 Elissa K. Deenick
Elissa K. Deenick Simon J. Pelham
Simon J. Pelham Alisa Kane1,2,3,4
Alisa Kane1,2,3,4 Cindy S. Ma
Cindy S. Ma- 1Immunology Division, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 2St Vincent’s Clinical School, UNSW Sydney, Darlinghurst, NSW, Australia
- 3Department of Immunology and Allergy, Liverpool Hospital, Liverpool, NSW, Australia
- 4South Western Sydney Clinical School, UNSW Sydney, Liverpool, NSW, Australia
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is activated downstream of many key cytokine receptors expressed by lymphocytes. As such, it plays a critical role in regulating B cells as well as CD4+ and CD8+ T cells. Patients with clinically significant immunodeficiency and immune dysregulation resulting from loss-of-function or gain-of-function mutations in STAT3 have been described. These individuals provide insight into the critical role of this transcription factor in the regulation of immune responses and the balance between effective immune protection and autoimmunity.
Introduction
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that is activated downstream of a large range of cell surface receptors. It forms part of a family of proteins that also includes STAT1, 2, 4, 5A, 5B, and 6, which are activated in a similar manner downstream of surface receptors. Binding of their ligand by these receptors, leads to the activation of receptor-associated Janus activating kinases (JAKs). The activated JAKs then phosphorylate the receptor providing docking sites for STATs, which in turn become tyrosine phosphorylated. This leads to the formation of homodimers or heterodimers, followed by translocation to the nucleus where the dimers bind to DNA and induce transcription of a broad range of target genes (1, 2).
Importantly, many of the cytokine receptors that lead to STAT3 activation are expressed by lymphocytes including those for IL-6, IL-10, IL-21, IL-23, and IFNs. The critical role of STAT3 in lymphocyte biology was highlighted by the discovery that loss-of-function (LOF) mutations in STAT3 cause the primary immunodeficiency autosomal dominant hyper IgE syndrome (AD-HIES), which is characterized by defects in both T and B cells (3, 4). More recently, gain-of-function (GOF) mutations in STAT3 were also identified, this time in patients who presented with early onset autoimmunity as well as immunodeficiency (5–7). These diseases demonstrate that STAT3 plays a central role in regulation of immune responses.
Immunodeficiency Caused by STAT3LOF Mutations
Autosomal dominant hyper IgE syndrome was first described about 50 years ago, but it was not until 2007 that two groups demonstrated that it is caused by heterozygous LOF mutations in STAT3 (3, 4). AD-HIES is characterized by a range of immunological manifestations including elevated IgE, eczema, chronic mucocutaneous candidiasis (CMC), recurrent staphylococcal infections, and pneumonias. Patients also display non-immunological manifestations such as joint hyperextensibility, facial dysmorphism, and retention of primary teeth (8). Since the initial description over 89 disease-causing mutations in STAT3 have been reported and are found distributed throughout the STAT3 molecule (9, 10). These mutations all lead to the same clinical phenotype, presumably because while different mutations impair signaling at different stages, they all impair the ability of STAT3 to bind to DNA and induce gene transcription (11). It should be noted that due to the dimerization step in the STAT3 signaling pathway these heterozygous mutations in STAT3 work in a dominant negative manner. That is, in patient cells, 75% of STAT3 dimers would contain at least one LOF STAT3 molecule and thus be dysfuctional, leaving only 25% of dimers functioning normally (3, 4). Thus, AD-HIES results in severely compromised, but not completely ablated, STAT3 signaling. This 25% of residual STAT3 function is presumably critical for survival as germline deletion of Stat3 in mice is embryonically lethal (12).
Immune Dysregulation Caused by STAT3GOF Mutations
More recently, patients with heterozygous GOF mutations in STAT3 have also been described (5–7). These patients present with early onset autoimmunity and/or lymphoproliferation. The range of autoimmune manifestations is broad and includes cytopenias, type I diabetes, enteropathy, scleroderma, arthritis, and thyroid disease (5–7). However, many of these patients were also reported to suffer from recurrent or severe infections as well as hypogammaglobulinemia (6, 7) suggesting concurrent immunodeficiency. Overall, the clinical phenotype of the patients has been found to be quite variable and unaffected family members who carried STAT3GOF mutations have also been identified suggesting there is incomplete disease penetrance and that other factors influence the pathogenicity of the mutations (7, 13).
The molecular mechanism that results in GOF from these germline mutations has not been extensively characterized; however, the varied patient phenotype suggests there may be more divergence in mechanism than is observed with LOF mutations. It has been observed that most disease causing GOF mutations do not alter phosphorylation; however, these mutations generally lead to increased transcriptional activity of STAT3 target genes in unstimulated and/or stimulated cells (6, 7). This in turn leads to upregulation of STAT3 target genes such as SOCS3 (7). Interestingly, SOCS3 can regulate the activation of STAT family members, and cells from these patients were found to have reduced STAT5 phosphorylation in response to IL-2, and STAT1 phosphorylation in response to IFNγ (7). Moreover, some of the symptoms of STAT3GOF patients are similar to those observed in STAT5b LOF patients (14) suggesting that reduced STAT5 activation may partially explain the phenotype (discussed below).
The Role of STAT3 in B Cells
Multiple findings in patients with dysregulated STAT3 function point to a role for STAT3 in regulating human B cells responses. For example, although patients with STAT3LOF mutations have relatively normal levels of total serum IgM, IgG, and IgA, they have elevated levels of serum IgE, defects in antigen specific antibody responses and reduced memory B cells (8, 15–19). Further, the STAT3-activating cytokines IL-21, and to a lesser extent IL-10, are potent B cell activators. In combination with CD40L, IL-21 and IL-10 are capable of inducing the proliferation, class switching, and differentiation of human B cells (19, 20). Interestingly, some, but not all, of the actions of IL-21 and IL-10 were found to be disrupted in B cells from AD-HIES patients. Specifically, STAT3LOF naïve B cells were unable to differentiate into antibody secreting cells in response to CD40L and IL-21 (19, 21) and failed to upregulate key transcriptional regulators of the plasma cell program such as BLIMP-1 and XBP-1 (19, 21). In contrast, IL-21 was able to induce normal levels of switching to IgG from naïve STAT3LOF B cells and could stimulate increased levels of proliferation from these cells compared to cultures with CD40L alone, albeit lower than what was observed in naive B cells from healthy controls responding to CD40L and IL-21 (19). This decreased proliferation/expansion could at least partially be attributed to an increase in cell death (19). It must be remembered, however, that these patient cells retain 25% STAT3 activity so it unclear whether the responses to IL-21 that are preserved reflect STAT3-independent effects of IL-21 or the function of the residual STAT3. Some insight can be gained from mouse models of B cell specific deletion of Stat3 in which all STAT3 function is ablated in these cells. These models showed relatively normal switching to IgG (22, 23) but decreased expansion and/or maintenance of B cells to a T cell-dependent antigen resulting in fewer germinal center B cells (23, 24). Further STAT3 deficiency also resulted in a defect in affinity maturation (23) although cells still underwent somatic hypermutation (19, 23). Together these studies reveal that STAT3 is required in naïve B cells to induce plasma cell formation, survival, and expansion of responding B cells but not for regulating switching to IgG. Interestingly, the small number of memory cells that do emerge in patients with STAT3LOF can respond normally to IL-21 to form antibody-secreting cells (21). This demonstrates that naïve and memory B cells have differential requirements for STAT3, which may have important implications for attempts to target the STAT3 pathway therapeutically.
Confirmation of the critical role of IL-21 upstream of STAT3 was provided by the identification of patients with LOF mutation in IL21R and IL21. These patients displayed similar B cell defects to those with LOF STAT3 mutations such as reduced memory B cells, poor responses to vaccination, and elevated levels of IgE (21, 25–28). This clearly demonstrates the importance of the IL-21/STAT3 signaling axis in human B cell function. However, the exact molecular mechanism that leads to decreased memory cells and increased IgE is still unclear. Interestingly, a study of a patient with somatic mosaicism of the STAT3 mutation has shown that STAT3LOF cells did not generate memory CD4+ or CD8+ T cells but the mutation was present in memory B cells, suggesting that there was an intrinsic requirement for STAT3 in T cells but not B cells for the generation of memory (29). Thus, it may be that the decrease in memory B cells is secondary to aberrant function of other cell types such as T follicular helper (Tfh) cells (discussed below).
B cells can influence immune responses through their role in antigen presentation and the production of cytokines. IL-10 produced by B cells has been implicated in a regulatory role in immune responses (30). Interestingly, STAT3LOF B cells have been shown to produce less IL-10 following stimulation, than normal controls (31) suggesting that this regulatory function of B cells may also be altered.
Given this critical role of IL-21/STAT3 in B cell differentiation, it might be predicted that STAT3GOF patients would exhibit increased B cell activity. Surprisingly, however, some of these patients seem to display hallmarks of B cell dysfunction such as hypogammaglobulinemia and decreased switched memory B cells (6, 7). Conversely, many of these patients seem to display antibody-mediated autoimmunity suggesting that B cell tolerance is disturbed (5–7). Unfortunately, little functional work has been done on B cells from these patients so it remains unclear if there are B cell intrinsic effects of STAT3 over activation or whether this is secondary to defects in other cells such as Tfh cells or regulatory T cells (Tregs) (discussed below).
The Role of STAT3 in CD4+ T Cells
Naive CD4+ T cells are able differentiate into distinct effector subsets that play specific roles in the immune response. These subsets include Th1, Th2, Th9, Th17, Th22, Tfh cells, and Tregs. The differentiation of CD4+ T cells is determined by the cytokine milieu at the time of activation and numerous STAT3-signaling cytokines have been implicated in this process (32, 33). Analysis of patients with STAT3 mutations has provided key insights into the role of STAT3 in controlling these processes.
Th17
The differentiation of human Th17 cells is controlled by the action of several STAT3-activating cytokines including IL-6, IL-21, and IL-23 (34–36). Analysis of AD-HIES patients revealed a deficiency in Th17 cells in the blood of these patients as measured by expression of CCR6 and the production of IL-17A, IL-17F, and IL-22 (28, 37–40). Furthermore, STAT3LOF naïve CD4+ T cells from these patients fail to differentiate into IL-17-expressing cells in vitro (38, 41). Together, this not only demonstrates an essential requirement for STAT3 signaling in the generation of human Th17 cells but also provides an explanation for the CMC observed in AD-HIES patients as IL-17-mediated immunity is crucial for control of candida infections (42, 43). Interestingly, patients with GOF mutations in STAT1 also display defects in the generation of Th17 cells and susceptibility to candida infections demonstrating that balanced STAT1/STAT3 signaling is required for generation of these cells (41, 44, 45). On the other hand, patients with STAT3GOF were not found to have increased IL-17-expressing CD4+ T cells suggesting that, while STAT3 is required for the generation of these cells, over activation alone is not sufficient to drive Th17 differentiation (6, 7). However, more detailed analysis of these STAT3GOF CD4+ T cells may be required to definitively conclude this as some patients may have increased Th17 cells (46).
Th1/Th2
In contrast to Th17 cells, the generation of human Th1 and Th2 is thought to act primarily through IL-12/STAT4 and IL-4/STAT6 signaling, respectively (32, 33). Consistent with this, generation of these populations was found to be largely STAT3-independent, as shown by normal frequencies of CXCR3+CCR6− and CXCR3−CCR6− and IFNγ-producing and IL-4, IL-5, IL-13-producing cells, respectively, in AD-HIES patients (28, 37, 38). Similarly, naïve CD4+ T cells from STAT3LOF patients could differentiate into Th1 or Th2 cells when cultured under the relevant polarizing conditions (41, 47). Interestingly, IFNγ expression tended to be increased in STAT3-deficient CD4+ T cells (28, 41), suggesting STAT3 signaling may inhibit Th1 cell differentiation.
Th9
Human Th9 cells develop in the presence of TGFβ and IL-4 (48, 49); however, they can also be induced by the addition of TGFβ to Th17 polarizing conditions (i.e., IL-1β/IL-6/IL-21/IL-23) (48), suggesting STAT3 may be involved in Th9 cell differentiation. Consistent with this, addition of IL-6, IL-10, or IL-21 to Th9 polarizing conditions enhanced IL-9 expression (50). In contrast, IL-27 partially suppressed TGFβ and IL-4-induced IL-9 expression (50). Since IL-6, IL-10, IL-21, and IL-27 are capable of activating both STAT1 and STAT3, further investigation is required to determine if the regulation of IL-9 production by these cytokines results from both STATs, or whether one STAT has a dominant function in regulating IL-9 production. However, one paper found that IL-9 production in responses to candida antigens was decreased in patients with STAT3LOF mutations (51) suggesting that STAT3 may be important, at least under some conditions, for the induction of IL-9.
Tfh
Like Th17 differentiation, the generation of human Tfh cells is driven by numerous STAT3-ativating cytokines, namely IL-6, IL-12, IL-21, and IL-27. Consistent with a requirement for STAT3 to induce this differentiation program, patients with STAT3LOF have a reduction in circulating CXCR5+ Tfh cells (28, 52, 53), and naïve STAT3LOF CD4+ T cells failed to differentiate in vitro into IL-21-producing Tfh-like cells (41, 52, 54). The role of STAT3 in differentiation and/or function of Tfh cells has also been demonstrated in mouse studies of Stat3-deficient T cells (55–59). However, the degree to which Stat3 is required seems to be dependent on the presence of other signals such as STAT1 and type 1 IFNs as well as the site of the immune response (57, 59, 60). This defect in Tfh cells in AD-HIES would contribute to impaired humoral immunity in these patients and potentially to the deficiency in memory B cells. In vitro, IL-12 was found to be the main driver of IL-21-producing cells (61, 62), but since patients with defects in IL-12R signaling do not present with impaired humoral immunity (63–65), it is likely that the other STAT3-dependent cytokines IL-6, IL-21, and/or IL-27 plays a redundant role in this process in vivo. Consistent with this, IL-6 and IL-21 were found to induce ICOS expression in cord blood CD4+ T cells in a STAT3-dependent manner (53, 66), and patients with mutations in IL21/IL21R show defects in Tfh cell development and function (28, 41).
Regulatory T Cell
Studies in mice have suggested that IL-6/STAT3 signaling inhibits Treg differentiation inasmuch as Stat3−/− CD4+ T cells stimulated under Th17 conditions (TGF-β+ IL-6 in mice) showed decreased Th17 differentiation and increased Treg differentiation (67–69). However, normal frequencies of Tregs (defined as CD25hiCD127lo or FoxP3+) were reported in AD-HIES patients (28, 38, 47) and Tregs from these patients displayed normal suppressive behavior in vitro (47), suggesting STAT3 was somewhat redundant in controlling the generation of human Tregs. On the other hand, a recent study demonstrated that naïve CD4+ T cells from STAT3LOF patients showed an increased propensity to develop into iTregs in culture (70). CD4+ T cells from these patients have also been reported to display a defect in IL-10 production (28, 38, 41) and DCs from STAT3-deficient patients failed to induce Tregs from naïve CD4+ T cells (47). Thus, it may be that in these patients the increased propensity of STAT3LOF CD4+ to form induced Tregs is compensated for by the reduced ability of STAT3LOF dendritic cells to induce them.
Indeed, patients with STAT3GOF mutations much more clearly demonstrate a role for STAT3 in the regulation of Tregs. As discussed above, these patients display early onset autoimmunity that is reminiscent of patients with Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, which is caused by mutations in FoxP3 leading to a loss of Treg function (71). These overlapping clinical phenotypes suggested that STAT3GOF patients also have dysfunctional Tregs. Consistent with this, they were found to have lower percentage of FoxP3+ cells in their blood and lower CD25 expression on their Tregs (7). This is thought to be due to increased SOCS3 levels that inhibit the activation of STAT5 downstream of IL-2 (7).
Studies in mice have now demonstrated that there are different populations of Tregs that seem to be specialized for inhibiting particular T-helper populations (72). Thus, a population of Treg cells that express CCR6 and are specialized for suppressing Th17 cells has been described. These “Treg17” cells, like the Th17 cells they suppress, were found to be dependent on STAT3 signaling (73). AD-HIES patients were also shown to have decreased CCR6+ Tregs suggesting that human “Treg17” cells may also exist and be dependent on STAT3 (74).
Taken together, these data suggest STAT3 plays a complex role in the regulation of Treg responses and care should be taken in targeting this pathway as a means of regulating Treg responses.
The Role of STAT3 in CD8+ T Cells
STAT3 activating cytokines such as IL-21 also play a role in regulating CD8+ T cells. Studies on STAT3LOF CD8+ T cells showed they had impaired induction of perforin and granzyme B in response to IL-21; however, this could be rescued by strong TCR ligation (75). In contrast, proliferation induced by IL-21 was not affected in naïve STAT3LOF CD8+ T cells (75). STAT3 deficiency, however, did result in reduced memory CD8+ T cells (29, 75), an effect that was shown to be cell intrinsic (29). IL-21R patients also showed decreases in memory CD8+ T cell populations suggesting that IL-21 may be a cytokine upstream of STAT3 that contributes to the maintenance of memory cells (75). Patients with AD-HIES also show increased susceptibility to reactivation of viruses such as EBV and VZV (29) indicating that STAT3LOF CD8+ T cells may be defective in their ability to control these chronic infections.
STAT3 in Autoimmunity
STAT3GOF patients provide a clear demonstration that STAT3 plays an important role in controlling autoimmunity. However, previous evidence from other disease states had already indicated that STAT3 played an important role in the regulation of autoimmunity. In particular, multiple studies have associated polymorphisms in STAT3 with various autoimmune conditions including Crohn’s disease, ulcerative colitis, psoriasis, and Behcet’s disease (76–78). Furthermore, many of the cell populations that STAT3 can induce (Figure 1) have been implicated in driving autoimmunity; this includes Tfh cells and B cells, which support autoantibody production and Th9 and Th17 cells, can produce potentially damaging cytokines (79, 80). Conversely, STAT3 can inhibit Tregs, which act to restrain destructive immune responses.
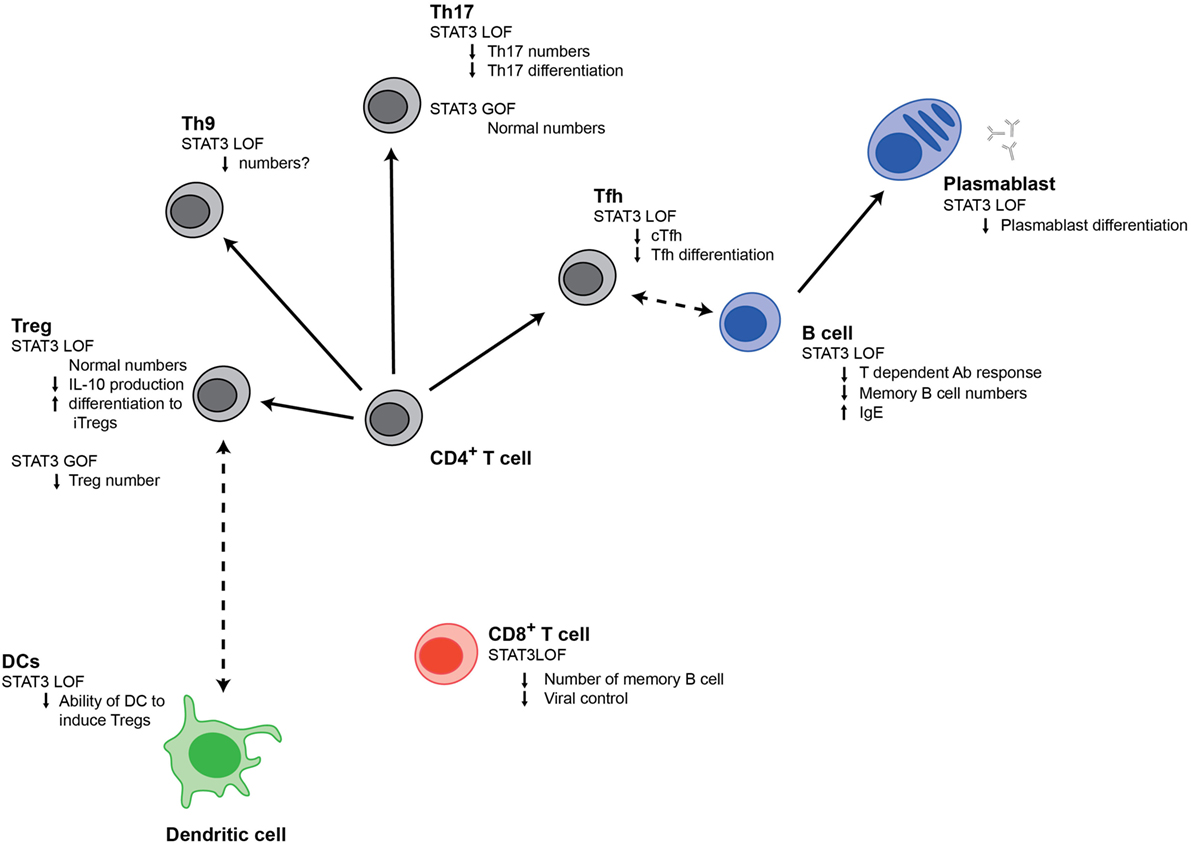
Figure 1. Signal transducer and activator of transcription 3 (STAT3) regulates multiple populations of immune cells. Changes in different immune cell populations that are observed in STAT3LOF or STAT3GOF patients are shown.
Interestingly, somatic STAT3GOF mutations have been reported in large granular lymphocytic (LGL) leukemia (81, 82) and are associated with higher rates of autoimmune complications such as rheumatoid arthritis and autoimmune cytopenias (81–83). As these LGL leukemias are of either CD8+ T cell or NK cell origin, this suggests that STAT3 overactivation in CD8+ T cells or NK cells alone may be sufficient to drive autoimmunity, possibly by inducing the production of cytokines/inflammatory mediators (84).
Taken together, these data are consistent with a role for STAT3 in promoting autoimmunity; however, STAT3 is also likely to play a role in inhibiting damaging responses downstream of IL-10. Indeed, germline LOF mutations in the genes coding for IL-10 or IL-10R are a major cause of early onset inflammatory bowel disease (85), demonstrating a critical requirement for IL-10 signaling (presumably through STAT3) to maintain tolerance in the bowel.
It remains unclear which of these many roles of STAT3 in multiple cell types underlies the phenotype in STAT3GOF patients. Certainly, the similarities with IPEX (71, 86) point to a clear role of Tregs in the phenotype; however, contributions from other pathways are also likely to make contributions. Further research will help clarify this and in turn pave the way for targeted treatments for both rare patients with STAT3GOF mutations as well as patients who suffer from autoimmunity more generally.
Author Contributions
ED, SP, AK, and CM wrote and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants awarded by the National Health and Medical Research Council (NHMRC) of Australia (grant nos. 1127157, 1108800, 1088215, and 1027400 to ED and CM). AK was supported by a National Health and Medical Research Council Postgraduate scholarship (1038881). CM is supported by a fellowship from NSW Ministry of Health under the NSW Health Early-Mid Career Fellowships Scheme. SP is supported by an Australian Postgraduate Award.
References
1. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med (2013) 368(2):161–70. doi:10.1056/NEJMra1202117
2. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol (2003) 3(11):900–11. doi:10.1038/nri1226
3. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med (2007) 357(16):1608–19. doi:10.1056/NEJMoa073687
4. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature (2007) 448(7157):1058–62. doi:10.1038/nature06096
5. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango Allen H, De Franco E, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi:10.1038/ng.3040
6. Haapaniemi EM, Kaustio M, Rajala HLM, van Adrichem AJ, Kainulainen L, Glumoff V, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood (2015) 125(4):639–48. doi:10.1182/blood-2014-04-570101
7. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood (2015) 125(4):591–9. doi:10.1182/blood-2014-09-602763
8. Chandesris M-O, Melki I, Natividad A, Puel A, Fieschi C, Yun L, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) (2012) 91(4):e1–19. doi:10.1097/MD.0b013e31825f95b9
9. Vogel TP, Milner JD, Cooper MA. The Ying and Yang of STAT3 in human disease. J Clin Immunol (2015) 35(7):615–23. doi:10.1007/s10875-015-0187-8
10. Chandrasekaran P, Zimmerman O, Paulson M, Sampaio EP, Freeman AF, Sowerwine KJ, et al. Distinct mutations at the same positions of STAT3 cause either loss or gain of function. J Allergy Clin Immunol (2016) 138(4):1222–4.e2. doi:10.1016/j.jaci.2016.05.007
11. Pelham SJ, Lenthall HC, Deenick EK, Tangye SG. Elucidating the effects of disease-causing mutations on STAT3 function in autosomal-dominant hyper-IgE syndrome. J Allergy Clin Immunol (2016) 138(4):1210–3.e5. doi:10.1016/j.jaci.2016.04.020
12. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A (1997) 94(8):3801–4. doi:10.1073/pnas.94.8.3801
13. Forbes LR, Milner J, Haddad E. Signal transducer and activator of transcription 3: a year in review. Curr Opin Hematol (2016) 23(1):23–7. doi:10.1097/MOH.0000000000000206
14. Hwa V. STAT5B deficiency: impacts on human growth and immunity. Growth Horm IGF Res (2016) 28:16–20. doi:10.1016/j.ghir.2015.12.006
15. Dreskin SC, Goldsmith PK, Gallin JI. Immunoglobulins in the hyperimmunoglobulin E and recurrent infection (Job’s) syndrome. Deficiency of anti-Staphylococcus aureus immunoglobulin A. J Clin Invest (1985) 75(1):26–34. doi:10.1172/JCI111683
16. Leung DY, Ambrosino DM, Arbeit RD, Newton JL, Geha RS. Impaired antibody responses in the hyperimmunoglobulin E syndrome. J Allergy Clin Immunol (1988) 81(6):1082–7. doi:10.1016/0091-6749(88)90873-1
17. Sheerin KA, Buckley RH. Antibody responses to protein, polysaccharide, and phi X174 antigens in the hyperimmunoglobulinemia E (hyper-IgE) syndrome. J Allergy Clin Immunol (1991) 87(4):803–11. doi:10.1016/0091-6749(91)90126-9
18. Meyer-Bahlburg A, Renner ED, Rylaarsdam S, Reichenbach J, Schimke LF, Marks A, et al. Heterozygous signal transducer and activator of transcription 3 mutations in hyper-IgE syndrome result in altered B-cell maturation. J Allergy Clin Immunol (2012) 129(2):559–62, 562.e1–2. doi:10.1016/j.jaci.2011.09.017
19. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med (2010) 207(1):155–71. doi:10.1084/jem.20091706
20. Bryant VL, Ma CS, Avery DT, Li Y, Good KL, Corcoran LM, et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J Immunol (2007) 179(12):8180–90. doi:10.4049/jimmunol.179.12.8180
21. Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med (2013) 210(12):2739–53. doi:10.1084/jem.20130323
22. Fornek JL, Tygrett LT, Waldschmidt TJ, Poli V, Rickert RC, Kansas GS. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood (2006) 107(3):1085–91. doi:10.1182/blood-2005-07-2871
23. Kane A, Lau A, Brink R, Tangye SG, Deenick EK. B-cell-specific STAT3 deficiency: insight into the molecular basis of autosomal-dominant hyper-IgE syndrome. J Allergy Clin Immunol (2016) 138(5):1455–8.e3. doi:10.1016/j.jaci.2016.05.018
24. Ding C, Chen X, Dascani P, Hu X, Bolli R, Zhang H-G, et al. STAT3 signaling in B cells is critical for germinal center maintenance and contributes to the pathogenesis of murine models of lupus. J Immunol (2016) 196(11):4477–86. doi:10.4049/jimmunol.1502043
25. Salzer E, Kansu A, Sic H, Májek P, Ikinciogullari A, Dogu FE, et al. Early-onset inflammatory bowel disease and common variable immunodeficiency-like disease caused by IL-21 deficiency. J Allergy Clin Immunol (2014) 133(6):1651–9.e12. doi:10.1016/j.jaci.2014.02.034
26. Kotlarz D, Ziętara N, Milner JD, Klein C. Human IL-21 and IL-21R deficiencies: two novel entities of primary immunodeficiency. Curr Opin Pediatr (2014) 26(6):704–12. doi:10.1097/MOP.0000000000000160
27. Kotlarz D, Ziętara N, Uzel G, Weidemann T, Braun CJ, Diestelhorst J, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med (2013) 210(3):433–43. doi:10.1084/jem.20111229
28. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1. doi:10.1016/j.jaci.2015.05.036
29. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity (2011) 35(5):806–18. doi:10.1016/j.immuni.2011.09.016
30. Tedder TF. B10 cells: a functionally defined regulatory B cell subset. J Immunol (2015) 194(4):1395–401. doi:10.4049/jimmunol.1401329
31. Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood (2013) 122(24):3940–50. doi:10.1182/blood-2013-06-506865
32. O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science (2010) 327(5969):1098–102. doi:10.1126/science.1178334
33. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly – TFH cells in human health and disease. Nat Rev Immunol (2013) 13(6):412–26. doi:10.1038/nri3447
34. Acosta-Rodríguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol (2007) 8(6):639–46. doi:10.1038/ni1467
35. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol (2007) 8(9):950–7. doi:10.1038/ni1497
36. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity (2008) 28(1):29–39. doi:10.1016/j.immuni.2007.11.016
37. Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature (2008) 452(7188):773–6. doi:10.1038/nature06764
38. Ma CS, Chew GYJ, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med (2008) 205(7):1551–7. doi:10.1084/jem.20080218
39. Renner ED, Rylaarsdam S, Aňover-Sombke S, Rack AL, Reichenbach J, Carey JC, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol (2008) 122(1):181–7. doi:10.1016/j.jaci.2008.04.037
40. de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med (2008) 205(7):1543–50. doi:10.1084/jem.20080321
41. Ma CS, Wong N, Rao G, Nguyen A, Avery DT, Payne K, et al. Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsets. J Exp Med (2016) 213(8):1589–608. doi:10.1084/jem.20151467
42. Cypowyj S, Picard C, Maródi L, Casanova J-L, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol (2012) 42(9):2246–54. doi:10.1002/eji.201242605
43. Lanternier F, Cypowyj S, Picard C, Bustamante J, Lortholary O, Casanova J-L, et al. Primary immunodeficiencies underlying fungal infections. Curr Opin Pediatr (2013) 25(6):736–47. doi:10.1097/MOP.0000000000000031
44. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LAB, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med (2011) 365(1):54–61. doi:10.1056/NEJMoa1100102
45. Liu L, Okada S, Kong X-F, Kreins AY, Cypowyj S, Abhyankar A, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med (2011) 208(8):1635–48. doi:10.1084/jem.20110958
46. Wienke J, Janssen W, Scholman R, Spits H, van Gijn M, Boes M, et al. A novel human STAT3 mutation presents with autoimmunity involving Th17 hyperactivation. Oncotarget (2015) 6(24):20037–42. doi:10.18632/oncotarget.5042
47. Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, et al. Defective IL-10 signaling in hyper-IgE syndrome results in impaired generation of tolerogenic dendritic cells and induced regulatory T cells. J Exp Med (2011) 208(2):235–49. doi:10.1084/jem.20100799
48. Beriou G, Bradshaw EM, Lozano E, Costantino CM, Hastings WD, Orban T, et al. TGF-beta induces IL-9 production from human Th17 cells. J Immunol (2010) 185(1):46–54. doi:10.4049/jimmunol.1000356
49. Chang H-C, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol (2010) 11(6):527–34. doi:10.1038/ni.1867
50. Wong MT, Ye JJ, Alonso MN, Landrigan A, Cheung RK, Engleman E, et al. Regulation of human Th9 differentiation by type I interferons and IL-21. Immunol Cell Biol (2010) 88(6):624–31. doi:10.1038/icb.2010.53
51. Becker KL, Rösler B, Wang X, Lachmandas E, Kamsteeg M, Jacobs CW, et al. Th2 and Th9 responses in patients with chronic mucocutaneous candidiasis and hyper-IgE syndrome. Clin Exp Allergy (2016) 46(12):1564–74. doi:10.1111/cea.12787
52. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood (2012) 119(17):3997–4008. doi:10.1182/blood-2011-11-392985
53. Mazerolles F, Picard C, Kracker S, Fischer A, Durandy A. Blood CD4+CD45RO+CXCR5+ T cells are decreased but partially functional in signal transducer and activator of transcription 3 deficiency. J Allergy Clin Immunol (2013) 131(4):1146–56, 1156.e1–5. doi:10.1016/j.jaci.2012.12.1519
54. Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J Exp Med (2010) 207(13):2895–906. doi:10.1084/jem.20100064
55. Eddahri F, Denanglaire S, Bureau F, Spolski R, Leonard WJ, Leo O, et al. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood (2009) 113(11):2426–33. doi:10.1182/blood-2008-04-154682
56. Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity (2008) 29(1):138–49. doi:10.1016/j.immuni.2008.05.009
57. Choi YS, Eto D, Yang JA, Lao C, Crotty S. Cutting edge: STAT1 is required for IL-6-mediated Bcl6 induction for early follicular helper cell differentiation. J Immunol (2013) 190(7):3049–53. doi:10.4049/jimmunol.1203032
58. McIlwain DR, Grusdat M, Pozdeev VI, Xu HC, Shinde P, Reardon C, et al. T-cell STAT3 is required for the maintenance of humoral immunity to LCMV. Eur J Immunol (2015) 45(2):418–27. doi:10.1002/eji.201445060
59. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40(3):367–77. doi:10.1016/j.immuni.2014.02.005
60. Wu H, Chen Y, Liu H, Xu L-L, Teuscher P, Wang S, et al. Follicular regulatory T cells repress cytokine production by follicular helper T cells and optimize IgG responses in mice. Eur J Immunol (2016) 46(5):1152–61. doi:10.1002/eji.201546094
61. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naïve human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol (2009) 87(8):590–600. doi:10.1038/icb.2009.64
62. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity (2009) 31(1):158–69. doi:10.1016/j.immuni.2009.04.016
63. Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science (1998) 280(5368):1432–5. doi:10.1126/science.280.5368.1432
64. de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency. Medicine (Baltimore) (2010) 89(6):381–402. doi:10.1097/MD.0b013e3181fdd832
65. de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science (1998) 280(5368):1435–8. doi:10.1126/science.280.5368.1435
66. Ysebrant de Lendonck L, Eddahri F, Delmarcelle Y, Nguyen M, Leo O, Goriely S, et al. STAT3 signaling induces the differentiation of human ICOS(+) CD4 T cells helping B lymphocytes. PLoS One (2013) 8(7):e71029. doi:10.1371/journal.pone.0071029
67. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26(3):371–81. doi:10.1016/j.immuni.2007.02.009
68. Nishihara M, Ogura H, Ueda N, Tsuruoka M, Kitabayashi C, Tsuji F, et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int Immunol (2007) 19(6):695–702. doi:10.1093/intimm/dxm045
69. Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature (2007) 448(7152):480–3. doi:10.1038/nature05969
70. Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med (2017) 214(3):669–80. doi:10.1084/jem.20161435
71. Bacchetta R, Barzaghi F, Roncarolo M-G. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci (2016) 100(2):1–18. doi:10.1111/nyas.13011
72. Josefowicz SZ, Lu L-F, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
73. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science (2009) 326(5955):986–91. doi:10.1126/science.1172702
74. Kluger MA, Luig M, Wegscheid C, Goerke B, Paust H-J, Brix SR, et al. Stat3 programs Th17-specific regulatory T cells to control GN. J Am Soc Nephrol (2014) 25(6):1291–302. doi:10.1681/ASN.2013080904
75. Ives ML, Ma CS, Palendira U, Chan A, Bustamante J, Boisson-Dupuis S, et al. Signal transducer and activator of transcription 3 (STAT3) mutations underlying autosomal dominant hyper-IgE syndrome impair human CD8(+) T-cell memory formation and function. J Allergy Clin Immunol (2013) 132(2):400–11.e9. doi:10.1016/j.jaci.2013.05.029
76. Ellinghaus D, Ellinghaus E, Nair RP, Stuart PE, Esko T, Metspalu A, et al. Combined analysis of genome-wide association studies for Crohn disease and psoriasis identifies seven shared susceptibility loci. Am J Hum Genet (2012) 90(4):636–47. doi:10.1016/j.ajhg.2012.02.020
77. Hu K, Hou S, Jiang Z, Kijlstra A, Yang P. JAK2 and STAT3 polymorphisms in a Han Chinese population with Behçet’s disease. Invest Ophthalmol Vis Sci (2012) 53(1):538–41. doi:10.1167/iovs.11-8440
78. de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet (2017) 49(2):256–61. doi:10.1038/ng.3760
79. Ma CS, Deenick EK. Human T follicular helper (Tfh) cells and disease. Immunol Cell Biol (2014) 92(1):64–71. doi:10.1038/icb.2013.55
80. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74(1):5–17. doi:10.1016/j.cyto.2014.09.011
81. Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med (2012) 366(20):1905–13. doi:10.1056/NEJMoa1114885
82. Jerez A, Clemente MJ, Makishima H, Koskela H, Leblanc F, Peng Ng K, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood (2012) 120(15):3048–57. doi:10.1182/blood-2012-06-435297
83. Teramo A, Barilà G, Calabretto G, Ercolin C, Lamy T, Moignet A, et al. STAT3 mutation impacts biological and clinical features of T-LGL leukemia. Oncotarget (2017) 8(37):61876–89. doi:10.18632/oncotarget.18711
84. Lamy T, Moignet A, Loughran TP. LGL leukemia: from pathogenesis to treatment. Blood (2017) 129(9):1082–94. doi:10.1182/blood-2016-08-692590
85. Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology (2012) 143(2):347–55. doi:10.1053/j.gastro.2012.04.045
Keywords: signal transducer and activator of transcription 3, T cells, B cells, immunodeficiency, immune dysregulation
Citation: Deenick EK, Pelham SJ, Kane A and Ma CS (2018) Signal Transducer and Activator of Transcription 3 Control of Human T and B Cell Responses. Front. Immunol. 9:168. doi: 10.3389/fimmu.2018.00168
Received: 06 December 2017; Accepted: 19 January 2018;
Published: 07 February 2018
Edited by:
Raffi Gugasyan, Burnet Institute, AustraliaReviewed by:
Alessandro Plebani, University of Brescia, ItalyAndrew L. Snow, Uniformed Services University of the Health Sciences, United States
Copyright: © 2018 Deenick, Pelham, Kane and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elissa K. Deenick, ZS5kZWVuaWNrQGdhcnZhbi5vcmcuYXU=