 Malte Bachmann
Malte Bachmann Josef Pfeilschifter
Josef Pfeilschifter Heiko Mühl
Heiko Mühl- Pharmazentrum Frankfurt/ZAFES, University Hospital Goethe-University Frankfurt am Main, Frankfurt am Main, Germany
Acetaminophen [paracetamol, N-acetyl-p-aminophenol (APAP)]-induced acute liver injury (ALI) not only remains a persistent clinical challenge but likewise stands out as well-characterized paradigmatic model of drug-induced liver damage. APAP intoxication associates with robust hepatic necroinflammation the role of which remains elusive with pathogenic but also pro-regenerative/-resolving functions being ascribed to leukocyte activation. Here, we shine a light on and put forward a unique role of the interleukin (IL)-1 family member IL-18 in experimental APAP-induced ALI. Indeed, amelioration of disease as previously observed in IL-18-deficient mice was further substantiated herein by application of the IL-18 opponent IL-18-binding protein (IL-18BPd:Fc) to wild-type mice. Data altogether emphasize crucial pathological action of this cytokine in APAP toxicity. Adding recombinant IL-22 to IL-18BPd:Fc further enhanced protection from liver injury. In contrast to IL-18, the role of prototypic pro-inflammatory IL-1 and tumor necrosis factor-α is controversially discussed with lack of effects or even protective action being repeatedly reported. A prominent detrimental function for IL-18 in APAP-induced ALI as proposed herein should relate to its pivotal role for hepatic expression of interferon-γ and Fas ligand, both of which aggravate APAP toxicity. As IL-18 serum levels increase in patients after APAP overdosing, targeting IL-18 may evolve as novel therapeutic option in those hard-to-treat patients where standard therapy with N-acetylcysteine is unsuccessful. Being a paradigmatic experimental model of ALI, current knowledge on ill-fated properties of IL-18 in APAP intoxication likewise emphasizes the potential of this cytokine to serve as therapeutic target in other entities of inflammatory liver diseases.
Introduction
Intended or unintended overdosing of acetaminophen [paracetamol, N-acetyl-p-aminophenol (APAP)] is regarded a major cause of acute liver failure provoking roughly 50,000 emergency room admissions, 2,500 hospitalizations, and 500 fatalities per year in the United States. The global burden on health-care systems that connects to APAP is based on a narrow therapeutic margin and supported by its broad over-the-counter availability. In fact, adverse consequences of APAP (self-)pharmacotherapy fuel a sustained discussion on safety issues and regulations regarding this fairly weak but frequently used analgesic drug (1–4).
Murine models of APAP-induced acute liver injury (ALI) are well established and adequately resemble key features of human intoxication (5). A crucial characteristic of APAP-induced ALI is centrilobular hepatocyte necrosis driven by N-acetyl-p-benzoquinone imine (NAPQI), an APAP metabolite generated by hepatic Cyp2e1 and Cyp1a2. Under the influence of NAPQI hepatocytes endure oxidative stress, malfunction of mitochondrial respiration, a drop in ATP, and predominantly necrotic cell death. Here, standard therapy with N-acetylcysteine interferes by providing NAPQI detoxifying glutathione (GSH) and by counteracting APAP-associated oxidative stress. Aforementioned noxious chain of events is amplified by cell intrinsic processes, among others activation of c-Jun N-terminal kinase (5–7). Since necrosis, by releasing danger-associated molecular patterns, notoriously connects to activation of innate immunity and inflammation (8), an additional immunological layer is considered a significant parameter determining APAP-induced ALI. Accordingly, several toll-like receptors (TLR) such as TLR4 (9, 10) and TLR3 (11) were reported to aggravate poisoning though conflicting reports impede straightforward interpretation (12, 13). Interestingly, TLR9 recognizing necrotic DNA has been identified consistently as pathogenic factor (12, 14, 15). Accordingly, TLR9 antagonism (16) or suppression of downstream type I interferon (IFN) function can ameliorate APAP toxicity (17, 18).
Whereas sterile liver inflammation is an undisputed consequence of APAP overdosing (5) its function is multifaceted and not fully understood. In this context, it must be appreciated that outcome of APAP poisoning is, to a significant degree, dependent on the capacity of the liver to activate repair and regeneration processes. Notably, in the later regenerative phase of intoxication, growth factors such as epidermal growth factor receptor ligands (19) but also pro-regenerative signal transducer and activator of transcription (STAT)-3-activating cytokines are regarded pivotal for efficient organ recovery (20). An interesting case in this context is interleukin (IL)-22 (21). Genetic models using IL22BP-deficient mice suggest a pathogenic role for endogenous IL-22 particularly during early intoxication (22). In contrast, administration of a single supra-physiological dose of recombinant IL-22 or its provision by liver-targeted IL-22 gene therapy mediates significant protection against APAP-induced ALI (23–25).
It adds to the overall complex nature of APAP-induced ALI that just sterile inflammation appears to be a prerequisite for activation of an efficient hepatic pro-regenerative program (20, 26).
A Complex Role for Paradigmatic Nuclear Factor (NF) κB-Activating IL-1 And TNFα In APAP-Induced ALI
Sterile inflammation is largely initiated by NF-κB-activating cytokines among which IL-1 and TNFα stand out as crucial (8, 27–29). Whereas hepatic upregulation of IL-1α/β during APAP-induced necroinflammation is undisputed (14, 30–32), the contribution of IL-1 to disease is undecided on every level of IL-1 biology. For example, inhibition of IL-1β maturation by lack of caspase-1 activity in C57BL/6 mice left APAP-induced ALI either unaffected (32, 33) or significantly bettered disease outcome (14). Notably, although IL-1α is not a caspase-1 substrate its protease activity is required for effective IL-1α release by monocytes (34). IL-1α/β-unresponsive IL-1 receptor-1 (IL-1R1)-deficient C57Bl/6 mice likewise displayed discordant behavior with either no effect (30) or amelioration of APAP intoxication (32, 35) put on record. Finally, administration of IL-1 receptor antagonist (IL-1Ra) (36) or neutralizing antibodies targeting either IL-1α (32) or IL-1β (14) improved APAP-induced ALI in C57Bl/6 mice. Surprisingly, IL-1Ra-deficient mice also displayed weakened intoxication (37), though BALB/c mice were used in that study. Alike IL-1, TNFα is evidently produced during APAP-induced ALI (11, 23, 38) and similarly puzzling with regard to function. Exemplarily, a report using TNFα-neutralizing antibody-treated or TNF receptor-1-deficient BALB/c mice proposed pathological action of this cytokine (39). Others found that TNFα-neutralization likewise inhibits (40) or is unable to influence (41) APAP toxicity in C57Bl/6 mice. Surprisingly, TNF receptor-1-deficiency actually aggravated disease in this mouse strain (42).
Differences in mouse characteristics, including the microbiome (43), as well as variations in APAP dosing may foster divergent conclusions regarding the role of IL-1 and TNFα in APAP-induced ALI. However, those observations may also echo an overlapping double-edged function of inflammation in the context of APAP overdosing. Specifically, while inflammation may initially promote early injury, hepatic repair and regeneration in a later phase of disease apparently rely on signals derived from innate immunity and associated cytokines. IL-1 and TNFα, for example, are able to upregulate pro-regenerative IL-6 as well as antioxidant pathways that enforce repair (20, 42, 44, 45). Some previous studies actually indicated a protective role of endogenous IL-1 (37) and TNFα (42) in APAP-induced ALI. Moreover, pro-inflammatory IL-36γ (46) was recently shown to promote regeneration in APAP toxicity, an observation that agrees with IL-36 supporting intestinal repair (47, 48). It is noteworthy that an early study reported on amelioration of APAP-induced ALI by application of recombinant IL-1α (49).
A Distinctive Role for IL-18 in APAP-Induced ALI
Due to some unique properties, IL-18 stands out among members of the IL-1 cytokine family (50, 51). IL-18 is constitutively expressed in a variety of cell types, for example, in hepatic Kupffer cells (52). Accordingly, IL-18 expression is detectable in healthy murine liver (53) where macrophages/Kupffer cells are a major source of bioactive IL-18 (50, 51). The active processed cytokine is usually (but not exclusively) generated by caspase-1 upon inflammatory stimulation (54). Besides being an inflammatory NF-κB-activating cytokine (55–57), two exceptional characteristics are key to the function of IL-18 in liver diseases. First of all, IL-18, initially coined IGIF for IFNγ-inducing factor (58), is pivotal for IFNγ production by T (58) and natural killer (NK) cells (59). In addition, IL-18 is a strong inducer of Fas ligand (FasL), particularly on NK cells (60). Both characteristics should be of significance for APAP-induced ALI because IFNγ (31) and Fas/FasL signaling (38, 61, 62) are crucial for the development of full APAP toxicity.
In accord with aforementioned characteristics, IL-18-deficient mice display strong protection from APAP-induced ALI (14). Since the pathogenic role of IL-18 in APAP intoxication has, best to our knowledge, not been confirmed in wild-type mice, we set out to determine consequences of IL-18 neutralization in this context. APAP (500 mg/kg) was applied intraperitoneally to fasted male C57Bl/6 mice as previously described (46). Where indicated, mice were i.v. cotreated with recombinant murine IL-18BPd:Fc (15 μg/mouse, R&D Systems, Wiesbaden, Germany). This genetically engineered agent corresponds to the neutralizing murine IL-18 opponent IL-18 binding protein d (IL-18BPd) (50, 63). Liver injury was quantified by determining serum alanine aminotransferase (ALT) activity 24 h after APAP administration, a time point coinciding with maximal hepatic damage in this protocol (46). In accord with data on IL-18-deficient mice, blockage of murine IL-18 biological activity by IL-18BPd:Fc indeed improved APAP-induced ALI (Figure 1A). As already alluded to, we and others have previously reported on amelioration of APAP intoxication by therapeutic provision of IL-22 (23–25). Interestingly, adding IL-22 (i.v. 3.5 μg/mouse, Immunotools, Friesoythe, Germany) to IL-18BPd:Fc further diminished serum ALT activity with an overall reduction by 69.5 ± 5.8% (Figure 1B). As further control, mice were i.v. treated with etanercept (75 μg/mouse, Pfizer, Karlsruhe, Germany), a clinically used TNFα blocker (TNFR2:Fc) known to likewise neutralize biological activity of the murine cytokine (64). As shown in Figure 1A, TNFα blockage did not affect APAP toxicity. Altogether, we confirm previous observations on a pathogenic role of IL-18 (14) and on lack of TNFα function (41) in APAP-induced ALI.
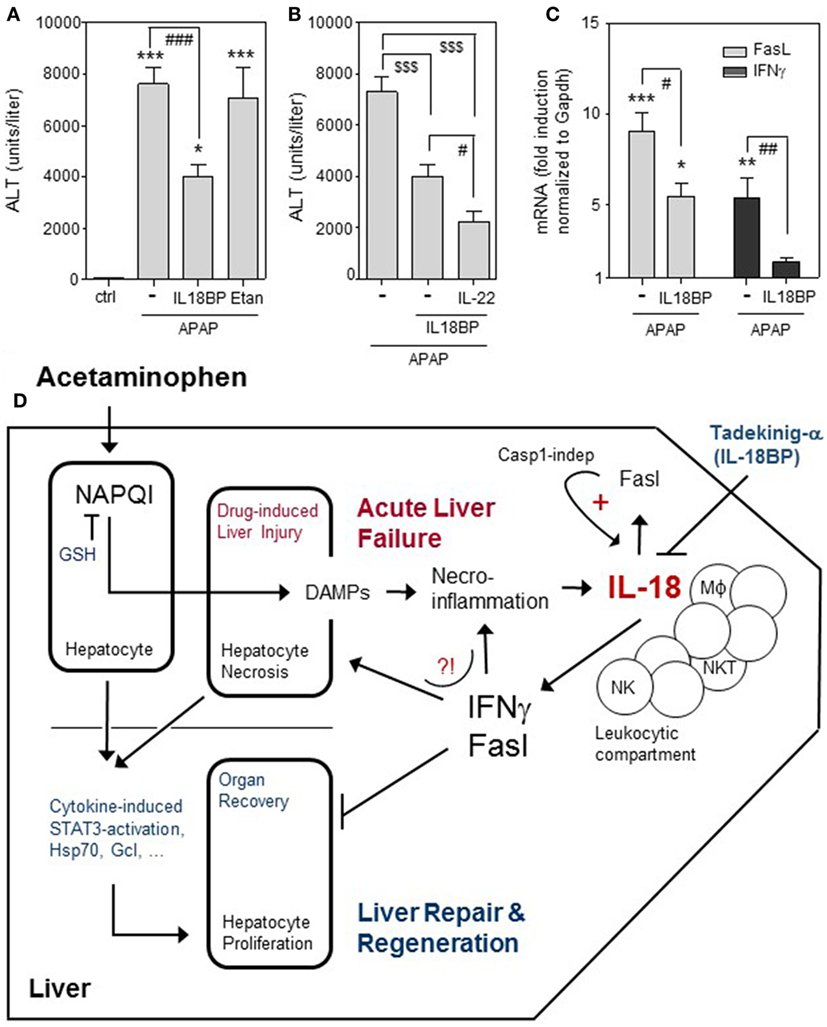
Figure 1. Effects of IL-18BPd:Fc on APAP-induced ALI. (A–C) All animal experiments (fasted male C57Bl/6 mice, 9–10 weeks old) were carried out in accordance with the recommendations of the Animal Protection Agency of the Federal State of Hessen (Regierungspräsidium Darmstadt, Germany). The protocol was approved by the Regierungspräsidium Darmstadt (Germany). The model of murine APAP (i.p. 500 mg/kg in 0.9% NaCl)-induced liver injury was performed as recently described (46). Where indicated, mice were i.v. cotreated with recombinant IL-18BPd:Fc (IL18BP), IL-22, or etanercept in PBS. (A) Mice received either APAP (n = 18), APAP/IL-18BPd:Fc (15 µg, n = 12), APAP/Etanercept (75 µg, n = 7), or 0.9% NaCl/PBS (ctrl, n = 6). After 24 h, serum alanine aminotransferase (ALT) activity was determined (Reflotron, Roche Diagnostics, Mannheim, Germany) and is depicted as units/liter (means ± SEM). *P < 0.05, ***P < 0.001 compared to ctrl; ###P < 0.001. (B) Mice received either APAP/PBS (n = 12), APAP/IL-18BPd:Fc (15 µg; n = 12), or APAP/IL-18BPd:Fc (15 µg) plus IL-22 (3.5 µg) (n = 13). After 24 h, serum ALT activity was determined and is depicted as units/liter (means ± SEM). $$$P < 0.001, #P < 0.05. (C) Mice received either APAP/PBS (n = 12) or APAP/IL-18BP (15 µg, n = 12) and were maintained for 16 h. For RNA analysis, liver tissue was snap frozen in liquid nitrogen and stored at −80°C. Total RNA was isolated as described (18). For real-time PCR, pre-developed reagents were used (Thermo Fisher Scientific, Darmstadt, Germany): GAPDH (VIC; 4352339E), Fas ligand (FasL) (FAM; Mm00438864_m1), and IFNγ (FAM; Mm01168134_m1). Assay mix was from Nippon Genetics (Düren, Germany). PCR: one initial step at 95°C (2 min) was followed by 40 cycles at 95°C (5 s) and 62°C (30 s). Detection of the dequenched probe, calculation of threshold cycles (CT values), and data analysis were performed by the Sequence Detector software (AbiPrism7500 Fast Sequence Detector, Thermo Fisher Scientific). Relative changes in hepatic FasL [(C), left panel] and IFNγ [(C), right panel] mRNA expression determined by real-time PCR were normalized to that of GAPDH and shown as fold-induction compared with untreated control mice (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 compared with untreated control; #P < 0.05, ##P < 0.01. (A–C) Data are shown as means ± SEM. Raw data were analyzed by one-way ANOVA with post hoc Bonferroni correction. (D) Graphical summary of processes affecting outcome of APAP-induced ALI with focus on the pathogenic role of IL-18. Detrimental pathways activated by APAP overdosing are counteracted by endogenous mechanisms supporting organ recovery through repair and regeneration [e.g., hepatocyte STAT3 activation; expression of heat shock protein (Hsp)70 and glutamate-cysteine ligase (Gcl)]. If therapeutic NAC intervention aiming at augmentation of hepatocyte glutathione (GSH) fails due a to an exceedingly high APAP dosage, a too late time point of intervention, and/or a pre-damaged liver parenchyma, acute liver failure may proceed to an ill-fated condition requiring transplantation for patient survival. Here, IL-18 may play a unique role by supporting hepatic expression of FasL and IFNγ. Application of recombinant IL-18 binding protein (Tadekinig-a) may evolve as a novel therapeutic option to intervene at this point.
The pathogenic role of IL-18 during APAP-induced ALI likely connects to the aforementioned potential to upregulate hepatic IFNγ and FasL. Both latter parameters are increased in liver tissues of APAP-challenged mice (38). Administration of IL-18BPd:Fc in fact suppressed hepatic expression of FasL (Figure 1C, left panel) and IFNγ (Figure 1C, right panel) in APAP-treated mice. Interestingly, IFNγ is known to support hepatocyte necrosis in response to APAP, possibly by enhancing nitric oxide formation (5, 31). IFNγ may additionally impair APAP-associated liver regeneration (45). This detrimental IFNγ activity has been shown to determine course of disease in experimental partial hepatectomy (65). The pathogenic role of Fas/FasL in APAP-induced ALI is likewise well established, detectable in Fas- or FasL-deficient (38, 62) as well as in wild-type mice (61), and apparently mediated by non-canonical Fas action. Specifically, apoptosis of hepatocytes is not regarded as relevant mechanism contributing to APAP-induced ALI. Accordingly, hepatocyte apoptosis by Fas/FasL is largely ruled out as relevant pathogenic mechanism in that context (26). Although Fas is famous for mediating apoptosis, it is noteworthy that this receptor can also activate classical signal transduction, e.g., via mitogen-activated protein kinases and NF-κB (66) which disconnects from pro-apoptotic signaling (67). Pathogenic action of Fas in APAP-induced ALI has been related to downregulation of glutamate-cysteine ligase and prolongation of GSH depletion as well as to reduction of heat shock protein (HSP)-70 (62). HSP70 is protective in APAP poisoning (68) and actually supports liver regeneration in murine partial hepatectomy (69). Moreover, Fas deficiency connects to impaired expression of STAT3-activating IL-6 and IL-10 (62), both are capable of ameliorating APAP-induced ALI (20). It is a further remarkable facet that interactions between hepatic macrophages and lymphocytes directed by Fas/FasL actually support production of bioactive IL-18 in caspase-1-independent but caspase-8-dependent manner (70, 71). Since IL-18 enhances FasL expression (60) which in turn enhances IL-18 (70, 71) this regulatory path represents a classical vicious cycle promoting liver pathology (54). Figure 1D provides a graphical summary of the complex events affecting outcome of APAP-induced ALI with focus on the pathogenic role of IL-18.
Concluding Remarks
The unresolved role of NF-κB-activating inflammatory cytokines including that of the caspase-1/IL-1β axis in APAP-induced ALI (20, 26, 72–74)—see Table 1—may reflect Janus-faced properties of theses mediators in the early injury and the later (partly overlapping) regeneration phase of intoxication. Herein, we confirm and put forward the perspective that IL-18 plays a unique pathogenic role in this model of sterile inflammation. Regardless of whether being activated by caspase-1, caspase-8, or by extracellular proteases such as proteinase-3 (50, 54), the potential of mature IL-18 to upregulate hepatic IFNγ and FasL appears decisive for its function during APAP-induced ALI. It is noteworthy that a detrimental role for hepatic IL-18 is not only conceivable for APAP intoxication. Specifically, administration of IL-18 neutralizing antibodies or recombinant IL-18 binding protein likewise ameliorates Propionibacterium acnes/lipopolysaccharide- (53, 75) as well as concanavalin A-, Fas-, and Pseudomonas aeruginosa exotoxin A-induced murine liver damage (75). Moreover, treatment with recombinant IL-18 binding protein protected from liver injury in murine experimental hemophagocytic lymphohistiocytosis (76). Current data also suggest an additional benefit of the combination IL-18BPd:Fc plus IL-22, an observation that deserves delineation in forthcoming experiments.

Table 1. Data on the role of IL-18, IL-1, caspase-1, and TNFα in experimental APAP-induced ALI as detected in C57Bl/6 and BALB/c mice.
Current data altogether advocate short-term blockage of IL-18 as therapeutic approach in acute liver diseases. A recent phase I/II clinical trial investigating application of recombinant IL-18BP (tadekinig-α) in adult onset still’s disease actually revealed an acceptable safety profile of this agent—besides specific therapeutic efficacy (77). Moreover, in human acute liver failure due to APAP overdosing, elevated levels of IL-18 are detectable in patients’ sera (78). It is thus tempting to speculate that provision of interleukin-18 binding protein therapy aids those unfortunate patients where standard therapy with N-acetylcysteine falls short.
Ethics Statement
All animal experiments using C57Bl/6 mice (male, 9–10 weeks old) were carried out in accordance with the recommendations of the Animal Protection Agency of the Federal State of Hessen (Regierungspräsidium Darmstadt, Germany). The protocol was approved by the Regierungspräsidium Darmstadt (Germany).
Author Contributions
MB: performed all experiments, analyzed the data, and contributed to manuscript writing and editing. JP: analyzed the data and contributed to manuscript editing. HM: analyzed the data, designed the study, wrote the paper, and performed manuscript editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by a grant from the DFG (MU 1284/6-1) to HM.
References
1. Bernal W, Auzinger G, Dhawan A, Wendon J. Acute liver failure. Lancet (2010) 376(9736):190–201. doi:10.1016/S0140-6736(10)60274–7
2. Lee WM. Acetaminophen and the U.S. acute liver failure study group: lowering the risks of hepatic failure. Hepatology (2004) 40(1):6–9. doi:10.1002/hep.20293
3. Lee WM. Acetaminophen (APAP) hepatotoxicity-isn’t it time for APAP to go away? J Hepatol (2017) 67(6):1324–31. doi:10.1016/j.jhep.2017.07.005
4. Brune K, Renner B, Tiegs G. Acetaminophen/paracetamol: a history of errors, failures and false decisions. Eur J Pain (2015) 19(7):953–65. doi:10.1002/ejp.621
5. Jaeschke H, Xie Y, McGill MR. Acetaminophen-induced liver injury: from animal models to humans. J Clin Transl Hepatol (2014) 2(3):153–61. doi:10.14218/JCTH.2014.00014
6. Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol (2010) 196:369–405. doi:10.1007/978-3-642-00663-0_12
7. Bantel H, Schulze-Osthoff K. Mechanisms of cell death in acute liver failure. Front Physiol (2012) 3:79. doi:10.3389/fphys.2012.00079
8. Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol (2012) 3:287. doi:10.3389/fimmu.2012.00287
9. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol (2011) 187(5):2626–31. doi:10.4049/jimmunol.1003930
10. Yohe HC, O’Hara KA, Hunt JA, Kitzmiller TJ, Wood SG, Bement JL, et al. Involvement of toll-like receptor 4 in acetaminophen hepatotoxicity. Am J Physiol Gastrointest Liver Physiol (2006) 290(6):G1269–79. doi:10.1152/ajpgi.00239.2005
11. Cavassani KA, Moreira AP, Habiel D, Ito T, Coelho AL, Allen RM, et al. Toll like receptor 3 plays a critical role in the progression and severity of acetaminophen-induced hepatotoxicity. PLoS One (2013) 8(6):e65899. doi:10.1371/journal.pone.0065899
12. Cai C, Huang H, Whelan S, Liu L, Kautza B, Luciano J, et al. Benzyl alcohol attenuates acetaminophen-induced acute liver injury in a toll-like receptor-4-dependent pattern in mice. Hepatology (2014) 60(3):990–1002. doi:10.1002/hep.27201
13. Ghaffari AA, Chow EK, Iyer SS, Deng JC, Cheng G. Polyinosinic-polycytidylic acid suppresses acetaminophen-induced hepatotoxicity independent of type I interferons and toll-like receptor 3. Hepatology (2011) 53(6):2042–52. doi:10.1002/hep.24316
14. Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. J Clin Invest (2009) 119(2):305–14. doi:10.1172/JCI35958
15. Marques PE, Oliveira AG, Pereira RV, David BA, Gomides LF, Saraiva AM, et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology (2015) 61(1):348–60. doi:10.1002/hep.27216
16. Hoque R, Farooq A, Malik A, Trawick BN, Berberich DW, McClurg JP, et al. A novel small-molecule enantiomeric analogue of traditional (-)-morphinans has specific TLR9 antagonist properties and reduces sterile inflammation-induced organ damage. J Immunol (2013) 190(8):4297–304. doi:10.4049/jimmunol.1202184
17. Bhattacharya S, Katlinski KV, Reichert M, Takano S, Brice A, Zhao B, et al. Triggering ubiquitination of IFNAR1 protects tissues from inflammatory injury. EMBO Mol Med (2014) 6:384–97. doi:10.1002/emmm.201303236
18. Bachmann M, Waibler Z, Pleli T, Pfeilschifter J, Mühl H. Type I interferon supports inducible nitric oxide synthase in murine hepatoma cells and hepatocytes and during experimental acetaminophen-induced liver damage. Front Immunol (2017) 8:890. doi:10.3389/fimmu.2017.00890
19. Bhushan B, Chavan H, Borude P, Xie Y, Du K, McGill MR, et al. Dual role of epidermal growth factor receptor in liver injury and regeneration after acetaminophen overdose in mice. Toxicol Sci (2017) 155(2):363–78. doi:10.1093/toxsci/kfw213
20. Mühl H. STAT3, a key parameter of cytokine-driven tissue protection during sterile inflammation—the case of experimental acetaminophen (paracetamol)-induced liver damage. Front Immunol (2016) 7:163. doi:10.3389/fimmu.2016.00163
21. Mühl H, Scheiermann P, Bachmann M, Härdle L, Heinrichs A, Pfeilschifter J. IL-22 in tissue-protective therapy. Br J Pharmacol (2013) 169(4):761–71. doi:10.1111/bph.12196
22. Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ, et al. A protective function of IL-22BP in ischemia reperfusion and acetaminophen-induced liver injury. J Immunol (2017) 199(12):4078–90. doi:10.4049/jimmunol.1700587
23. Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Mühl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol (2013) 182(4):1107–13. doi:10.1016/j.ajpath.2012.12.010
24. Feng D, Wang Y, Wang H, Weng H, Kong X, Martin-Murphy BV, et al. Acute and chronic effects of IL-22 on acetaminophen-induced liver injury. J Immunol (2014) 193(5):2512–8. doi:10.4049/jimmunol.1400588
25. Chen W, Zhang X, Fan J, Zai W, Luan J, Li Y, et al. Tethering interleukin-22 to apolipoprotein A-I ameliorates mice from acetaminophen-induced liver injury. Theranostics (2017) 7(17):4135–48. doi:10.7150/thno.20955
26. Jaeschke H, Williams CD, Ramachandran A, Bajt ML. Acetaminophen hepatotoxicity and repair: the role of sterile inflammation and innate immunity. Liver Int (2012) 32(1):8–20. doi:10.1111/j.1478-3231.2011.02501.x
27. Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol (2011) 41(5):1203–17. doi:10.1002/eji.201141550
28. Kono H, Onda A, Yanagida T. Molecular determinants of sterile inflammation. Curr Opin Immunol (2014) 26:147–56. doi:10.1016/j.coi.2013.12.004
29. Allam R, Kumar SV, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) (2014) 92(5):465–72. doi:10.1007/s00109-014-1148-z
30. Williams CD, Farhood A, Jaeschke H. Role of caspase-1 and interleukin-1beta in acetaminophen-induced hepatic inflammation and liver injury. Toxicol Appl Pharmacol (2010) 247(3):169–78. doi:10.1016/j.taap.2010.07.004
31. Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J (2002) 16(10):1227–36. doi:10.1096/fj.02-0046com
32. Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, et al. Macrophage-derived IL-1α promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol Immunol (2017). doi:10.1038/cmi.2017.22
33. Williams CD, Antoine DJ, Shaw PJ, Benson C, Farhood A, Williams DP, et al. Role of the Nalp3 inflammasome in acetaminophen-induced sterile inflammation and liver injury. Toxicol Appl Pharmacol (2011) 252(3):289–97. doi:10.1016/j.taap.2011.03.001
34. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (1995) 267(5206):2000–3. doi:10.1126/science.7535475
35. Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med (2007) 13(7):851–6. doi:10.1038/nm1603
36. Hu J, Yan D, Gao J, Xu C, Yuan Y, Zhu R, et al. rhIL-1Ra reduces hepatocellular apoptosis in mice with acetaminophen-induced acute liver failure. Lab Invest (2010) 90(12):1737–46. doi:10.1038/labinvest.2010.127
37. Ishibe T, Kimura A, Ishida Y, Takayasu T, Hayashi T, Tsuneyama K, et al. Reduced acetaminophen-induced liver injury in mice by genetic disruption of IL-1 receptor antagonist. Lab Invest (2009) 89(1):68–79. doi:10.1038/labinvest.2008.110
38. Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology (2004) 127(6):1760–74. doi:10.1053/j.gastro.2004.08.053
39. Ishida Y, Kondo T, Tsuneyama K, Lu P, Takayasu T, Mukaida N. The pathogenic roles of tumor necrosis factor receptor p55 in acetaminophen-induced liver injury in mice. J Leukoc Biol (2004) 75(1):59–67. doi:10.1189/jlb.0403152
40. Chastre A, Bélanger M, Beauchesne E, Nguyen BN, Desjardins P, Butterworth RF. Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications. PLoS One (2012) 7(11):e49670. doi:10.1371/journal.pone.0049670
41. Yee SB, Bourdi M, Masson MJ, Pohl LR. Hepatoprotective role of endogenous interleukin-13 in a murine model of acetaminophen-induced liver disease. Chem Res Toxicol (2007) 20(5):734–44. doi:10.1021/tx600349f
42. Chiu H, Gardner CR, Dambach DM, Brittingham JA, Durham SK, Laskin JD, et al. Role of p55 tumor necrosis factor receptor 1 in acetaminophen-induced antioxidant defense. Am J Physiol Gastrointest Liver Physiol (2003) 285(5):G959–66. doi:10.1152/ajpgi.00219.2003
43. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology (2014) 146(6):1513–24. doi:10.1053/j.gastro.2014.01.020
44. Böhm F, Köhler UA, Speicher T, Werner S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol Med (2010) 2(8):294–305. doi:10.1002/emmm.201000085
45. Li N, Hua J. Immune cells in liver regeneration. Oncotarget (2017) 8(2):3628–39. doi:10.18632/oncotarget.12275
46. Scheiermann P, Bachmann M, Härdle L, Pleli T, Piiper A, Zwissler B, et al. Application of IL-36 receptor antagonist weakens CCL20 expression and impairs recovery in the late phase of murine acetaminophen-induced liver injury. Sci Rep (2015) 5:8521. doi:10.1038/srep08521
47. Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, et al. Cutting edge: IL-36 receptor promotes resolution of intestinal damage. J Immunol (2016) 196(1):34–8. doi:10.4049/jimmunol.1501312
48. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut (2017) 66(5):823–38. doi:10.1136/gutjnl-2015-310374
49. Renić M, Culo F, Bilić A, Bukovec Z, Sabolović D, Zupanović Z. The effect of interleukin 1 alpha on acetaminophen-induced hepatotoxicity. Cytokine (1993) 5(3):192–7. doi:10.1016/1043-4666(93)90004-O
50. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi:10.3389/fimmu.2013.00289
51. Mühl H, Pfeilschifter J. Interleukin-18 bioactivity: a novel target for immunopharmacological anti-inflammatory intervention. Eur J Pharmacol (2004) 500(1–3):63–71. doi:10.1016/j.ejphar.2004.07.012
52. Matsui K, Yoshimoto T, Tsutsui H, Hyodo Y, Hayashi N, Hiroishi K, et al. Propionibacterium acnes treatment diminishes CD4+ NK1.1+ T cells but induces type I T cells in the liver by induction of IL-12 and IL-18 production from kupffer cells. J Immunol (1997) 159(1):97–106.
53. Tsutsui H, Matsui K, Kawada N, Hyodo Y, Hayashi N, Okamura H, et al. IL-18 accounts for both TNF-alpha- and fas ligand-mediated hepatotoxic pathways in endotoxin-induced liver injury in mice. J Immunol (1997) 159(8):3961–7.
54. Tsutsui H, Nishiguchi S. Importance of kupffer cells in the development of acute liver injuries in mice. Int J Mol Sci (2014) 15(5):7711–30. doi:10.3390/ijms15057711
55. Matsumoto S, Tsuji-Takayama K, Aizawa Y, Koide K, Takeuchi M, Ohta T, et al. Interleukin-18 activates NF-kappaB in murine T helper type 1 cells. Biochem Biophys Res Commun (1997) 234(2):454–7. doi:10.1006/bbrc.1997.6665
56. Puren AJ, Fantuzzi G, Gu Y, Su MS, Dinarello CA. Interleukin-18 (IFNgamma-inducing factor) induces IL-8 and IL-1beta via TNFalpha production from non-CD14+ human blood mononuclear cells. J Clin Invest (1998) 101(3):711–21. doi:10.1172/JCI1379
57. Bachmann M, Dragoi C, Poleganov MA, Pfeilschifter J, Mühl H. Interleukin-18 directly activates T-bet expression and function via p38 mitogen-activated protein kinase and nuclear factor-kappaB in acute myeloid leukemia-derived predendritic KG-1 cells. Mol Cancer Ther (2007) 6(2):723–31. doi:10.1158/1535-7163.MCT-06-0505
58. Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature (1995) 378(6552):88–91. doi:10.1038/378088a0
59. Zhang T, Kawakami K, Qureshi MH, Okamura H, Kurimoto M, Saito A. Interleukin-12 (IL-12) and IL-18 synergistically induce the fungicidal activity of murine peritoneal exudate cells against Cryptococcus neoformans through production of gamma interferon by natural killer cells. Infect Immun (1997) 65(9):3594–9.
60. Tsutsui H, Nakanishi K, Matsui K, Higashino K, Okamura H, Miyazawa Y, et al. IFN-gamma-inducing factor up-regulates fas ligand-mediated cytotoxic activity of murine natural killer cell clones. J Immunol (1996) 157(9):3967–73.
61. Zhang H, Cook J, Nickel J, Yu R, Stecker K, Myers K, et al. Reduction of liver fas expression by an antisense oligonucleotide protects mice from fulminant hepatitis. Nat Biotechnol (2000) 18(8):862–7. doi:10.1038/78475
62. Williams CD, McGill MR, Farhood A, Jaeschke H. Fas receptor-deficient lpr mice are protected against acetaminophen hepatotoxicity due to higher glutathione synthesis and enhanced detoxification of oxidant stress. Food Chem Toxicol (2013) 58:228–35. doi:10.1016/j.fct.2013.04.031
63. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity (1999) 10(1):127–36. doi:10.1016/S1074-7613(00)80013-8
64. Popivanova BK, Kitamura K, Wu Y, Kondo T, Kagaya T, Kaneko S, et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest (2008) 118(2):560–70. doi:10.1172/JCI32453
65. Sun R, Gao B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma). Gastroenterology (2004) 127(5):1525–39. doi:10.1053/j.gastro.2004.08.055
66. Le Gallo M, Poissonnier A, Blanco P, Legembre P. CD95/Fas, non-apoptotic signaling pathways, and kinases. Front Immunol (2017) 8:1216. doi:10.3389/fimmu.2017.01216
67. Imamura R, Konaka K, Matsumoto N, Hasegawa M, Fukui M, Mukaida N, et al. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J Biol Chem (2004) 279(45):46415–23. doi:10.1074/jbc.M403226200
68. Tolson JK, Dix DJ, Voellmy RW, Roberts SM. Increased hepatotoxicity of acetaminophen in Hsp70i knockout mice. Toxicol Appl Pharmacol (2006) 210(1–2):157–62. doi:10.1016/j.taap.2005.10.001
69. Wolf JH, Bhatti TR, Fouraschen S, Chakravorty S, Wang L, Kurian S, et al. Heat shock protein 70 is required for optimal liver regeneration after partial hepatectomy in mice. Liver Transpl (2014) 20(3):376–85. doi:10.1002/lt.23813
70. Tsutsui H, Kayagaki N, Kuida K, Nakano H, Hayashi N, Takeda K, et al. Caspase-1-independent, fas/fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity (1999) 11(3):359–67. doi:10.1016/S1074-7613(00)80111-9
71. Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ, Rathinam VA, et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J Immunol (2012) 189(12):5508–12. doi:10.4049/jimmunol.1202121
72. Woolbright BL, Jaeschke H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol (2017) 66(4):836–48. doi:10.1016/j.jhep.2016.11.017
73. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology (2012) 143(5):1158–72. doi:10.1053/j.gastro.2012.09.008
75. Faggioni R, Cattley RC, Guo J, Flores S, Brown H, Qi M, et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of fas/fas ligand-mediated models of liver disease in mice. J Immunol (2001) 167(10):5913–20. doi:10.4049/jimmunol.167.10.5913
76. Chiossone L, Audonnet S, Chetaille B, Chasson L, Farnarier C, Berda-Haddad Y, et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front Immunol (2012) 3:239. doi:10.3389/fimmu.2012.00239
77. Gabay C, Fautrel B, Rech J, Spertini F, Feist E, Koetter I, et al. Open-label, multicenter, dose-escalating phase ii clinical trial on the safety and efficacy of tadekinig alpha in adult onset still’s disease. Ann Rheum Dis (2017) 76:709. doi:10.1136/annrheumdis-2017-eular.2336
Keywords: interleukin-18, IL-18BP, interleukin-1, acetaminophen, liver injury, inflammation
Citation: Bachmann M, Pfeilschifter J and Mühl H (2018) A Prominent Role of Interleukin-18 in Acetaminophen-Induced Liver Injury Advocates Its Blockage for Therapy of Hepatic Necroinflammation. Front. Immunol. 9:161. doi: 10.3389/fimmu.2018.00161
Received: 02 December 2017; Accepted: 18 January 2018;
Published: 08 February 2018
Edited by:
Detlef Neumann, Hannover Medical School, GermanyCopyright: © 2018 Bachmann, Pfeilschifter and Mühl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heiko Mühl, aC5tdWVobEBlbS51bmktZnJhbmtmdXJ0LmRl