 John Eppensteiner
John Eppensteiner Robert Patrick Davis
Robert Patrick Davis Andrew S. Barbas
Andrew S. Barbas Jean Kwun
Jean Kwun Jaewoo Lee
Jaewoo Lee- Department of Surgery, Duke University, Durham, NC, United States
Despite significant improvements in injury prevention and emergency response, injury-related death and morbidity continues to increase in the US and worldwide. Patients with trauma, invasive operations, anti-cancer treatment, and organ transplantation produce a host of danger signals and high levels of pro-inflammatory and pro-thrombotic mediators, such as damage-associated molecular patterns (DAMPs) and extracellular vesicles (EVs). DAMPs (e.g., nucleic acids, histone, high-mobility group box 1 protein, and S100) are molecules released from injured, stressed, or activated cells that act as endogenous ligands of innate immune receptors, whereas EVs (e.g., microparticle and exosome) are membranous vesicles budding off from plasma membranes and act as messengers between cells. DAMPs and EVs can stimulate multiple innate immune signaling pathways and coagulation cascades, and uncontrolled DAMP and EV production causes systemic inflammatory and thrombotic complications and secondary organ failure (SOF). Thus, DAMPs and EVs represent potential therapeutic targets and diagnostic biomarkers for SOF. High plasma levels of DAMPs and EVs have been positively correlated with mortality and morbidity of patients or animals with trauma or surgical insults. Blocking or neutralizing DAMPs using antibodies or small molecules has been demonstrated to ameliorate sepsis and SOF in animal models. Furthermore, a membrane immobilized with nucleic acid-binding polymers captured and removed multiple DAMPs and EVs from extracellular fluids, thereby preventing the onset of DAMP- and EV-induced inflammatory and thrombotic complications in vitro and in vivo. In this review, we will summarize the current state of knowledge of DAMPs, EVs, and SOF and discuss potential therapeutics and preventive intervention for organ failure secondary to trauma, surgery, anti-cancer therapy, and allogeneic transplantation.
Key Concepts
• Secondary Organ Failure: Dysfunction or injury of organs remote from the primary injury site is often the sequelae of the host’s dysregulated immune response.
• Immunothrombotic Agents: Certain cellular components released from stressed, damaged, or dead cells can activate both innate immune receptors and coagulation cascades, leading to inflammatory response and blood coagulation, respectively.
• Feed-Forward Loop: In solid organ transplantation, graft injury from infiltrating inflammatory cells leads to further DAMP release, intensified inflammation, and exacerbation of graft injury.
• Vicious Cycle of Injury and DAMP/EV Production: Polytrauma or invasive surgery will produce circulating pro-inflammatory and pro-thrombotic mediators that cause microinjury and de novo release of the pro-inflammatory and pro-thrombotic mediators in remote organs, thereby developing SOF.
Introduction
About five million people die from injuries worldwide every year (1). Most injury deaths are immediate or early death, occurring within 2–3 days as a result of primary injuries, while 10–20% of injury deaths occur in the late phase (2). Secondary damage in organs remote from the primary site of injury causes 50–60% of late injury deaths (3). Secondary organ failure (SOF) is often caused by systemic, overwhelming inflammatory response following hemorrhage and reperfusion injury (3). Although SOF is most prevalent in patients with traumatic injuries, SOF also occurs in patients with sterile insults such as invasive surgery or anti-cancer treatment (4, 5). Injuries induce significant immune and thrombotic consequences at local and remote organ sites, as well as systemic circulatory changes. After injury, tissues release various cellular components into the extracellular space or bloodstream. These components play a key role in hemostasis, repair of damaged tissue, and initiation of host immune response against infection (6, 7). On the other hand, they are directly and indirectly involved in the pathogenesis of systemic inflammatory and thrombotic complications that cause multiple organ failure (MOF) (8, 9).
Damage-associated molecular patterns (DAMPs) are a broad array of molecules or molecular complexes released from damaged, stressed, or activated cells. DAMPs are recognized by various innate immune receptors called pattern recognition receptors (PRRs), e.g., toll-like receptors (TLRs), C-type lectin receptors, nucleotide-binding oligomerization domain-like receptors, retinoic acid-inducible gene I-like receptors, and receptors for advanced glycation end products (RAGE), which are expressed on both immune and non-immune cells (10). Each PRR recognizes a particular molecular pattern presented in DAMPs (Table 1). Upon binding to DAMPs, PRRs trigger intracellular signaling cascades that lead to the expression of inflammation-associated genes that have pleiotropic effects on host immune defense and pathogeneses (11).
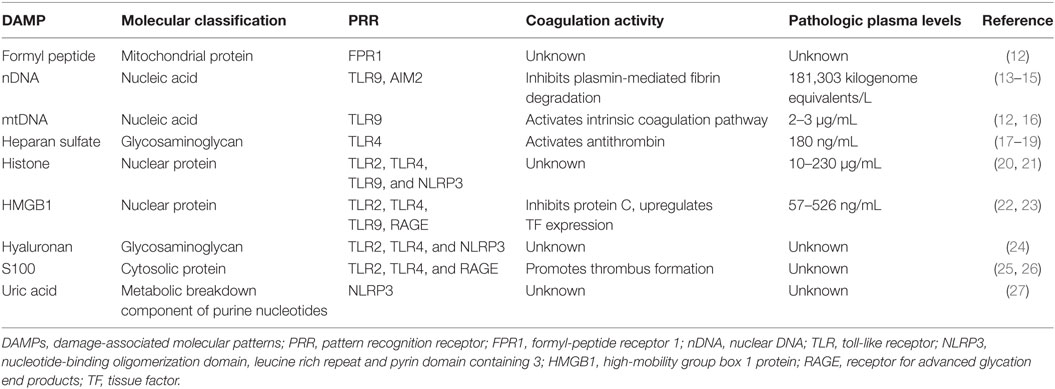
Table 1. Immunothrombotic activity of DAMPs released after trauma and sepsis.
Extracellular vesicles (EVs) are small membranous vesicles released from cells. EVs contain various cellular contents, such as proteins, DNA, and RNA and represent their parental cells. Thus, EVs may play a fundamental role in the communication between cells (28). On the other hand, EVs carry various pro-inflammatory and pro-coagulative mediators [e.g., mitochondrial DNA (mtDNA), high-mobility group box 1 protein (HMGB1), heat shock protein (HSP), tissue factor (TF), and phosphatidylserine] that modulate inflammatory response and coagulation (29, 30). This review will focus on discussing the pro-inflammatory and pro-thrombotic DAMPs and EVs in the pathogenesis of SOFs after trauma, invasive therapy, and organ transplantation. This review will also introduce potential therapeutics and preventive approaches for SOF.
Secondary Organ Failure
The concept of MOF [also called multiple organ dysfunction syndrome (MODS)] following surgical or traumatic insult dates back to 1970s as Tilney et al. first described the phenomena as sequential organ failure after ruptured abdominal aneurysms (31). Eiseman et al. first used MOF in 1977 to describe a clinical presentation after trauma-initiated hospitalization (32). The spectrum of physiological dysfunction prior to MOF was subsequently refined with the advent of systemic inflammatory response syndrome (SIRS) in 1990s (33), which provides a systematic way to identify the systemic hyperinflammatory state and potential reversibility, prior to overt organ failure. Regardless of the etiology of insult, MOF is ultimately attributed to the loss of homeostatic host immune function resulting in irreversible tissue and organ damage.
Multiple organ failure can be further classified as primary or secondary. Primary organ failure refers to organ dysfunction directly attributed to the principal insult. SOF, however, is not necessarily the direct result of traumatic tissue or organ injury, but rather the sequelae of the host’s dysregulated immune response, and it may not be appreciated until days after the primary insult. While early organ failure as a result of polytraumatic injury or invasive surgical intervention may be intuitive, the exact pathogenesis of SOF is less clear. Multiple studies have attempted to elucidate the multimodal distribution of SOF following traumatic or surgical insult. Deitch introduced the “gut hypothesis” in 1989, which hypothesized that translocation of bacteria across the intestinal mucosal barrier occurs more easily following trauma or hemorrhagic shock, leading to a condition that mirrors septicemia (34). Meakins proposed the “two-hit model,” purporting that an initial surgical or traumatic insult primes the immune system for a second hit (infection or surgical intervention) (35). This second insult propagates an exaggerated SIRS response, causing SOF.
Multiple organ failure is the leading cause of mortality in late death after traumatic injury (36, 37). However, because a singular definition does not exist for post-traumatic MOF, the variance in its epidemiologic impact, time course, and pattern remains broad. Two commonly used scoring systems are the Marshall MODS score (38) and the Denver postinjury MOF score (39). These clinical adjuncts attempt to further stratify risk and categorize critically ill patients by combining objective measures of continuous variables over multiple organ systems for a quantifiable threshold of MOF (Table 2). A recent paper aimed to examine the predictive properties of the Marshall MODS and Denver postinjury MOF score to better understand, synthesize, and prognosticate MOF in trauma patients (40). Hutchings et al. showed that the incidence of post-trauma MOF varies between 22.8 and 58.5% of patients, depending on which scoring system was used, as the Denver MOF score tends to be more stringent than the Marshall MODS in categorizing organ failure (40). Although the overall incidence of MOF has decreased with the improvement of prehospital and early inhospital resuscitative strategies, the rate of MOF-related morbidity and mortality has not significantly changed over time (37). It is clear that the impact of MOF remains significant, and further studies to clarify the complex pathologic interplay are needed.
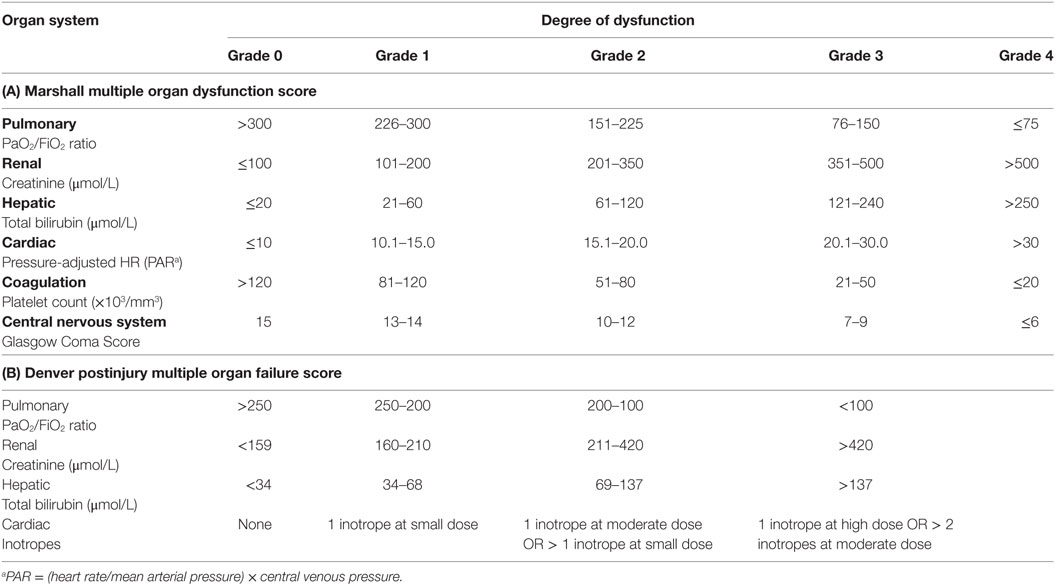
Table 2. Multiple organ failure scoring system.
Immunothrombotic DAMPs and EVs
Extracellular DNA
Elevated levels of extracellular DNA (exDNA) in the form of nuclear DNA (nDNA), mtDNA, or neutrophil extracellular trap (NETs) are often found in patients with sepsis (41, 42), traumatic injury (14), cancer (43), autoimmune disease (44), cardiopulmonary bypass surgery (45), and solid organ transplantation (46) and are correlated with morbidity and mortality of these patients. The exDNAs released from mammalian cells and bacteria are known as potent innate immune stimulators. The unique cellular location of nucleic acid-sensing PRR probably explains how nucleic acid-sensing PRR in immune cells can distinguish dangerous nucleic acids from safe counterparts (47, 48). Barton et al. demonstrated that a chimeric TLR9, engineered for expression on the cell surface, could be activated by self-DNA that could not stimulate wild-type TLR9 in the endosomal compartment (49). Thus, free DNA without delivery into endosomal compartments may not stimulate TLR9. Prikhodko et al. demonstrated that the levels of circulating mtDNA significantly increased in trauma patients compared to those in healthy volunteers, but purified mtDNA could not stimulate innate immune cells (50). These data suggest that the level of exDNA in the blood may be a suboptimal marker for human disease. Development of new approaches to detect circulating exDNA that actually activate innate immune cells would be beneficial.
In addition to pro-inflammatory activity, exDNA may have potent pro-thrombotic activity. A prospective cohort study demonstrated that elevated plasma DNA was detected in 19 of 23 patients with pulmonary embolism and in none of the 49 patients with other diagnoses (pneumonia, myocardial infarction, thrombophlebitis, or normal lung scans) (51). Moreover, the levels of plasma mtDNA and nDNA were much higher in patients with massive pulmonary embolism than in patients with submassive pulmonary embolism or healthy controls (52). Fuchs et al. demonstrated that NETs, a meshwork of DNA fibers comprising histones and antimicrobial proteins, stimulated thrombus formation in vitro and in vivo, and treatment with deoxyribonuclease (DNase) or anticoagulant heparin prevented NET-mediated thrombus formation (53). Thus, exDNA acts as potent immunothrombotic agents.
High-Mobility Group Box 1 Protein
High-mobility group box 1 protein is a non-histone nuclear protein composed of two positively charged DNA-binding motifs and a C-terminal acidic tail (54). HMGB1 is known to be passively released from dead and dying cells or actively released from live cells (55, 56). Circulating HMGB1 was markedly elevated in patients after traumatic injuries (22), ischemic injuries (57), severe acute pancreatitis (58), organ transplantation (59, 60), and arthritis (61, 62). Elevated plasma HMGB1 was significantly correlated with poor clinical outcome of these patients. In innate immune response, HMGB1 acts as an endogenous ligand of TLRs 2, 4, 9 and RAGE (63, 64). The innate immune stimulatory activity of HMGB1 has been determined by the redox state of cysteine residues C23, C45, and C106 (65). A disulfide bond between C23 and C45 is required for HMGB1 to activate innate immune cells and produce inflammatory cytokines (65), while reduction of all cysteine residues makes HMGB1 a chemoattractant rather than a cytokine inducer (66). By contrast, oxidization of the cysteine residues using reactive oxygen species abrogated both activities (66). Therefore, the level of total HMGB1 may be proximal in the innate immune response and may not accurately reflect the complex clinical nature of patients with inflammatory complications, as well as relevant clinical inflammation downstream signaling.
High-mobility group box 1 protein is also a potent pro-coagulant. HMGB1 directly stimulated and recruited platelets through TLR4 and RAGE (67–69). In a rat model, combined administration of thrombin and HMGB1 resulted in excessive fibrin deposition in glomeruli, prolonged plasma clotting times, and increased mortality (23). Mice with HMGB1-deficient platelets exhibited increased bleeding times and reduced thrombus formation, platelet aggregation, inflammation, and organ damage during experimental trauma/hemorrhagic shock (67). In a mouse venous thromboembolism (VTE) model, disulfide HMGB1 played a critical role in the development of venous thrombosis through facilitation of RAGE-dependent NET formation and platelet activation (56). Thus, HMGB1 is an important pathogenic factor of inflammatory and thrombotic complications.
Histone
Histone is a cationic nuclear protein that packages DNA into nucleosomes. Extracellular histones are found in three different forms (free, DNA-bound nucleosome, and a part of NET) in the blood of patients with sepsis, trauma, ischemia and reperfusion injury (IRI), and autoimmune disease (70). In mouse models of concanavalin A-induced inflammatory complication and toxin-induced liver injury, extracellular histones contributed to mortality after inflammatory and cellular injuries through TLRs 2 and 4 (71). Moreover, extracellular histones exacerbated IRI through TLR9-medicated cytotoxic effects in a mouse hepatic IRI model (72).
On the other hand, recombinant human histones H3 and H4 directly triggered thrombin generation in vitro in a platelet-dependent manner (73). Moreover, extracellular histones upregulated the expression of TF on endothelial cells and macrophages through TLRs 2 and 4 (74). Extracellular histones activated platelets to aggregate through fibrinogen-mediated cross-linking of platelet integrin αIIbβ3, leading to profound thrombocytopenia and tissue damage in mice (75). Treatment with heparin could prevent histone-mediated thrombocytopenia and tissue damage in vivo (75). Clinically, elevated levels of circulating histones and histone-DNA complexes were associated with the incidence of MOF, disseminated intravascular coagulation, cardiac injury, arrhythmia, and ventricular dysfunction in patients with sepsis (76, 77). Therefore, the extracellular histone also acts as an immunothrombotic agent.
S100
S100 proteins are a family of intracellular low-molecular weight, calcium-binding proteins. At least 25 distinct S100 proteins have been identified, and each S100 protein exerts diverse cellular functions in cell proliferation, differentiation, migration, calcium homeostasis, inflammation, and cell death (78). The S100 proteins are known to be either passively released from damaged cells or actively secreted from activated cells, and they have been detected in various body fluids, such as serum, urine, sputum, cerebrospinal fluid and feces of patients with cancer, inflammatory and autoimmune disease, or cardiovascular complications (79).
Extracellular S100 proteins act as potent pro-inflammatory and pro-thrombotic mediators. S100A1 released from damaged cardiomyocytes during myocardial infarction triggers TLR4-dependent pro-inflammatory responses, leading to induction of myocardial damage (80). S100A8, S100A9, and S100A12 induced TLR4-mediated inflammatory cytokine production by human peripheral blood mononuclear cells (81). In contrast, S100A9 induced RAGE-dependent cell migration of human monocytes and leukocytes (81), and S100B mediates neuronal damages in a RAGE- and NF-κB-dependent manner (82). Furthermore, S100A8/A9 heterodimeric proteins released from neutrophils induced RAGE-dependent activation of hepatic Kupffer cells, leading to the development of inflammatory thrombocytosis and atherogenesis in diabetic mice (83). Platelet-secreted S100A9 and S100A8/A9 proteins facilitated thrombus formation and occlusive thrombosis in mice with carotid artery injury (25).
IL-1α and IL-33
IL-1α and IL-33 are expressed in precursor form in the nucleus of various hematopoietic cells, and these nuclear proteins play important roles in regulation of gene expression (79). IL-1α and IL-33 do not contain a secretory signal peptide, and thus they are released into extracellular space through either non-canonical vesicular secretion pathway or passive necrotic release (79). Proteolytic cleavage is required for the release and pro-inflammatory activity of IL-1α and IL-33 (79). Extracellular IL-1α interacts with ubiquitously expressed IL-1 receptor-1 and IL-1 receptor accessory protein (IL-1RAcP) that activates downstream signaling proteins, such as myeloid differentiation primary response gene 88 (MyD88) and interleukin-1 receptor-activated protein kinase 4, and induces an inflammatory response (84). IL-1α has been considered a potential pathogenic factor involved in the development and progression of diabetes (85), inflammatory bowel disease (86), myocardial inflammation (87), and cancer (88). Active IL-33 binds to the heterodimeric plasma membrane receptor complex, consisting of ST2 and IL-1RAcP, inducing NF-κB and mitogen-activated protein kinase (MAPK) activation and Th2 maturation (89). Elevated IL-33 expression in lung tissue and blood was correlated with the severity of asthma (89) and chronic obstructive pulmonary disease (90). IL-33 upregulated TF expression on endothelial cells in ST2 and NF-κB-dependent manner, promoting arterial thrombus formation after plaque rupture (91).
Extracellular Vesicles
Extracellular vesicles comprise various membranous particles that originate from different intracellular origins and have different sizes. Thus, different types of EVs have been isolated by differential ultracentrifugations (92). Organelle size ranges from 0.5 to 10 µm. Microparticles (MPs) range from 0.1 to 1 µm in diameter, while apoptotic bodies range from 0.5 to 4 µm in diameter; both of these originate from plasma membranes. Exosomes are smaller than 0.1 µm and originate from multivesicular bodies (MVBs). Different types of EVs are generated by different mechanisms of biogenesis (30). For example, exosomes form by inward budding of MVB membranes while MPs and apoptotic bodies are generated by outward budding of plasma membrane (30). The biogenesis of MPs requires cytoskeletal reformations, such as redistribution of phospholipids, repositioning of phosphatidylserine, and contraction of the actin–myosin machinery (93, 94). The release of exosomes requires sequential assembly of the endosomal sorting complex on the MVB membrane (95). Upon release, EVs transfer their cargo by multiple mechanisms, such as endocytosis, phagocytosis, micropinocytosis, and membrane fusion (96, 97).
Depending on their origin, EVs carry various immune modulators and pro-coagulants (29, 30). Dendritic cell (DC)-derived exosomes express major histocompatibility complex (MHC) I, MHC II, and costimulatory molecules, and they can induce antigen-specific T cell responses (98). Interestingly, exosomes released from organ donor-derived DCs presented alloantigen and activated alloreactive T cells (99, 100). Exosomes and MPs released from cancer cells contributed to the suppression of host immune surveillance, cancer progression and metastasis, and angiogenesis (101–103). Like cancer-derived EVs, EVs released from mesenchymal stem cells attenuated host inflammatory responses and facilitated tissue regeneration, and are therefore being developed as therapeutic agents to treat graft-versus-host disease (104), chronic kidney disease (105), and acute radiation injury (106). On the other hand, MPs express various pro-coagulants, such as phosphatidylserine and TF (107), that promote vascular thrombosis in cancer patients (108). Moreover, endothelial and circulating cells after sepsis-induced microvascular injury released pro-thrombotic MPs into circulation (109). Thus, EVs may play dual roles in tissue repair and damage.
Immunothrombotic Factors in Post-Trauma Organ Failures
The biologic response to traumatic injury is a complex physiological phenomena that involves a host of inflammatory and thrombotic mediators including cytokines, chemokines, complement, oxygen free radicals, inflammatory cells (neutrophils, monocytes, and macrophages), and endothelial cells (110). Immediately after traumatic injury, the immune response is mounted in reaction to cellular stress and tissue damage. Matzinger first introduced the “Danger Theory” in 1994 to explain how endogenous mediators released from damaged tissues can stimulate the innate immune response and elicit a nearly identical exaggerated SIRS response to that of infectious insult (111). This relatively new perspective of the immunostimulatory effects of self-molecules in sterile inflammation of a trauma model has challenged researchers to identify and categorize specific DAMPs.
The library of known DAMPs and their respective immunostimulatory consequences is constantly evolving as researchers continue to elucidate the complex signaling pathways involved. The release of DAMPs following traumatic injury promotes local inflammation and tissue repair, though when left unchecked leads to systemically injurious effects. One of the earliest characterized and well-known DAMPs in post-traumatic MOF is HMGB1, whose pathogenic effects were first outlined in experimental sepsis models. HMGB1 was found in the extracellular space after IRI and hemorrhagic shock (112) and led to inflammatory responses and microthrombotic effects through TLR4 and RAGE (23, 26). Given the multitude of inflammatory and coagulopathic effects, not surprisingly, elevated plasma levels of HMGB1 in critically ill trauma patients have been shown to be a negative prognostic indicator early in the course of traumatic disease (113).
Mitochondrial DNA is a well-studied endogenous signal for systemic inflammatory response after traumatic injury. Originally postulated from endosymbiotic theory, mtDNA shares many evolutionarily conserved molecular motifs with bacterial DNA (114), and human mtDNA is mostly unmethylated similar to bacterial DNAs (115). More recently, Zhang et al. have demonstrated that not only are mtDNA levels markedly elevated in trauma patients but also that in vivo shock-induced cell damage stimulates neutrophils to produce cytokines and cause end-organ damage via TLR9 (12, 116). 20–30% of CpG DNA across the genome of mammalian somatic tissues is unmethylated and may act as potential ligands of TLR9 (117). However, pathologic roles of unmethylated nDNA and mtDNA are debated in trauma and sepsis. Elevated mtDNA levels in the blood are positively correlated with the mortality of patients with sepsis or SOF in the intensive care unit (118). Furthermore, trauma patients who developed SIRS or MOF had elevated levels of mtDNA in their blood compared to those who did not (119). By contrast, the level of circulating nDNA, but not mtDNA was profoundly elevated in patients immediately after trauma, and the elevated nDNA was associated with immune suppression in these patients (120).
Another important pro-inflammatory mediator typically found in the mitochondrial matrix is N-formyl peptide, a well-established leukocyte chemoattractant (121). When released from necrotic or damaged cells, these proteins have been shown to aid in chemotaxis of neutrophils to sites of sterile inflammation (122). In addition, circulating histones caused direct cytotoxicity to epithelial and endothelial tissues by altering membrane permeability, which was associated with the incidence of acute lung injury after severe trauma (20). While an in-depth characterization is beyond the scope of this review, other well-known DAMPs mediating inflammation and tissue injury following trauma include but are not limited to HSP, uric acid, adenosine triphosphate, hyaluronan, galectins, and thioredoxin.
Organ Damage and Failure after Solid Organ Transplantation
Damage-associated molecular pattern-mediated processes are increasingly recognized as important drivers of pathophysiology in solid organ transplantation (123, 124). The IRI process inherent to solid organ transplantation produces significant cellular injury with the concomitant release of multiple DAMPs. In turn, DAMPs are robust activators of the innate immune system, inciting inflammatory and thrombotic cascades that contribute to graft injury. More recently, the role of DAMPs in the subsequent activation of the alloimmune response has become an area of active investigation. Below, we review key aspects of DAMP biology in solid organ transplantation.
Damage-associated molecular patterns bind to PRRs on leukocytes and endothelial cells to initiate intracellular signaling cascades that lead to activation of the transcription factor NF-κB and increased gene expression of inflammatory response elements, particularly inflammatory cytokines. This cytokine milieu generates a sterile inflammatory environment and promotes infiltration of graft tissues with neutrophils and macrophages. Graft injury from infiltrating inflammatory cells contributes to a feed-forward loop leading to further DAMP release, intensified inflammation, and exacerbation of graft injury (123, 124). The clinical manifestation of these cellular events is the development of early allograft dysfunction (125). While the clinical definition of early allograft dysfunction differs by organ type, the common phenotype is insufficient physiologic function of the transplanted organ. In kidney transplantation, the immediate consequences of graft dysfunction are less severe, given the availability of renal replacement therapy by dialysis. However, for liver, lung, and heart transplantation, post-transplant graft dysfunction can be life-threatening.
Inflammation and thrombosis have a strong link, with significant interplay between elements of the inflammatory and coagulation cascades. Extracellular nucleic acid DAMPs (RNA and DNA) activate factors XII and XI in the coagulation cascade, inducing a pro-thrombotic state (126). The observation that such a pro-thrombotic state can be reversed through the actions of nucleic acid scavengers further supports the mechanistic link between DAMPs and thrombosis (127). The development of a pro-thrombotic state following solid organ transplantation has important clinical implications. One of the most severe complications following solid organ transplantation is the development of graft thrombosis. While important technical factors contribute to this complication (size and quality of blood vessels, surgical technique, etc.), there is growing recognition that the pro-thrombotic milieu generated by DAMP signaling may be an important risk factor warranting further investigation.
While DAMPs have been viewed predominantly as activators of the innate immune system, there is growing recognition that DAMPs represent an important link between the innate and adaptive immune response following solid organ transplantation (128). The initial inflammatory response generated by DAMP signaling leads to infiltration of the graft with host immune cells, including DCs and macrophages. These host antigen-presenting cells then traffic to local lymph nodes, presenting graft antigens to host T cells and initiating the adaptive immune response. Additionally, DAMP-mediated signaling potentiates the alloimmune response via increased expression of costimulatory and MHC molecules on antigen-presenting cells (129).
The clinical consequence of this array of molecular events is the development of acute allograft rejection. Allograft rejection is broadly categorized as either acute cellular rejection (T cell-mediated process) or antibody-mediated rejection (B cell-mediated process). The frequency and severity of rejection episodes vary greatly by organ type and by characteristics of the individual recipient. While acute rejection is usually reversible with high-dose steroid treatment, the clinical management of these complex patients continues to be a significant challenge routinely encountered by transplant physicians. Perhaps, an even more vexing and unsolved clinical problem in transplantation is the development of chronic allograft rejection. Chronic rejection is characterized by an inexorable decline in graft function over months or years, ultimately resulting in complete graft loss. Although chronic rejection manifests differently by organ type, this poorly understood process is frequently characterized by the development of graft fibrosis. Growing evidence demonstrates that DAMP signaling in response to subclinical immune injury over a prolonged time frame may contribute to the development of graft fibrosis (129). This exciting hypothesis warrants further study and may yield significant progress in combating one of the major clinical problems still limiting solid organ transplantation.
DAMPs and EVs: Cancer Therapy-Induced Favorable and Adverse Effects
Thromboembolism is the obstruction of a blood vessel by abnormal clot formation in the circulation and a common fatal disease. Interestingly, VTE was 22-fold higher in patients with recent surgery, more than 12-fold higher in patients with recent trauma, 4.1-fold higher in patients with cancer, and 6.5-fold higher in patients undergoing anticancer therapies compared with healthy people (130). It is still unclear why VTE incidence elevates in such patients. Moreover, a precise prediction marker of VTE is indecisive. A growing body of evidence has demonstrated that increased levels of cancer-released MPs in the blood are highly correlated with the incidence of VTE in various types of cancers, e.g., malignant melanoma (131), pancreatic cancer (132), breast cancer (133), and glioblastoma (134). Furthermore, the level of transmembrane coagulation initiator TF in the blood was positively correlated with the recurrence of VTE in cancer patients (132, 135). Anticancer therapies increased the release of MPs from cancer cells (136–139) and upregulated the expression of TF on malignant and non-malignant cells (140, 141).
Depending on its mode of action, certain anticancer therapies, such as anthracycline chemotherapy (142, 143), radiation therapy (142, 144), transfection with PRR agonist polyriboinosinic:polyribocytidylic acid (145, 146), oncolytic virotherapy (147), and focused ultrasound ablation therapy (148), can induce substantial T cell-mediated antitumor responses. Unlike other cancer therapies, these anticancer therapies are known to induce immunogenic cancer cell death characterized by the release of tumor antigens and high levels of immune stimulators (DAMPs, adenosine triphosphate and cell-surface calreticulin) (142, 144, 149–151). DAMPs activate innate and adaptive immune cells via TLR and PRR signaling (152) while adenosine triphosphate and calreticulin act as “find-me” and “eat-me” signals, respectively, to recruit DCs and promote uptake and clearance of dead cells (153, 154). These DAMPs, find-me/eat-me signals, and tumor antigens cooperate to induce adaptive antitumor immune responses after treatment with immunogenic cancer cell death-inducing anticancer agents (155). On the other hand, uncontrolled DAMPs released from cancer cells with antineoplastic treatments are known as a risk factor of SOF in cancer patients (156–159). Therefore, it is evident that DAMPs and EVs released from cancer cells treated with anticancer therapeutic agents are double-edged swords in cancer therapy. Further studies are needed to elucidate mechanisms by which DAMPs and EVs produced by anticancer therapies contribute to the development of adverse and favorable responses in cancer patients.
Therapeutics Targeting Uncontrolled DAMPs, EVs, and Downstream Signals
TLR and TLR Signaling Inhibitors
Small Molecules
Eritoran, a synthetic lipid A antagonist, binds to and blocks the TLR4–MD2 complex. Intravenous administration of eritoran decreased lung injury and pulmonary inflammation and increased survival of mice infected with influenza (160). However, treatments with eritoran did not reduce mortality in patients with sepsis in a randomized, double-blind phase 3 clinical trial (161). 2-Acetamidopyranoside (C34) is a small molecule that tightly binds to the hydrophobic internal pocket of the TLR4–MD2 complex (162). C34 inhibited endotoxin-simulated TLR4 in enterocytes and macrophages in vitro and reduced systemic inflammation in the mouse models of endotoxemia and enterocolitis (162). Furthermore, C34 protected mice against acute lung injury after trauma/hemorrhagic shock (163). C34 has not been studied in humans.
TAK-242 is a small molecule TLR4 signaling inhibitor. TAK-242 as monotherapy failed to significantly reduce systemic inflammatory response, organ dysfunction, or survival in patients with sepsis, shock, or respiratory failure compared to a placebo control (164). TLR signaling pathways involve multiple transcription factors, e.g., NF-κB, c-Jun N-terminal kinase, and p38 MAPK. Inhibition of MAPK using a small molecule FR167653 suppressed lung heme oxygenase 1 expression and attenuated acute lung injury after hemorrhagic shock in mice (165). Furthermore, postburn treatments with MAPK inhibitor SB203580 prevented cardiac dysfunction after burn injury and resuscitation in rats (166). Interestingly, the SB203580 improved cardiac function but worsened lung injury and overall survival of mice with Escherichia coli-induced pneumonia, probably because of reduced innate immune response and bacteria clearance (167).
Anti-TLR Antibodies
Systemic blockade of TLR2 or TLR4 using monoclonal antibodies reduced systemic and pulmonary inflammation and mortality after polymicrobial sepsis in mice with cecal ligation and puncture (168). Furthermore, treatment with anti-TLR3 antibody attenuated ischemic gut injury and SOFs in mice with cecal ligation without puncture (169). A humanized anti-TLR2 antibody, OPN-305, reduced infarct size and myocardial necrosis and improved cardiac function in pigs after IRI (170).
Endosomal Acidification Inhibitor
Chloroquine and its derivative hydroxychloroquine are the prototype antimalaria drug. They also act as an endosomal acidification inhibitor, thus inhibiting endosomal TLRs 3, 7, and 9 (171). Administration of chloroquine after cecal ligation and puncture attenuated sepsis-induced MOFs and mortality in mice (172). In a rat hepatic IRI model, chloroquine treatment ameliorated acute liver injury at the early phase (0–6 h after reperfusion) but worsened liver injury at the late phase of reperfusion (24–48 h after reperfusion) (173). The mechanism of early protective action of chloroquine involved the modulation of MAPK activation and HMGB1 release, whereas chloroquine inhibited autophagy and induced hepatic apoptosis at the late phase (173). In addition to injury prevention, chloroquine and hydroxychloroquine have been used as anticancer therapeutic agents (174) and immunosuppressive agent for rheumatic diseases, lupus erythematosus, skin diseases, and graft-versus-host disease after bone marrow transplantation (175).
Oligonucleotide
Small oligonucleotides that bind to and inhibit TLR9 protected mice against sepsis-induced death (176). HMGB1 is an endogenous ligand of TLR4 and RAGE. In addition, HMGB1 can bind to immune stimulatory nucleic acids and facilitate nucleic acid-mediated innate immune stimulation (177). Non-immunogenic oligonucleotides were screened to bind to HMGB1 with high affinity but did not activate TLR9, and treatment with these non-immunogenic oligonucleotides protected mice against endotoxin-induced septic shock (177).
DAMP and EV Inhibitors
Anti-HMGB1 and DNase
Because of the interconnectedness and redundancy of TLR and PRR signaling, the inhibition of single TLR and PRR signal pathways may be ineffective in ameliorating disease progress. Targeting upstream of TLR and PRR signal pathways would be more therapeutically effective than targeting downstream. Treatment with anti-HMGB1 antibody protected mice against acute lung injury after fracture and hemorrhagic shock (163). DNase I inhibited NET formation, and treatment with DNase I protected hepatocytes from cell death after IRI and significantly reduced IRI-induced liver injury in mice (178). Furthermore, intrathecal injection of DNase I prevented pulmonary endothelial dysfunction in rats with ventilator-associated pneumonia (179).
EV Inhibitors
Circulating exosomes after septic shock induced a statistically significant decrease in in vitro myocardial contractility compared with normal exosomes (180). Furthermore, exosomes isolated from septic patients induced vascular dysfunction by inducing reactive oxygen species generation and endothelial cell apoptosis (181). GW4869 is a neutral sphingomyelinase inhibitor. GW4869 inhibits sphingolipid ceramide-dependent release of exosomes from MVBs (182). Treatment with GW4869 prior to endotoxin challenge or cecal ligation and puncture in mice significantly reduced the levels of circulating exosomes and diminished sepsis-induced cardiac inflammation, myocardial dysfunction, and mortality (183).
Cationic Polymer-Based DAMP and EV Scavengers
Cationic polymers have been broadly used as nucleic acid transfection or drug delivery agents over the last few decades (184, 185). We have demonstrated that certain nucleic acid-binding cationic polymers (NABPs), e.g., polyamidoamine dendrimer, hexadimethrine bromide, and β-cyclodextrin-containing polymer, neutralized the ability of free DNA, RNA, and inorganic polyphosphate to activate nucleic acid-sensing TLRs (TLRs 3, 7, 8, and 9) (186) and intrinsic coagulation cascade (127). These NABPs were systemically administered to prevent TLR-mediated lethal liver injury (186). Furthermore, NABP-immobilized membranes simultaneously inhibited nucleic acid and non-nucleic acid DAMPs released from dead or dying cells or circulating in the blood of trauma patients, thereby preventing DAMP-induced inflammation and occlusive thrombosis in mice (187). NABP-immobilized hemoperfusion has a potential use during extracorporeal membrane oxygenation, continuous veno-venous hemofiltration, and continuous renal replacement therapy in intensive care units. Removing pro-inflammatory and pro-coagulative mediators from circulation is an unmet need in the treatment of critically ill patients.
Conclusion
Patients with sterile insults produce a host of danger signals and high levels of pro-inflammatory and pro-thrombotic mediators after cellular injury and tissue damage. We postulate that circulating pro-inflammatory and pro-thrombotic mediators will cause the microinjury of organs remote from the primary site of injuries and de novo release of the pro-inflammatory and pro-thrombotic mediators, thereby developing a vicious cycle of the release of pro-coagulative and pro-inflammatory mediators and local tissue damage and subsequent development of SOFs (Figure 1). Breaking the vicious cycle will prevent SOF. Furthermore, pro-coagulative and pro-inflammatory mediators will develop as early prediction marker(s) for late-onset SOF in patients exposed to sterile insults.
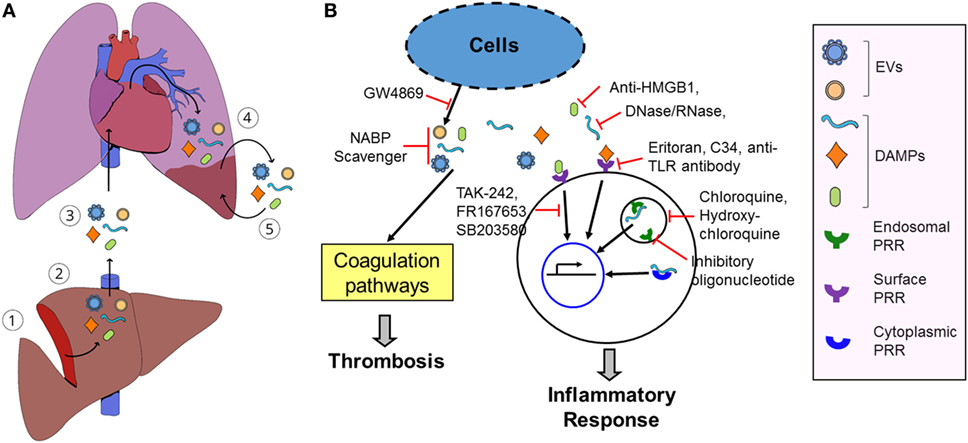
Figure 1. Model of damage-associated molecular pattern (DAMP)- and extracellular vesicle (EV)-induced secondary organ failure (SOF) and potential therapeutics. (A), (1) Sterile insults cause primary tissue damage. (2) The damaged tissue releases various pro-inflammatory and pro-coagulative mediators, such as DAMPs and EVs. (3) Some mediators may not be cleared in the local damaged tissue and are released into the blood and circulated into remote organs. (4) These mediators will induce microthrombosis and local inflammation in the remote tissues, causing microinjuries. (5) The microinjured tissue subsequently releases de novo DAMPs and EVs, aggravating local tissue damages. Release of DAMPs and EVs and microinjuries in the remote tissues develops a vicious cycle and induces SOF. (B) Inhibition of inflammation and thrombosis using pattern recognition receptor (PRR) antagonists, PRR signaling inhibitor, DAMP inhibitor, EV biosynthesis inhibitors, and nucleic acid-binding cationic polymer (NABP) scavengers, thereby ameliorating and preventing SOF after tissue injury.
Author Contributions
Conceived and designed the concept: JL, JK, and AB. Wrote the manuscript: JL, JE, AB, JK, and RD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by Duke Department of Surgery (Clarence Gardner Award) (JL), a pilot grant from the Opportunity Funds Management Core of the Centers for Medical Countermeasures against Radiation, and the National Institute of Allergy and Infectious Diseases (U19AI067773) (JL). We thank Ms. Ashley Morgan and Ms. Megan Llewellyn for editing and illustrations.
Abbreviations
DAMP, damage-associated molecular pattern; DC, dendritic cell; DNase, deoxyribonuclease; EV, extracellular vesicle; exDNA, extracellular DNA; HMGB1, high-mobility group box 1 protein; IRI, ischemia and reperfusion injury; MAPK, mitogen-activated protein kinase; MHC, major histocompatibility complex; MODS, multiple organ dysfunction syndrome; MOF, multiple organ failure; mtDNA, mitochondrial DNA; NABPs, nucleic acid-binding cationic polymers; nDNA, nuclear DNA; NET, neutrophil extracellular trap; PRR, pattern recognition receptor; RAGE, receptor for advanced glycation end products; SIRS, systemic inflammatory response syndrome; SOF, secondary organ failure; TF, tissue factor; TLR, toll-like receptor; VTE, venous thromboembolism; FPR1, formyl-peptide receptor 1; AIM2, absent in melanoma 2; NLRP3, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing 3.
References
1. Haagsma JA, Graetz N, Bolliger I, Naghavi M, Higashi H, Mullany EC, et al. The global burden of injury: incidence, mortality, disability-adjusted life years and time trends from the Global Burden of Disease study 2013. Inj Prev (2016) 22(1):3–18. doi:10.1136/injuryprev-2015-041616
2. Moore K. Trauma mortality: understanding mortality distribution to improve outcomes. J Emerg Nurs (2014) 40(4):405–6. doi:10.1016/j.jen.2014.04.006
3. Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury (2009) 40(9):912–8. doi:10.1016/j.injury.2009.05.024
4. Perl M, Lomas-Neira J, Venet F, Chung CS, Ayala A. Pathogenesis of indirect (secondary) acute lung injury. Expert Rev Respir Med (2011) 5(1):115–26. doi:10.1586/ers.10.92
5. Feltracco P, Carollo C, Barbieri S, Pettenuzzo T, Ori C. Early respiratory complications after liver transplantation. World J Gastroenterol (2013) 19(48):9271–81. doi:10.3748/wjg.v19.i48.9271
6. Kawai T, Akira S. TLR signaling. Semin Immunol (2007) 19(1):24–32. doi:10.1016/j.smim.2006.12.004
7. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol (2015) 6:422. doi:10.3389/fimmu.2015.00422
8. Pugin J. How tissue injury alarms the immune system and causes a systemic inflammatory response syndrome. Ann Intensive Care (2012) 2(1):27. doi:10.1186/2110-5820-2-27
9. Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev (2016) 30(4):257–61. doi:10.1016/j.blre.2015.12.004
10. Raymond SL, Holden DC, Mira JC, Stortz JA, Loftus TJ, Mohr AM, et al. Microbial recognition and danger signals in sepsis and trauma. Biochim Biophys Acta (2017) 1863(10 Pt B):2564–73. doi:10.1016/j.bbadis.2017.01.013
11. Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. Induction of cytokines and chemokines by toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol (2010) 30(1):53–67. doi:10.1615/CritRevImmunol.v30.i1.40
12. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
13. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature (2009) 458(7237):509–13. doi:10.1038/nature07710
14. Lo YMD, Rainer TH, Chan LYS, Hjelm NM, Cocks RA. Plasma DNA as a prognostic marker in trauma patients. Clin Chem (2000) 46(3):319–23.
15. Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, et al. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol (2015) 35(12):2544–53. doi:10.1161/ATVBAHA.115.306035
16. Bhagirath VC, Dwivedi DJ, Liaw PC. Comparison of the proinflammatory and procoagulant properties of nuclear, mitochondrial, and bacterial DNA. Shock (2015) 44(3):265–71. doi:10.1097/SHK.0000000000000397
17. Rahbar E, Cardenas JC, Baimukanova G, Usadi B, Bruhn R, Pati S, et al. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J Transl Med (2015) 13:117. doi:10.1186/s12967-015-0481-5
18. Liu J, Pedersen LC. Anticoagulant heparan sulfate: structural specificity and biosynthesis. Appl Microbiol Biotechnol (2007) 74(2):263–72. doi:10.1007/s00253-006-0722-x
19. Goodall KJ, Poon IK, Phipps S, Hulett MD. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS One (2014) 9(10):e109596. doi:10.1371/journal.pone.0109596
20. Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med (2013) 187(2):160–9. doi:10.1164/rccm.201206-1037OC
21. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, et al. Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol (2012) 23(8):1375–88. doi:10.1681/ASN.2011111077
22. Peltz ED, Moore EE, Eckels PC, Damle SS, Tsuruta Y, Johnson JL, et al. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock (2009) 32(1):17–22. doi:10.1097/SHK.0b013e3181997173
23. Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost (2007) 5(1):109–16. doi:10.1111/j.1538-7836.2006.02255.x
24. Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. J Biol Chem (2007) 282(25):18265–75. doi:10.1074/jbc.M606352200
25. Wang Y, Fang C, Gao H, Bilodeau ML, Zhang Z, Croce K, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest (2014) 124(5):2160–71. doi:10.1172/JCI70966
26. Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell (1999) 97(7):889–901. doi:10.1016/S0092-8674(00)80801-6
27. Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum (2005) 52(9):2936–46. doi:10.1002/art.21238
28. Turturici G, Tinnirello R, Sconzo G, Geraci F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am J Physiol Cell Physiol (2014) 306(7):C621–33. doi:10.1152/ajpcell.00228.2013
29. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol (2011) 29:139–62. doi:10.1146/annurev-immunol-030409-101323
30. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi:10.1146/annurev-cellbio-101512-122326
31. Tilney NL, Bailey GL, Morgan AP. Sequential system failure after rupture of abdominal aortic aneurysms: an unsolved problem in postoperative care. Ann Surg (1973) 178(2):117–22. doi:10.1097/00000658-197308000-00001
33. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest (1992) 101(6):1644–55. doi:10.1378/chest.101.6.1644
34. Deitch EA. Simple intestinal obstruction causes bacterial translocation in man. Arch Surg (1989) 124(6):699–701. doi:10.1001/archsurg.1989.01410060065013
35. Meakins JL. Etiology of multiple organ failure. J Trauma (1990) 30(12 Suppl):S165–8. doi:10.1097/00005373-199012001-00033
36. Regel G, Grotz M, Weltner T, Sturm JA, Tscherne H. Pattern of organ failure following severe trauma. World J Surg (1996) 20(4):422–9. doi:10.1007/s002689900067
37. Sauaia A, Moore EE, Johnson JL, Chin TL, Banerjee A, Sperry JL, et al. Temporal trends of postinjury multiple-organ failure: still resource intensive, morbid, and lethal. J Trauma Acute Care Surg (2014) 76(3):582–92. doi:10.1097/TA.0000000000000147
38. Marshall JC, Cook DJ, Christou NV, Bernard GR, Sprung CL, Sibbald WJ. Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med (1995) 23(10):1638–52. doi:10.1097/00003246-199510000-00007
39. Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. J Trauma (1996) 40(4):501–10. doi:10.1097/00005373-199604000-00001
40. Hutchings L, Watkinson P, Young JD, Willett K. Defining multiple organ failure after major trauma: a comparison of the Denver, sequential organ failure assessment, and Marshall scoring systems. J Trauma Acute Care Surg (2017) 82(3):534–41. doi:10.1097/TA.0000000000001328
41. Kung C-T, Hsiao S-Y, Tsai T-C, Su C-M, Chang W-N, Huang C-R, et al. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J Transl Med (2012) 10:130. doi:10.1186/1479-5876-10-130
42. Itagaki K, Kaczmarek E, Lee YT, Tang IT, Isal B, Adibnia Y, et al. Mitochondrial DNA released by trauma induces neutrophil extracellular traps. PLoS One (2015) 10(3):e0120549. doi:10.1371/journal.pone.0120549
43. Cedervall J, Zhang Y, Olsson AK. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res (2016) 76(15):4311–5. doi:10.1158/0008-5472.CAN-15-3051
44. Lee KH, Kronbichler A, Park DD, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev (2017) 16(11):1160–73. doi:10.1016/j.autrev.2017.09.012
45. Sandler N, Kaczmarek E, Itagaki K, Zheng Y, Otterbein L, Khabbaz K, et al. Mitochondrial DAMPs are released during cardiopulmonary bypass surgery and are associated with postoperative atrial fibrillation. Heart Lung Circ (2018) 27(1):122–9. doi:10.1016/j.hlc.2017.02.014
46. Zou J, Duffy B, Slade M, Young AL, Steward N, Hachem R, et al. Rapid detection of donor cell free DNA in lung transplant recipients with rejections using donor-recipient HLA mismatch. Hum Immunol (2017) 78(4):342–9. doi:10.1016/j.humimm.2017.03.002
47. Deane JA, Bolland S. Nucleic acid-sensing TLRs as modifiers of autoimmunity. J Immunol (2006) 177(10):6573–8. doi:10.4049/jimmunol.177.10.6573
48. Saitoh S, Miyake K. Regulatory molecules required for nucleotide-sensing toll-like receptors. Immunol Rev (2009) 227(1):32–43. doi:10.1111/j.1600-065X.2008.00729.x
49. Barton GM, Kagan JC, Medzhitov R. Intracellular localization of toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol (2006) 7(1):49–56. doi:10.1038/ni1280
50. Prikhodko AS, Shabanov AK, Zinovkina LA, Popova EN, Aznauryan MA, Lanina NO, et al. Pure mitochondrial DNA does not activate human neutrophils in vitro. Biochemistry (Mosc) (2015) 80(5):629–35. doi:10.1134/S0006297915050168
51. Sipes JN, Suratt PM, Teates CD, Barada FA, Davis JS, Tegtmeyer CJ. A prospective study of plasma DNA in the diagnosis of pulmonary embolism. Am Rev Respir Dis (1978) 118(3):475–8. doi:10.1164/arrd.1978.118.3.475
52. Arnalich F, Maldifassi MC, Ciria E, Codoceo R, Renart J, Fernandez-Capitan C, et al. Plasma levels of mitochondrial and nuclear DNA in patients with massive pulmonary embolism in the emergency department: a prospective cohort study. Crit Care (2013) 17(3):R90. doi:10.1186/cc12735
53. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A (2010) 107(36):15880–5. doi:10.1073/pnas.1005743107
54. Belgrano FS, de Abreu da Silva IC, Bastos de Oliveira FM, Fantappie MR, Mohana-Borges R. Role of the acidic tail of high mobility group protein B1 (HMGB1) in protein stability and DNA bending. PLoS One (2013) 8(11):e79572. doi:10.1371/journal.pone.0079572
55. Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol (2015) 6:93. doi:10.3389/fphys.2015.00093
56. Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood (2016) 128(20):2435–49. doi:10.1182/blood-2016-04-710632
57. Goldstein RS, Gallowitsch-Puerta M, Yang L, Rosas-Ballina M, Huston JM, Czura CJ, et al. Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock (2006) 25(6):571–4. doi:10.1097/01.shk.0000209540.99176.72
58. Yasuda T, Ueda T, Takeyama Y, Shinzeki M, Sawa H, Nakajima T, et al. Significant increase of serum high-mobility group box chromosomal protein 1 levels in patients with severe acute pancreatitis. Pancreas (2006) 33(4):359–63. doi:10.1097/01.mpa.0000236741.15477.8b
59. Itoh T, Iwahashi S, Kanak MA, Shimoda M, Takita M, Chujo D, et al. Elevation of high-mobility group box 1 after clinical autologous islet transplantation and its inverse correlation with outcomes. Cell Transplant (2014) 23(2):153–65. doi:10.3727/096368912X658980
60. Volz HC, Laohachewin D, Schellberg D, Wienbrandt AR, Nelles M, Zugck C, et al. HMGB1 is an independent predictor of death and heart transplantation in heart failure. Clin Res Cardiol (2012) 101(6):427–35. doi:10.1007/s00392-011-0409-x
61. Li ZC, Cheng GQ, Hu KZ, Li MQ, Zang WP, Dong YQ, et al. Correlation of synovial fluid HMGB-1 levels with radiographic severity of knee osteoarthritis. Clin Invest Med (2011) 34(5):E298. doi:10.25011/cim.v34i5.15673
62. Ke X, Jin G, Yang Y, Cao X, Fang R, Feng X, et al. Synovial fluid HMGB-1 levels are associated with osteoarthritis severity. Clin Lab (2015) 61(7):809–18. doi:10.7754/Clin.Lab.2015.141205
63. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A (2010) 107(26):11942–7. doi:10.1073/pnas.1003893107
64. Yu L, Wang L, Chen S. Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med (2010) 14(11):2592–603. doi:10.1111/j.1582-4934.2010.01127.x
65. Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med (2012) 18:250–9. doi:10.2119/molmed.2011.00389
66. Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med (2012) 209(9):1519–28. doi:10.1084/jem.20120189
67. Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest (2015) 125(12):4638–54. doi:10.1172/JCI81660
68. Yang X, Wang H, Zhang M, Liu J, Lv B, Chen F. HMGB1: a novel protein that induced platelets active and aggregation via toll-like receptor-4, NF-kappaB and cGMP dependent mechanisms. Diagn Pathol (2015) 10:134. doi:10.1186/s13000-015-0348-3
69. Ahrens I, Chen YC, Topcic D, Bode M, Haenel D, Hagemeyer CE, et al. HMGB1 binds to activated platelets via the receptor for advanced glycation end products and is present in platelet rich human coronary artery thrombi. Thromb Haemost (2015) 114(5):994–1003. doi:10.1160/TH14-12-1073
70. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis (2017) 8(5):e2812. doi:10.1038/cddis.2017.52
71. Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol (2011) 187(5):2626–31. doi:10.4049/jimmunol.1003930
72. Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through toll-like receptor 9 in mice. Hepatology (2011) 54(3):999–1008. doi:10.1002/hep.24501
73. Noubouossie DF, Whelihan MF, Yu YB, Sparkenbaugh E, Pawlinski R, Monroe DM, et al. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood (2017) 129(8):1021–9. doi:10.1182/blood-2016-06-722298
74. Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-kappaB and AP-1. Thromb Res (2016) 137:211–8. doi:10.1016/j.thromres.2015.10.012
75. Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood (2011) 118(13):3708–14. doi:10.1182/blood-2011-01-332676
76. Toh CH, Alhamdi Y, Abrams ST. Current pathological and laboratory considerations in the diagnosis of disseminated intravascular coagulation. Ann Lab Med (2016) 36(6):505–12. doi:10.3343/alm.2016.36.6.505
77. Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med (2015) 43(10):2094–103. doi:10.1097/CCM.0000000000001162
78. Granowska M, Mather SJ, Jobling T, Naeem M, Burchell J, Taylor-Papadimitriou J, et al. Radiolabelled stripped mucin, SM3, monoclonal antibody for immunoscintigraphy of ovarian tumours. Int J Biol Markers (1990) 5(2):89–96.
79. Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol (2017) 14(1):43–64. doi:10.1038/cmi.2016.34
80. Rohde D, Schon C, Boerries M, Didrihsone I, Ritterhoff J, Kubatzky KF, et al. S100A1 is released from ischemic cardiomyocytes and signals myocardial damage via toll-like receptor 4. EMBO Mol Med (2014) 6(6):778–94. doi:10.15252/emmm.201303498
81. Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, et al. S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One (2015) 10(2):e0115828. doi:10.1371/journal.pone.0115828
82. Angelo MF, Aguirre A, Aviles Reyes RX, Villarreal A, Lukin J, Melendez M, et al. The proinflammatory RAGE/NF-kappaB pathway is involved in neuronal damage and reactive gliosis in a model of sleep apnea by intermittent hypoxia. PLoS One (2014) 9(9):e107901. doi:10.1371/journal.pone.0107901
83. Kraakman MJ, Lee MK, Al-Sharea A, Dragoljevic D, Barrett TJ, Montenont E, et al. Neutrophil-derived S100 calcium-binding proteins A8/A9 promote reticulated thrombocytosis and atherogenesis in diabetes. J Clin Invest (2017) 127(6):2133–47. doi:10.1172/JCI92450
84. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal (2010) 3(105):cm1. doi:10.1126/scisignal.3105cm1
85. Ballak DB, Stienstra R, Tack CJ, Dinarello CA, van Diepen JA. IL-1 family members in the pathogenesis and treatment of metabolic disease: focus on adipose tissue inflammation and insulin resistance. Cytokine (2015) 75(2):280–90. doi:10.1016/j.cyto.2015.05.005
86. Scarpa M, Kessler S, Sadler T, West G, Homer C, McDonald C, et al. The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol (2015) 185(6):1624–37. doi:10.1016/j.ajpath.2015.02.018
87. Lugrin J, Parapanov R, Rosenblatt-Velin N, Rignault-Clerc S, Feihl F, Waeber B, et al. Cutting edge: IL-1alpha is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction. J Immunol (2015) 194(2):499–503. doi:10.4049/jimmunol.1401948
88. Di Paolo NC, Shayakhmetov DM. Interleukin 1alpha and the inflammatory process. Nat Immunol (2016) 17(8):906–13. doi:10.1038/ni.3503
89. Milovanovic M, Volarevic V, Radosavljevic G, Jovanovic I, Pejnovic N, Arsenijevic N, et al. IL-33/ST2 axis in inflammation and immunopathology. Immunol Res (2012) 52(1–2):89–99. doi:10.1007/s12026-012-8283-9
90. Xia J, Zhao J, Shang J, Li M, Zeng Z, Zhao J, et al. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol (2015) 308(7):L619–27. doi:10.1152/ajplung.00305.2014
91. Stojkovic S, Kaun C, Basilio J, Rauscher S, Hell L, Krychtiuk KA, et al. Tissue factor is induced by interleukin-33 in human endothelial cells: a new link between coagulation and inflammation. Sci Rep (2016) 6:25171. doi:10.1038/srep25171
92. Li P, Kaslan M, Lee SH, Yao J, Gao Z. Progress in exosome isolation techniques. Theranostics (2017) 7(3):789–804. doi:10.7150/thno.18133
93. Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, et al. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol (2009) 19(22):1875–85. doi:10.1016/j.cub.2009.09.059
94. Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett (2009) 283(2):168–75. doi:10.1016/j.canlet.2009.03.041
95. Iraci N, Leonardi T, Gessler F, Vega B, Pluchino S. Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci (2016) 17(2):171. doi:10.3390/ijms17020171
96. Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles (2014) 3(1):24641. doi:10.3402/jev.v3.24641
97. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood (2012) 119(3):756–66. doi:10.1182/blood-2011-02-338004
98. Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med (1998) 4(5):594–600. doi:10.1038/nm0598-594
99. Liu Q, Rojas-Canales DM, Divito SJ, Shufesky WJ, Stolz DB, Erdos G, et al. Donor dendritic cell-derived exosomes promote allograft-targeting immune response. J Clin Invest (2016) 126(8):2805–20. doi:10.1172/JCI84577
100. Marino J, Babiker-Mohamed MH, Crosby-Bertorini P, Paster JT, LeGuern C, Germana S, et al. Donor exosomes rather than passenger leukocytes initiate alloreactive T cell responses after transplantation. Sci Immunol (2016) 1(1):aaf8759. doi:10.1126/sciimmunol.aaf8759
101. Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Cancer Res (2011) 17(5):959–64. doi:10.1158/1078-0432.CCR-10-1489
102. Rak J. Microparticles in cancer. Semin Thromb Hemost (2010) 36(8):888–906. doi:10.1055/s-0030-1267043
103. Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res (2011) 71(11):3792–801. doi:10.1158/0008-5472.CAN-10-4455
104. Kordelas L, Rebmann V, Ludwig AK, Radtke S, Ruesing J, Doeppner TR, et al. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia (2014) 28(4):970–3. doi:10.1038/leu.2014.41
105. Nassar W, El-Ansary M, Sabry D, Mostafa MA, Fayad T, Kotb E, et al. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater Res (2016) 20:21. doi:10.1186/s40824-016-0068-0
106. Chang PY, Qu YQ, Wang J, Dong LH. The potential of mesenchymal stem cells in the management of radiation enteropathy. Cell Death Dis (2015) 6:e1840. doi:10.1038/cddis.2015.189
107. Muhsin-Sharafaldine MR, Saunderson SC, Dunn AC, Faed JM, Kleffmann T, McLellan AD. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget (2016) 7(35):56279–94. doi:10.18632/oncotarget.10783
108. Gardiner C, Harrison P, Belting M, Boing A, Campello E, Carter BS, et al. Extracellular vesicles, tissue factor, cancer and thrombosis – discussion themes of the ISEV 2014 educational day. J Extracell Vesicles (2015) 4:26901. doi:10.3402/jev.v4.26901
109. Souza AC, Yuen PS, Star RA. Microparticles: markers and mediators of sepsis-induced microvascular dysfunction, immunosuppression, and AKI. Kidney Int (2015) 87(6):1100–8. doi:10.1038/ki.2015.26
110. Tsukamoto T, Chanthaphavong RS, Pape HC. Current theories on the pathophysiology of multiple organ failure after trauma. Injury (2010) 41(1):21–6. doi:10.1016/j.injury.2009.07.010
111. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi:10.1146/annurev.iy.12.040194.005015
112. Levy RM, Mollen KP, Prince JM, Kaczorowski DJ, Vallabhaneni R, Liu S, et al. Systemic inflammation and remote organ injury following trauma require HMGB1. Am J Physiol Regul Integr Comp Physiol (2007) 293(4):R1538–44. doi:10.1152/ajpregu.00272.2007
113. Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care (2009) 13(6):R174. doi:10.1186/cc8152
114. Wallin IE. A note on the morphology of bacteria symbiotic in the tissues of higher organisms. J Bacteriol (1922) 7(5):471–4.
115. Liu B, Du Q, Chen L, Fu G, Li S, Fu L, et al. CpG methylation patterns of human mitochondrial DNA. Sci Rep (2016) 6:23421. doi:10.1038/srep23421
116. Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock (2010) 34(1):55–9. doi:10.1097/SHK.0b013e3181cd8c08
117. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol (2014) 6(5):a019133. doi:10.1101/cshperspect.a019133
118. Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med (2013) 10(12):e1001577. doi:10.1371/journal.pmed.1001577
119. Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg (2013) 258(4):591–6. doi:10.1097/SLA.0b013e3182a4ea46
120. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med (2016) 42(4):551–61. doi:10.1007/s00134-015-4205-3
121. Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci U S A (1975) 72(3):1059–62. doi:10.1073/pnas.72.3.1059
122. McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science (2010) 330(6002):362–6. doi:10.1126/science.1195491
123. Braza F, Brouard S, Chadban S, Goldstein DR. Role of TLRs and DAMPs in allograft inflammation and transplant outcomes. Nat Rev Nephrol (2016) 12(5):281–90. doi:10.1038/nrneph.2016.41
124. Land WG, Agostinis P, Gasser S, Garg AD, Linkermann A. Transplantation and damage-associated molecular patterns (DAMPs). Am J Transplant (2016) 16(12):3338–61. doi:10.1111/ajt.13963
125. Olthoff KM, Kulik L, Samstein B, Kaminski M, Abecassis M, Emond J, et al. Validation of a current definition of early allograft dysfunction in liver transplant recipients and analysis of risk factors. Liver Transpl (2010) 16(8):943–9. doi:10.1002/lt.22091
126. Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, et al. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A (2007) 104(15):6388–93. doi:10.1073/pnas.0608647104
127. Jain S, Pitoc GA, Holl EK, Zhang Y, Borst L, Leong KW, et al. Nucleic acid scavengers inhibit thrombosis without increasing bleeding. Proc Natl Acad Sci U S A (2012) 109(32):12938–43. doi:10.1073/pnas.1204928109
128. Land WG, Agostinis P, Gasser S, Garg AD, Linkermann A. DAMP-induced allograft and tumor rejection: the circle is closing. Am J Transplant (2016) 16(12):3322–37. doi:10.1111/ajt.14012
129. Todd JL, Palmer SM. Danger signals in regulating the immune response to solid organ transplantation. J Clin Invest (2017) 127(7):2464–72. doi:10.1172/JCI90594
130. Heit JA, Silverstein MD, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ III. Risk factors for deep vein thrombosis and pulmonary embolism: a population-based case-control study. Arch Intern Med (2000) 160(6):809–15. doi:10.1001/archinte.160.6.809
131. Laresche C, Pelletier F, Garnache-Ottou F, Lihoreau T, Biichle S, Mourey G, et al. Increased levels of circulating microparticles are associated with increased procoagulant activity in patients with cutaneous malignant melanoma. J Invest Dermatol (2014) 134(1):176–82. doi:10.1038/jid.2013.288
132. Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, et al. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J Thromb Haemost (2015) 13(7):1310–9. doi:10.1111/jth.13002
133. Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S. Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost (2007) 5(3):520–7. doi:10.1111/j.1538-7836.2007.02369.x
134. Sartori MT, Della Puppa A, Ballin A, Saggiorato G, Bernardi D, Padoan A, et al. Prothrombotic state in glioblastoma multiforme: an evaluation of the procoagulant activity of circulating microparticles. J Neurooncol (2011) 104(1):225–31. doi:10.1007/s11060-010-0462-8
135. Khorana AA, Kamphuisen PW, Meyer G, Bauersachs R, Janas MS, Jarner MF, et al. Tissue factor as a predictor of recurrent venous thromboembolism in malignancy: biomarker analyses of the CATCH trial. J Clin Oncol (2017) 35(10):1078–85. doi:10.1200/JCO.2016.67.4564
136. Boles JC, Williams JC, Hollingsworth RM, Wang JG, Glover SL, Owens AP III, et al. Anthracycline treatment of the human monocytic leukemia cell line THP-1 increases phosphatidylserine exposure and tissue factor activity. Thromb Res (2012) 129(2):197–203. doi:10.1016/j.thromres.2011.06.022
137. Lechner D, Kollars M, Gleiss A, Kyrle PA, Weltermann A. Chemotherapy-induced thrombin generation via procoagulant endothelial microparticles is independent of tissue factor activity. J Thromb Haemost (2007) 5(12):2445–52. doi:10.1111/j.1538-7836.2007.02788.x
138. Fu Y, Zhou J, Li H, Cao F, Su Y, Fan S, et al. Daunorubicin induces procoagulant activity of cultured endothelial cells through phosphatidylserine exposure and microparticles release. Thromb Haemost (2010) 104(6):1235–41. doi:10.1160/TH10-02-0102
139. Zhou J, Zheng Y, Shi J, Lu C, Hou J, Yu H, et al. Daunorubicin induces procoagulant response through phosphatidylserine exposure in red blood cells. Thromb Res (2010) 125(2):178–83. doi:10.1016/j.thromres.2009.09.006
140. Goldin-Lang P, Niebergall F, Antoniak S, Szotowski B, Rosenthal P, Pels K, et al. Ionizing radiation induces upregulation of cellular procoagulability and tissue factor expression in human peripheral blood mononuclear cells. Thromb Res (2007) 120(6):857–64. doi:10.1016/j.thromres.2007.01.008
141. Lysov Z, Swystun LL, Kuruvilla S, Arnold A, Liaw PC. Lung cancer chemotherapy agents increase procoagulant activity via protein disulfide isomerase-dependent tissue factor decryption. Blood Coagul Fibrinolysis (2015) 26(1):36–45. doi:10.1097/MBC.0000000000000145
142. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi:10.1038/nm1622
143. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202(12):1691–701. doi:10.1084/jem.20050915
144. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RR, et al. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res (2011) 71(7):2488–96. doi:10.1158/0008-5472.CAN-10-2820
145. Duewell P, Beller E, Kirchleitner SV, Adunka T, Bourhis H, Siveke J, et al. Targeted activation of melanoma differentiation-associated protein 5 (MDA5) for immunotherapy of pancreatic carcinoma. Oncoimmunology (2015) 4(10):e1029698. doi:10.1080/2162402X.2015.1029698
146. Duewell P, Steger A, Lohr H, Bourhis H, Hoelz H, Kirchleitner SV, et al. RIG-I-like helicases induce immunogenic cell death of pancreatic cancer cells and sensitize tumors toward killing by CD8(+) T cells. Cell Death Differ (2014) 21(12):1825–37. doi:10.1038/cdd.2014.96
147. De Munck J, Binks A, McNeish IA, Aerts JL. Oncolytic virus-induced cell death and immunity: a match made in heaven? J Leukoc Biol (2017) 102(3):631–43. doi:10.1189/jlb.5RU0117-040R
148. Bandyopadhyay S, Quinn TJ, Scandiuzzi L, Basu I, Partanen A, Tome WA, et al. Low-intensity focused ultrasound induces reversal of tumor-induced T cell tolerance and prevents immune escape. J Immunol (2016) 196(4):1964–76. doi:10.4049/jimmunol.1500541
149. Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol (2005) 6(7):722–9. doi:10.1038/ni1213
150. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med (2011) 208(10):1989–2003. doi:10.1084/jem.20101158
151. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med (2011) 208(10):2005–16. doi:10.1084/jem.20101159
152. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12(12):860–75. doi:10.1038/nrc3380
153. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature (2009) 461(7261):282–6. doi:10.1038/nature08296
154. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell (2005) 123(2):321–34. doi:10.1016/j.cell.2005.08.032
155. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi:10.1146/annurev-immunol-032712-100008
156. McBride A, Westervelt P. Recognizing and managing the expanded risk of tumor lysis syndrome in hematologic and solid malignancies. J Hematol Oncol (2012) 5:75. doi:10.1186/1756-8722-5-75
157. Sammon JD, Klett DE, Sood A, Olugbade K Jr, Schmid M, Kim SP, et al. Sepsis after major cancer surgery. J Surg Res (2015) 193(2):788–94. doi:10.1016/j.jss.2014.07.046
158. Taira F, Horimoto Y, Saito M. Tumor lysis syndrome following trastuzumab for breast cancer: a case report and review of the literature. Breast Cancer (2015) 22(6):664–8. doi:10.1007/s12282-013-0448-4
159. Macaluso A, Genova S, Maringhini S, Coffaro G, Ziino O, D’Angelo P. Acute respiratory distress syndrome associated with tumor lysis syndrome in a child with acute lymphoblastic leukemia. Pediatr Rep (2015) 7(1):5760. doi:10.4081/pr.2015.5760
160. Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature (2013) 497(7450):498–502. doi:10.1038/nature12118
161. Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA (2013) 309(11):1154–62. doi:10.1001/jama.2013.2194
162. Neal MD, Jia H, Eyer B, Good M, Guerriero CJ, Sodhi CP, et al. Discovery and validation of a new class of small molecule toll-like receptor 4 (TLR4) inhibitors. PLoS One (2013) 8(6):e65779. doi:10.1371/journal.pone.0065779
163. Sodhi CP, Jia H, Yamaguchi Y, Lu P, Good M, Egan C, et al. Intestinal epithelial TLR-4 Activation Is required for the development of acute lung injury after trauma/hemorrhagic shock via the release of HMGB1 from the gut. J Immunol (2015) 194(10):4931–9. doi:10.4049/jimmunol.1402490
164. Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med (2010) 38(8):1685–94. doi:10.1097/CCM.0b013e3181e7c5c9
165. Chen C, Wang Y, Zhang Z, Wang C, Peng M. Toll-like receptor 4 regulates heme oxygenase-1 expression after hemorrhagic shock induced acute lung injury in mice: requirement of p38 mitogen-activated protein kinase activation. Shock (2009) 31(5):486–92. doi:10.1097/SHK.0b013e318188f7e1
166. Ballard-Croft C, White DJ, Maass DL, Hybki DP, Horton JW. Role of p38 mitogen-activated protein kinase in cardiac myocyte secretion of the inflammatory cytokine TNF-alpha. Am J Physiol Heart Circ Physiol (2001) 280(5):H1970–81. doi:10.1152/ajpheart.2001.280.5.H1970
167. Su J, Cui X, Li Y, Mani H, Ferreyra GA, Danner RL, et al. SB203580, a p38 inhibitor, improved cardiac function but worsened lung injury and survival during Escherichia coli pneumonia in mice. J Trauma (2010) 68(6):1317–27. doi:10.1097/TA.0b013e3181bb9cd3
168. Lima CX, Souza DG, Amaral FA, Fagundes CT, Rodrigues IP, Alves-Filho JC, et al. Therapeutic effects of treatment with anti-TLR2 and anti-TLR4 monoclonal antibodies in polymicrobial sepsis. PLoS One (2015) 10(7):e0132336. doi:10.1371/journal.pone.0132336
169. Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med (2008) 205(11):2609–21. doi:10.1084/jem.20081370
170. Arslan F, Houtgraaf JH, Keogh B, Kazemi K, de Jong R, McCormack WJ, et al. Treatment with OPN-305, a humanized anti-toll-like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv (2012) 5(2):279–87. doi:10.1161/CIRCINTERVENTIONS.111.967596
171. Lee J, Chuang TH, Redecke V, She L, Pitha PM, Carson DA, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of toll-like receptor 7. Proc Natl Acad Sci U S A (2003) 100(11):6646–51. doi:10.1073/pnas.0631696100
172. Yasuda H, Leelahavanichkul A, Tsunoda S, Dear JW, Takahashi Y, Ito S, et al. Chloroquine and inhibition of toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol (2008) 294(5):F1050–8. doi:10.1152/ajprenal.00461.2007
173. Fang H, Liu A, Dahmen U, Dirsch O. Dual role of chloroquine in liver ischemia reperfusion injury: reduction of liver damage in early phase, but aggravation in late phase. Cell Death Dis (2013) 4:e694. doi:10.1038/cddis.2013.225
174. Manic G, Obrist F, Kroemer G, Vitale I, Galluzzi L. Chloroquine and hydroxychloroquine for cancer therapy. Mol Cell Oncol (2014) 1(1):e29911. doi:10.4161/mco.29911
175. Al-Bari MA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother (2015) 70(6):1608–21. doi:10.1093/jac/dkv018
176. Plitas G, Burt BM, Nguyen HM, Bamboat ZM, DeMatteo RP. Toll-like receptor 9 inhibition reduces mortality in polymicrobial sepsis. J Exp Med (2008) 205(6):1277–83. doi:10.1084/jem.20080162
177. Yanai H, Chiba S, Ban T, Nakaima Y, Onoe T, Honda K, et al. Suppression of immune responses by nonimmunogenic oligodeoxynucleotides with high affinity for high-mobility group box proteins (HMGBs). Proc Natl Acad Sci U S A (2011) 108(28):11542–7. doi:10.1073/pnas.1108535108
178. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology (2015) 62(2):600–14. doi:10.1002/hep.27841
179. Simmons JD, Freno DR, Muscat CA, Obiako B, Lee YL, Pastukh VM, et al. Mitochondrial DNA damage associated molecular patterns in ventilator-associated pneumonia: prevention and reversal by intratracheal DNase I. J Trauma Acute Care Surg (2017) 82(1):120–5. doi:10.1097/TA.0000000000001269
180. Azevedo LC, Janiszewski M, Pontieri V, Pedro Mde A, Bassi E, Tucci PJ, et al. Platelet-derived exosomes from septic shock patients induce myocardial dysfunction. Crit Care (2007) 11(6):R120. doi:10.1186/cc6176
181. Janiszewski M, Do Carmo AO, Pedro MA, Silva E, Knobel E, Laurindo FR. Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: a novel vascular redox pathway. Crit Care Med (2004) 32(3):818–25. doi:10.1097/01.CCM.0000114829.17746.19
182. Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (2008) 319(5867):1244–7. doi:10.1126/science.1153124
183. Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, et al. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta (2015) 1852(11):2362–71. doi:10.1016/j.bbadis.2015.08.010
184. Merkle HP. Drug delivery’s quest for polymers: where are the frontiers? Eur J Pharm Biopharm (2015) 97(Pt B):293–303. doi:10.1016/j.ejpb.2015.04.038
185. Wagner E. Polymers for nucleic acid transfer-an overview. Adv Genet (2014) 88:231–61. doi:10.1016/B978-0-12-800148-6.00008-0
186. Lee J, Sohn JW, Zhang Y, Leong KW, Pisetsky D, Sullenger BA. Nucleic acid-binding polymers as anti-inflammatory agents. Proc Natl Acad Sci U S A (2011) 108(34):14055–60. doi:10.1073/pnas.1105777108
Keywords: damage-associated molecular pattern, extracellular vesicle, thrombosis, inflammation, polymer, trauma, cancer, transplantation
Citation: Eppensteiner J, Davis RP, Barbas AS, Kwun J and Lee J (2018) Immunothrombotic Activity of Damage-Associated Molecular Patterns and Extracellular Vesicles in Secondary Organ Failure Induced by Trauma and Sterile Insults. Front. Immunol. 9:190. doi: 10.3389/fimmu.2018.00190
Received: 08 November 2017; Accepted: 22 January 2018;
Published: 08 February 2018
Edited by:
Julien Pottecher, Hôpitaux Universitaires de Strasbourg, FranceReviewed by:
Michael Thomas Lotze, University of Pittsburgh Cancer Institute, United StatesJames Harris, Monash University, Australia
Copyright: © 2018 Eppensteiner, Davis, Barbas, Kwun and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaewoo Lee, amFld29vLmxlZUBkdWtlLmVkdQ==