 Elizabeth A. Mills
Elizabeth A. Mills Yang Mao-Draayer
Yang Mao-Draayer- 1Department of Neurology, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Graduate Program in Immunology, Program in Biomedical Sciences, University of Michigan Medical School, Ann Arbor, MI, United States
The increased use of newer potent immunomodulatory therapies for multiple sclerosis (MS), including natalizumab, fingolimod, and dimethyl fumarate, has expanded the patient population at risk for developing progressive multifocal leukoencephalopathy (PML). These MS therapies shift the profile of lymphocytes within the central nervous system (CNS) leading to increased anti-inflammatory subsets and decreased immunosurveillance. Similar to MS, PML is a demyelinating disease of the CNS, but it is caused by the JC virus. The manifestation of PML requires the presence of an active, genetically rearranged form of the JC virus within CNS glial cells, coupled with the loss of appropriate JC virus-specific immune responses. The reliability of metrics used to predict risk for PML could be improved if all three components, i.e., viral genetic strain, localization, and host immune function, were taken into account. Advances in our understanding of the critical lymphocyte subpopulation changes induced by these MS therapies and ability to detect viral mutation and reactivation will facilitate efforts to develop these metrics.
Introduction
Progressive multifocal leukoencephalopathy (PML) is a rare polyomavirus-associated disease involving progressive damage to brain white matter that often results in permanent disability or death. PML was first characterized in the 1950s in immunocompromised patients with lymphoproliferative disorders (1) and has come to be understood as an opportunistic infection associated with immunosuppression. The disease is caused by the infection, and subsequent loss of glial cells, especially myelin-producing oligodendrocytes, by a mutated form of the John Cunningham virus (JCV). Most people acquire JCV, usually in childhood, as it is found in 70–90% of the population. The initial infection is thought to occur in the tonsils or gastrointestinal tract, and then the virus remains latent, often in the kidneys or lymphoid organs, in an archetypal form that is incapable of productively infecting glial cells (2). Immunosuppression can lead to the reactivation of the latent virus and may promote viral mutation, thereby facilitating infection of glia by viral strains with mutated regulatory regions (3). Continued immunosuppression then prevents clearance of the virus from infected glia, resulting in demyelination and neurodegeneration.
The AIDS epidemic of the 1980s led to a dramatic increase in new cases of PML, which helped fuel research toward the mechanism, risk factors, diagnostic markers, and possible therapeutic interventions for this devastating disease (4). Unfortunately, while survival rates have increased, there has not been enough progress in the prevention of PML, and many immunocompromised individuals remain at risk. The increased availability of antiretroviral therapies for HIV patients has not decreased rates of HIV-associated PML as much as anticipated (5). Furthermore, the recent widespread use of next-generation immunomodulatory treatments for autoimmune diseases such as multiple sclerosis (MS) has expanded the patient population at an increased risk for the development of PML.
The immunomodulatory drugs natalizumab, fingolimod, and dimethyl fumarate (DMF) have greater efficacy in reducing relapses for many relapsing-remitting MS (RRMS) patients than older treatments, such as interferon-β and glatiramer acetate, but have also led some patients to develop PML (6). Natalizumab was the first of these therapies to be tied to PML in MS patients, and, of all MS therapies, it has the highest global incidence for PML (7). The inability to accurately predict risk can lead to difficult treatment decisions, particularly in patients with highly active MS, which need to switch therapies. The off-label use of the chimeric anti-CD20 monoclonal antibody, rituximab, has not been associated with PML in MS patients. Consequently, MS patients who develop PML while taking other therapies, particularly natalizumab, are often switched to rituximab to prevent worsening of either MS or PML (8). However, cases of PML have been linked to the use of rituximab in other autoimmune conditions such as rheumatoid arthritis (9). The reasons for the discrepancy are not fully understood but may be related to the combinatorial use of rituximab with other immunosuppressive agents. The risk of PML for MS patients taking anti-CD20 therapy with a history of taking natalizumab remains unclear and merits further study. This is likely to become an increasingly important factor in future treatment decisions, as the humanized anti-CD20 antibody, ocrelizumab, has recently been approved by the FDA for RRMS and primary progressive MS.
The most commonly used clinical metric for identifying patients at risk for developing PML is absolute lymphocyte count. Unfortunately, this metric has not been a reliable predictor. Indeed due to its mechanism of action, fingolimod generally lowers lymphocyte counts to a greater extent than natalizumab or DMF (10–12), but has a lower incidence of PML (7). Efforts have been made to determine the factors associated with increased risk, but thus far there are no truly predictive measures available.
Three factors identified to help stratify the pool of natalizumab-treated MS patients at greatest risk for developing PML include the presence of JCV antibodies in patient’s serum, previous use of immunosuppressive drugs, and use of natalizumab exceeding 24 months (13). A patient with all three factors has a 2.3% risk of developing PML, while a patient with none of the risk factors has a 0.002% risk (14). JCV blood antibody index can further stratify this risk, with an antibody index above 1.5 associated with a higher risk (15). Determining whether a patient is seropositive for JCV prior to the start of treatment is recommended; however, the high rate of false negatives in these assays can skew risk assessments (16). In addition, patients seroconvert at higher frequencies during treatment (17, 18). Therefore, it will be necessary to monitor additional parameters throughout the course of treatment to more accurately determine risk.
Since PML has occurred in the context of several MS disease-modifying therapies, a better understanding of how PML develops under each of these conditions could lead to the identification of universal predictive risk factors, as well as those factors that are unique. Recent work suggests that efforts toward the combined monitoring of specific lymphocyte subsets, JCV reactivation, and viral genomic rearrangements within the central nervous system (CNS) may offer better insight toward predicting risk (see Figure 1).
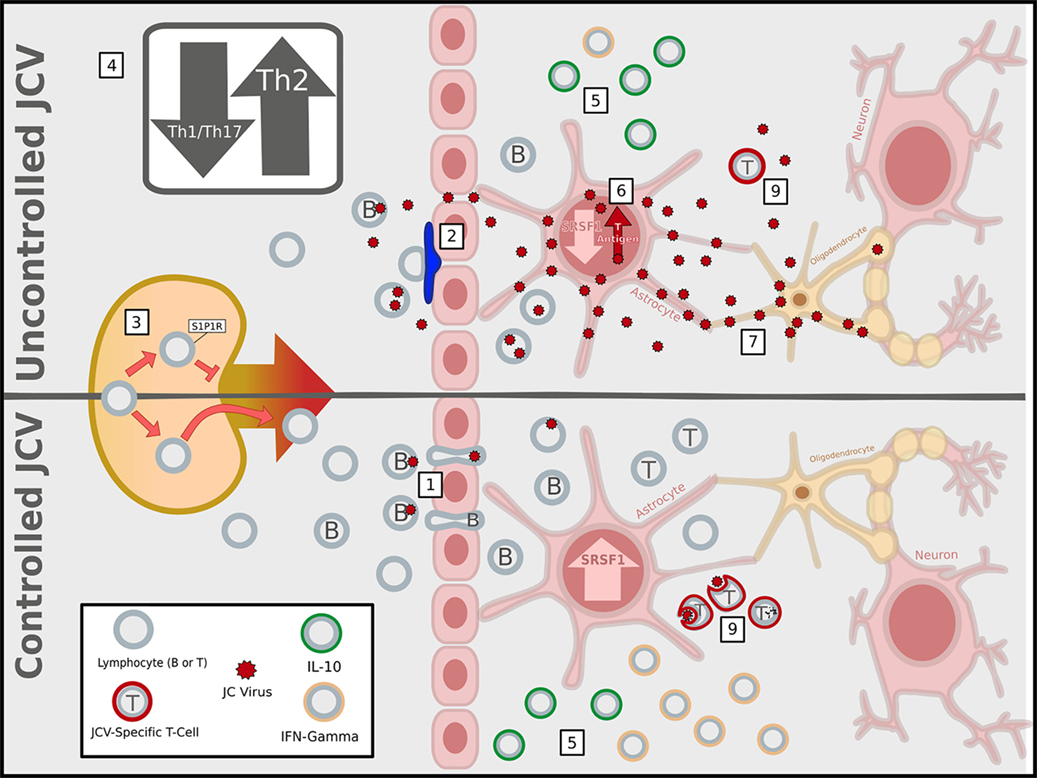
Figure 1. Immunosuppressive activities of multiple sclerosis (MS) therapies facilitate John Cunningham virus (JCV) infection and replication in the central nervous system (CNS). MS immunomodulatory therapies associated with progressive multifocal leukoencephalopathy have different mechanisms of action, but ultimately lead to an immunosuppressed state within the CNS that increases the likelihood of a productive infection of glial cells by JCV, represented as the uncontrolled JCV state. (1) In a healthy immune system (or the absence of immunomodulatory therapy), lymphocytes can enter the CNS via the blood–brain barrier, blood–meningeal barrier, or blood–cerebrospinal fluid barrier (latter two not shown), and JCV infected B-cells have been proposed as carriers of the virus into the CNS. In contrast, MS therapies block CNS entry of specific lymphocyte subsets: (2) natalizumab prevents CNS access of α4 integrin expressing lymphocytes primarily across the BBB by blocking α4β1/vascular cell adhesion molecule 1 (VCAM-1) adhesion interactions; (3) fingolimod traps within lymph nodes the lymphocytes that utilize sphingosine-1-phosphate 1 (S1P1) receptors for homing; (4) dimethyl fumarate interferes with the maturation of Th1 T-lymphocytes, tipping the balance in favor of anti-inflammatory Th2 cells. (5) Within the CNS, the net effect reduced the entry of conventional lymphocytes secreting pro-inflammatory IFN-γ and a relatively higher percentage of interleukin-10 (IL-10)-producing anti-inflammatory regulatory lymphocytes, compared to the healthy/untreated state. (6) The altered cytokine profile affects cross-talk between lymphocytes and CNS-resident astrocytes leading to transcriptional changes, such as the suppression of SRSF1, which can promote viral T-antigen expression, reactivation, and replication. (7) The JCV-infected astrocytes could then pass on the virus to oligodendrocytes and (8) fail to properly recruit the subset of lymphocytes necessary to clear the virus. (9) While most are blocked, the JCV specific T-cells that are present fail to adequately clear JCV (Image copyright: Caitlyn Fisher and Yang Mao-Draayer, reprints use with permission).
Immune Cell Function
Lymphopenia is a PML risk factor, but simply monitoring absolute lymphocyte count does not accurately convey risk, because it does not take into account the diversity and complexity of the immune system. MS therapies have been designed to ameliorate the inflammatory overresponse to autoantigens (19). This inflammatory response toward myelin-associated proteins results in the demyelinating lesions that are hallmarks of the disease. Consequently, MS treatments do not affect all immune cell types equally, but rather attempt to restore the balance toward a more anti-inflammatory state (20). This can lead to alterations in specific critical immune cell subtypes without dramatic changes to the overall lymphocyte count. Since the immune system is composed of a multitude of cell types, which have unique roles in maintaining proper immune function, the loss of particular subsets of lymphocytes can disproportionately elevate PML risk. For instance, CD4+ T-cell lymphocytopenia has been associated with PML in the context of HIV (21); however, simply monitoring absolute changes in a single specific subset is also insufficient. Instead, changes in the relative distribution and function of specific immune cells are critical to both the therapeutic benefit toward MS and the potential risk of developing PML.
The shift toward a more anti-inflammatory environment is driven, in part, by changes in the balance between conventional and regulatory immune cells in number and/or function. Regulatory cells can dampen the response of conventional immune cells, thereby suppressing the immune system (22). In some cases, the balance may be shifted too far, resulting in chronic immunosuppression and increasing the risk for opportunistic infections. In MS patients, the function of regulatory cells is thought to be compromised, leading to an exaggerated inflammatory response (23–25). The cytokine interleukin-10 (IL-10) is a critical anti-inflammatory mediator, which is decreased in RRMS patients (26). Many effective RRMS therapies have been found to increase the number or response of regulatory cells (20) and levels of IL-10 (27). It is important to note, however, that there is no correlation between MS therapies, which affect IL-10 and the subset associated with PML. Consequently, changes in IL-10 levels alone are not prognostically useful in risk assessments for PML.
Many pathogens, particularly those which can remain latent within B-cells, exploit the immunosuppressive properties of IL-10 to promote their persistence (28). These mechanisms are especially prevalent among viruses that are associated with chronic infections and immune exhaustion. Some viruses, such as human cytomegalovirus and Epstein–Barr virus (EBV), encode for an IL-10 ortholog within their viral genome (29), whereas others, such as hepatitis B virus, increase the expression of cellular IL-10 (30). The net effect of these strategies is to alter the balance of the endogenous cytokine secretions of immune cells toward an anti-inflammatory state. In some strains of mice, polyomaviruses are associated with tumor formation, and susceptibility appears to be related to the aberrant production of IL-10 in response to the virus in affected strains (31). A similar aberrant IL-10 response to JCV antigens by JCV-specific T-cells has been found in some PML patients, which facilitates the maintenance rather than the clearance of the virus (32, 33). This inappropriate response is typically a feature of exhausted immune cells in a chronically immunosuppressed environment (34). Furthermore, elevated cerebrospinal fluid (CSF) levels of IL-10 have been detected in about half of early stage PML patients (33). Consequently, treatments that increase levels of IL-10 are therapeutically beneficial in the context of MS, but elevated IL-10, particularly in the CNS, may also prevent antigen-specific T-cells from appropriately responding to JCV.
Chronic infections involving the continued presence of antigen lead to immune exhaustion, resulting in the eventual inability of T-cells to mount an effective response to the persisting antigen (35). In some cases, the development of an exhausted T-cell response can represent a “compromise” between pathogen and host, which allows for maintenance of the virus without incurring widespread tissue damage (36). Under these circumstances, attempts to restore T-cell responses can inflict great harm to the host, but in the context of other pathogens, restoration of immune responses can be beneficial. Thus, it is crucial to understand the relationship between immune exhaustion and pathogenesis for a given virus when developing or administering potential immune boosting therapies. Achieving the correct balance is a particularly challenging endeavor in the context of autoimmune diseases, since immune suppression promotes exhaustion both toward autoantigens and foreign antigens.
Sustained surface expression of the inhibitory receptor programmed death-1 (PD-1) is associated with T-cell exhaustion, and blockade of PD-1 can help restore immune responsiveness (37). Relative to the total population of CD8+ T-cells, elevated levels of PD-1 have been found on the JCV-specific CD8+ T-cells of PML patients and are associated with the lack of functional response to JCV peptides (38). The results of an in vitro assay suggest that blocking PD-1 could help enhance JCV-specific CD8+ T-cell responses in a subset of PML patients, especially those at early stages (38). However, this particular experiment involved patients with HIV-associated PML, rather than immunomodulatory therapy-induced PML, and the use of anti-PD-1 therapy may exacerbate autoimmune diseases. Similar to IL-10, dysfunction of PD-1 is associated with MS, such that polymorphisms that decrease PD-1 function are linked to disease progression and severity (39, 40). Furthermore, these data suggest that targeting PD-1 may only be clinically useful in a specific subset of early PML patients and that monitoring PD-1 expression alone is unlikely to be a reliable predictive or diagnostic metric. As PD-1 is only one of several co-stimulatory receptors associated with immune exhaustion, a reliable assay will likely require the combined analysis of multiple markers.
Notably, in the context of murine polyomavirus, both susceptible and resistant mouse strains produce a similar antibody response, but differ in the strength of their antigen-specific cytotoxic T-lymphocyte (CTL) responses (41). In the process of co-opting B-cells as a host, EBV promotes B-cell proliferation and antibody production, with high antibody titers associated with chronic uncontrolled infection, rather than immunity (42). A similar situation may occur in JCV-infected B-cells, thereby explaining the seemingly paradoxical clinical finding that higher levels of anti-JCV antibodies are associated with greater risk in the context of PML (15), as there is a reason to believe that the infection of B-cells could be a significant risk factor for PML.
B-cells have been implicated as carriers of JCV from the periphery into the CNS, since they can be non-productively infected by JCV and serve as reservoirs of latent virus (43). The form of JCV associated with PML has a rearranged non-coding regulatory region (NCCR) and often contains binding sites for the transcription factor Spi-B, which promotes expression of early genes in the JCV genome (44). Similar to EBV, JCV may induce pathogenic mutations within its viral genome through activation of class-switch recombinases in B-cells, as the JCV NCCR contains putative recombination sites (45). Viral homology-based recombination can also occur in B-cells co-infected with EBV and JCV and provides another potential avenue for pathogenic rearrangement of JCV (46). Furthermore, because Spi-B is also important for proper B-cell maturation and function (47), therapies that affect the distribution or expression profile of B-cells could facilitate viral reactivation and spread. Indeed, natalizumab promotes B-cell differentiation-associated gene expression (48), including increased expression of Spi-B in CD19+ B-cells (49). It is likely that the subpopulation of B-cells infected by the virus is also of critical consequence, with some stages having a more detrimental effect on subsequent immune responses and viral transmission. The rearrangement of immunoglobulin genes through V(D)J recombination in pre-B-cells makes this population an attractive candidate to facilitate JCV NCCR rearrangement (50). Notably, circulating pre-B-cells also increase in response to natalizumab treatment (51, 52). Therefore, the use of diagnostic tests for JCV specifically within B-cells may help further stratify patients at greatest risk for PML.
As a disease of the CNS, there needs to be a correlation between peripheral and central changes for peripheral lymphocyte monitoring to have predicative and diagnostic merit for PML. Since each drug has a unique mechanism of action, the lymphocyte populations affected, and the cell subtype ratio changes that are most predictive of PML will also likely vary based on the treatment history of the patient. In addition, changes in the relative frequencies and function of lymphocyte subsets may vary overtime with a given therapy. Although accurate predictive metrics for PML are still lacking, recent advances in our understanding of how these next-generation MS therapies differentially affect lymphocyte populations may allow for the development of novel assays and monitoring guidelines with increased predictive power.
Natalizumab
Mechanism
Natalizumab is a recombinant humanized monoclonal immunoglobulin G4 antibody targeting α-4 integrin (CD49d) that has been approved for the treatment of RRMS and Crohn’s disease (53). Natalizumab treatment leads to CD49d downregulation in CD49d-expressing cells (54). Very late antigen-4 (VLA-4) is a heterodimer composed of CD49d and β-1 integrin (α4β1 integrin) found on several immune cell populations, which interacts with vascular cell adhesion molecule 1 (VCAM-1) on endothelial cells and facilitates migration across endothelial barriers, including the blood–brain barrier (BBB) (55). VLA-4/VCAM-1 is one of the several adhesive molecule interactions that mediate CNS entry, and thus loss of this interaction will diminish, but not abolish, lymphocyte migration into the CNS. The other most prominent interaction is between lymphocyte function-associated antigen 1 (LFA-1) and endothelial intercellular adhesion molecule 1 (ICAM-1) (56). Some subsets utilize specialized mechanisms, such as melanoma cell adhesion molecule (MCAM)-expressing T helper 17 (Th17) cells, which interact with the vascular ligand lamin-411 to facilitate CNS entry (57). Furthermore, there are multiple routes of entry for lymphocytes into the CNS, including the BBB, blood–meningeal barrier (BMB), and blood–CSF barrier (BCSFB) within the choroid plexus. The latter two barriers utilize additional adhesive molecules, and entry is less stringently regulated. The BMB depends on P-selectin, which is expressed in a constitutive manner, as opposed to dynamically regulated VCAM-1 (58). The chemokine CCL20 produced by the choroid plexus can bind to the CCR6 receptor on lymphocytes, allowing entry across the BCSFB into the ventricular space (59). This provides for the preferential CNS access of CCR6+ subsets, such as Th17 and T regulatory (Treg) cells in the absence of the VLA-4/VCAM-1 interaction (60, 61). Since CD4+ anti-JCV responses in MS patients have been shown to depend primarily on Th1 cells, the reliance of this population on VLA-4/VCAM-1 contributes to the inability of natalizumab-treated patients to effectively clear JCV from the CNS (62).
Migration into the CNS is a two-step process, in which immune cells first migrate into CSF-drained perivascular, leptomeningeal, and ventricular spaces for the purpose of immunosurveillance. Interaction with antigen-presenting cells within these compartments then allows for the α6β1 integrin-mediated migration of activated lymphocytes across the glial limitans barrier into the CNS parenchyma in a cytokine-dependent manner (63). This two-step process allows for blockade of surveilling lymphocytes from the CNS parenchyma in the absence of inflammation or infection. Effective immunosurveillance, then, relies on both productive antigen presentation and activation of the appropriate lymphocyte subpopulation.
Natalizumab impairs immunosurveillance on both of these fronts through its effects on the migration and activation of T-lymphocytes and antigen-producing cells. Mature dendritic cells within perivascular spaces, meninges, and choroid plexus are important for re-stimulating peripherally activated CD4+ T-cells and initiating a T-lymphocyte response in the CNS (64). CD49d expression on dendritic cells varies in a maturation-dependent manner (65), such that a loss of CD49d interferes with the ability of mature dendritic cells to cross the BBB and activate T-lymphocytes (66). Furthermore, co-stimulatory signals, such as the VLA-4/VCAM-1 interaction, are necessary for effector memory T-cells to function optimally (67). Although the full extent to which natalizumab influences co-stimulatory molecule expression has not been delineated, natalizumab treatment has been shown to negatively impact surface expression of CD49d and OX40 (CD134) (68), thereby limiting T-cell activation capacity. The loss of this CNS immunosurveillance capacity is thought to contribute to the increased risk of PML for MS patients treated with natalizumab.
Peripheral Lymphocytes
In contrast to other MS treatments associated with PML, natalizumab leads to increases in absolute lymphocyte counts in the blood (11). This change in distribution is expected from a therapy that primarily targets the migratory capacity of lymphocytes into the CNS. However, the increased peripheral levels are not necessarily an indication that these particular populations are blocked from the CNS, since CD49d is important for adhesive interactions that mediate entry into other organs systems as well. Indeed, a direct correlation between changes in immune cells subsets between the periphery and CNS in response to natalizumab treatment has not been found. Instead, the elevated lymphocyte levels can be attributed to a selective release of lymphoid precursor cells in the hematopoietic precursor population from the bone marrow and B-lymphocytes from the spleen (69). Since natalizumab decreases surface expression of CD49d, longitudinal studies have indicated that lymphocyte subsets most dependent on CD49d for localization or trafficking are most affected, resulting in increased levels of conventional memory B-cells (51, 69, 70) and activated pro-inflammatory T-cells (68, 70–73) in the peripheral blood. This imbalance of T-cells likely occurs because certain subsets of CD4+ T-cells, such as Th1 cells, are particularly dependent on CD49d, whereas FoxP3+ regulatory T-cells express low levels of CD49d and are thus relatively unaffected by natalizumab (74). Disruption to the peripheral homeostasis of CD4+ T-cell populations has been proposed to contribute to the manifestation of both MS and PML, with natalizumab treatment disrupting the balance in a manner that favors the expansion of autoreactive T-cells over virus-specific T-cells (75).
A notable feature of these longitudinal studies is that the percentages of the activated subpopulations varied with duration of treatment, such that some of these changes occurred transiently at early time points, whereas others were only apparent after 1 year of treatment. This suggests that subpopulations of lymphocytes are dynamically changing in response to continued exposure to natalizumab. Since the risk of PML is typically associated with exposure to natalizumab for more than 2 years, short-term studies may not provide much insight into the relevant players conferring the increased risk, and a better understanding of the critical changes may be facilitated by long-term studies. Indeed, short-term studies have failed to detect significant changes in the peripheral CD4+/CD8+ ratio, whereas studies examining patients treated for at least 2 years found decreases in the CD4+/CD8+ ratio (76, 77). This is likely explained by the finding that while remaining within normal range, the peripheral CD4+/CD8+ ratio decreases in accordance with the number of doses of natalizumab (78). At this point, until a clear connection can be established between peripheral and CNS changes, the most meaningful predictive markers are likely to come from within the CNS.
CNS Lymphocytes
As expected, natalizumab treatment leads to a dramatic decline in lymphocytes within the CNS, particularly in CD19+ B-cells and CD4+ T-cells, due to their high dependency on CD49d for CNS entry (79, 80). While absolute numbers of CD8+ T-cells also decrease, the elevated expression of ICAM-1 and LFA-1 on these cells allows for preferential CNS access relative to CD4+ T-cells (81). Consequently, the CD4+/CD8+ ratio decreases toward levels seen in patients with HIV within a single dose (78), and low CD4+ T-cell counts have been implicated in HIV-associated PML (21). Furthermore, due to the loss of CD49d, the remaining CD4+ T-cells are forced to use alternative adhesion molecules, such as P-selectin glycoprotein ligand 1 and MCAM, for CNS entry (82), which will impact subset distribution and may negatively impact their functional and activation status.
In contrast to the peripheral blood, levels of pro-inflammatory chemokines and cytokines in CSF are decreased within 1 year of natalizumab treatment at the protein (83) and mRNA levels (84). This suggests that the overall milieu in the CNS is biased toward a less inflammatory state. This imbalance could be mediated by a disruption in the balance between conventional and regulatory lymphocyte subsets; however, an analysis of CSF regulatory subsets has not been performed in this context, but could be highly informative. Overall, the significant loss of CD4+ T-cells within the CNS coupled with the imbalances of the remaining subpopulations is likely to be a primary driver of PML risk.
JCV-Specific Response
The inability of natalizumab-treated PML patients to effectively clear JCV from the CNS likely stems from both the decreased migration of lymphocytes and the loss of functional capacity in the small subset of immune cells that do enter the CNS. Cellular immune response assays testing the functional capacity of JCV specific T-cells have been shown to hold predictive power for disease control in HIV-associated PML (85, 86). Consequently, some efforts have been made to determine whether measurements of the JCV-specific T-cell response also hold predictive or diagnostic power in natalizumab-associated PML. A potential caveat is the specific loss of immunosurveillance and immune function within the CNS, compared to the periphery, in natalizumab-treated patients. Unlike in HIV patients, the composition of immune cells in the CSF cannot be directly correlated to that of the peripheral blood, thus the usefulness of assays reporting the response of peripherally derived JCV specific T-cells remains unclear.
Most of this JCV-specific response testing involves a combination of intracellular cytokine staining and enzyme-linked immunosorbent assays in peripheral blood mononuclear cells (PBMCs). Consequently, the results of these studies are affected by the selection of cytokines assayed, and the ex vivo stimulation paradigm, particularly the JCV viral strain or peptide library, used for antigen activation. Furthermore, they are unable to assess prospective differences between patients who will eventually develop PML with those who will not and thus cannot determine the predictive value of their findings.
One pilot study found that peripheral JCV viremia was associated with decreased IFN-γ responsiveness to a JCV VP1 peptide library over the course of natalizumab treatment (87). Another study looking at JCV-specific effector memory T-cell responses found that purified JCV was a more potent antigen than VP1 alone using ELISPOT and that increased duration of treatment was associated with a stronger response (88). The probability that patients would show a positive IFN-γ response to JCV also increased the longer the patients were treated with natalizumab. Notably, stimulated PBMCs from the two PML patients in this study also exhibited robust IFN-γ responses to JCV. This suggests that while viremia and the presence of IFN-γ-producing JCV-specific effector memory T-cells in PBMCs may be indicative of peripheral JCV reactivation, they are not on their own reliable predictors of which patients will eventually develop PML. The use of more comprehensive JCV peptide libraries and cytokine assays could improve the utility of this type of testing. Indeed, another study using a more comprehensive JCV peptide library and cytokine profiling found that PML patients had aberrant responses to JCV compared to natalizumab-treated patients without PML (33). These PML patients either failed to produce a response to JCV or produced an atypical cytokine response. The CD4+ T-cells in two of the PML patients produced IL-10 in response to JCV, and increased levels of IL-10 were detected in the CSF from 50% of early stage PML patients. The increased CSF IL-10 may be related to the loss of CD49d+CD4+ T-cells within the CNS.
Due to differences in CD49d dependency for CNS entry, natalizumab may produce a bias toward more Treg cells and less conventional T-cells, thereby skewing the CNS cytokine milieu in a manner that facilitates viral reactivation and persistence. In a murine model of another chronic virus, y-herpesvirus, CD4+ T-cell deficiency led to the development of a population of IL-10-producing PD-1+ CD8+ Treg cells during viral reactivation (89). Furthermore, the loss of CD49d expression on T-cells can also impair their functional capacity. In the context of lymphatic choriomeningitis virus (LCMV) infection, virus-specific CD4+ T-cells have been shown to be characterized by surface expression of CDllahi and CD49d (90). Moreover, it was the CD11ahi CD49d+ CD4+ memory T-cells which productively responded to a secondary challenge of LCMV, suggesting that the loss of CD49d expression on CD4+ T-cells could prevent an adequate immune response to JCV reactivation. Therefore, the monitoring of CD49d expression on peripheral CD4+ and CD8+ JCV-specific T-cells may provide another way to help determine PML risk.
Overall, the aberrant production of IL-10, expression of PD-1, and decreased expression of CD49d by JCV-specific T-cells may provide relevant PML risk-associated metrics although more work is needed to validate the reliability of these potential markers.
Fingolimod
Mechanism
Fingolimod is an immunomodulatory drug that targets the sphingosine-1-phosphate (S1P) receptor. The loss of S1P receptors prevents lymphocytes from traversing the endothelial barriers of lymphatic compartments, effectively trapping them (91). This leads to decreases in circulating lymphocytes available to enter the CNS. The retention within lymph nodes is dependent on expression of the chemokine receptor CCR7, thus lymphocyte subsets that lack or downregulate this receptor maintain the ability to egress (92). Therefore, fingolimod disproportionally affects cell populations that express both S1P and CCR7 receptors (93). This sparing of CCR7− populations was expected to mitigate the risk for PML by permitting the free migration of JCV-specific effector memory T-cells. However, PML has developed in MS patients treated with fingolimod, which may stem from recent findings that fingolimod affects effector T-cell function (94) and that CCR5−CCR7+ central memory Th1 cells play a role in the antiviral response to JCV (62). The initial cases of fingolimod-associated PML were found in MS patients who had had been previously treated with natalizumab and thus could not be conclusively linked to fingolimod (95). In recent years, there have been 15 additional MS patients where PML could be attributed to fingolimod alone, and it is important to note that there was no correlation between absolute lymphocyte count and PML onset or severity in these patients (96).
Peripheral Lymphocytes
In accordance with its mechanism of action, the absolute lymphocyte count in the peripheral blood decreases within 2 days of initiating fingolimod treatment, and stabilizes within 2–4 weeks, in MS patients (97). The most drastic decreases take place in the CD19+ B-lymphocyte and CD4+ T-lymphocyte populations, which have been reported to reach a steady state by 3 months of treatment (98). The distribution of the remaining B-lymphocytes is shifted toward less pro-inflammatory memory B-cells and more IL-10-producing naive B-cells and regulatory B-cells defined as CD38+CD24+ or CD38+CD27-CD24+CD5+ (99–101). This skewed cell type distribution produces an overall cytokine profile with an anti-inflammatory bias (101).
In contrast to the B-lymphocytes, the peripheral T-lymphocyte population shifts with fingolimod treatment toward a lower proportion of naive T-cells and greater proportion of memory T-cells, specifically effector memory T-cells, in both conventional and regulatory subsets (93, 98, 102), consistent with their differential expression of CCR7. Absolute levels of circulating CD4+ and CD8+ T-lymphocytes decrease in MS patients treated with fingolimod; however, CD4+ T-cells are affected to a greater degree, leading to a decrease in the CD4+/CD8+ ratio (93), which worsens over the course of treatment (103). The percentage of CD4+ and CD8+ T-cells that produce IFN-γ and IL-17 also decreases, primarily within the Th1 or Th1/Th17 subsets (102). Since these IFN-γ-producing populations have been shown to be critical for mounting an antiviral response to JCV (62), the fingolimod-induced decreases in these subsets has important implications for PML risk.
In addition, the relative percentage of CD25+CD127− Treg cells within the CD4+ population is increased peripherally in MS patients treated with fingolimod (97, 98, 102, 104). This loss of critical IFN-γ+ CD4+ T-cells coupled with the increase in Tregs may result in the failure of the immune system to appropriately respond to pathogens in some patients.
CNS Lymphocytes
If the effects of fingolimod were restricted to its ability to trap particular lymphocyte populations within lymph nodes, the relative composition of lymphocytes within peripheral and CNS compartments should be similar. However, studies specifically aimed at assessing the composition of immune cells in the CNS reveal discrepancies, which suggest that fingolimod also influences migration of lymphocytes into and out of the CNS. Fingolimod-treated patients exhibit an increased CSF/blood ratio for B-lymphocytes. Interestingly, the level of IL-10-producing Breg cells (CD38+ CD27− CD24+ CD5+) within the CSF was found not to change, despite decreases in the blood (100). The authors of this study used an in vitro assay to determine that B-lymphocytes, especially Breg cells, from fingolimod-treated patients have increased transendothelial migratory capacity. If B-cells are the primary carriers of JCV, this augmented ability of B-cells to cross the BBB may facilitate the entry of JCV into the CNS.
The increased migratory capacity was not found to extend to T-lymphocytes, so differences in peripheral and CNS compartments for this population may involve a different mechanism. Similar to the blood, CD4+ T-lymphocytes are disproportionally affected in the CSF by fingolimod treatment, but to a lesser degree (105), resulting in a decreased CD4+/CD8+ ratio, similar to what is seen in the CSF of natalizumab-treated MS patients (106). Overall, an increase in the percentage of IL-10-producing Breg cells and decrease in CD4+ T-cells could result in the CNS becoming an immunosuppressive environment with reduced immunosurveillance capacity, thereby allowing JCV to evade immune-mediated elimination.
JCV-Specific Response
Treatment with fingolimod has been associated with an increased risk for infection (107), indicating that the ability of these patients to mount an appropriate and effective immune response is compromised. Although there is currently no data regarding the effect of fingolimod on JCV-specific CTL responses, fingolimod treatment has been shown to produce a suboptimal response to another chronic latent virus, varicella zoster virus (VZV), with decreased cell proliferation and IFN-γ production (108). Notably, most fingolimod-treated MS patients exhibit a robust response to VZV, and only those who lack this response experience viral reactivation (109) The lack of response may be related to the finding that fingolimod can also impact CD4+ effector T-cell function through the upregulation of T-cell factor 1 (94). In this study, activated CD4+ T-cells were shown to produce lower levels of IFN-γ and granzyme B in the presence of fingolimod. More studies are needed to address whether a similar CTL impairment is found toward JCV in these patients.
Dimethyl Fumarate
Mechanism
DMF-associated PML usually occurs in the context of severe lymphopenia, leading to the establishment of lymphocyte count guidelines for treatment discontinuation (110, 111). Five cases of PML have been reported in MS patients taking DMF (112–115). In one of these patients, lymphocyte counts did not drop below the guideline threshold for over 6 months (114). PML has also been reported in a DMF-treated psoriasis patient without severe lymphopenia (116). As a result, outside of absolute lymphocyte count, additional monitoring measures are needed.
The therapeutic mechanism of action for DMF in MS is not completely understood, but it is believed to be related to its established roles in protecting cells facing oxidative stress and blocking NF-κB-mediated pro-inflammatory cytokine production (117). The clinical benefit of fumarates in autoimmune disease is associated with their ability to disrupt the production of functional Th1 cells (117). Stimulated T-cells treated with DMF downregulate the expression of antiapoptotic Bcl-2, ultimately resulting in the loss of activated T-lymphocytes (118). The decreased differentiation of Th1 cells also appears to involve the inhibition of dendritic cell maturation, which thwarts the process of antigen presentation to T-cells (119). Furthermore, the cytokine profile of T-lymphocytes treated with DMF is altered, leading to a polarization shift away from IFN-γ and toward IL-10 (120).
Peripheral Lymphocytes
In a longitudinal study, the peripheral blood leukocyte and lymphocyte counts of DMF-treated MS patients decreased over a 12-month period (121). Significant loss of both central and effector memory CD4+ and CD8+ T-cells was apparent by 6 months of treatment, but CD8+ T-cells were more profoundly affected, leading to an increase in the CD4+/CD8+ ratio (121–124). Although among total CD4+ T-cells, both Th1 and Th17 classes are decreased, there is a shift away from pro-inflammatory cytokine-producing Th1 and toward anti-inflammatory cytokine-producing Tregs and Th2 cells specifically within the memory T-cell subset (123, 124). Once again, the preferential loss of Th1 cells likely impedes the production of an effective anti-JCV response, thereby increasing the risk for PML.
Similarly, DMF alters the profile of circulating B-lymphocytes in MS patients toward an anti-inflammatory state. The total levels of CD19+ B-lymphocytes are decreased, with the most profound loss occurring in the mature CD27+ B-lymphocyte subset (125–127). The proportion of Breg subsets (CD24highCD38high and CD43+CD27+) has been found to be significantly increased after 12 months of treatment with DMF (125). Correspondingly, there is a shift in the B-cell cytokine profile toward more IL-10, relative to pro-inflammatory cytokines (125–127).
CNS Lymphocytes
Information on how DMF affects the composition of immune cells within the CNS is still needed. Since DMF works directly on the function and survival of the immune cells themselves, there has been an underlying assumption of a direct correlation between peripheral assays and central immune cell composition. This assumption may not be valid, based on in vitro studies indicating that DMF can alter cytokine-induced adhesion molecule expression in endothelial cells (128–130). DMF decreases transendothelial migration due to the downregulation of the adhesion molecules E-selectin, VCAM-1 and ICAM-1 (128). Since these adhesion molecules are important for lymphocyte trafficking into the CNS, the CNS lymphocyte composition of DMF-treated patients would be expected to differ from the periphery. In the context of experimental autoimmune encephalomyelitis (EAE), DMF also decreases the expression of CD49d on circulating lymphocytes (131), and similar to natalizumab, the loss of CD49d may prevent adequate JCV antigen presentation and clearance. If this finding applies to human MS patients, then the monitoring of CD49d expression may be prognostically useful in DMF-treated patients as well. Similar to the other PML-associated MS therapies, the ability of DMF to shift the balance between IFN-γ- and IL-10-producing cells likely inhibits the effective clearance of JCV from the CNS.
JCV-Specific Response
Although there is currently no information about the JCV-specific CTL response, the CD8+ T-cell lymphocytopenia associated with DMF suggests that the loss of JCV-specific CD8+ T-cells may be a contributing factor in the development of PML in lymphopenic patients. Information regarding the activation profile and responsiveness of JCV-specific CD8+ T-cells from DMF-treated patients will be needed to determine how this population varies over the course of treatment and whether it can be used to assay risk for PML.
JCV Entry and Reactivation in the CNS
Viral Entry into CNS
As a disease of the CNS, changes in lymphocyte subsets cannot lead to the development of PML if JCV is not located in the CNS. The mechanism by which JCV enters the CNS is currently unknown, but it has been proposed that it could be transferred as free virus or carried by B-cells (43). Since B-cells utilize multiple ports of entry into the CNS, there are also several avenues by which JCV could enter. JCV could be transferred directly to glial cells from B-cells that have entered the CNS parenchyma, or it could be transferred indirectly via the BBB, BMB, or BCSFB.
Susceptibility to infection by polyomaviruses is primarily mediated by lactoseries tetrasaccharide c (LSTc), which serves as the attachment receptor for the virus via the VP1 capsid protein (132, 133), and the 5-HT2 serotonin receptors, which facilitate entry (134). However, these are likely not the only mechanisms for viral entry, as some cell types prone to infection lack at least one of these receptors. Notably, glial cells, even in individuals with PML, lack expression of LSTc (135). Similarly, the LSTc expressing brain vascular endothelial cells of the BBB have been shown to be capable of supporting viral entry in vitro, but lack 5-HT2 receptors (136). The epithelial cells of the choroid plexus in the BCSFB express both LSTc and 5-HT2 and are thus also capable of being infected by JCV (135). Furthermore, there is clinical evidence for the productive JCV infection of the cells of the leptomeninges and choroid, resulting in meningitis (137). Interestingly, the regulatory region associated with JCV-related meningitis is not the rearranged form associated with PML, but rather the archetypal version typically found in the periphery. Thus, the NCCR rearrangements that facilitate the productive infection of oligodendrocytes are not required for JCV to enter the CNS but may play a role in the subsequent manifestation of PML.
Based on the NCCR sequence structure, four classes of JCV variants have been characterized. While particular variants are preferentially found in certain tissues, they have all been detected to some extent within both the CNS and the periphery (138). Correspondingly, latent virus has been found in the brains of healthy individuals, including both rearranged and archetypal regulatory region forms (139–141). Evidence of peripheral I-R forms, which are associated with PML and enriched in the brain, could indicate that JCV has already reached the CNS and that these patients have higher risk for PML. Therefore, prescreening of MS patients for JCV should focus not simply on seropositivity but on the detection of rearranged variants.
JCV Reactivation Markers
Many viruses have been shown to modify the contents of host cellular secretions to promote their own spread and survival (142). Exosomes are used as a form of communication and molecular transfer between cells types and used extensively by immune cells to modulate inflammatory responses (143). The viral manipulation of exosome contents involves both altered secretion of host-derived factors and incorporation of viral-derived components (142). MicroRNAs (miRNAs) are key immunomodulatory components of exosomes. The circulating miRNA expression profile of patients with active viral infections has been shown to be altered in some cases (144). In the context of JCV infection, however, the profile of circulating host-derived miRNAs does not appear to be diagnostically useful (145). Viral-derived miRNAs, on the other hand, offer a potentially more relevant source of biomarkers, since they provide a direct readout of a specific virus. JCV encodes the miRNA jcv-miR-J1. jcv-miR-J1-5p, which is derived from the 5p arm and specific to JCV, has been detected in the urine, plasma, and CSF of JCV-seropositive individuals (146, 147). Furthermore, the presence of jcv-miR-J1-5p is indicative of latent viral infection, which may be clinically useful in the stratification of risk for PML, particularly in regards to the CSF. The miRNA derived from the 3p arm of JCV pre-miRNA, jcv-miR-J1-3p, is identical with the 3p-derived miRNA from the BK polyomavirus, so is less diagnostically useful, but important for viral immune evasion (148). JCV-miR-3p downregulates the expression of ULBP3, thereby inhibiting the clearance of virus-infected cells by natural killer cells (149). Interestingly, the results of one study suggest that due to viral homology or cross-talk, the production of antibodies toward type 1 BK polyomavirus may be protective against JCV-induced PML (150). A better understanding of JCV’s ability to modify the content and release of exosomes may allow for the development of viral reactivation diagnostic biomarkers and therapeutic interventions to prevent CNS transmission.
Astrocytes as Gatekeepers of the CNS
Within the CNS, astrocytes are the best positioned conduit of JCV. In a chimeric mouse model, in which glia were derived from human glial progenitors, intracerebral delivery led to the preferential accumulation of JCV within astrocytes, suggesting that astrocytes are the most susceptible to JCV infection within the CNS (151). This susceptibility may stem from the vast interconnectedness of astrocytes, both with each other and with other cells in the CNS. Moreover, astrocytes play a pivotal role as the interface between the CNS and the periphery, with bidirectional communication between astrocytes and vascular endothelial cells (152). The BMB is a likely avenue of viral transmission, since meningeal cells can be productively infected by JCV in vivo (137), are coupled to astrocytes via gap junctions (153), and can influence the gap junction coupling of neighboring astrocytes (154).
Gap junction channels allow for the rapid intercellular transport of ions and small molecules, including miRNAs (155). Many pathogens have developed strategies to take advantage of gap channel-based intercellular communication to facilitate their spread (156). The release of pro-inflammatory factors results in the decreased expression of gap channel proteins, connexins, thereby disrupting networks of communication mediating essential functions, such as ionic balance, which can lead to CNS injury (157). The loss of connexins can also impair the phagocytic clearance of infected cellular debris and antigen cross-presentation at immunological synapses (156). Therefore, in addition to exosomes, viruses could potentially use gap junction channels or hemichannels to traffic viral miRNAs or proteins to neighboring uninfected cells for immunomodulation or to prime glial cells for subsequent infection.
The perivascular endfeet of astrocytes are involved in neurovascular regulation and permeability of the BBB (152). In this capacity, astrocytes serve as the gate keepers of immune cells into the CNS. The profile of chemokines released by astrocytes is tailored to the nature of the specific invading pathogen to coordinate the most effective and least damaging immune response (158). The typical response involves an initial release of pro-inflammatory molecules to recruit leukocytes into the CNS, followed by the release of more immunosuppressive factors, such as IL-10, to prevent inflammatory damage, particularly in the context of chronic infections (159, 160). Consequently, virus-mediated modification of astrocytes could impact their ability to recruit and control the T-cells necessary to clear the virus. Perhaps more importantly is the manner in which immunomodulatory therapies can disrupt the interactions between the immune system and glial cells, resulting in viral reactivation and ineffective clearance.
JCV Infection and Reactivation within Glia
Immune cells can affect the ability of glial cells to be productively infected by JCV through cytokine-mediated changes in gene expression. JCV contains a capsid-enclosed double-stranded DNA genome that encodes early and late genes (161). The early transcript encodes the large T- and small t-antigens, which are critical for viral replication. The NCCR controls the expression of early and late viral genes through a bidirectional promoter, thus the availability of relevant transcription and mRNA-processing factors within a host cell influences the ability of the virus to propagate (162). Since T-antigen plays such a vital role in viral replication and autoregulation of the viral promoter, factors that interact or interfere with T-antigen can impact viral reactivation (163, 164).
The alternative slicing factor SRSF1 (SF2/ASF) and T-antigen are antagonistic to each other. SRSF1 inhibits the splicing of JCV pre-mRNA transcripts, which is necessary for the production of the individual viral proteins (165). Furthermore, SRSF1 also binds to the NCCR to inhibit transcription of both early and late viral genes, including T-antigen, and suppression of SRSF1 in glial cells facilitates their infection by JCV (165, 166). T-antigen, in turn, can suppress the expression of SRSF1 through interactions with the SRSF1 promoter (167). SRSF1 suppression can also be mediated by the DNA- and RNA-binding protein, Pur-alpha, through its activation of T-antigen expression and direct inhibition of SRSF1 gene expression (168). Soluble factors secreted by immune cells, such as cytokines, can tip the balance in favor of SRSF1 and suppress JCV replication. IFN-γ, specifically, has been found to inhibit T-antigen translation in a Jak/Stat-dependent manner (169). Although, the mechanism directly linking cytokines with glial SRSF1 expression is still unclear, these studies suggest that alterations in cytokine output or glial gene expression by immunomodulatory therapies could confer risk in the development of PML. Indeed, a recent 2-year longitudinal study following patients treated with natalizumab or fingolimod indicates differences in SRSF1 expression levels (170). While SRSF1 expression levels remained stable in fingolimod-treated patients, they transiently increased within a year of natalizumab and then decreased with longer duration therapy. This is consistent with the higher incidence of PML with natalizumab and increased the risk associated with long-term use. In addition, the transcription factor Spi-B, which is increased in CD19+ B-cells (49) and CD8+ T-cells (171) following natalizumab treatment, is also important for the expression of T-antigen in astrocytes (172).
The disparity of PML incidence may also be related to direct treatment-specific changes within the astrocytes themselves. While the secretion of pro-inflammatory cytokines by immune cells may be beneficial to the containment of JCV, the expression of pro-inflammatory-associated factors by astrocytes could actually contribute to JCV reactivation. A microarray analysis comparing glial cells lines resistant to JCV with those that are susceptible to JCV infection revealed that resistant cells expressed lower levels of pro-inflammatory cytokine and chemokine gene transcripts (173). This difference is related to decreased expression of the transcription factors NF-κB and NFAT4, which can modulate expression of both pro-inflammatory cytokines and JCV transcripts (174, 175). NF-κB and NFAT4 are also themselves activated by pro-inflammatory cytokines, such as TNFα, and regulated by calcium signaling/calcineurin activity in a context-dependent manner (176). These factors promote JCV early gene expression through interactions with the KB element of the NCCR, which is negatively regulated by C/EBPb (177). In addition, histone acetylation at the KB element can activate viral gene expression, and this epigenetic modification appears to involve NF-κB (178).
Therefore, decreases in pro-inflammatory signaling within astrocytes by some immunomodulatory therapies, including fingolimod and DMF, could potentially lower the risk of JCV reactivation with the CNS. Fingolimod treatment suppresses TNFα-induced pro-inflammatory genes, primarily cytokines, in human astrocytes, which confers neuroprotection (179). Critically, NF-κB nuclear translocation and signaling are also reduced (180). The stimulation of calcium-signaling/calcineurin activity by fingolimod (181) may help mediate these effects, as calcineurin has been implicated in the inhibition of NF-κB and NFAT in reactive astrocytes (176). DMF inhibits NF-κB nuclear translocation and associated pro-inflammatory cytokine expression (182–184); however, it also decreases histone deacetylase activity (185). It remains to be determined whether these or other MS therapies actually influence JCV reactivation in the astrocytes of MS patients.
Reactivation of latent JCV within astrocytes is a potential gateway to PML. Astrocytes are actively involved in oligodendrocyte survival, differentiation, and myelination through extensive cross-talk between the two glial subtypes (186). Viral components could also be transferred in the process of this communication. Due to this reliance on astrocytes, the loss or dysfunction of infected astrocytes can negatively impact oligodendrocytes and contribute to demyelinating pathology (187). Furthermore, the uptake of infected myelin and cellular debris from JCV-induced cell lysis, by phagocytic astrocytes, could facilitate the spread of the virus within the CNS (188). Overall, more studies are needed to uncover the mechanisms used by JCV to enter the CNS and the conditions governing infection and reactivation within astrocytes. The presence or absence of JCV within the CNS at the onset of immunomodulatory treatment could help further stratify patients according to which parameters are most relevant to their individual risk of developing PML. In patients with latent JCV in the CNS, the strategy should involve preventing viral reactivation. Accordingly, the type of MS therapy used should be tailored to minimize both symptoms of MS and risk of PML.
Genetic Factors
Viral Mutations
The development of JCV-induced CNS pathology involves genetic mutations within the viral genome. It is the nature of the mutation that determines the manner in which the infection and subsequent pathology will manifest. The mutations can affect the virus on a variety of fronts, from controlling which cell types are susceptible to infection to virulence, depending on the region of the viral genome alteration. PML is associated with complex, but highly variable, rearrangements of the NCCR, which not only facilitates infection of glial cells, but also leads to increased expression of early genes, namely T-antigen, which then boosts the replication rate (189). About half of PML patients also have mutations in the VP1 capsid protein, which can change the affinity of binding to the sialic acid containing receptors that mediate viral entry, such as LSTc, thereby increasing virulence (190). In contrast, JCV-associated diseases affecting neurons are more dependent on deletions within the late viral genes. Granule cell neuronopathy is a demyelinating disease that results from JCV infection of cerebellar granule cell neurons (191). It is associated with mutations in the C terminus of the JCV VP1 gene, which are believed to enhance replication in cerebellar granule cells and provide for the evasion of immune detection (192). JCV encephalitis is linked to mutations in agnoprotein (193). At this point, only three cases of JCV-induced meningitis have been reported in the literature, and the relevant mutations have not been identified (137, 194).
While PML-associated JCV may share similar features, the variability of mutations found in PML patients suggests that the viral trajectory toward a pathogenic form is unique in each individual (195). Consequently, while the loss of JCV-specific CTL and localization of JCV within the CNS are also essential features, the single greatest risk factor for PML is likely the genetic variant(s) of JCV residing within a patient (see Figure 2). This explanation best accounts for why most immunosuppressed individuals do not develop PML despite the high prevalence of (archetypal) JCV in the general population. Although individuals typically host several classes of JCV within their body, it is unclear whether the form associated with PML can be acquired through a primary infection or if it requires de novo mutation within the host. The rarity of PML likely reflects the complexity of the mutations needed to induce a productive infection within oligodendrocytes. The transition of the NCCR from archetypal to rearranged requires a series of deletion and duplication events, such that even if the probability of any one mutation is high, the likelihood of the combination is very low (45). However, the hijacking of host recombinase activity could bypass the requirement for several independent mutagenic events and increase the probability of pathogenic rearrangement. Currently, it is unclear if outside of immunosuppression, MS immunomodulatory therapies can directly affect the replication and mutation rates of JCV.
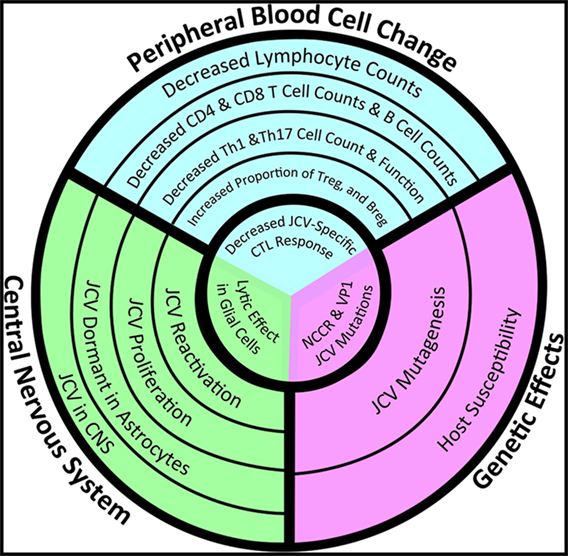
Figure 2. Convergence of peripheral immune changes, central nervous system (CNS) glial infectivity, and genetic susceptibility in the development of progressive multifocal leukoencephalopathy (PML). Whether a multiple sclerosis (MS) patient treated with immunomodulatory therapy will develop PML depends on changes in the peripheral immune system (blue) that lead to immunosuppression, particularly within the CNS (green), and in a genetic background (purple) that increases the susceptibility of glial cells to productive infection by John Cunningham virus (JCV). Risk increases as the circles get smaller, but a patient will not develop PML until they experience all three events located within the innermost circle: the loss of number/function of JCV-specific T-cells within the CNS in the context of a lytic JCV infection within oligodendrocytes by a version of JCV with a mutated non-coding regulatory region (NCCR). The failure of current assessments to accurately predict risk lies in the failure of these metrics to take into account all three facets. New analytics need to take into account JCV-specific lymphocyte function, JCV genetic variants, and the location of latent JCV within the body of a particular MS patient (Image copyright: Caitlyn Fisher and Yang Mao-Draayer, reprints with permission).
While immunosuppression increases viral mutation rates by facilitating viral reactivation and unchecked replication, the majority of mutations will be detrimental for the virus. Indeed, within PML patients, multiple genetic variants can be found, many of which actually make the virus less virulent (196). However, the persistence of multiple versions of the virus, even those with suboptimal adaptation, could be important for immune evasion. Recent work aimed at designing an effective vaccine for JCV suggests that the mutation of capsid proteins impairs the ability of anti-JCV antibodies to neutralize the infection (197). The presence of viral protein variants not targeted by anti-JCV antibodies could also help explain the disparity between antibody production and effective JCV clearance in PML patients. Following reactivation, continued immunosuppression further prevents the immune system from containing the mutated virus once it successfully infects oligodendrocytes. This genetic variability can also hamper analyses that rely on the detection of a specific sequence, thereby contributing to false negatives and inaccuracy of risk assessments. Efforts aimed at monitoring changes within the JCV genome in immunosuppressed patients over time could reveal the presence of a pattern of changes or cohort of mutations associated with a higher probability of developing PML. Newer assays such as high-resolution melting analysis (198) and multiplex qPCR (199) are improving the accuracy and availability of testing for NCCR rearrangements and could become a standard for monitoring risk.
Host Genetic Susceptibility
In addition to the composition of the viral genome, the genetic makeup of the host can influence the susceptibility and response to infection. Genome-wide association studies have indicated that many single-nucleotide polymorphism (SNP) variants that confer risk toward MS and other autoimmune diseases are involved in the function of the immune system (200, 201). Consequently, some of these SNPs could also increase susceptibility or offer protection against PML. For example, SNPs that lead to the augmentation of NF-κB-mediated signaling (202) could increase the risk of viral reactivation in glial cells. In patients with these SNPs, the use of immunotherapies that specifically target NF-κB could help mitigate this risk. Human leukocyte antigen (HLA) genes encode major histocompatibility complex cell surface proteins involved in antigen presentation, which are critical for immune system regulation. The presence or absence of particular HLA alleles has been shown to confer protection or susceptibility for autoimmune disorders and infectious diseases (203). HLA-DRB1*15 is the most well-established susceptibility allele for MS (204), but may confer protection against JCV. In a study of Scandinavian and German cohorts, MS patients with the DRB1*15-DQA1*01:02-DQB1*06:02 haplotype had a negative association with JCV serostatus (205). Since elevated JCV antibody index levels are associated with increased risk for PML (15), HLA alleles associated with lower levels of JCV antibody production are predicted to minimize risk. Interestingly, there appears to be an inverse relation between HLA alleles associated with risk for autoimmune diseases and PML. HLA-DRB1*13 is negatively associated with several autoimmune diseases and is hypothesized to play a protective role (206). However, the DRB1*13-DQA1*01:03-DQB1*06:03 haplotype was positively associated with JCV serostatus (205). These findings suggest that HLA typing may be useful in the stratification of patients at risk for PML.
Variation in IL-10 signaling could also contribute to PML risk. Polymorphisms in the IL-10 promoter, which mitigate the severity of MS (207), may also increase the susceptibility for PML. In contrast, many MS patients have increased levels of miR-98 (208), which negatively regulates IL-10, and could be protective in this context. miR-98 can also inhibit IL-10 production induced by toll-like receptor 4 (TLR4) (209). In polyomavirus-susceptible mice, viral activation of B-cells by TLR4 induces an IL-10 response, whereas TLR2 activation of dendritic cells in resistant mice leads to an IL-12 response (41). This may stem from differences in the posttranscriptional stability of IL-10 mRNA downstream of the TLRs, as TLR4 has been found to protect IL-10 mRNA from degradation (210). Although TL4 is typically expressed at very low levels in human B-cells, it is increased in the context of inflammatory disease (211). Furthermore, some viruses, such as hepatitis C virus (HCV), have been shown to increase TLR4 expression on B-cells (212). In addition, TLR4 expression is downstream of IL-10, and a correlation between IL-10 promoter SNPs, TLR4 expression, and response to therapy has been found in a cohort of HCV patients (213). TLR SNPs have been hypothesized to influence infection and persistence of a variety of viruses (214), which may be related to the interaction between TLRs and IL-10. Therefore, it is possible that differences in expression of TLR4 or other TLRs on antigen-presenting cells could influence susceptibility to productive polyomavirus infection, such as JCV, in humans. However, it is unclear what effect any single SNP has on disease, which currently makes it difficult to glean reliable diagnostic metrics from host genetic data.
Case reports of supposedly immunocompetent individuals who developed PML offer some insight into which genes may be most relevant. The ability of IFN-γ to inhibit T-antigen in a Jak/Stat-dependent manner (169), suggests that its presence can help prevent JCV reactivation. Gain-of-function mutations in Stat1 increase the susceptibility of infections due to the aberrant regulation of IFN-γ-mediated inflammation (215). These mutations enhance IFN-γ-induced gene expression but impair response to IFN-γ re-stimulation. Stat1 mutations have been found in three patients who succumbed to an aggressive form of PML, and these mutations are now understood to be a major risk genetic factor for PML (216). In addition, gene sequencing from another immunocompetent PML patient revealed a genetic mutation conferring a deficit in the production of IFN-γ (217). Although even less well understood, SNPs that influence a patient’s response to immunotherapy could also impact risk. Genetic testing works best in the context of single genes with well-characterized mutations, but, for the majority of patients, susceptibility for PML will involve minor variants in a variety of genes, which may be difficult to tease out. Overall, it is likely the changes induced by a therapy in combination with the genetic makeup of the individual that determines risk.
Conclusion
Ultimately, more studies are needed to determine why some patients develop PML. There appears to be a general scheme of immunosuppression leading to JCV reactivation and mutation toward a form that infects glia. Continued immunosuppression then inhibits effective viral clearance and culminates in PML. However, there are still many open questions regarding the details. Not all types of immunosuppression induce PML, and clearly not all immunosuppressed patients develop PML. Current studies suggest that CD4+ T-cell lymphocytopenia coupled with increases in IL-10-producing regulatory subsets specifically within the CNS may be most relevant. It is also unclear whether having latent JCV within the brain prior to immunosuppression confers greater risk than having it in the periphery, as well as how the mutant form of the virus develops. New biomarkers of reactivation and assays to monitor viral genomic sequences may help address these questions and provide for better monitoring strategies to reduce the future incidence of PML in MS patients.
Author Contributions
EM and YM-D contributed to the concept, design, and writing of this review.
Conflict of Interest Statement
EM has no conflict of interest. No commercial funding was received to support this work. YM-D has served as a consultant and/or received grant support from Acorda, Bayer Pharmaceutical, Biogen Idec, EMD Serono, Genzyme, Novartis, Questor, Chugai, and Teva Neuroscience.
Acknowledgments
We thank Caitlyn Fisher for assistance with the graphic design of the figures.
Funding
YM-D is currently supported by grants from NIH NIAID Autoimmune Center of Excellence: UM1-AI110557; NIH NINDS R01-NS080821 and the University of Michigan Neurology Department. EM is supported by a Kirschstein-NRSA 2T32HD007505-21.
References
1. Astrom KE, Mancall EL, Richardson EP Jr. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin’s disease. Brain (1958) 81(1):93–111.
2. Brew BJ, Davies NW, Cinque P, Clifford DB, Nath A. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol (2010) 6(12):667–79. doi:10.1038/nrneurol.2010.164
3. Bellizzi A, Nardis C, Anzivino E, Rodio D, Fioriti D, Mischitelli M, et al. Human polyomavirus JC reactivation and pathogenetic mechanisms of progressive multifocal leukoencephalopathy and cancer in the era of monoclonal antibody therapies. J Neurovirol (2012) 18(1):1–11. doi:10.1007/s13365-012-0080-7
4. Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol (2010) 9(4):425–37. doi:10.1016/S1474-4422(10)70040-5
5. Engsig FN, Hansen AB, Omland LH, Kronborg G, Gerstoft J, Laursen AL, et al. Incidence, clinical presentation, and outcome of progressive multifocal leukoencephalopathy in HIV-infected patients during the highly active antiretroviral therapy era: a nationwide cohort study. J Infect Dis (2009) 199(1):77–83. doi:10.1086/595299
6. Vargas DL, Tyor WR. Update on disease-modifying therapies for multiple sclerosis. J Investig Med (2017) 65(5):883–91. doi:10.1136/jim-2016-000339
7. Berger JR. Classifying PML risk with disease modifying therapies. Mult Scler Relat Disord (2017) 12:59–63. doi:10.1016/j.msard.2017.01.006
8. Asztely F, Gilland E, Wattjes MP, Lycke J. Rituximab treatment did not aggravate ongoing progressive multifocal leukoencephalopathy in a patient with multiple sclerosis. J Neurol Sci (2015) 353(1–2):155–7. doi:10.1016/j.jns.2015.04.010
9. Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports Project. Blood (2009) 113(20):4834–40. doi:10.1182/blood-2008-10-186999
10. Francis G, Kappos L, O’Connor P, Collins W, Tang D, Mercier F, et al. Temporal profile of lymphocyte counts and relationship with infections with fingolimod therapy. Mult Scler (2014) 20(4):471–80. doi:10.1177/1352458513500551
11. Bridel C, Beauverd Y, Samii K, Lalive PH. Hematologic modifications in natalizumab-treated multiple sclerosis patients: an 18-month longitudinal study. Neurol Neuroimmunol Neuroinflamm (2015) 2(4):e123. doi:10.1212/NXI.0000000000000123
12. Longbrake EE, Naismith RT, Parks BJ, Wu GF, Cross AH. Dimethyl fumarate-associated lymphopenia: risk factors and clinical significance. Mult Scler J Exp Transl Clin (2015) 1. doi:10.1177/2055217315596994
13. Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med (2012) 366(20):1870–80. doi:10.1056/NEJMoa1107829
14. Berger JR, Fox RJ. Reassessing the risk of natalizumab-associated PML. J Neurovirol (2016) 22(4):533–5. doi:10.1007/s13365-016-0427-6
15. Plavina T, Subramanyam M, Bloomgren G, Richman S, Pace A, Lee S, et al. Anti-JC virus antibody levels in serum or plasma further define risk of natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol (2014) 76(6):802–12. doi:10.1002/ana.24286
16. Major EO, Frohman E, Douek D. JC viremia in natalizumab-treated patients with multiple sclerosis. N Engl J Med (2013) 368(23):2240–1. doi:10.1056/NEJMc1214233
17. Correia I, Jesus-Ribeiro J, Batista S, Martins AI, Nunes C, Macario MC, et al. Anti-JCV antibody serostatus and longitudinal evaluation in a Portuguese multiple sclerosis population. J Clin Neurosci (2017) 45:257–60. doi:10.1016/j.jocn.2017.08.006
18. Schwab N, Schneider-Hohendorf T, Hoyt T, Gross CC, Meuth SG, Klotz L, et al. Anti-JCV serology during natalizumab treatment: review and meta-analysis of 17 independent patient cohorts analyzing anti-John Cunningham polyoma virus sero-conversion rates under natalizumab treatment and differences between technical and biological sero-converters. Mult Scler (2017). doi:10.1177/1352458517728814
19. Castro-Borrero W, Graves D, Frohman TC, Flores AB, Hardeman P, Logan D, et al. Current and emerging therapies in multiple sclerosis: a systematic review. Ther Adv Neurol Disord (2012) 5(4):205–20. doi:10.1177/1756285612450936
20. Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm (2013) 2013:963748. doi:10.1155/2013/963748
21. Berger JR, Pall L, Lanska D, Whiteman M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol (1998) 4:59–68. doi:10.3109/13550289809113482
22. Arce-Sillas A, Alvarez-Luquin DD, Tamaya-Dominguez B, Gomez-Fuentes S, Trejo-Garcia A, Melo-Salas M, et al. Regulatory T cells: molecular actions on effector cells in immune regulation. J Immunol Res (2016) 2016:1720827. doi:10.1155/2016/1720827
23. Venken K, Hellings N, Liblau R, Stinissen P. Disturbed regulatory T cell homeostasis in multiple sclerosis. Trends Mol Med (2010) 16(2):58–68. doi:10.1016/j.molmed.2009.12.003
24. Dalla Libera D, Di Mitri D, Bergami A, Centonze D, Gasperini C, Grasso MG, et al. T regulatory cells are markers of disease activity in multiple sclerosis patients. PLoS One (2011) 6(6):e21386. doi:10.1371/journal.pone.0021386
25. Dhaeze T, Peelen E, Hombrouck A, Peeters L, Van Wijmeersch B, Lemkens N, et al. Circulating follicular regulatory T cells are defective in multiple sclerosis. J Immunol (2015) 195(3):832–40. doi:10.4049/jimmunol.1500759
26. Salmaggi A, Dufour A, Eoli M, Corsini E, La Mantia L, Massa G, et al. Low serum interleukin-10 levels in multiple sclerosis: further evidence for decreased systemic immunosuppression? J Neurol (1996) 243(1):13–7. doi:10.1007/BF00878525
27. Trenova A, Slavov G. Cytokines in multiple sclerosis – possible targets for immune therapies. J Neurol Exp Neurosci (2016) 1(2):25–9. doi:10.177561/jnen.2016-006
28. Rojas JM, Avia M, Martin V, Sevilla N. IL-10: a multifunctional cytokine in viral infections. J Immunol Res (2017) 2017:6104054. doi:10.1155/2017/6104054
29. Slobedman B, Barry PA, Spencer JV, Avdic S, Abendroth A. Virus-encoded homologs of cellular interleukin-10 and their control of host immune function. J Virol (2009) 83(19):9618–29. doi:10.1128/JVI.01098-09
30. Das A, Ellis G, Pallant C, Lopes AR, Khanna P, Peppa D, et al. IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J Immunol (2012) 189(8):3925–35. doi:10.4049/jimmunol.1103139
31. Velupillai P, Carroll JP, Benjamin TL. Susceptibility to polyomavirus-induced tumors in inbred mice: role of innate immune responses. J Virol (2002) 76(19):9657–63. doi:10.1128/JVI.76.19.9657-9663.2002
32. Weber F, Goldmann C, Kramer M, Kaup FJ, Pickhardt M, Young P, et al. Cellular and humoral immune response in progressive multifocal leukoencephalopathy. Ann Neurol (2001) 49(5):636–42. doi:10.1002/ana.1004
33. Perkins MR, Ryschkewitsch C, Liebner JC, Monaco MCG, Himelfarb D, Ireland S, et al. Changes in JC virus-specific T Cell responses during natalizumab treatment and in natalizumab-associated progressive multifocal leukoencephalopathy. PLoS Pathog (2012) 8(11):e1003014. doi:10.1371/journal.ppat.1003014
34. Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology (2010) 129(4):474–81. doi:10.1111/j.1365-2567.2010.03255.x
35. Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology (2015) 47(9–480):180–93. doi:10.1016/j.virol.2014.12.033
36. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi:10.1038/nri3862
37. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515(7528):568–71. doi:10.1038/nature13954
38. Tan CS, Bord E, Broge TAJ, Glotzbecker B, Mills H, Gheuens S, et al. Increased program cell death–1 expression on T lymphocytes of patients with progressive multifocal leukoencephalopathy. J Acquir Immune Defic Syndr (2012) 60(3):244–8. doi:10.1097/QAI.0b013e31825a313c
39. Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, Maurer M, et al. A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol (2005) 58(1):50–7. doi:10.1002/ana.20514
40. Pawlak-Adamska E, Nowak O, Karabon L, Pokryszko-Dragan A, Partyka A, Tomkiewicz A, et al. PD-1 gene polymorphic variation is linked with first symptom of disease and severity of relapsing-remitting form of MS. J Neuroimmunol (2017) 305:115–27. doi:10.1016/j.jneuroim.2017.02.006
41. Velupillai P, Garcea RL, Benjamin TL. Polyoma virus-like particles elicit polarized cytokine responses in APCs from tumor-susceptible and -resistant mice. J Immunol (2006) 176(2):1148–53. doi:10.4049/jimmunol.176.2.1148
42. Hatton OL, Harris-Arnold A, Schaffert S, Krams SM, Martinez OM. The interplay between Epstein-Barr virus and B lymphocytes: implications for infection, immunity, and disease. Immunol Res (2014) 58(2–3):268–76. doi:10.1007/s12026-014-8496-1
43. Chapagain ML, Nerurkar VR. Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J Infect Dis (2010) 202(2):184–91. doi:10.1086/653823
44. Marshall LJ, Dunham L, Major EO. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer of JC virus and supports viral activity. J Gen Virol (2010) 91(Pt 12):3042–52. doi:10.1099/vir.0.023184-0
45. Johnson EM, Wortman MJ, Dagdanova AV, Lundberg PS, Daniel DC. Polyomavirus JC in the context of immunosuppression: a series of adaptive, DNA replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin Dev Immunol (2013) 2013:197807. doi:10.1155/2013/197807
46. Wortman MJ, Lundberg PS, Dagdanova AV, Venkataraman P, Daniel DC, Johnson EM. Opportunistic DNA recombination with Epstein-Barr virus at sites of control region rearrangements mediating JC virus neurovirulence. J Infect Dis (2016) 213(9):1436–43. doi:10.1093/infdis/jiv755
47. Garrett-Sinha LA, Su GH, Rao S, Kabak S, Hao Z, Clark MR, et al. PU.1 and Spi-B are required for normal B cell receptor-mediated signal transduction. Immunity (1999) 10(4):399–408. doi:10.1016/S1074-7613(00)80040-0
48. Lindberg RL, Achtnichts L, Hoffmann F, Kuhle J, Kappos L. Natalizumab alters transcriptional expression profiles of blood cell subpopulations of multiple sclerosis patients. J Neuroimmunol (2008) 194(1–2):153–64. doi:10.1016/j.jneuroim.2007.11.007
49. Marshall LJ, Ferenczy MW, Daley EL, Jensen PN, Ryschkewitsch CF, Major EO. Lymphocyte gene expression and JC virus noncoding control region sequences are linked with the risk of progressive multifocal leukoencephalopathy. J Virol (2014) 88(9):5177–83. doi:10.1128/JVI.03221-13
50. Houff SA, Berger J. The curious incident of the dog in the nighttime: does the absence of virus replication in Epstein-Barr virus-transformed B cells point to an important feature of JC virus biology? J Infect Dis (2010) 202(2):181–3. doi:10.1086/653824
51. Krumbholz M, Meinl I, Kumpfel T, Hohlfeld R, Meinl E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology (2008) 71(17):1350–4. doi:10.1212/01.wnl.0000327671.91357.96
52. Saraste M, Penttilä T-L, Airas L. Natalizumab treatment leads to an increase in circulating CXCR3-expressing B cells. Neurol Neuroimmunol Neuroinflamm (2016) 3(6):e292. doi:10.1212/nxi.0000000000000292
53. Hutchinson M. Natalizumab: a new treatment for relapsing remitting multiple sclerosis. Ther Clin Risk Manag (2007) 3(2):259–68. doi:10.2147/tcrm.2007.3.2.259
54. Rice GP, Hartung HP, Calabresi PA. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology (2005) 64(8):1336–42. doi:10.1212/01.WNL.0000158329.30470.D0
55. Schwab N, Schneider-Hohendorf T, Wiendl H. Therapeutic uses of anti-α4-integrin (anti-VLA-4) antibodies in multiple sclerosis. Int Immunol (2015) 27(1):47–53. doi:10.1093/intimm/dxu096
56. Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol (1986) 137(4):1270–4.
57. Flanagan K, Fitzgerald K, Baker J, Regnstrom K, Gardai S, Bard F, et al. Laminin-411 is a vascular ligand for MCAM and facilitates TH17 cell entry into the CNS. PLoS One (2012) 7(7):e40443. doi:10.1371/journal.pone.0040443
58. Carrithers MD, Visintin I, Kang SJ, Janeway CA Jr. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain (2000) 123(Pt 6):1092–101. doi:10.1093/brain/123.6.1092
59. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol (2009) 10(5):514–23. doi:10.1038/ni.1716
60. Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol (2008) 181(12):8391–401. doi:10.4049/jimmunol.181.12.8391
61. Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B, et al. Th17 lymphocytes traffic to the central nervous system independently of alpha4 integrin expression during EAE. J Exp Med (2011) 208(12):2465–76. doi:10.1084/jem.20110434
62. Paroni M, Maltese V, De Simone M, Ranzani V, Larghi P, Fenoglio C, et al. Recognition of viral and self-antigens by TH1 and TH1/TH17 central memory cells in patients with multiple sclerosis reveals distinct roles in immune surveillance and relapses. J Allergy ClinImmunol (2017) 140(3):797–808. doi:10.1016/j.jaci.2016.11.045
63. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol (2012) 33(12):579–89. doi:10.1016/j.it.2012.07.004
64. Sagar D, Lamontagne A, Foss CA, Khan ZK, Pomper MG, Jain P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood-brain barrier through paracellular transmigration and ERK activation. J Neuroinflammation (2012) 9:245. doi:10.1186/1742-2094-9-245
65. Puig-Kroger A, Sanz-Rodriguez F, Longo N, Sanchez-Mateos P, Botella L, Teixido J, et al. Maturation-dependent expression and function of the CD49d integrin on monocyte-derived human dendritic cells. J Immunol (2000) 165(8):4338–45. doi:10.4049/jimmunol.165.8.4338
66. de Andres C, Teijeiro R, Alonso B, Sanchez-Madrid F, Martinez ML, Guzman de Villoria J, et al. Long-term decrease in VLA-4 expression and functional impairment of dendritic cells during natalizumab therapy in patients with multiple sclerosis. PLoS One (2012) 7(4):e34103. doi:10.1371/journal.pone.0034103
67. Mo RR, Eisenbraun JK, Sonstein J, Craig RA, Curtis JL, Stoolman LM, et al. CD49d overexpression and T cell autoimmunity. J Immunol (2003) 171(2):745–53. doi:10.4049/jimmunol.171.2.745
68. Bornsen L, Christensen JR, Ratzer R, Oturai AB, Sorensen PS, Sondergaard HB, et al. Effect of natalizumab on circulating CD4+ T-cells in multiple sclerosis. PLoS One (2012) 7(11):e47578. doi:10.1371/journal.pone.0047578
69. Planas R, Jelcic I, Schippling S, Martin R, Sospedra M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur J Immunol (2012) 42(3):790–8. doi:10.1002/eji.201142108
70. Mellergård J, Edström M, Jenmalm MC, Dahle C, Vrethem M, Ernerudh J. Increased B cell and cytotoxic NK cell proportions and increased T cell responsiveness in blood of natalizumab-treated multiple sclerosis patients. PLoS One (2013) 8(12):e81685. doi:10.1371/journal.pone.0081685
71. Khademi M, Stol D, Olsson T, Wallstrom E. Induction of systemic TNFalpha in natalizumab-treated multiple sclerosis. Eur J Neurol (2008) 15(3):309–12. doi:10.1111/j.1468-1331.2007.02037.x
72. Kivisakk P, Healy BC, Viglietta V, Quintana FJ, Hootstein MA, Weiner HL, et al. Natalizumab treatment is associated with peripheral sequestration of proinflammatory T cells. Neurology (2009) 72(22):1922–30. doi:10.1212/WNL.0b013e3181a8266f
73. Frisullo G, Iorio R, Plantone D, Marti A, Nociti V, Patanella AK, et al. CD4+T-bet+, CD4+pSTAT3+ and CD8+T-bet+ T cells accumulate in peripheral blood during NZB treatment. Mult Scler (2011) 17(5):556–66. doi:10.1177/1352458510392263
74. Kimura K, Nakamura M, Sato W, Okamoto T, Araki M, Lin Y, et al. Disrupted balance of T cells under natalizumab treatment in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3(2):e210. doi:10.1212/NXI.0000000000000210
75. Geginat J, Paroni M, Pagani M, Galimberti D, De Francesco R, Scarpini E, et al. The enigmatic role of viruses in multiple sclerosis: molecular mimicry or disturbed immune surveillance? Trends Immunol (2017) 38(7):498–512. doi:10.1016/j.it.2017.04.006
76. Marousi S, Karkanis I, Kalamatas T, Travasarou M, Paterakis G, Karageorgiou CE. Immune cells after prolonged natalizumab therapy: implications for effectiveness and safety. Acta Neurol Scand (2013) 128(1):e1–5. doi:10.1111/ane.12080
77. Carotenuto A, Scalia G, Ausiello F, Moccia M, Russo CV, Sacca F, et al. CD4/CD8 ratio during natalizumab treatment in multiple sclerosis patients. J Neuroimmunol (2017) 309:47–50. doi:10.1016/j.jneuroim.2017.05.006
78. Stuve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol (2006) 63(10):1383–7. doi:10.1001/archneur.63.10.1383
79. Stuve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol (2006) 59(5):743–7. doi:10.1002/ana.20858
80. Warnke C, Stettner M, Lehmensiek V, Dehmel T, Mausberg AK, von Geldern G, et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult Scler (2014) 21(8):1036–44. doi:10.1177/1352458514556296
81. Harrer A, Pilz G, Wipfler P, Oppermann K, Sellner J, Hitzl W, et al. High interindividual variability in the CD4/CD8 T cell ratio and natalizumab concentration levels in the cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol (2015) 180(3):383–92. doi:10.1111/cei.12590
82. Schneider-Hohendorf T, Rossaint J, Mohan H, Böning D, Breuer J, Kuhlmann T, et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J Exp Med (2014) 211(9):1833–46. doi:10.1084/jem.20140540
83. Mellergard J, Edstrom M, Vrethem M, Ernerudh J, Dahle C. Natalizumab treatment in multiple sclerosis: marked decline of chemokines and cytokines in cerebrospinal fluid. Mult Scler (2010) 16(2):208–17. doi:10.1177/1352458509355068
84. Khademi M, Bornsen L, Rafatnia F, Andersson M, Brundin L, Piehl F, et al. The effects of natalizumab on inflammatory mediators in multiple sclerosis: prospects for treatment-sensitive biomarkers. Eur J Neurol (2009) 16(4):528–36. doi:10.1111/j.1468-1331.2009.02532.x
85. Du Pasquier RA, Kuroda MJ, Zheng Y, Jean-Jacques J, Letvin NL, Koralnik IJ. A prospective study demonstrates an association between JC virus-specific cytotoxic T lymphocytes and the early control of progressive multifocal leukoencephalopathy. Brain (2004) 127(9):1970–8. doi:10.1093/brain/awh215
86. Gheuens S, Bord E, Kesari S, Simpson DM, Gandhi RT, Clifford DB, et al. Role of CD4+ and CD8+ T-cell responses against JC virus in the outcome of patients with progressive multifocal leukoencephalopathy (PML) and PML with immune reconstitution inflammatory syndrome. J Virol (2011) 85(14):7256–63. doi:10.1128/jvi.02506-10
87. Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP, et al. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med (2009) 361(11):1067–74. doi:10.1056/NEJMoa0904267
88. Hendel-Chavez H, de Goer de Herve MG, Giannesini C, Mazet AA, Papeix C, Louapre C, et al. Immunological hallmarks of JC virus replication in multiple sclerosis patients on long-term natalizumab therapy. J Virol (2013) 87(10):6055–9. doi:10.1128/JVI.00131-13
89. Molloy MJ, Zhang W, Usherwood EJ. Suppressive CD8+ T cells arise in the absence of CD4 help and compromise control of persistent virus. J Immunol (2011) 186(11):6218–26. doi:10.4049/jimmunol.1003812
90. McDermott DS, Varga SM. Quantifying antigen-specific CD4 T cells during a viral infection: CD4 T cell responses are larger than we think. J Immunol (2011) 187(11):5568–76. doi:10.4049/jimmunol.1102104
91. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature (2004) 427(6972):355–60. doi:10.1038/nature02284
92. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401(6754):708–12. doi:10.1038/44385
93. Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology (2008) 71(16):1261–7. doi:10.1212/01.wnl.0000327609.57688.ea
94. Mazzola MA, Raheja R, Murugaiyan G, Rajabi H, Kumar D, Pertel T, et al. Identification of a novel mechanism of action of fingolimod (FTY720) on human effector T cell function through TCF-1 upregulation. J Neuroinflammation (2015) 12:245. doi:10.1186/s12974-015-0460-z
95. Killestein J, Vennegoor A, van Golde AEL, Bourez RLJH, Wijlens MLB, Wattjes MP. PML-IRIS during fingolimod diagnosed after natalizumab discontinuation. Case Rep Neurol Med (2014) 2014:4. doi:10.1155/2014/307872
97. Sato DK, Nakashima I, Bar-Or A, Misu T, Suzuki C, Nishiyama S, et al. Changes in Th17 and regulatory T cells after fingolimod initiation to treat multiple sclerosis. J Neuroimmunol (2014) 268(1–2):95–8. doi:10.1016/j.jneuroim.2014.01.008
98. Claes N, Dhaeze T, Fraussen J, Broux B, Van Wijmeersch B, Stinissen P, et al. Compositional changes of B and T cell subtypes during fingolimod treatment in multiple sclerosis patients: a 12-month follow-up study. PLoS One (2014) 9(10):e111115. doi:10.1371/journal.pone.0111115
99. Nakamura M, Matsuoka T, Chihara N, Miyake S, Sato W, Araki M, et al. Differential effects of fingolimod on B-cell populations in multiple sclerosis. Mult Scler (2014) 20(10):1371–80. doi:10.1177/1352458514523496
100. Grützke B, Hucke S, Gross CC, Herold MVB, Posevitz-Fejfar A, Wildemann BT, et al. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann Clin Transl Neurol (2015) 2(2):119–30. doi:10.1002/acn3.155
101. Blumenfeld S, Staun-Ram E, Miller A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL-10 and TGFbeta in patients with multiple sclerosis. J Autoimmun (2016) 70:40–51. doi:10.1016/j.jaut.2016.03.012
102. Serpero LD, Filaci G, Parodi A, Battaglia F, Kalli F, Brogi D, et al. Fingolimod modulates peripheral effector and regulatory T cells in MS patients. J Neuroimmune Pharmacol (2013) 8(5):1106–13. doi:10.1007/s11481-013-9465-5
103. Rudnicka J, Czerwiec M, Grywalska E, Siwicka-Gieroba D, Walankiewicz M, Grafka A, et al. Influence of fingolimod on basic lymphocyte subsets frequencies in the peripheral blood of multiple sclerosis patients – preliminary study. Cent Eur J Immunol (2015) 40(3):354–9. doi:10.5114/ceji.2015.54599
104. Muls N, Dang HA, Sindic CJ, van Pesch V. Fingolimod increases CD39-expressing regulatory T cells in multiple sclerosis patients. PLoS One (2014) 9(11):e113025. doi:10.1371/journal.pone.0113025
105. Kowarik MC, Pellkofer HL, Cepok S, Korn T, Kumpfel T, Buck D, et al. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology (2011) 76(14):1214–21. doi:10.1212/WNL.0b013e3182143564
106. Yokoseki A, Saji E, Arakawa M, Hokari M, Ishiguro T, Yanagimura F, et al. Relapse of multiple sclerosis in a patient retaining CCR7-expressing T cells in CSF under fingolimod therapy. Mult Scler (2013) 19(9):1230–3. doi:10.1177/1352458513481395
107. Novartis. Gilenya (Novartis Pharmaceuticals Corporation): FDA Package Insert. (2016). Available from: http://Medlibrary.org
108. Ricklin ME, Lorscheider J, Waschbisch A, Paroz C, Mehta SK, Pierson DL, et al. T-cell response against varicella-zoster virus in fingolimod-treated MS patients. Neurology (2013) 81(2):174–81. doi:10.1212/WNL.0b013e31829a3311
109. Mathias A, Perriard G, Canales M, Vuilleumier F, Perrotta G, Schluep M, et al. The VZV/IE63-specific T cell response prevents herpes zoster in fingolimod-treated patients. Neurol Neuroimmunol Neuroinflamm (2016) 3(2):e209. doi:10.1212/NXI.0000000000000209
110. EMA. Updated Recommendations to Minimise the Risk of the Rare Brain Infection PML with Tecfidera. London: European Medicines Agency (2015).
111. Biogen. Tecfidera (Biogen Inc.): FDA Package Insert. (2017). Available from: http://MedLibrary.org
112. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med (2015) 372(15):1476–8. doi:10.1056/NEJMc1415408
113. Baharnoori M, Lyons J, Dastagir A, Koralnik I, Stankiewicz JM. Nonfatal PML in a patient with multiple sclerosis treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm (2016) 3(5):e274. doi:10.1212/nxi.0000000000000274
114. Lehmann-Horn K, Penkert H, Grein P, Leppmeier U, Teuber-Hanselmann S, Hemmer B, et al. PML during dimethyl fumarate treatment of multiple sclerosis: how does lymphopenia matter? Neurology (2016) 87(4):440–1. doi:10.1212/WNL.0000000000002900
115. Gieselbach RJ, Muller-Hansma AH, Wijburg MT, de Bruin-Weller MS, van Oosten BW, Nieuwkamp DJ, et al. Progressive multifocal leukoencephalopathy in patients treated with fumaric acid esters: a review of 19 cases. J Neurol (2017) 264(6):1155–64. doi:10.1007/s00415-017-8509-9
116. Nieuwkamp DJ, Murk JL, van Oosten BW, Cremers CH, Killestein J, Viveen MC, et al. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med (2015) 372(15):1474–6. doi:10.1056/NEJMc1413724
117. Al-Jaderi Z, Maghazachi AA. Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer, and other diseases. Front Immunol (2016) 7:278. doi:10.3389/fimmu.2016.00278
118. Treumer F, Zhu K, Glaser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol (2003) 121(6):1383–8. doi:10.1111/j.1523-1747.2003.12605.x
119. Linker RA, Lee DH, Stangel M, Gold R. Fumarates for the treatment of multiple sclerosis: potential mechanisms of action and clinical studies. Expert Rev Neurother (2008) 8(11):1683–90. doi:10.1586/14737175.8.11.1683
120. Ockenfels HM, Schultewolter T, Ockenfels G, Funk R, Goos M. The antipsoriatic agent dimethylfumarate immunomodulates T-cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br J Dermatol (1998) 139(3):390–5. doi:10.1046/j.1365-2133.1998.02400.x
121. Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BAC, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm (2015) 2(3):e76. doi:10.1212/nxi.0000000000000076
122. Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Mult Scler (2015) 22(8):1061–70. doi:10.1177/1352458515608961
123. Gross CC, Schulte-Mecklenbeck A, Klinsing S, Posevitz-Fejfár A, Wiendl H, Klotz L. Dimethyl fumarate treatment alters circulating T helper cell subsets in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3(1):e183. doi:10.1212/NXI.0000000000000183
124. Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao-Draayer Y. Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J Immunol (2017) 198(8):3069–80. doi:10.4049/jimmunol.1601532
125. Lundy SK, Wu Q, Wang Q, Dowling CA, Taitano SH, Mao G, et al. Dimethyl fumarate treatment of relapsing-remitting multiple sclerosis influences B-cell subsets. Neurol Neuroimmunol Neuroinflamm (2016) 3(2):e211. doi:10.1212/NXI.0000000000000211
126. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol (2017) 198(2):691–8. doi:10.4049/jimmunol.1601649
127. Smith MD, Martin KA, Calabresi PA, Bhargava P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann Clin Transl Neurol (2017) 4(5):351–5. doi:10.1002/acn3.411
128. Vandermeeren M, Janssens S, Borgers M, Geysen J. Dimethylfumarate is an inhibitor of cytokine-induced E-selectin, VCAM-1, and ICAM-1 expression in human endothelial cells. Biochem Biophys Res Commun (1997) 234(1):19–23. doi:10.1006/bbrc.1997.6570
129. Rubant SA, Ludwig RJ, Diehl S, Hardt K, Kaufmann R, Pfeilschifter JM, et al. Dimethylfumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J Invest Dermatol (2008) 128(2):326–31. doi:10.1038/sj.jid.5700996
130. Wallbrecht K, Drick N, Hund AC, Schon MP. Downregulation of endothelial adhesion molecules by dimethylfumarate, but not monomethylfumarate, and impairment of dynamic lymphocyte-endothelial cell interactions. Exp Dermatol (2011) 20(12):980–5. doi:10.1111/j.1600-0625.2011.01376.x
131. Kihara Y, Groves A, Rivera RR, Chun J. Dimethyl fumarate inhibits integrin alpha4 expression in multiple sclerosis models. Ann Clin Transl Neurol (2015) 2(10):978–83. doi:10.1002/acn3.251
132. Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, et al. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe (2010) 8(4):309–19. doi:10.1016/j.chom.2010.09.004
133. Stroh LJ, Maginnis MS, Blaum BS, Nelson CD, Neu U, Gee GV, et al. The greater affinity of JC polyomavirus capsid for alpha2,6-linked lactoseries tetrasaccharide c than for other sialylated glycans is a major determinant of infectivity. J Virol (2015) 89(12):6364–75. doi:10.1128/JVI.00489-15
134. Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, et al. 5-HT2 receptors facilitate JC polyomavirus entry. J Virol (2013) 87(24):13490–8. doi:10.1128/JVI.02252-13
135. Haley SA, O’Hara BA, Nelson CD, Brittingham FL, Henriksen KJ, Stopa EG, et al. Human polyomavirus receptor distribution in brain parenchyma contrasts with receptor distribution in kidney and choroid plexus. Am J Pathol (2015) 185(8):2246–58. doi:10.1016/j.ajpath.2015.04.003
136. Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology (2007) 364(1):55–63. doi:10.1016/j.virol.2007.02.018
137. Agnihotri SP, Wuthrich C, Dang X, Nauen D, Karimi R, Viscidi R, et al. A fatal case of JC virus meningitis presenting with hydrocephalus in a human immunodeficiency virus-seronegative patient. Ann Neurol (2014) 76(1):140–7. doi:10.1002/ana.24192
138. Jensen PN, Major EO. A classification scheme for human polyomavirus JCV variants based on the nucleotide sequence of the noncoding regulatory region. J Neurovirol (2001) 7(4):280–7. doi:10.1080/13550280152537102
139. White FA III, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol (1992) 66(10):5726–34.
140. Elsner C, Dorries K. Human polyomavirus JC control region variants in persistently infected CNS and kidney tissue. J Gen Virol (1998) 79(Pt 4):789–99. doi:10.1099/0022-1317-79-4-789
141. Tan CS, Ellis LC, Wuthrich C, Ngo L, Broge TA Jr, Saint-Aubyn J, et al. JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. J Virol (2010) 84(18):9200–9. doi:10.1128/JVI.00609-10
142. Petrik J. Immunomodulatory effects of exosomes produced by virus-infected cells. Transfus Apher Sci (2016) 55(1):84–91. doi:10.1016/j.transci.2016.07.014
143. Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol (2014) 14(3):195–208. doi:10.1038/nri3622
144. Verma P, Pandey RK, Prajapati P, Prajapati VK. Circulating MicroRNAs: potential and emerging biomarkers for diagnosis of human infectious diseases. Front Microbiol (2016) 7:1274. doi:10.3389/fmicb.2016.01274
145. Lagatie O, Van Loy T, Tritsmans L, Stuyver LJ. Circulating human microRNAs are not linked to JC polyomavirus serology or urinary viral load in healthy subjects. Virol J (2014) 11:41. doi:10.1186/1743-422X-11-41
146. Lagatie O, Van Loy T, Tritsmans L, Stuyver LJ. Viral miRNAs in plasma and urine divulge JC polyomavirus infection. Virol J (2014) 11:158. doi:10.1186/1743-422X-11-158
147. Pietila T, Nummi M, Auvinen P, Mannonen L, Auvinen E. Expression of BKV and JCV encoded microRNA in human cerebrospinal fluid, plasma and urine. J Clin Virol (2015) 65:1–5. doi:10.1016/j.jcv.2015.01.019
148. Seo GJ, Fink LH, O’Hara B, Atwood WJ, Sullivan CS. Evolutionarily conserved function of a viral microRNA. J Virol (2008) 82(20):9823–8. doi:10.1128/JVI.01144-08
149. Bauman Y, Nachmani D, Vitenshtein A, Tsukerman P, Drayman N, Stern-Ginossar N, et al. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe (2011) 9(2):93–102. doi:10.1016/j.chom.2011.01.008
150. Rossi F, Prosperini L, Rossi N, Capra R, Rivanera D, Li X, et al. Association between BKPyV serotype I antibody level and natalizumab-associated progressive multifocal leukoencephalopathy. Viral Immunol (2017) 8:622–6. doi:10.1089/vim.2017.0039
151. Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, et al. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest (2014) 124(12):5323–36. doi:10.1172/JCI76629
152. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
153. Grafstein B, Liu S, Cotrina ML, Goldman SA, Nedergaard M. Meningeal cells can communicate with astrocytes by calcium signaling. Ann Neurol (2000) 47(1):18–25. doi:10.1002/1531-8249(200001)47:1<18::AID-ANA6>3.0.CO;2-N
154. Anders JJ, Salopek M. Meningeal cells increase in vitro astrocytic gap junctional communication as measured by fluorescence recovery after laser photobleaching. J Neurocytol (1989) 18(2):257–64. doi:10.1007/BF01206666
155. Lemcke H, Steinhoff G, David R. Gap junctional shuttling of miRNA – a novel pathway of intercellular gene regulation and its prospects in clinical application. Cell Signal (2015) 27(12):2506–14. doi:10.1016/j.cellsig.2015.09.012
156. Castellano P, Eugenin EA. Regulation of gap junction channels by infectious agents and inflammation in the CNS. Front Cell Neurosci (2014) 8:122. doi:10.3389/fncel.2014.00122
157. Kielian T. Glial connexins and gap junctions in CNS inflammation and disease. J Neurochem (2008) 106(3):1000–16. doi:10.1111/j.1471-4159.2008.05405.x
158. McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun (2010) 394(4):1006–11. doi:10.1016/j.bbrc.2010.03.111
159. Barcia C Sr, Mitxitorena I, Carrillo-de Sauvage MA, Gallego JM, Perez-Valles A, Barcia C Jr. Imaging the microanatomy of astrocyte-T-cell interactions in immune-mediated inflammation. Front Cell Neurosci (2013) 7:58. doi:10.3389/fncel.2013.00058
160. Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M. Balancing the immune response in the brain: IL-10 and its regulation. J Neuroinflammation (2016) 13(1):297. doi:10.1186/s12974-016-0763-8
161. Frisque RJ. Nucleotide sequence of the region encompassing the JC virus origin of DNA replication. J Virol (1983) 46(1):170–6.
162. White MK, Safak M, Khalili K. Regulation of gene expression in primate polyomaviruses. J Virol (2009) 83(21):10846–56. doi:10.1128/JVI.00542-09
163. Tavis JE, Frisque RJ. Altered DNA binding and replication activities of JC virus T-antigen mutants. Virology (1991) 183(1):239–50. doi:10.1016/0042-6822(91)90136-Y
164. Kim HS, Goncalves NM, Henson JW. Glial cell-specific regulation of the JC virus early promoter by large T antigen. J Virol (2000) 74(2):755–63. doi:10.1128/JVI.74.2.755-763.2000
165. Sariyer IK, Khalili K. Regulation of human neurotropic JC virus replication by alternative splicing factor SF2/ASF in glial cells. PLoS One (2011) 6(1):e14630. doi:10.1371/journal.pone.0014630
166. Uleri E, Regan P, Dolei A, Sariyer IK. SF2/ASF binding region within JC virus NCCR limits early gene transcription in glial cells. Virol J (2013) 10:147. doi:10.1186/1743-422X-10-147
167. Craigie M, Regan P, Otalora Y-L, Sariyer IK. Molecular interplay between T-antigen and splicing factor, arginine/serine-rich 1 (SRSF1) controls JC virus gene expression in glial cells. Virol J (2015) 12:196. doi:10.1186/s12985-015-0426-x
168. Sariyer IK, Sariyer R, Otte J, Gordon J. Pur-alpha induces JCV gene expression and viral replication by suppressing SRSF1 in glial cells. PLoS One (2016) 11(6):e0156819. doi:10.1371/journal.pone.0156819
169. De-Simone FI, Sariyer R, Otalora Y-L, Yarandi S, Craigie M, Gordon J, et al. IFN-gamma inhibits JC virus replication in glial cells by suppressing T-antigen expression. PLoS One (2015) 10(6):e0129694. doi:10.1371/journal.pone.0129694
170. Uleri E, Ibba G, Piu C, Caocci M, Leoni S, Arru G, et al. JC polyomavirus expression and bell-shaped regulation of its SF2/ASF suppressor during the follow-up of multiple sclerosis patients treated with natalizumab. J Neurovirol (2017) 23(2):226–38. doi:10.1007/s13365-016-0492-x
171. Meira M, Sievers C, Hoffmann F, Haghikia A, Rasenack M, Decard BF, et al. Natalizumab-induced POU2AF1/Spi-B upregulation: a possible route for PML development. Neurol Neuroimmunol Neuroinflamm (2016) 3(3):e223. doi:10.1212/NXI.0000000000000223
172. Marshall LJ, Moore LD, Mirsky MM, Major EO. JC virus promoter/enhancers contain TATA box-associated Spi-B-binding sites that support early viral gene expression in primary astrocytes. J Gen Virol (2012) 93(Pt 3):651–61. doi:10.1099/vir.0.035832-0
173. Manley K, Gee GV, Simkevich CP, Sedivy JM, Atwood WJ. Microarray analysis of glial cells resistant to JCV infection suggests a correlation between viral infection and inflammatory cytokine gene expression. Virology (2007) 366(2):394–404. doi:10.1016/j.virol.2007.05.016
174. Ranganathan PN, Khalili K. The transcriptional enhancer element, kappa B, regulates promoter activity of the human neurotropic virus, JCV, in cells derived from the CNS. Nucleic Acids Res (1993) 21(8):1959–64. doi:10.1093/nar/21.8.1959
175. Manley K, O’Hara BA, Gee GV, Simkevich CP, Sedivy JM, Atwood WJ. NFAT4 is required for JC virus infection of glial cells. J Virol (2006) 80(24):12079–85. doi:10.1128/JVI.01456-06
176. Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci (2007) 27(33):8745–56. doi:10.1523/JNEUROSCI.1002-07.2007
177. Wollebo HS, Melis S, Khalili K, Safak M, White MK. Cooperative roles of NF-kappaB and NFAT4 in polyomavirus JC regulation at the KB control element. Virology (2012) 432(1):146–54. doi:10.1016/j.virol.2012.06.010
178. Wollebo HS, Woldemichaele B, Khalili K, Safak M, White MK. Epigenetic regulation of polyomavirus JC. Virol J (2013) 10:264. doi:10.1186/1743-422X-10-264
179. Hoffmann FS, Hofereiter J, Rubsamen H, Melms J, Schwarz S, Faber H, et al. Fingolimod induces neuroprotective factors in human astrocytes. J Neuroinflammation (2015) 12:184. doi:10.1186/s12974-015-0393-6
180. Rothhammer V, Kenison JE, Tjon E, Takenaka MC, de Lima KA, Borucki DM, et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc Natl Acad Sci U S A (2017) 114(8):2012–7. doi:10.1073/pnas.1615413114
181. Hagihara K, Kita A, Mizukura A, Yao M, Kitai Y, Kunoh T, et al. Fingolimod (FTY720) stimulates Ca(2+)/calcineurin signaling in fission yeast. PLoS One (2013) 8(12):e81907. doi:10.1371/journal.pone.0081907
182. Loewe R, Holnthoner W, Groger M, Pillinger M, Gruber F, Mechtcheriakova D, et al. Dimethylfumarate inhibits TNF-induced nuclear entry of NF-kappa B/p65 in human endothelial cells. J Immunol (2002) 168(9):4781–7. doi:10.4049/jimmunol.168.9.4781
183. Gillard GO, Collette B, Anderson J, Chao J, Scannevin RH, Huss DJ, et al. DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J Neuroimmunol (2015) 283:74–85. doi:10.1016/j.jneuroim.2015.04.006
184. Galloway DA, Williams JB, Moore CS. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann Clin Transl Neurol (2017) 4(6):381–91. doi:10.1002/acn3.414
185. Kalinin S, Polak PE, Lin SX, Braun D, Guizzetti M, Zhang X, et al. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J Neuroimmunol (2013) 263(1–2):13–9. doi:10.1016/j.jneuroim.2013.07.007
186. Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol (2016) 4:71. doi:10.3389/fcell.2016.00071
187. De Keyser J, Mostert JP, Koch MW. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J Neurol Sci (2008) 267(1–2):3–16. doi:10.1016/j.jns.2007.08.044
188. Wharton KA Jr, Quigley C, Themeles M, Dunstan RW, Doyle K, Cahir-McFarland E, et al. JC polyomavirus abundance and distribution in progressive multifocal leukoencephalopathy (PML) brain tissue implicates myelin sheath in intracerebral dissemination of infection. PLoS One (2016) 11(5):e0155897. doi:10.1371/journal.pone.0155897
189. Gosert R, Kardas P, Major EO, Hirsch HH. Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J Virol (2010) 84(20):10448–56. doi:10.1128/JVI.00614-10
190. Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). PLoS Genet (2009) 5(2):e1000368. doi:10.1371/journal.pgen.1000368
191. Koralnik IJ, Wuthrich C, Dang X, Rottnek M, Gurtman A, Simpson D, et al. JC virus granule cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol (2005) 57(4):576–80. doi:10.1002/ana.20431
192. Dang X, Vidal JE, Oliveira AC, Simpson DM, Morgello S, Hecht JH, et al. JC virus granule cell neuronopathy is associated with VP1 C terminus mutants. J Gen Virol (2012) 93(Pt 1):175–83. doi:10.1099/vir.0.037440-0
193. Dang X, Wuthrich C, Gordon J, Sawa H, Koralnik IJ. JC virus encephalopathy is associated with a novel agnoprotein-deletion JCV variant. PLoS One (2012) 7(4):e35793. doi:10.1371/journal.pone.0035793
194. Miskin DP, Koralnik IJ. Novel syndromes associated with JC virus infection of neurons and meningeal cells: no longer a gray area. Curr Opin Neurol (2015) 28(3):288–94. doi:10.1097/WCO.0000000000000201
195. Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, et al. Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J Infect Dis (2011) 204(2):237–44. doi:10.1093/infdis/jir256
196. Takahashi K, Sekizuka T, Fukumoto H, Nakamichi K, Suzuki T, Sato Y, et al. Deep-sequence identification and role in virus replication of a JC virus quasispecies in patients with progressive multifocal leukoencephalopathy. J Virol (2017) 91(1). doi:10.1128/JVI.01335-16
197. Jelcic I, Combaluzier B, Jelcic I, Faigle W, Senn L, Reinhart BJ, et al. Broadly neutralizing human monoclonal JC polyomavirus VP1-specific antibodies as candidate therapeutics for progressive multifocal leukoencephalopathy. Sci Transl Med (2015) 7(306):306ra150. doi:10.1126/scitranslmed.aac8691
198. Nakamichi K, Tajima S, Lim CK, Saijo M. High-resolution melting analysis for mutation scanning in the non-coding control region of JC polyomavirus from patients with progressive multifocal leukoencephalopathy. Arch Virol (2014) 159(7):1687–96. doi:10.1007/s00705-014-1988-4
199. Ryschkewitsch CF, Jensen PN, Major EO. Multiplex qPCR assay for ultra sensitive detection of JCV DNA with simultaneous identification of genotypes that discriminates non-virulent from virulent variants. J Clin Virol (2013) 57(3):243–8. doi:10.1016/j.jcv.2013.03.009
200. Patsopoulos NA, Bayer Pharma MS; Genetics Working Group; Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium, GeneMSA, International Multiple Sclerosis Genetics Consortium, et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann Neurol (2011) 70(6):897–912. doi:10.1002/ana.22609
201. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature (2015) 518(7539):337–43. doi:10.1038/nature13835
202. Hussman JP, Beecham AH, Schmidt M, Martin ER, McCauley JL, Vance JM, et al. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun (2016) 17(5):305–12. doi:10.1038/gene.2016.23
203. Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol (2017) 18(1):76. doi:10.1186/s13059-017-1207-1
204. Schmidt H, Williamson D, Ashley-Koch A. HLA-DR15 haplotype and multiple sclerosis: a HuGE review. Am J Epidemiol (2007) 165(10):1097–109. doi:10.1093/aje/kwk118
205. Sundqvist E, Buck D, Warnke C, Albrecht E, Gieger C, Khademi M, et al. JC polyomavirus infection is strongly controlled by human leucocyte antigen class II variants. PLoS Pathog (2014) 10(4):e1004084. doi:10.1371/journal.ppat.1004084
206. Bettencourt A, Carvalho C, Leal B, Bras S, Lopes D, Martins da Silva A, et al. The protective role of HLA-DRB1(*)13 in autoimmune diseases. J Immunol Res (2015) 2015:948723. doi:10.1155/2015/948723
207. Luomala M, Lehtimaki T, Huhtala H, Ukkonen M, Koivula T, Hurme M, et al. Promoter polymorphism of IL-10 and severity of multiple sclerosis. Acta Neurol Scand (2003) 108(6):396–400. doi:10.1034/j.1600-0404.2003.00165.x
208. Sondergaard HB, Hesse D, Krakauer M, Sorensen PS, Sellebjerg F. Differential microRNA expression in blood in multiple sclerosis. Mult Scler (2013) 19(14):1849–57. doi:10.1177/1352458513490542
209. Liu Y, Chen Q, Song Y, Lai L, Wang J, Yu H, et al. MicroRNA-98 negatively regulates IL-10 production and endotoxin tolerance in macrophages after LPS stimulation. FEBS Lett (2011) 585(12):1963–8. doi:10.1016/j.febslet.2011.05.029
210. Teixeira-Coelho M, Guedes J, Ferreirinha P, Howes A, Pedrosa J, Rodrigues F, et al. Differential post-transcriptional regulation of IL-10 by TLR2 and TLR4-activated macrophages. Eur J Immunol (2014) 44(3):856–66. doi:10.1002/eji.201343734
211. Ganley-Leal LM, Liang Y, Jagannathan-Bogdan M, Farraye FA, Nikolajczyk BS. Differential regulation of TLR4 expression in human B cells and monocytes. Mol Immunol (2010) 48(1–3):82–8. doi:10.1016/j.molimm.2010.09.008
212. Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol (2006) 80(2):866–74. doi:10.1128/JVI.80.2.866-874.2006
213. Sadik NAH, Shaker OG, Ghanem HZ, Hassan HA, Abdel-Hamid A-HZ. Single-nucleotide polymorphism of toll-like receptor 4 and interleukin-10 in response to interferon-based therapy in Egyptian chronic hepatitis C patients. Arch Virol (2015) 160(9):2181–95. doi:10.1007/s00705-015-2493-0
214. Skevaki C, Pararas M, Kostelidou K, Tsakris A, Routsias JG. Single nucleotide polymorphisms of toll-like receptors and susceptibility to infectious diseases. Clin Exp Immunol (2015) 180(2):165–77. doi:10.1111/cei.12578
215. Sampaio EP, Hsu AP, Pechacek J, Bax HI, Dias DL, Paulson ML, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol (2013) 131(6):1624–34. doi:10.1016/j.jaci.2013.01.052
216. Zerbe CS, Marciano BE, Katial RK, Santos CB, Adamo N, Hsu AP, et al. Progressive multifocal leukoencephalopathy in primary immune deficiencies: Stat1 gain of function and review of the literature. Clin Infect Dis (2016) 62(8):986–94. doi:10.1093/cid/civ1220
Keywords: John Cunningham virus, natalizumab, fingolimod, dimethyl fumarate, glia
Citation: Mills EA and Mao-Draayer Y (2018) Understanding Progressive Multifocal Leukoencephalopathy Risk in Multiple Sclerosis Patients Treated with Immunomodulatory Therapies: A Bird’s Eye View. Front. Immunol. 9:138. doi: 10.3389/fimmu.2018.00138
Received: 29 September 2017; Accepted: 16 January 2018;
Published: 02 February 2018
Edited by:
Jens Geginat, Istituto Nazionale Genetica Molecolare (INGM), ItalyReviewed by:
Jorge Correale, Fundación para la Lucha contra las Enfermedades Neurológicas de la Infancia, ArgentinaTomas Per Olsson, Karolinska Institute (KI), Sweden
Copyright: © 2018 Mills and Mao-Draayer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Mao-Draayer, bWFvZHJhYXlAdW1pY2guZWR1