Abstract
Metabolism, including catabolism and anabolism, is a basic cellular process necessary for cell survival. T lymphocytes have a distinct metabolism that can determine both fate and function. T-cell activation depends on glycolysis to obtain materials and energy for proliferation and effector function. Importantly, T cells utilize different metabolic processes under different conditions and diseases. Allogeneic hematopoietic cell transplantation (allo-HCT) is a classic immunotherapy for hematological malignancies; however, the development of graft-versus-host disease (GVHD) is a major factor limiting the success of allo-HCT. T cells in the donor graft drive GVHD by mounting a robust immunological attack against recipient normal tissues. Hence, understanding T-cell metabolism after allo-HCT would provide potential metabolic targets for the control of GVHD and primary tumor relapse. The purpose of the current review is to highlight the key metabolic pathways involved in alloantigen-activated T cells and to discuss how manipulating these pathways can serve as potential new therapeutic strategies to induce immune tolerance after allo-transplantation. We will also summarize the recent progress in regulating T-cell metabolism in bone marrow transplantation by targeting novel metabolic regulators or immune checkpoint molecules.
Introduction
Allogeneic bone marrow transplantation [BMT; allogeneic hematopoietic stem-cell transplantation (allo-HCT)] is a curative option to treat hematological malignancies. However, graft-versus-host disease (GVHD) limits the success of allo-HCT (1). GVHD pathogenesis is characterized by a robust immunological attack by donor T cells against normal tissues of transplanted recipients (2). As donor T cells are the driving force in GVHD, suppressing T-cell responses is a standard therapeutic approach for the treatment of GVHD. However, these broadly immunosuppressive drugs, including corticosteroids and inhibitors of calcineurin or mammalian target of rapamycin (mTOR), leave patients highly susceptible to infections and induce remission in <50% of patients. The mortality rate of patients with steroid-refractory aGVHD is close to 90% (3). Hence, understanding T-cell pathobiology is critical to the development of effective therapies to prevent GVHD. Cell metabolism impacts the fate and function of T cells (4). Targeting T-cell metabolism is a viable therapeutic strategy in other immunological disorders, including systemic lupus erythematosus, rheumatoid arthritis, and experimental autoimmune encephalomyelitis (5–7). A growing body of evidence from multiple studies suggests T-cell metabolism is a promising target for controlling GVHD. Recently, our group and others attempted to characterize the metabolic profile of donor T cells following allo-HCT, yet a consensus on the data has not been reached (2, 8, 9). In this review, we will detail the recent findings in the evolving field of immuno-metabolism with a focus on T-cell metabolism in the context of allo-HCT and discuss how this knowledge can help us reevaluate our current understanding of immune activation and suppression after allo-HCT, and promising immunotherapeutic strategies to archive long-term transplantation tolerance in transplanted recipients aiming to prevent allograft rejection and GVHD.
Overview of T-Cell Metabolism
Glycolysis and oxidative phosphorylation (OXPHOS) are fundamental cellular processes in generating energy, or adenosine triphosphate (ATP) (10, 11). Naïve T cells rely primarily on OXPHOS to meet their energy demands (12). Upon antigen recognition, naïve T cells clonally expand into T effector cells (Teffs). Upon antigen clearance, most of these effector T cells die, but a subset of long-lived memory T cells (Tm) persist with an enhanced mitochondrial capacity relying on fatty acid oxidation (FAO) to fuel OXPHOS (13). OXPHOS can generate up to 36 molecules of ATP. The transition from resting naïve T-cells into activated Teffs requires substantial metabolic reprogramming (12, 13). A Teff’s metabolic profile is characterized by a shift to aerobic glycolysis as a main energy source (12, 14). Aerobic glycolysis involves the mitochondrion-independent metabolism of glucose into pyruvate and provides only two molecules of ATP per glucose (15). While glycolysis is less efficient than OXPHOS at yielding an abundance of ATP per molecule of glucose, aerobic glycolysis supplies metabolic intermediates for cell growth and proliferation as well as induces the pentose phosphate pathway (PPP), which produces nucleotides and amino acids that subsequently generate reducing power in the form of NADH to maintain cellular redox balance (NAD+/NADH) (15). Teffs also use glutamine as a carbon source to fuel the tricarboxylic acid (TCA) cycle via α-ketoglutarate (α-KG) through the process of glutaminolysis (16, 17).
Metabolism and CD4+T Cell Differentiation
Depending on the nature of antigen and cytokine signal, CD4+ T cells differentiate into Th1, Th2, Th9, Th17, T follicular helper cells (Tfh), Tr-1, or Treg. While Th1, Th2, and Th17 are pathogenic, Tr-1 and Treg are suppressive in acute GVHD (18–20). Metabolism plays a critical role in CD4+ T-cell differentiation (12). While Th1, Th2, and Th17 lineages preferentially use glycolysis to meet energetic demand though activation of PI3K/Akt/mTOR pathway, CD4+ Tregs use mitochondrial-dependent FAO (4). Therefore, enhanced FAO via inhibiting mTOR leads to increased Treg generation (21). Hypoxia-inducible factor 1 is the key regulator of anabolic metabolism in Th17 cells (22). Meanwhile, Tfh, a pathogenic T-cell subset in chronic GVHD, depend on glycolysis and lipogenesis to meet energy demands required for differentiation (23). The metabolic profiles of Th9 and Tr1 remain unclear.
Metabolism of Allogeneic T Cells
Glucose Metabolism
Using MHC-mismatched or haploidentical murine models of BMT, we uncovered that upon alloantigen activation, donor T cells increase both glycolysis and OXPHOS to obtain energetic materials necessary for activation and proliferation (2, 9). Albeit, they preferentially rely on glycolysis to maintain their capacity to induce GVHD (2, 9, 24). While OXPHOS of donor T cells isolated from syngeneic (no GVHD) and allogeneic (GVHD) recipients were similar, the glycolytic activity of donor T cells was significantly higher in allogeneic than syngeneic recipients, indicating an escalation of T-cell glucose metabolism correlated with GVHD development (2) (Figure 1). Furthermore, T cells isolated from livers of allogeneic recipients exhibited higher glycolytic activity compared to those of syngeneic recipients 14 days after allo-HCT, implying an enduring glycolytic response by allogeneic T cells in GVHD target organs. While in vitro activated T cells upregulate and maintain expression of Glut1 for sufficient glucose uptake (17), allo-activated T cells also increase Glut 3 to fulfill their extremely high demand for glucose (2). In addition, alloantigen-activated T cells upregulate both hexokinase 1 (HK1) and HK2 to facilitate induction of glycolysis (2). To maintain sufficient glycolytic activity, allogeneic CD4+ T cells activate mTOR and increase differentiation into Th1 and Th17 (2, 25) while decreasing Treg generation (24). Inhibition of glycolysis by genetic depletion or pharmacological blockade of mTORC1 (2, 26) or glycolytic checkpoints, including glut-1 (24), HK-2, PFKB3 (2), or PKM2 (unpublished study), reduces alloreactive T-cell generation and subsequently ameliorates GVHD severity. Alternatively, enhancing FAO to inhibit mTOR using PI3K/AKT or AMPK inhibitors (27, 28) effectively prevents GVHD development.
Figure 1
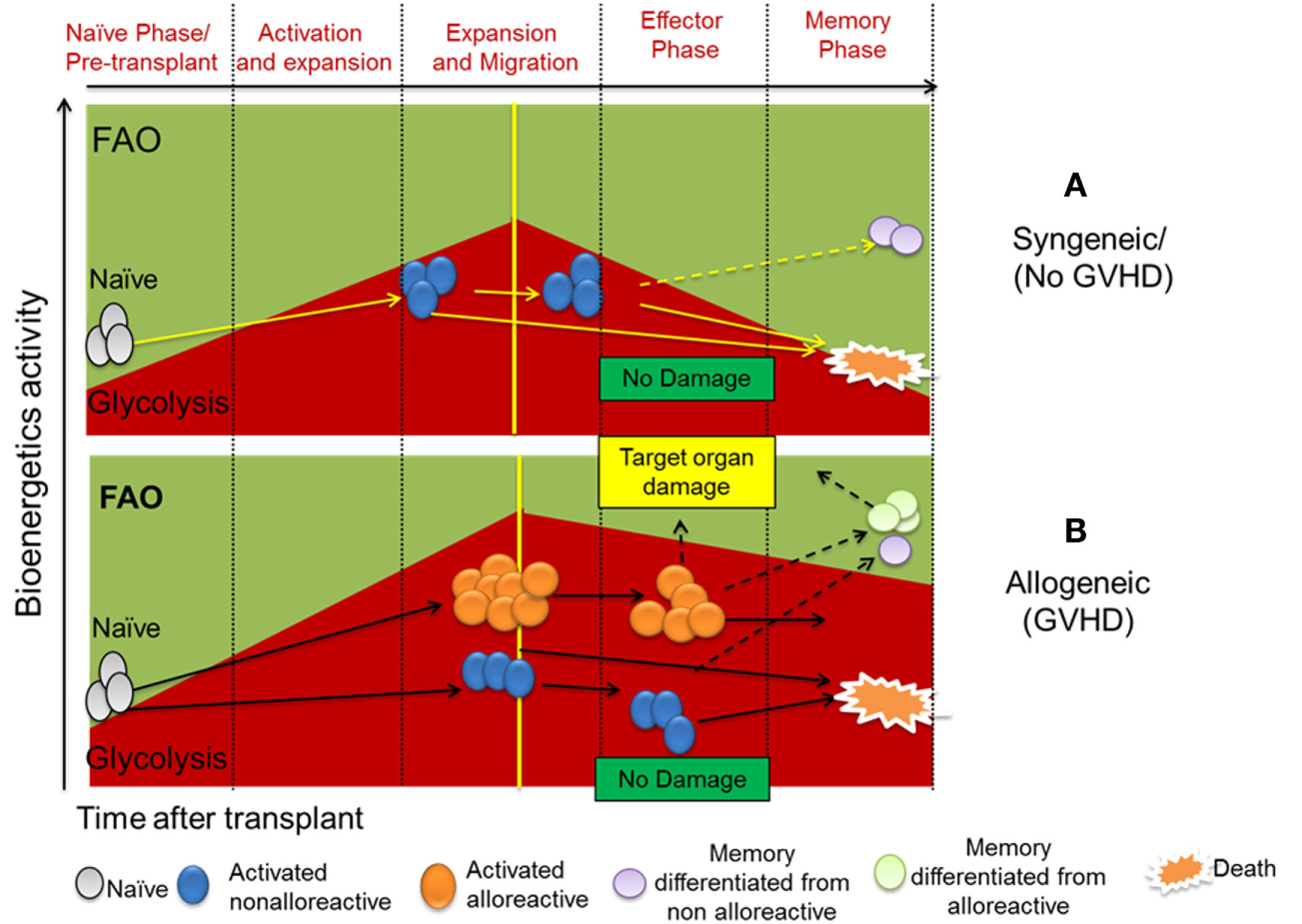
(A) Naïve/resting T cells are dependent on oxidative phosphorylation with fatty acid oxidation (FAO) as a major material resource. Upon activation by self-antigens under homeostatic state, naïve/resting T cells reprogram their metabolic phenotype to become partially activated T cells (29), which possess glycolytic metabolic phenotype. Due to lack of specific TCR stimulation, a large proportion of non-alloreactive T cells gradually die. However, specific self-epitopes of T cells can become memory T cells (Tm) which depend upon FAO for their metabolism. (B) Upon activation by alloantigen in transplant recipients, naïve/resting T cells proliferate and their memory differentiate to activate T cells both alloreactive and non-alloreactive. Alloreactive T cells and their differentiated memory cells are capable of causing target organ damage. Alloreactive T cells have much higher glycolytic activity compared to non-alloreactive counterpart. Both alloreactive and non-alloreactive T cells can die or differentiate into Tms accordingly. Glucose retention and glycolytic activity decide survival and alloreactivity of alloreactive T cells to induce graft-versus-host disease (GVHD) after allogeneic hematopoietic cell transplantation.
OXPHOS and Oxidative Stress in Allogeneic T Cells
Allogeneic T cells in lymphoid or target organs of recipients significantly increase OXPHOS compared to resting T cells after allo-HCT (2, 9). Since OXPHOS activity was comparable in allogeneic and syngeneic T cells (2), increased OXPHOS may not be a direct mechanism by which pathogenic T cells are generated. However, due to increased non-mitochondrial oxygen consumption rate (OCR), allogeneic T cells had higher levels of oxidative stress yet lower levels of antioxidants (2, 9). As reactive oxygen species (ROS) are required for T-cell activation (30), this indicates chronic allo-activation of donor T cells after transplant. Increased ROS generation in allogeneic T cells may be the result of a hyperpolarized mitochondrial membrane potential (ΔΨm), subsequently making alloreactive CD4+ and CD8+T cells highly susceptible to small-molecule inhibitors of mitochondrial F1F0 adenosine triphosphate synthase in haploidentical BMT model (9, 31).
The Pentose Phosphate Pathway
In murine models of GVHD, alloantigen-activated T cells have increased PPP activity (2, 31). Intracellular glucose metabolized by HK forms glucose 6-phospate (G-6P), which then enters the PPP to generate ribose-5 phosphate (R-5P); the carbon donor during nucleotide biogenesis (32). The conversion of G-6P to R-5P is regulated by glucose-6-phosphate dehydrogenase in the oxidative arm of the PPP (33), which is significantly increased in allogeneic T cells (2, 31). The oxidative arm of the PPP is crucial for the formation of NADPH, which plays a critical role in reductive biosynthesis of antioxidant molecules, such as GSH (34). GSH promotes T-cell expansion by driving glycolysis and glutaminolysis, and supporting mTORC1 and c-Myc signaling in inflammation (35). Due to chronic stimulation by alloantigens, nucleotide biosynthesis is sustained to support anabolic growth of T cells during allogeneic responses; leading to a deficit in purine and pyrimidine catabolism (2) and exhaustion of GS and GSH (9).
Glutamine Metabolism
Glutamine uptake and metabolism are crucial for normal T-cell function (36). Donor T cells require the rapid synthesis of macromolecules for their growth, proliferation, and for energy after allo-HCT (11). Glutamine converted to glutamate can support the progression of the TCA cycle, ultimately leading to production of α-KG, a citrate precursor. To generate new lipids, citrate is secreted into the cytosol and metabolized to form acetyl-CoA, the backbone for lipid synthesis (34). In addition to the PPP, glutaminolysis can provide NADPH to support lipid and nucleotide biosynthesis as well as maintenance of GSH (37). In vitro-activated T cells utilize the transcription factor Myc to incorporate glutamine into metabolic pathways (17). Allogeneic T cells increase glutamine uptake by upregulating glutamine transport channels, such as glutamine-fructose-6-phosphate transaminase, phosphoribosyl pyrophosphate amidotransferase, and glutaminase 2 post allo-HCT (2). While the level of glutamine was increased in allogeneic T cells, the level of glutamate was lower. Moreover, the levels of aspartate and ornithine, products of glutamate conversion to α-KG by ornithine aminotransferase and glutamate oxaloacetate transaminase, respectively, were increased in allogenic T cells after allo-HCT (2, 31). These data suggest that alloantigen-activated T cells further increase glutaminolysis to replenish intermediate metabolites of the TCA cycle that are depleted in proliferating T cells after allo-HCT. Studies using radioactive tracers indicate that alloreactive CD4+ and CD8+ T cells preferentially use glutamine to provide substrates for ribose synthesis (31).
Fatty Acid Metabolism
Alloantigen-activated T cells accumulate various types of FAs and lysophospholipids after allo-HCT (2). In addition to glucose and glutamine, lipids are an effective energy source as well as biosynthetic intermediates (38). FAs can be generated through three different pathways: environmental uptake, synthesis, or hydrolysis of membrane or lipid droplets (39). FAs are classified according to (a) to their backbone lengths (short-, medium-, long-, and very long-chain), (b) saturation, i.e., the number of double bonds (unsaturated, mono-, poly-unsaturated), and (c) position of the double bonds (37). During activation, in vitro activated T cells augment fatty acid synthase (FAS) while decreasing FAO, thus enhancing the accumulation of FA metabolites needed for the membrane (17). The effect of lipids on T-cell function seems to be mediated by a complex network dependent on the type of lipids (40).
Fatty Acid Synthesis
FAs have an important role in Teff function and differentiation. Acetyl-CoA carboxylases 1 (ACC1), ACC2, and FAS are recognized as key rate-limiting enzymes in this process (41). Inhibition of FAS limits development of Th1, Th2, and Th17 subsets (42, 43). Blockade of the enzyme ACC1 enhances the formation of Tregs during Th17 differentiation (43). In vitro, induction of FAS after TCR stimulation is regulated via the mTORC1–SREBP pathway (14, 44). Moreover, Myc is essential for activation of glucose-metabolizing genes and also for FA synthesis, linking glycolysis to de novo FAS (45). Recent studies showed that FAS is required for maintaining glycolytic activity in allogeneic T cells (46). Disruption of FAS at ACC1 effectively ameliorates GVHD development (46, 47). This study emphasizes the relationship between glycolysis and FAS in allogeneic T cells.
Fatty Acid Oxidation
Fatty acid oxidation is a multistep energetic process by which FAs are broken down in the mitochondria via sequential removal of 2-carbon units at the β-carbon position of a fatty acyl-CoA molecule (39, 48). A given long-chain acyl-CoA that enters the FAO yields one molecule of acetyl-CoA from each cycle of FAO. This acetyl-CoA can be directly shuttled into TCA cycle. The NADH and FADH2 produced during FAO and the TCA cycle are then available to be used. While saturated short long-chain FA (SCFAs) and medium chain FA are almost exclusively oxidized in the mitochondria, long-chain FA and very long-chain fatty acids (>14 carbons) can also be oxidized in peroxisomes (49). Previous studies have indicated that alloreactive T cells increase FAO, and that targeting FAO could arrest GVHD in haploidentical allo-HCT (8, 9). Although they reported substantial increases in FA transport and intracellular acylcarnitines, suggesting changes in FA metabolism, it was not determined if FAO was directly responsible for the increase in OXPHOS (31, 34). Also, no improvement in survival of recipients treated with FAO inhibitors was shown. By contrast, our recent study showed intracellular carnitine-derived metabolites were diminished in alloantigen-activated T cells after MHC-mismatched or haploidentical allo-HCT (2). Allogeneic T cells dramatically decreased mitochondrial-dependent FAO and pyruvate oxidation through the TCA cycle. Therefore, it is possible that FAO is downregulated in allogeneic T cells after allo-HCT. These inconsistent observations likely result from the different controls used in these two studies. While studies from Ferrara’s group compared bioenergetic parameters of allogeneic T cells to naïve/resting T cells (9), we used those isolated from syngeneic recipients as controls (2); intended to account for homeostatic proliferation of T cells under an inflammatory environment (29). In addition, we observed both Glut1 and Glut3 expression could serve as indicators of glycolytic activity (9), as alloreactive T cells increase Glut3 to an even larger extent than Glut1 in allogeneic recipients (2). Taken together, with study from by Rathmell’s group (24), we speculate that FAO might not be the major material resource fueling the TCA cycle and OXPHOS in alloreactive T cells.
Sphingolipids (SLs) in Allogeneic T-Cell Metabolism
Sphingolipids represent a major class of lipids important for cell membrane formation (50). S1P is emerging as a key regulator of proliferation, inflammation, vasculogenesis, and resistance to apoptotic cell death (51). Recently, a report demonstrated that S1P1 regulates T cell metabolism through activation of mTOR-Akt, which suppressed Treg function (52). Blockade of the S-1P receptor effectively prevents GVHD by modulating the migration of allogeneic T cells. Ceramide plays a central role in the metabolism of SL (53, 54). Ceramide can be generated via de novo synthesis or by degradation of complex SLs, especially sphingomyelin (51). The key rate-limiting step in the biosynthesis of ceramide is the attachment of various acyl-CoA side chains to a sphingoid base by ceramide synthases (CerS) (55). The CerS show substrate preferences for specific chain lengths of fatty acyl CoAs. Briefly, CerS1 shows significant preference for C18-FA CoA, CerS4 for C18-/C20-FA CoA, CerS5 and CerS6 for C16- FA CoA, CerS2 for C22/C24- FA CoA, and CerS3 for ultra-long-chain FA CoA (51, 56). Recent work from our lab showed that CerS6 regulates SL metabolism in alloantigen-activated CD4+ and CD8+ T cells and required for alloreactive T cells to induce GVHD (57).
The Role of PD-1 and Check Point Blockade on Allogeneic T Cell Metabolism
The coinhibitory receptor programmed death 1 (PD-1; CD279) has key roles in modulating T-cell responses in both normal and antitumor immunity (58). PD-1 binds to PD-L1 (B7-H1; CD274), which is expressed by macrophages, DCs and non-hematopoietic cells, and PD-L2 (B7-DC; CD273), which is primarily expressed by monocytes and inflammatory macrophages in GVHD target organs (59, 60). Donor T cells significantly upregulate PD-1 expression, which can increase in response to FAO, superoxide, hyperpolarized mitochondrial membrane potential, and ROS formation which subsequently induces T-cell death following allo-HCT. In the absence of PD-1/PD-L1 ligation, donor T cells displayed higher glycolytic activity and OCR. Hence, PD-L1/PD-1 ligation, versus that of PD-L2/PD-1, plays a predominant role in downregulating GVHD (59).
Microbiota Regulates T Cell Metabolism
The composition, or diversity, of intestinal microbiota shapes the innate and adaptive immune responses (61). The onset of GVHD is associated with a progressive reduction in microbiota diversity, with an increase in Lactobacillales and Blautia and a decrease in Clostridiales species (62–64). The microbiota metabolome, which consists of products generated by host metabolism, microbial metabolism, and mammalian–microbial co-metabolism in the intestines, influences the development of GVHD (65, 66). SCFA-bacterial metabolites, derived from carbohydrate fermentation and include acetate, propionate, isobutyrate, and butyrate, increase histone H3 acetylation in the locus of Foxp3; thereby increasing the numbers of Tregs directly, yet also indirectly through increasing the production of TGFβ in the intestinal epithelium (67). The effect of SCFAs on T cells is also related to mTOR activation (68). SCFAs induce the expression of receptor GPR15, which is responsible for the recruitment of Tregs to the large intestine (69–71). Restoration of butyrate, which is diminished in intestinal epithelial cells (IECs) after allo-HCT, improved IEC junctional integrity, decreased apoptosis, and mitigated GVHD (66). Aryl hydrocarbon receptor (AhR) is a cellular metabolic sensor (72). AhR ligands are derived from intestinal microbiota metabolism. AhR ligand, indole-3-aldehyde, produced by Lactobacilli through tryptophan breakdown (73), modulates the development of GVHD through inducing Tregs and Tr1 cells (74).
Targeting T-Cell Metabolism to Separate GVHD and the Graft-Versus-Tumor Effect
Given that tumors and alloreactive T cells share a glycolytic phenotype, pharmacological glycolysis inhibition could prevent both GVHD and tumor relapse, a primary complication after allo-HCT. Inhibition of glucose-metabolizing enzymes could reduce allogeneic T activation and function (2, 17) and, further, lower levels of glycolysis would support the generation of long-lived CD8 Tm (3) which are required for maintaining the graft-versus-tumor (GVT) effect. Moreover, in vivo activated CD4+T cells are more dependent on glycolysis than CD8+T cells (75), which are critically important for maintaining GVT activity in allo-HCT. Increasing evidence indicates that CD8+ T cells with lower rates of glycolytic activity have better antitumor efficacy in eradicating established tumor in adoptive T cell transfer (ACT) models (76). Blocking glucose metabolism at HK2 by 2-deoxyglucose improves antitumor efficacy of ACT therapy (40). The aforementioned evidence suggests a valid possibility of targeting glycolysis to treat GVHD while preserving the GVT effect after allo-HCT.
Impact of Current Immunosuppressive Drugs on T-Cell Metabolism in Allo-HCT
Corticosteroids inhibit glycolysis and endogenous respiration in donor lymphocytes and impair GVL activity (77). Inhibiting mTOR with rapamycin decreases glycolysis and enhances FAO in donor T cells; this is expected to reduce alloreactive T cells and enhance Treg function (27). However, attempts to conceptually translate this into patients have proven difficult. This challenge may be because rapamycin can promote CD8 memory T-cell responses by enhancing FAO and hence be detrimental in establishing tolerance (78). Alternatively, inhibition of calcineurin with cyclosporine diminishes glycolytic activity of donor T cells by decreasing glycolytic enzymes and the expression of glut1/3 (79); which support Treg expansion and GVHD attenuation (80).
Concluding Remarks
Current immunosuppressive regimens, including steroids and calcineurin inhibitors, help to prevent allograft rejection and GVHD. Consequently, patients are vulnerable to complications, such as opportunistic infections and tumor relapse. Therefore, bioenergetic signatures of immune cells at different stages of tolerance induction after transplant could serve as a promising clinical therapeutic strategy. Metabolism inhibitors, in concert with cancer immunotherapies, highlight an avenue by which to achieve better antitumor efficacy and functional tolerance to allografts. Hence, distinguishing metabolic signatures between allogeneic T cells and tumor cells is critical to truly fulfilling this goal.
Statements
Author contributions
HN and XZ-Y wrote manuscript and HN, SK, DB, and XZ-Y revised manuscript.
Funding
This work is partially supported by NIH grants R01 AI118305, CA118116, CA169116, and R21 CA192202 (to XZ-Y).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
FerraraJLLevineJEReddyPHollerE. Graft-versus-host disease. Lancet (2009) 373:1550–61.10.1016/S0140-6736(09)60237-3
2
NguyenHDChatterjeeSHaarbergKMWuYBastianDHeinrichsJet alMetabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest (2016) 126:1337–52.10.1172/JCI82587
3
BlazarBRMurphyWJAbediM. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol (2012) 12:443–58.10.1038/nri3212
4
BuckMDSowellRTKaechSMPearceEL. Metabolic instruction of immunity. Cell (2017) 169:570–86.10.1016/j.cell.2017.04.004
5
TsokosGC. Metabolic control of arthritis: switch pathways to treat. Sci Transl Med (2016) 8:331fs8.10.1126/scitranslmed.aaf4953
6
YinYChoiSCXuZPerryDJSeayHCrokerBPet alNormalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med (2015) 7:274ra18.10.1126/scitranslmed.aaa0835
7
ChangCHPearceEL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17:364–8.10.1038/ni.3415
8
ByersdorferCATkachevVOpipariAWGoodellSSwansonJSandquistSet alEffector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood (2013) 122:3230–7.10.1182/blood-2013-04-495515
9
GatzaEWahlDROpipariAWSundbergTBReddyPLiuCet alManipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med (2011) 3:67ra8.10.1126/scitranslmed.3001975
10
CameronAMLawlessSJPearceEJ. Metabolism and acetylation in innate immune cell function and fate. Semin Immunol (2016) 28:408–16.10.1016/j.smim.2016.10.003
11
BuckMDO’SullivanDPearceEL. T cell metabolism drives immunity. J Exp Med (2015) 212:1345–60.10.1084/jem.20151159
12
GerrietsVARathmellJC. Metabolic pathways in T cell fate and function. Trends Immunol (2012) 33:168–73.10.1016/j.it.2012.01.010
13
PearceELPoffenbergerMCChangCHJonesRG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454.10.1126/science.1242454
14
YangKShresthaSZengHKarmausPWNealeGVogelPet alT cell exit from quiescence and differentiation into Th2 cells depend on raptor-mTORC1-mediated metabolic reprogramming. Immunity (2013) 39:1043–56.10.1016/j.immuni.2013.09.015
15
Vander HeidenMGCantleyLCThompsonCB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33.10.1126/science.1160809
16
MetalloCMGameiroPABellELMattainiKRYangJHillerKet alReductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481:380–4.10.1038/nature10602
17
WangRDillonCPShiLZMilastaSCarterRFinkelsteinDet alThe transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82.10.1016/j.immuni.2011.09.021
18
ZengHZhangRJinBChenL. Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol (2015) 12:566–71.10.1038/cmi.2015.44
19
ZhangPLeeJSGartlanKHSchusterISComerfordIVareliasAet alEomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci Immunol (2017) 2:eaah7152.10.1126/sciimmunol.aah7152
20
CoghillJMSarantopoulosSMoranTPMurphyWJBlazarBRSerodyJS. Effector CD4+ T cells, the cytokines they generate, and GVHD: something old and something new. Blood (2011) 117:3268–76.10.1182/blood-2010-12-290403
21
ZengHYangKCloerCNealeGVogelPChiH. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499:485–90.10.1038/nature12297
22
ShiLZWangRHuangGVogelPNealeGGreenDRet alHIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76.10.1084/jem.20110278
23
ZengHCohenSGuyCShresthaSNealeGBrownSAet almTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45:540–54.10.1016/j.immuni.2016.08.017
24
MacintyreANGerrietsVANicholsAGMichalekRDRudolphMCDeoliveiraDet alThe glucose transporter glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab (2014) 20:61–72.10.1016/j.cmet.2014.05.004
25
ParkMJLeeSHLeeSHLeeEJKimEKChoiJYet alIL-1 receptor blockade alleviates graft-versus-host disease through downregulation of an interleukin-1beta-dependent glycolytic pathway in Th17 cells. Mediators Inflamm (2015) 2015:631384.10.1155/2015/631384
26
Herrero-SanchezMCRodriguez-SerranoCAlmeidaJSan-SegundoLInogesSSantos-BrizAet alEffect of mTORC1/mTORC2 inhibition on T cell function: potential role in graft-versus-host disease control. Br J Haematol (2016) 173:754–68.10.1111/bjh.13984
27
Herrero-SanchezMCRodriguez-SerranoCAlmeidaJSan SegundoLInogesSSantos-BrizAet alTargeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol Oncol (2016) 9:113.10.1186/s13045-016-0343-5
28
ParkMJLeeSYMoonSJSonHJLeeSHKimEKet alMetformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl Res (2016) 173:115–30.10.1016/j.trsl.2016.03.006
29
MirzaNZierhutMKornABornemannAVogelWSchmid-HorchBet alGraft versus self (GvS) against T-cell autoantigens is a mechanism of graft-host interaction. Proc Natl Acad Sci U S A (2016) 113:13827–32.10.1073/pnas.1609118113
30
MurphyMPSiegelRM. Mitochondrial ROS fire up T cell activation. Immunity (2013) 38:201–2.10.1016/j.immuni.2013.02.005
31
GlickGDRossignolRLyssiotisCAWahlDLeschCSanchezBet alAnaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther (2014) 351:298–307.10.1124/jpet.114.218099
32
WeyandCMGoronzyJJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol (2017) 13:291–301.10.1038/nrrheum.2017.49
33
WangRGreenDR. Metabolic checkpoints in activated T cells. Nat Immunol (2012) 13:907–15.10.1038/ni.2386
34
van der WindtGJPearceEL. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol Rev (2012) 249:27–42.10.1111/j.1600-065X.2012.01150.x
35
MakTWGrusdatMDuncanGSDostertCNonnenmacherYCoxMet alGlutathione primes T cell metabolism for inflammation. Immunity (2017) 46:1089–90.10.1016/j.immuni.2017.03.019
36
FrauwirthKARileyJLHarrisMHParryRVRathmellJCPlasDRet alThe CD28 signaling pathway regulates glucose metabolism. Immunity (2002) 16:769–77.10.1016/S1074-7613(02)00323-0
37
PearceELPearceEJ. Metabolic pathways in immune cell activation and quiescence. Immunity (2013) 38:633–43.10.1016/j.immuni.2013.04.005
38
AngelaMEndoYAsouHKYamamotoTTumesDJTokuyamaHet alFatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun (2016) 7:13683.10.1038/ncomms13683
39
LochnerMBerodLSparwasserT. Fatty acid metabolism in the regulation of T cell function. Trends Immunol (2015) 36:81–91.10.1016/j.it.2014.12.005
40
SukumarMLiuJJiYSubramanianMCromptonJGYuZet alInhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123:4479–88.10.1172/JCI69589
41
DelgoffeGMKoleTPZhengYZarekPEMatthewsKLXiaoBet alThe mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44.10.1016/j.immuni.2009.04.014
42
PearceELWalshMCCejasPJHarmsGMShenHWangLSet alEnhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7.10.1038/nature08097
43
BerodLFriedrichCNandanAFreitagJHagemannSHarmrolfsKet alDe novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20:1327–33.10.1038/nm.3704
44
KidaniYElsaesserHHockMBVergnesLWilliamsKJArgusJPet alSterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol (2013) 14:489–99.10.1038/ni.2570
45
DuvelKYeciesJLMenonSRamanPLipovskyAISouzaALet alActivation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell (2010) 39:171–83.10.1016/j.molcel.2010.06.022
46
RahaSRaudBOberdorferLCastroCNSchrederAFreitagJet alDisruption of de novo fatty acid synthesis via acetyl-CoA carboxylase 1 inhibition prevents acute graft-versus-host disease. Eur J Immunol (2016) 46:2233–8.10.1002/eji.201546152
47
LeeJWalshMCHoehnKLJamesDEWherryEJChoiY. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol (2014) 192:3190–9.10.4049/jimmunol.1302985
48
LopaschukGDUssherJRFolmesCDJaswalJSStanleyWC. Myocardial fatty acid metabolism in health and disease. Physiol Rev (2010) 90:207–58.10.1152/physrev.00015.2009
49
WandersRJKomenJKempS. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J (2011) 278:182–94.10.1111/j.1742-4658.2010.07947.x
50
SimonsKIkonenE. Functional rafts in cell membranes. Nature (1997) 387:569–72.10.1038/42408
51
OgretmenBHannunYA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer (2004) 4:604–16.10.1038/nrc1411
52
LiuGBurnsSHuangGBoydKProiaRLFlavellRAet alThe receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol (2009) 10:769–77.10.1038/ni.1743
53
BartkeNHannunYA. Bioactive sphingolipids: metabolism and function. J Lipid Res (2009) 50(Suppl):S91–6.10.1194/jlr.R800080-JLR200
54
HannunYA. Functions of ceramide in coordinating cellular responses to stress. Science (1996) 274:1855–9.10.1126/science.274.5294.1855
55
JiangWOgretmenB. Autophagy paradox and ceramide. Biochim Biophys Acta (2014) 1841:783–92.10.1016/j.bbalip.2013.09.005
56
Pewzner-JungYBen-DorSFutermanAH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)? Insights into the regulation of ceramide synthesis. J Biol Chem (2006) 281:25001–5.10.1074/jbc.R600010200
57
SofiMHHeinrichsJDanyMNguyenHDaiMBastianDet alCeramide synthesis regulates T cell activity and GVHD development. JCI Insight (2017) 2:91701.10.1172/jci.insight.91701
58
FranciscoLMSagePTSharpeAH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42.10.1111/j.1600-065X.2010.00923.x
59
SahaAAoyamaKTaylorPAKoehnBHVeenstraRGPanoskaltsis-MortariAet alHost programmed death ligand 1 is dominant over programmed death ligand 2 expression in regulating graft-versus-host disease lethality. Blood (2013) 122:3062–73.10.1182/blood-2013-05-500801
60
Al-ChaqmaqchiHSadeghiBAbedi-ValugerdiMAl-HashmiSFaresMKuiperRet alThe role of programmed cell death ligand-1 (PD-L1/CD274) in the development of graft versus host disease. PLoS One (2013) 8:e60367.10.1371/journal.pone.0060367
61
RoundJLMazmanianSK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol (2009) 9:313–23.10.1038/nri2515
62
JenqRRUbedaCTaurYMenezesCCKhaninRDudakovJAet alRegulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med (2012) 209:903–11.10.1084/jem.20112408
63
TaurYJenqRRPeralesMALittmannERMorjariaSLingLet alThe effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood (2014) 124:1174–82.10.1182/blood-2014-02-554725
64
JenqRRTaurYDevlinSMPonceDMGoldbergJDAhrKFet alIntestinal blautia is associated with reduced death from graft-versus-host disease. Biol Blood Marrow Transplant (2015) 21:1373–83.10.1016/j.bbmt.2015.04.016
65
Gaboriau-RouthiauVRakotobeSLecuyerEMulderILanABridonneauCet alThe key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity (2009) 31:677–89.10.1016/j.immuni.2009.08.020
66
MathewsonNDJenqRMathewAVKoenigsknechtMHanashAToubaiTet alGut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol (2016) 17:505–13.10.1038/ni.3400
67
Correa-OliveiraRFachiJLVieiraASatoFTVinoloMA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol (2016) 5:e73.10.1038/cti.2016.17
68
ParkJKimMKangSGJannaschAHCooperBPattersonJet alShort-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol (2015) 8(1):80–93.10.1038/mi.2014
69
SmithPMHowittMRPanikovNMichaudMGalliniCABohloolyYMet alThe microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73.10.1126/science.1241165
70
FurusawaYObataYFukudaSEndoTANakatoGTakahashiDet alCommensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature (2013) 504:446–50.10.1038/nature12721
71
ArpaiaNCampbellCFanXDikiySvan der VeekenJdeRoosPet alMetabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5.10.1038/nature12726
72
CasadoFL. The aryl hydrocarbon receptor relays metabolic signals to promote cellular regeneration. Stem Cells Int (2016) 2016:4389802.10.1155/2016/4389802
73
ZelanteTIannittiRGCunhaCDe LucaAGiovanniniGPieracciniGet alTryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39:372–85.10.1016/j.immuni.2013.08.003
74
QuintanaFJSherrDH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev (2013) 65:1148–61.10.1124/pr.113.007823
75
WahlDRPetersenBWarnerRRichardsonBCGlickGDOpipariAW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus (2010) 19:1492–501.10.1177/0961203310373109
76
SukumarMKishtonRJRestifoNP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol (2017) 46:14–22.10.1016/j.coi.2017.03.011
77
BlecherMWhiteA. Effects of various steroids and metabolic inhibitors on the incorporation of glycine-2-C 14 into total proteins and nucleic acids of normal and malignant lymphocytes in vitro. J Biol Chem (1958) 233:1161–8.
78
ArakiKTurnerAPShafferVOGangappaSKellerSABachmannMFet almTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12.10.1038/nature08155
79
VaethMMausMKlein-HesslingSFreinkmanEYangJEcksteinMet alStore-operated Ca(2+) entry controls clonal expansion of T cells through metabolic reprogramming. Immunity (2017) 47:664–679e6.10.1016/j.immuni.2017.09.003
80
SatakeASchmidtAMNomuraSKambayashiT. Inhibition of calcineurin abrogates while inhibition of mTOR promotes regulatory T cell expansion and graft-versus-host disease protection by IL-2 in allogeneic bone marrow transplantation. PLoS One (2014) 9:e92888.10.1371/journal.pone.0092888
Summary
Keywords
T cell, metabolism, hematopoietic stem cell transplantation, graft-versus-host disease, glycolysis
Citation
Nguyen HD, Kuril S, Bastian D and Yu X-Z (2018) T-Cell Metabolism in Hematopoietic Cell Transplantation. Front. Immunol. 9:176. doi: 10.3389/fimmu.2018.00176
Received
13 November 2017
Accepted
19 January 2018
Published
09 February 2018
Volume
9 - 2018
Edited by
Claudio Mauro, Queen Mary University of London, United Kingdom
Reviewed by
Lianjun Zhang, University of Lausanne, Switzerland; Harley Y. Tse, Wayne State University, United States
Updates
Copyright
© 2018 Nguyen, Kuril, Bastian and Yu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xue-Zhong Yu, yux@musc.edu
Specialty section: This article was submitted to Immunological Tolerance and Regulation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.