta D, Shankar S, Srivastava RK. Biomolecular characterization of exosomes released from cancer stem cells: possible implications for biomarker and treatment of cancer. Oncotarget (2015) 6:3280–91. doi:10.18632/oncotarget.2462
90. Zöller M. Exosomes in cancer disease. Methods Mol Biol (2016) 1381:111–49. doi:10.1007/978-1-4939-3204-7_7
91. Pinto R, De Summa S, Petriella D, Tudoran O, Danza K, Tommasi S. The value of new high-throughput technologies for diagnosis and prognosis in solid tumors. Cancer Biomark (2014) 14:103–17. doi:10.3233/CBM-130328
92. Iwakawa HO, Tomari Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol (2015) 25:651–65. doi:10.1016/j.tcb.2015.07.011
93. Sato-Kuwabara Y, Melo SA, Soares FA, Calin GA. The fusion of two worlds: non-coding RNAs and extracellular vesicles – diagnostic and therapeutic implications. Int J Oncol (2015) 46:17–27. doi:10.3892/ijo.2014.2712
94. Garg M. Targeting microRNAs in epithelial-to-mesenchymal transition-induced cancer stem cells: therapeutic approaches in cancer. Expert Opin Ther Targets (2015) 19:285–97. doi:10.1517/14728222.2014.975794
95. Garofalo M, Croce CM. Role of microRNAs in maintaining cancer stem cells. Adv Drug Deliv Rev (2015) 81C:53–61. doi:10.1016/j.addr.2014.11.014
96. Katoh M. Therapeutics targeting angiogenesis: genetics and epigenetics, extracellular miRNAs and signaling networks. Int J Mol Med (2013) 32:763–77. doi:10.3892/ijmm.2013.1444
97. Pan X, Wang ZX, Wang R. MicroRNA-21: a novel therapeutic target in human cancer. Cancer Biol Ther (2010) 10:1224–32. doi:10.4161/cbt.10.12.14252
98. Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med (2011) 17:211–5. doi:10.7554/eLife.06434
99. Wu ZS, Wu Q, Wang CQ, Wang XN, Huang J, Zhao JJ, et al. miR-340 inhibition of breast cancer cell migration and invasion through targeting of oncoprotein c-Met. Cancer (2011) 117:2842–52. doi:10.1002/cncr.25860
100. Simpson LJ, Ansel KM. MicroRNA regulation of lymphocyte tolerance and autoimmunity. J Clin Invest (2015) 125:2242–9. doi:10.1172/JCI78090
101. Zhu S, Pan W, Qian Y. MicroRNA in immunity and autoimmunity. J Mol Med (Berl) (2013) 91:1039–50. doi:10.1007/s00109-013-1043-z
102. Singh RP, Massachi I, Manickavel S, Singh S, Rao NP, Hasan S, et al. The role of miRNA in inflammation and autoimmunity. Autoimmun Rev (2013) 12:1160–5. doi:10.1016/j.autrev.2013.07.003
103. O’Carroll D, Mecklenbrauker I, Das PP, Santana A, Koenig U, Enright AJ, et al. A slicer independent role for argonaute 2 in hematopoiesis and the microRNA pathway. Genes Dev (2007) 21:1999–2004. doi:10.1101/gad.1565607
104. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med (2005) 201:1367–73. doi:10.1084/jem.20050572
105. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med (2005) 202:261–9. doi:10.1084/jem.20050678
106. Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell (2008) 132:860–74. doi:10.1016/j.cell.2008.02.020
107. Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med (2008) 205:2005–17. doi:10.1084/jem.20081219
108. Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med (2008) 205:1993–2004. doi:10.1084/jem.20081062
109. Goh W, Huntington ND. Regulation of murine natural killer cell development. Front Immunol (2017) 8:130. doi:10.3389/fimmu.2017.00130
110. Liu M, Zhou J, Chen Z, Cheng AS. Understanding the epigenetic regulation of tumours and their microenvironments: opportunities and problems for epigenetic therapy. J Pathol (2017) 241:10–24. doi:10.1002/path.4832
111. Ma H, Guo S, Luo Y, Wang Y, Wang H, He J, et al. MicroRNA-20b promotes the accumulation of CD11b+Ly6G+Ly6Clow myeloid-derived suppressor cells in asthmatic mice. Cent Eur J Immunol (2017) 42:30–8. doi:10.5114/ceji.2017.67316
112. Cantoni C, Cignarella F, Ghezzi L, Mikesell B, Bollman B, Berrien-Elliott MM, et al. miR-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol (2017) 133:61–77. doi:10.1007/s00401-016-1621-6
113. Silva A, Bullock M, Calin G. The clinical relevance of long non-coding RNAs in cancer. Cancers (Basel) (2015) 7:2169–82. doi:10.3390/cancers7040884
114. Zhang R, Xia Y, Wang Z, Zheng J, Chen Y, Li X, et al. Serum long non coding RNA MALAT-1 protected by exosomes is up-regulated and promotes cell proliferation and migration in non-small cell lung cancer. Biochem Biophys Res Commun (2017) 490:406–14. doi:10.1016/j.bbrc.2017.06.055
115. Lang HL, Hu GW, Chen Y, Liu Y, Tu W, Lu YM, et al. Glioma cells promote angiogenesis through the release of exosomes containing long non-coding RNA POU3F3. Eur Rev Med Pharmacol Sci (2017) 21:959–72.
116. Pan L, Liang W, Fu M, Huang ZH, Li X, Zhang W, et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J Cancer Res Clin Oncol (2017) 143:991–1004. doi:10.1007/s00432-017-2361-2
117. Deng H, Zhang J, Shi J, Guo Z, He C, Ding L, et al. Role of long non-coding RNA in tumor drug resistance. Tumour Biol (2016) 37:11623–31. doi:10.1007/s13277-016-5125-8
118. Chen M, Xu R, Ji H, Greening DW, Rai A, Izumikawa K, et al. Transcriptome and long noncoding RNA sequencing of three extracellular vesicle subtypes released from the human colon cancer LIM1863 cell line. Sci Rep (2016) 6:38397. doi:10.1038/srep38397
119. Chen L, Yang W, Guo Y, Chen W, Zheng P, Zeng J, et al. Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS One (2017) 12:e0185406. doi:10.1371/journal.pone.0185406
120. Carpenter S. Long noncoding RNA: novel links between gene expression and innate immunity. Virus Res (2016) 212:137–45. doi:10.1016/j.virusres.2015.08.019
121. Satpathy AT, Chang HY. Long noncoding RNA in hematopoiesis and immunity. Immunity (2015) 42:792–804. doi:10.1016/j.immuni.2015.05.004
122. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles (2014) 3:1–14. doi:10.3402/jev.v3.24641
123. Mu W, Rana S, Zöller M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia (2013) 15:875–87. doi:10.1593/neo.13786
124. Sung BH, Ketova T, Hoshino D, Zijlstra A, Weaver AM. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun (2015) 6:7164. doi:10.1038/ncomms8164
125. Lai CP, Mardini O, Ericsson M, Prabhakar S, Maguire C, Chen JW, et al. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano (2014) 8:483–94. doi:10.1021/nn404945r
126. Wiklander OP, Nordin JZ, O’Loughlin A, Gustafsson Y, Corso G, Mäger I, et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles (2015) 4:26316. doi:10.3402/jev.v4.26316
127. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature (2015) 527:329–35. doi:10.1038/nature15756
128. Nolte-’t Hoen EN, Buschow SI, Anderton SM, Stoorvogel W, Wauben MH. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood (2009) 113:1977–81. doi:10.1182/blood-2008-08-174094
129. Segura E, Guérin C, Hogg N, Amigorena S, Théry C. CD8+ dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in vivo. J Immunol (2007) 179:1489–96. doi:10.4049/jimmunol.179.3.1489
130. Gesierich S, Berezovskiy I, Ryschich E, Zöller M. Systemic induction of the angiogenesis switch by the tetraspanin D6.1A/CO-029. Cancer Res (2006) 66:7083–94. doi:10.1158/0008-5472.CAN-06-0391
131. Christianson HC, Svensson KJ, van Kuppevelt TH, Li JP, Belting M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc Natl Acad Sci U S A (2013) 110:17380–5. doi:10.1073/pnas.1304266110
132. Fei F, Joo EJ, Tarighat SS, Schiffer I, Paz H, Fabbri M, et al. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through galectin-3. Oncotarget (2015) 6:11378–94. doi:10.18632/oncotarget.3409
133. Gomes J, Gomes-Alves P, Carvalho SB, Peixoto C, Alves PM, Altevogt P, et al. Extracellular vesicles from ovarian carcinoma cells display specific glycosignatures. Biomolecules (2015) 5:1741–61. doi:10.3390/biom5031741
134. Saunderson SC, Dunn AC, Crocker PR, McLellan AD. CD169 mediates the capture of exosomes in spleen and lymph node. Blood (2014) 123:208–16. doi:10.1182/blood-2013-03-489732
135. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood (2012) 119:756–66. doi:10.1182/blood-2011-02-338004
136. Del Conde I, Shrimpton CN, Thiagarajan P, López JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood (2005) 106:1604–11. doi:10.1182/blood-2004-03-1095
137. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood (2004) 104:3257–66. doi:10.1182/blood-2004-03-0824
138. Tian T, Zhu YL, Hu FH, Wang YY, Huang NP, Xiao ZD. Dynamics of exosome internalization and trafficking. J Cell Physiol (2013) 228:1487–95. doi:10.1002/jcp.24304
139. Frühbeis C, Fröhlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, et al. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol (2013) 11:e1001604. doi:10.1371/journal.pbio.1001604
140. Feng D, Zhao WL, Ye YY, Bai XC, Liu RQ, Chang LF, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic (2010) 11:675–87. doi:10.1111/j.1600-0854.2010.01041.x
141. Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev (2014) 262:193–215. doi:10.1111/imr.12212
142. Lim JP, Gleeson PA. Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol (2011) 89:836–43. doi:10.1038/icb.2011.20
143. Nakase I, Kobayashi NB, Takatani-Nakase T, Yoshida T. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep (2015) 5:10300. doi:10.1038/srep10300
144. Thuma F, Zöller M. Outsmart tumor exosomes to steal the cancer initiating cell its niche. Semin Cancer Biol (2014) 28:39–50. doi:10.1016/j.semcancer.2014.02.011
145. Nanbo A, Kawanishi E, Yoshida R, Yoshiyama H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J Virol (2013) 87:10334–47. doi:10.1128/JVI.01310-13
146. Buschow SI, Nolte-’t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic (2009) 10:1528–42. doi:10.1111/j.1600-0854.2009.00963
147. Lakkaraju A, Rodriguez-Boulan E. Itinerant exosomes: emerging roles in cell and tissue polarity. Trends Cell Biol (2008) 18:199–209. doi:10.1016/j.tcb.2008.03.002
148. Nakayama M. Antigen presentation by MHC-dressed cells. Front Immunol (2015) 5:672. doi:10.3389/fimmu.2014.00672
149. Leone DA, Peschel A, Brown M, Schachner H, Ball MJ, Gyuraszova M, et al. Surface LAMP-2 is an endocytic receptor that diverts antigen internalized by human dendritic cells into highly immunogenic exosomes. J Immunol (2017) 199:531–46. doi:10.4049/jimmunol.1601263
150. Holder B, Jones T, Sancho Shimizu V, Rice TF, Donaldson B, Bouqueau M, et al. Macrophage exosomes induce placental inflammatory cytokines: a novel mode of maternal-placental messaging. Traffic (2016) 17:168–78. doi:10.1111/tra.12352
151. Heusermann W, Hean J, Trojer D, Steib E, von Bueren S, Graff-Meyer A, et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J Cell Biol (2016) 213:173–84. doi:10.1083/jcb.201506084
152. Fedele C, Singh A, Zerlanko BJ, Iozzo RV, Languino LR. The αvβ6 integrin is transferred intercellularly via exosomes. J Biol Chem (2015) 290:4545–51. doi:10.1074/jbc.C114.617662
153. Gu X, Erb U, Büchler MW, Zöller M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int J Cancer (2015) 136:E74–84. doi:10.1002/ijc.29100
154. Lamichhane TN, Jeyaram A, Patel DB, Parajuli B, Livingston NK, Arumugasaamy N, et al. Oncogene knockdown via active loading of small RNAs into extracellular vesicles by sonication. Cell Mol Bioeng (2016) 9:315–24. doi:10.1007/s12195-016-0457-4
155. Saari H, Lázaro-Ibáñez E, Viitala T, Vuorimaa-Laukkanen E, Siljander P, Yliperttula M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of paclitaxel in autologous prostate cancer cells. J Control Release (2015) 220(Pt B):727–37. doi:10.1016/j.jconrel.2015.09.031
156. Kapustin AN, Schoppet M, Schurgers LJ, Reynolds JL, McNair R, Heiss A, et al. Prothrombin loading of vascular smooth muscle cell-derived exosomes regulates coagulation and calcification. Arterioscler Thromb Vasc Biol (2017) 37:e22–32. doi:10.1161/ATVBAHA.116.308886
157. Théry C, Duban L, Segura E, Véron P, Lantz O, Amigorena S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat Immunol (2002) 3:1156–62. doi:10.1038/ni854
158. Matsumoto K, Morisaki T, Kuroki H, Kubo M, Onishi H, Nakamura K, et al. Exosomes secreted from monocyte-derived dendritic cells support in vitro naive CD4+ T cell survival through NF-(kappa)B activation. Cell Immunol (2004) 231:20–9. doi:10.1016/j.cellimm.2004.11.002
159. Burrello J, Monticone S, Gai C, Gomez Y, Kholia S, Camussi G. Stem cell-derived extracellular vesicles and immune-modulation. Front Cell Dev Biol (2016) 4:83. doi:10.3389/fcell.2016.00083
160. Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res (2005) 65:5238–47. doi:10.1158/0008-5472.CAN-04-3804
161. Simhadri VR, Reiners KS, Hansen HP, Topolar D, Simhadri VL, Nohroudi K, et al. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS One (2008) 3:e3377. doi:10.1371/journal.pone.0003377
162. Viaud S, Terme M, Flament C, Taieb J, André F, Novault S, et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: a role for NKG2D ligands and IL-15Ralpha. PLoS One (2009) 4(3):e4942. doi:10.1371/journal.pone.0004942
163. Vulpis E, Cecere F, Molfetta R, Soriani A, Fionda C, Peruzzi G, et al. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: role of HSP70/TLR2/NF-kB axis. Oncoimmunology (2017) 6:e1279372. doi:10.1080/2162402X.2017.1279372
164. Korkut C, Ataman B, Ramachandran P, Ashley J, Barria R, Gherbesi N, et al. Trans-synaptic transmission of vesicular Wnt signals through Evi/Wntless. Cell (2009) 139:393–404. doi:10.1016/j.cell.2009.07.051
165. Korkut C, Li Y, Koles K, Brewer C, Ashley J, Yoshihara M, et al. Regulation of postsynaptic retrograde signaling by presynaptic exosome release. Neuron (2013) 77:1039–46. doi:10.1016/j.neuron.2013.01.013
166. Budnik V, Ruiz-Cañada C, Wendler F. Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci (2016) 17:160–72. doi:10.1038/nrn.2015.29
167. Gong J, Körner R, Gaitanos L, Klein R. Exosomes mediate cell contact-independent ephrin-Eph signaling during axon guidance. J Cell Biol (2016) 214:35–44. doi:10.1083/jcb.201601085
168. Takahashi Y, Nishikawa M, Shinotsuka H, Matsui Y, Ohara S, Imai T, et al. Visualization and in vivo tracking of the exosomes of murine melanoma B16-BL6 cells in mice after intravenous injection. J Biotechnol (2013) 165:77–84. doi:10.1016/j.jbiotec.2013.03.013
169. Erb U, Zhao K, Wang Z, Xiao L, Zöller M. Murine and human pancreatic tumor exosome recovery in mouse serum: diagnostic and prognostic potential and target cell delivery. Cancer Lett (2017) 403:1–12. doi:10.1016/j.canlet.2017.06.005
170. Choi H, Lee DS. Illuminating the physiology of extracellular vesicles. Stem Cell Res Ther (2016) 7:55. doi:10.1186/s13287-016-0316-1
171. Hyenne V, Lefebvre O, Goetz JG. Going live with tumor exosomes and microvesicles. Cell Adh Migr (2017) 11:173–86. doi:10.1080/19336918.2016.1276694
172. Di Rocco G, Baldari S, Toietta G. Towards therapeutic delivery of extracellular vesicles: strategies for in vivo tracking and biodistribution analysis. Stem Cells Int (2016) 2016:5029619. doi:10.1155/2016/5029619
173. Betzer O, Perets N, Angel A, Motiei M, Sadan T, Yadid G, et al. In vivo neuroimaging of exosomes using gold nanoparticles. ACS Nano (2017) 11:10883–93. doi:10.1021/acsnano.7b04495
174. Xitong D, Xiaorong Z. Targeted therapeutic delivery using engineered exosomes and its applications in cardiovascular diseases. Gene (2016) 575(2 Pt 2):377–84. doi:10.1016/j.gene.2015.08.067
175. Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release (2015) 219:396–405. doi:10.1016/j.jconrel.2015.07.030
176. Wang J, Li W, Lu Z, Zhang L, Hu Y, Li Q, et al. The use of RGD-engineered exosomes for enhanced targeting ability and synergistic therapy toward angiogenesis. Nanoscale (2017) 9:15598–605. doi:10.1039/c7nr04425a
177. Tian T, Zhang HX, He CP, Fan S, Zhu YL, Qi C, et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials (2018) 150:137–49. doi:10.1016/j.biomaterials.2017.10.012
178. Kim MS, Haney MJ, Zhao Y, Yuan D, Deygen I, Klyachko NL, et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine (2018) 14:195–204. doi:10.1016/j.nano.2017.09.011
179. Kim OY, Lee J, Gho YS. Extracellular vesicle mimetics: novel alternatives to extracellular vesicle-based theranostics, drug delivery, and vaccines. Semin Cell Dev Biol (2017) 67:74–82. doi:10.1016/j.semcdb.2016.12.001
180. Burke M, Choksawangkarn W, Edwards N, Ostrand-Rosenberg S, Fenselau C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J Proteome Res (2014) 13:836–43. doi:10.1021/pr400879c
181. Tanno H, Komada M. The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem (2013) 153:497–504. doi:10.1093/jb/mvt028
182. Burke MC, Oei MS, Edwards NJ, Ostrand-Rosenberg S, Fenselau C. Ubiquitinated proteins in exosomes secreted by myeloid-derived suppressor cells. J Proteome Res (2014) 13:5965–72. doi:10.1021/pr500854x
183. Adams KR, Chauhan S, Patel DB, Clements VK, Wang Y, Jay SM, et al. Ubiquitin conjugation probed by inflammation in myeloid-derived suppressor cell extracellular vesicles. J Proteome Res (2018) 17:315–24. doi:10.1021/acs.jproteome.7b00585
184. Geis-Asteggiante L, Dhabaria A, Edwards N, Ostrand-Rosenberg S, Fenselau C. Top-down analysis of low mass proteins in exosomes shed by murine myeloid-derived suppressor cells. Int J Mass Spectrom (2015) 378:264–9. doi:10.1016/j.ijms.2014.08.035
185. Chauhan S, Danielson S, Clements V, Edwards N, Ostrand-Rosenberg S, Fenselau C. Surface glycoproteins of exosomes shed by myeloid-derived suppressor cells contribute to function. J Proteome Res (2017) 16:238–46. doi:10.1021/acs.jproteome.6b00811
186. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi:10.4049/jimmunol.181.7.4666
187. Malik P, Chaudhry N, Mittal R, Mukherjee TK. Role of receptor for advanced glycation end products in the complication and progression of various types of cancers. Biochim Biophys Acta (2015) 1850:1898–904. doi:10.1016/j.bbagen.2015.05.020
188. El Gazzar M. MicroRNAs as potential regulators of myeloid-derived suppressor cell expansion. Innate Immun (2014) 20:227–38. doi:10.1177/1753425913489850
189. Zhang C, Wang S, Liu Y, Yang C. Epigenetics in myeloid derived suppressor cells: a sheathed sword towards cancer. Oncotarget (2016) 7:57452–63. doi:10.18632/oncotarget.10767
190. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, et al. MicroRNA-155 and microRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol (2014) 192:1034–43. doi:10.4049/jimmunol.1301309
191. McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, et al. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun (2014) 82:3816–25. doi:10.1128/IAI.01495-14
192. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 regulates the differentiation and function of myeloid-derived suppressor cells via targeting Runx1. J Immunol (2015) 195:1301–11. doi:10.4049/jimmunol.1500209
193. Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene (2017) 36:639–51. doi:10.1038/onc.2016.229
194. Xiang X, Poliakov A, Liu C, Liu Y, Deng ZB, Wang J, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer (2009) 124:2621–33. doi:10.1002/ijc.24249
195. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest (2010) 120:457–71. doi:10.1172/JCI40483
196. Liu Y, Xiang X, Zhuang X, Zhang S, Liu C, Cheng Z, et al. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol (2010) 176:2490–9. doi:10.2353/ajpath.2010.090777
197. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, et al. Multiple myeloma exosomes establish a favourable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol (2016) 239:162–73. doi:10.1002/path.4712
198. Wen SW, Sceneay J, Lima LG, Wong CS, Becker M, Krumeich S, et al. The biodistribution and immune suppressive effects of breast cancer-derived exosomes. Cancer Res (2016) 76:6816–27. doi:10.1158/0008-5472.CAN-16-0868
199. Wang J, De Veirman K, De Beule N, Maes K, De Bruyne E, Van Valckenborgh E, et al. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget (2015) 6:43992–4004. doi:10.18632/oncotarget.6083
200. Bruschi M, Petretto A, Bertelli R, Galetti M, Bonanni A, Pratesi F, et al. Post-translational modified proteins are biomarkers of autoimmune-processes: NETosis and the inflammatory-autoimmunity connection. Clin Chim Acta (2017) 464:12–6. doi:10.1016/j.cca.2016.11.006
201. Yang R, Zou X, Tenhunen J, Tønnessen TI. HMGB1 and extracellular histones significantly contribute to systemic inflammation and multiple organ failure in acute liver failure. Mediators Inflamm (2017) 2017:5928078. doi:10.1155/2017/5928078
202. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) (2014) 92:465–72. doi:10.1007/s00109-014-1148-z
203. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol (2012) 8:195–202. doi:10.1038/nrrheum.2011.222
204. Parker KH, Sinha P, Horn LA, Clements VK, Yang H, Li J, et al. HMGB1 enhances immune suppression by facilitating the differentiation and suppressive activity of myeloid-derived suppressor cells. Cancer Res (2014) 74:5723–33. doi:10.1158/0008-5472.CAN-13-2347
205. Su Z, Ni P, She P, Liu Y, Richard SA, Xu W, et al. Bio-HMGB1 from breast cancer contributes to M-MDSC differentiation from bone marrow progenitor cells and facilitates conversion of monocytes into MDSC-like cells. Cancer Immunol Immunother (2017) 66:391–401. doi:10.1007/s00262-016-1942-2
206. Lai RC, Yeo RW, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol (2015) 40:82–8. doi:10.1016/j.semcdb.2015.03.001
207. Ohno S, Drummen GP, Kuroda M. Focus on extracellular vesicles: development of extracellular vesicle-based therapeutic systems. Int J Mol Sci (2016) 17:172. doi:10.3390/ijms17020172
208. Schupp J, Krebs FK, Zimmer N, Trzeciak E, Schuppan D, Tuettenberg A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell Immunol (2017). doi:10.1016/j.cellimm.2017.10.013
209. Di Mitri D, Toso A, Alimonti A. Molecular pathways: targeting tumor-infiltrating myeloid-derived suppressor cells for cancer therapy. Clin Cancer Res (2015) 21:3108–12. doi:10.1158/1078-0432.CCR-14-2261
210. Albeituni SH, Ding C, Yan J. Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J (2013) 19:490–501. doi:10.1097/PPO.0000000000000006
211. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67:11021–8. doi:10.1158/0008-5472.CAN-07-2593
212. Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res (2008) 14:8270–8. doi:10.1158/1078-0432.CCR-08-0165
213. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother (2013) 62:909–18. doi:10.1007/s00262-013-1396-8
214. Lei GS, Zhang C, Shao S, Jung HW, Durant PJ, Lee CH. All-trans retinoic acid in combination with primaquine clears pneumocystis infection. PLoS One (2013) 8:e53479. doi:10.1371/journal.pone.0053479
215. Sasso MS, Lollo G, Pitorre M, Solito S, Pinton L, Valpione S, et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials (2016) 96:47–62. doi:10.1016/j.biomaterials.2016.04.010
216. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15:2148–57. doi:10.1158/1078-0432.CCR-08-1332
217. Guislain A, Gadiot J, Kaiser A, Jordanova ES, Broeks A, Sanders J, et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol Immunother (2015) 64:1241–50. doi:10.1007/s00262-015-1735-z
218. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology (2015) 4:e989764. doi:10.4161/2162402X.2014.989764
219. Song X, Guo W, Cui J, Qian X, Yi L, Chang M, et al. A tritherapy combination of a fusion protein vaccine with immune-modulating doses of sequential chemotherapies in an optimized regimen completely eradicates large tumors in mice. Int J Cancer (2011) 128:1129–38. doi:10.1002/ijc.25451
220. Chen HM, Ma G, Gildener-Leapman N, Eisenstein S, Coakley BA, Ozao J, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res (2015) 21:4073–85. doi:10.1158/1078-0432.CCR-14-2742
221. Nishida-Aoki N, Tominaga N, Takeshita F, Sonoda H, Yoshioka Y, Ochiya T. Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol Ther (2017) 25:181–91. doi:10.1016/j.ymthe.2016.10.009
222. Wang H, Rana S, Giese N, Büchler MW, Zöller M. Tspan8, CD44v6 and alpha6beta4 are biomarkers of migrating pancreatic cancer-initiating cells. Int J Cancer (2013) 133:416–26. doi:10.1002/ijc.28044
223. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver’s seat. Nat Rev Cancer (2017) 17:577–93. doi:10.1038/nrc.2017.77
224. Numico G, Fusco V, Franco P, Roila F. Proton pump inhibitors in cancer patients: how useful they are? A review of the most common indications for their use. Crit Rev Oncol Hematol (2017) 111:144–51. doi:10.1016/j.critrevonc.2017.01.014
225. Eisenblaetter M, Flores-Borja F, Lee JJ, Wefers C, Smith H, Hueting R, et al. Visualization of tumor-immune interaction – target-specific imaging of S100A8/A9 reveals pre-metastatic niche establishment. Theranostics (2017) 7:2392–401. doi:10.7150/thno.17138
226. Marleau AM, Chen CS, Joyce JA, Tullis RH. Exosome removal as a therapeutic adjuvant in cancer. J Transl Med (2012) 10:134. doi:10.1186/1479-5876-10-134
227. Muller L, Hong CS, Stolz DB, Watkins SC, Whiteside TL. Isolation of biologically-active exosomes from human plasma. J Immunol Methods (2014) 411:55–65. doi:10.1016/j.jim.2014.06.007
228. Yim N, Ryu SW, Choi K, Lee KR, Lee S, Choi H, et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat Commun (2016) 7:12277. doi:10.1038/ncomms12277
229. El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, et al. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc (2012) 7:2112–26. doi:10.1038/nprot.2012.131
230. Yuyama K, Sun H, Sakai S, Mitsutake S, Okada M, Tahara H, et al. Decreased amyloid-β pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J Biol Chem (2014) 289:24488–98. doi:10.1074/jbc.M114.577213
231. Srikanthan S, Li W, Silverstein RL, McIntyre TM. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro-atherothombotic cellular functions. J Thromb Haemost (2014) 12:1906–17. doi:10.1111/jth.12712
232. Lai RC, Yeo RW, Tan KH, Lim SK. Exosomes for drug delivery – a novel application for the mesenchymal stem cell. Biotechnol Adv (2013) 31:543–51. doi:10.1016/j.biotechadv.2012.08.008
233. Wang J, Yu F, Jia X, Iwanowycz S, Wang Y, Huang S, et al. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int J Cancer (2015) 136:E602–13. doi:10.1002/ijc.29151
234. Torres Andón F, Alonso MJ. Nanomedicine and cancer immunotherapy – targeting immunosuppressive cells. J Drug Target (2015) 23:656–71. doi:10.3109/1061186X.2015.1073295
235. Da Silva CG, Rueda F, Löwik CW, Ossendorp F, Cruz LJ. Combinatorial prospects of nano-targeted chemoimmunotherapy. Biomaterials (2016) 83:308–20. doi:10.1016/j.biomaterials.2016.01.006
236. Klauber TCB, Laursen JM, Zucker D, Brix S, Jensen SS, Andresen TL. Delivery of TLR7 agonist to monocytes and dendritic cells by DCIR targeted liposomes induces robust production of anti-cancer cytokines. Acta Biomater (2017) 53:367–77. doi:10.1016/j.actbio.2017.01.072
237. Ban Y, Mai J, Li X, Mitchell-Flack M, Zhang T, Zhang L, et al. Targeting autocrine CCL5-CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res (2017) 77:2857–68. doi:10.1158/0008-5472.CAN-16-2913
238. Nakano I, Garnier D, Minata M, Rak J. Extracellular vesicles in the biology of brain tumour stem cells – implications for inter-cellular communication, therapy and biomarker development. Semin Cell Dev Biol (2015) 40:17–26. doi:10.1016/j.semcdb.2015.02.011
239. Zeng AL, Yan W, Liu YW, Wang Z, Hu Q, Nie E, et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene (2017) 36:5369–81. doi:10.1038/onc.2017.134
240. Nagy G, Huszthy PC, Fossum E, Konttinen Y, Nakken B, Szodoray P. Selected aspects in the pathogenesis of autoimmune diseases. Mediators Inflamm (2015) 2015:351732. doi:10.1155/2015/351732
241. Lee JH, Wang LC, Lin YT, Yang YH, Lin DT, Chiang BL. Inverse correlation between CD4+ regulatory T-cell population and autoantibody levels in paediatric patients with systemic lupus erythematosus. Immunology (2006) 117:280–6. doi:10.1111/j.1365-2567.2005.02306.x
242. Azizi G, Pouyani MR, Abolhassani H, Sharifi L, Dizaji MZ, Mohammadi J, et al. Cellular and molecular mechanisms of immune dysregulation and autoimmunity. Cell Immunol (2016) 310:14–26. doi:10.1016/j.cellimm.2016.08.012
243. Papp G, Horvath IF, Barath S, Gyimesi E, Vegh J, Szodoray P, et al. Immunomodulatory effects of extracorporeal photochemotherapy in systemic sclerosis. Clin Immunol (2012) 142:150–9. doi:10.1016/j.clim.2011.09.014
244. Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS. Experimental autoimmune encephalomyelitis (EAE) in CCR2(-/-) mice: susceptibility in multiple strains. Am J Pathol (2003) 162:139–50. doi:10.1016/S0002-9440(10)63805-9
245. Cripps JG, Gorham JD. MDSC in autoimmunity. Int Immunopharmacol (2011) 11(7):789–93. doi:10.1016/j.intimp.2011.01.026
246. Liberal R, Grant CR, Longhi MS, Mieli-Vergani G, Vergani D. Regulatory T cells: mechanisms of suppression and impairment in autoimmune liver disease. IUBMB Life (2015) 67:88–97. doi:10.1002/iub.1349
247. Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin Ther Targets (2015) 19:1091–103. doi:10.1517/14728222.2015.1037282
248. Iborra M, Bernuzzi F, Invernizzi P, Danese S. MicroRNAs in autoimmunity and inflammatory bowel disease: crucial regulators in immune response. Autoimmun Rev (2012) 11:305–14. doi:10.1016/j.autrev.2010.07.002
249. Chen JQ, Papp G, Szodoray P, Zeher M. The role of microRNAs in the pathogenesis of autoimmune diseases. Autoimmun Rev (2016) 15:1171–80. doi:10.1016/j.autrev.2016.09.003
250. Gysler SM, Mulla MJ, Guerra M, Brosens JJ, Salmon JE, Chamley LW, et al. Antiphospholipid antibody-induced miR-146a-3p drives trophoblast interleukin-8 secretion through activation of toll-like receptor 8. Mol Hum Reprod (2016) 22:465–74. doi:10.1093/molehr/gaw027
251. Seddiki N, Brezar V, Draenert R. Cell exhaustion in HIV-1 infection: role of suppressor cells. Curr Opin HIV AIDS (2014) 9:452–8. doi:10.1097/COH.0000000000000087
252. Ish-Shalom E, Meirow Y, Sade-Feldman M, Kanterman J, Wang L, Mizrahi O, et al. Impaired SNX9 expression in immune cells during chronic inflammation: prognostic and diagnostic implications. J Immunol (2016) 196:156–67. doi:10.4049/jimmunol.1402877
253. Finlay CM, Walsh KP, Mills KH. Induction of regulatory cells by helminth parasites: exploitation for the treatment of inflammatory diseases. Immunol Rev (2014) 259:206–30. doi:10.1111/imr.12164
254. Weinstock JV, Elliott DE. Helminth infections decrease host susceptibility to immune-mediated diseases. J Immunol (2014) 193:3239–47. doi:10.4049/jimmunol.1400927
255. Besnard AG, Guabiraba R, Niedbala W, Palomo J, Reverchon F, Shaw TN, et al. IL-33-mediated protection against experimental cerebral malaria is linked to induction of type 2 innate lymphoid cells, M2 macrophages and regulatory T cells. PLoS Pathog (2015) 11:e1004607. doi:10.1371/journal.ppat.1004607
256. Ren JP, Zhao J, Dai J, Griffin JW, Wang L, Wu XY, et al. Hepatitis C virus-induced myeloid-derived suppressor cells regulate T-cell differentiation and function via the signal transducer and activator of transcription 3 pathway. Immunology (2016) 148:377–86. doi:10.1111/imm.12616
257. Wang L, Zhao J, Ren JP, Wu XY, Morrison ZD, Elgazzar MA, et al. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. AIDS (2016) 30:1521–31. doi:10.1097/QAD.0000000000001083
258. Knaul JK, Jörg S, Oberbeck-Mueller D, Heinemann E, Scheuermann L, Brinkmann V, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med (2014) 190:1053–66. doi:10.1164/rccm.201405-0828OC
259. Hill ND, Bunata K, Hebert AA. Treatment of alopecia areata with squaric acid dibutylester. Clin Dermatol (2015) 33:300–4. doi:10.1016/j.clindermatol.2014.12.001
260. Yoshimasu T, Furukawa F. Modified immunotherapy for alopecia areata. Autoimmun Rev (2016) 15:664–7. doi:10.1016/j.autrev.2016.02.021
261. Singh V, Mueller U, Freyschmidt-Paul P, Zöller M. Delayed type hypersensitivity-induced myeloid-derived suppressor cells regulate autoreactive T cells. Eur J Immunol (2011) 41:2871–82. doi:10.1002/eji.201141696
262. Singh V, Erb U, Zöller M. Cooperativity of CD44 and CD49d in leukemia cell homing, migration, and survival offers a means for therapeutic attack. J Immunol (2013) 191:5304–16. doi:10.4049/jimmunol.1301543
263. Peón AN, Ledesma-Soto Y, Olguín JE, Bautista-Donis M, Sciutto E, Terrazas LI. Helminth products potently modulate experimental autoimmune encephalomyelitis by downregulating neuroinflammation and promoting a suppressive microenvironment. Mediators Inflamm (2017) 2017:8494572. doi:10.1155/2017/8494572
264. Lei A, Yang Q, Li X, Chen H, Shi M, Xiao Q, et al. Atorvastatin promotes the expansion of myeloid-derived suppressor cells and attenuates murine colitis. Immunology (2016) 149:432–46. doi:10.1111/imm.12662
265. Zhang D, Tu E, Kasagi S, Zanvit P, Chen Q, Chen W. Manipulating regulatory T cells: a promising strategy to treat autoimmunity. Immunotherapy (2015) 7:1201–11. doi:10.2217/imt.15.79
266. Dai YD, Sheng H, Dias P, Jubayer Rahman M, Bashratyan R, Regn D, et al. Autoimmune responses to exosomes and candidate antigens contribute to type 1 diabetes in non-obese diabetic mice. Curr Diab Rep (2017) 17:130. doi:10.1007/s11892-017-0962-4
267. Bai L, Shao H, Wang H, Zhang Z, Su C, Dong L, et al. Effects of mesenchymal stem cell-derived exosomes on experimental autoimmune uveitis. Sci Rep (2017) 7:4323. doi:10.1038/s41598-017-04559-y
268. Yin W, Ouyang S, Luo Z, Zeng Q, Hu B, Xu L, et al. Immature exosomes derived from microRNA-146a overexpressing dendritic cells act as antigen-specific therapy for myasthenia gravis. Inflammation (2017) 40:1460–73. doi:10.1007/s10753-017-0589-2
269. Turpin D, Truchetet ME, Faustin B, Augusto JF, Contin-Bordes C, Brisson A, et al. Role of extracellular vesicles in autoimmune diseases. Autoimmun Rev (2016) 15:174–83. doi:10.1016/j.autrev.2015.11.004
270. Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest (2016) 126:1173–80. doi:10.1172/JCI81131
271. Tan L, Wu H, Liu Y, Zhao M, Li D, Lu Q. Recent advances of exosomes in immune modulation and autoimmune diseases. Autoimmunity (2016) 49:357–65. doi:10.1080/08916934.2016.1191477
272. Kouwaki T, Fukushima Y, Daito T, Sanada T, Yamamoto N, Mifsud EJ, et al. Extracellular vesicles including exosomes regulate innate immune responses to hepatitis B virus infection. Front Immunol (2016) 7:335. doi:10.3389/fimmu.2016.00335
273. Wang Y, Tian J, Tang X, Rui K, Tian X, Ma J, et al. Exosomes released by granulocytic myeloid-derived suppressor cells attenuate DSS-induced colitis in mice. Oncotarget (2016) 7:15356–68. doi:10.18632/oncotarget.7324
274. Marhaba R, Vitacolonna M, Hildebrand D, Baniyash M, Freyschmidt-Paul P, Zöller M. The importance of myeloid-derived suppressor cells in the regulation of autoimmune effector cells by a chronic contact eczema. J Immunol (2007) 179:5071–81. doi:10.4049/jimmunol.179.8.5071
275. Greening DW, Gopal SK, Xu R, Simpson RJ, Chen W. Exosomes and their roles in immune regulation and cancer. Semin Cell Dev Biol (2015) 40:72–81. doi:10.1016/j.semcdb.2015.02.009
276. Ibáñez-Vea M, Zuazo M, Gato M, Arasanz H, Fernández-Hinojal G, Escors D, et al. Myeloid-derived suppressor cells in the tumor microenvironment: current knowledge and future perspectives. Arch Immunol Ther Exp (Warsz) (2017). doi:10.1007/s00005-017-0492-4
277. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res (2015) 128:95–139. doi:10.1016/bs.acr.2015.04.002
278. Van Ginderachter JA, Beschin A, De Baetselier P, Raes G. Myeloid-derived suppressor cells in parasitic infections. Eur J Immunol (2010) 40:2976–85. doi:10.1002/eji.201040911
279. Crook KR, Liu P. Role of myeloid-derived suppressor cells in autoimmune disease. World J Immunol (2014) 4:26–33. doi:10.5411/wji.v4.i1.26
280. Derkus B, Emregul KC, Emregul E. A new approach in stem cell research-exosomes: their mechanism of action via cellular pathways. Cell Biol Int (2017) 41:466–75. doi:10.1002/cbin.10742
281. Wilcockson SG, Sutcliffe C, Ashe HL. Control of signaling molecule range during developmental patterning. Cell Mol Life Sci (2017) 74:1937–56. doi:10.1007/s00018-016-2433-5
282. Saadeldin IM, Oh HJ, Lee BC. Embryonic-maternal cross-talk via exosomes: potential implications. Stem Cells Cloning (2015) 8:103–7. doi:10.2147/SCCAA.S84991
283. Basu J, Ludlow JW. Exosomes for repair, regeneration and rejuvenation. Expert Opin Biol Ther (2016) 16:489–506. doi:10.1517/14712598.2016.1131976
284. Iraci N, Leonardi T, Gessler F, Vega B, Pluchino S. Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci (2016) 17:171. doi:10.3390/ijms17020171
285. Fatima F, Ekstrom K, Nazarenko I, Maugeri M, Valadi H, Hill AF, et al. Non-coding RNAs in mesenchymal stem cell-derived extracellular vesicles: deciphering regulatory roles in stem cell potency, inflammatory resolve, and tissue regeneration. Front Genet (2017) 8:161. doi:10.3389/fgene.2017.00161
286. Phinney DG, Pittenger MF. Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells (2017) 35:851–8. doi:10.1002/stem.2575
287. Sabapathy V, Kumar S. hiPSC-derived iMSCs: NextGen MSCs as an advanced therapeutically active cell resource for regenerative medicine. J Cell Mol Med (2016) 20:1571–88. doi:10.1111/jcmm.12839
288. Fakoya AO. New delivery systems of stem cells for vascular regeneration in ischemia. Front Cardiovasc Med (2017) 4:7. doi:10.3389/fcvm.2017.00007
289. Wang L, Gu Z, Zhao X, Yang N, Wang F, Deng A, et al. Extracellular vesicles released from human umbilical cord-derived mesenchymal stromal cells prevent life-threatening acute graft-versus-host disease in a mouse model of allogeneic hematopoietic stem cell transplantation. Stem Cells Dev (2016) 25:1874–83. doi:10.1089/scd.2016.0107
290. Wang X, Zhang H, Yang H, Bai M, Ning T, Li S, et al. Cell-derived exosomes as promising carriers for drug delivery and targeted therapy. Curr Cancer Drug Targets (2017). doi:10.2174/1568009617666170710120311
Keywords: myeloid-derived suppressor cells, exosomes, cancer, autoimmune disease, therapy
Citation: Zöller M (2018) Janus-Faced Myeloid-Derived Suppressor Cell Exosomes for the Good and the Bad in Cancer and Autoimmune Disease. Front. Immunol. 9:137. doi: 10.3389/fimmu.2018.00137
Received: 26 November 2017; Accepted: 16 January 2018;
Published: 02 February 2018
Edited by:
Miroslaw Kornek, Universitätsklinikum des Saarlandes, GermanyReviewed by:
Alain Le Moine, Université libre de Bruxelles, BelgiumKarl Bechter, University of Ulm, Germany
Copyright: © 2018 Zöller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margot Zöller, bS56b2VsbGVyQHVuaS1oZC5kZQ==