 Swadhinya Arjunaraja
Swadhinya Arjunaraja Pamela Angelus
Pamela Angelus Helen C. Su
Helen C. Su Andrew L. Snow
Andrew L. Snow- 1Department of Pharmacology and Molecular Therapeutics, Uniformed Services University of the Health Sciences, Bethesda, MD, United States
- 2Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
- 3Clinical Research Directorate/Clinical Monitoring Research Program, Leidos Biomedical Research, Inc., National Cancer Institute at Frederick, Frederick, MD, United States
B-cell expansion with NF-κB and T-cell anergy (BENTA) disease is a B-cell-specific lymphoproliferative disorder caused by germline gain-of-function mutations in CARD11. These mutations force the CARD11 scaffold into an open conformation capable of stimulating constitutive NF-κB activation in lymphocytes, without requiring antigen receptor engagement. Many BENTA patients also suffer from recurrent infections, with 7 out of 16 patients exhibiting chronic, low-grade Epstein–Barr virus (EBV) viremia. In this mini-review, we discuss EBV infection in the pathogenesis and clinical management of BENTA disease, and speculate on mechanisms that could explain inadequate control of viral infection in BENTA patients.
Introduction
Epstein–Barr virus (EBV) is a ubiquitous human herpesvirus that establishes life-long infection in ~90% of individuals (1). Primary EBV infection in childhood is usually asymptomatic in immunocompetent hosts, while acquisition of EBV during adolescence can result in infectious mononucleosis (IM) that usually resolves within days to weeks. However, EBV infections can also trigger lymphoproliferative disease, lymphoma, fulminant infectious mononucleosis (FIM), and/or hemophagocytic lymphohistiocytosis (HLH) in genetically or iatrogenically immunocompromised patients (2–4). These conditions clearly suggest that EBV has co-evolved with its host under constant immune surveillance to ensure that virus–host homeostasis is maintained (5, 6).
The transmission, circulation, and persistence of EBV in the human host have been reviewed extensively (1, 4, 7–9). EBV initially establishes lytic infection in both B lymphocytes and epithelial cells of the oropharynx. The EBV lytic gene program ensures both viral replication and evasion from early detection by either natural killer (NK) cells or CD8+ T cells (10). EBV then switches to a latent infection program that expands the pool of infected B cells considerably (11). This is achieved in part through the expression of key latent membrane proteins 1 and 2A, which mimic constitutive CD40 and B cell receptor signaling, respectively. Evidence suggests that this “growth” program of latency (i.e., latency III) provides the proliferative and pro-survival signals necessary to drive the EBV-infected B cell through a germinal center-like reaction and eventually into the memory B cell pool, all without requiring cognate antigen recognition. At the same time, latent proteins such as LMP2 and EBNA3A/B/C contain potent immunodominant class I MHC peptide epitopes for CD8+ T cell recognition, ensuring the destruction of most EBV-infected B cells at this stage (3). CD8+ T (and NK) cell-mediated killing is also aided by the robust upregulation of ligands for both NKG2D and signaling lymphocyte activation molecule (SLAM) family receptors on EBV-infected B cells, which participate in signaling for cytotoxic functions (see below) (5). Surviving EBV+ memory B cells remain quiescently infected without expressing viral antigens (latency 0) for the lifetime of the individual, with occasional rounds of viral reactivation thought to occur as these cells traffic back through the oropharynx (12). EBV-specific cellular immunity maintains tight control throughout these cycles (5).
In this manner, EBV has evolved an elegant strategy for ensuring initial colonization followed by persistent, benign infection in the immunocompetent host, in which a sizable portion of the memory CD8+ T cell pool (~2–5%) is dedicated to maintaining EBV-specific immunosurveillance (3, 13). Indeed, cytolytic killing of EBV-infected B cells by effector CD8+ T cells occurs during acute IM, comprising up to 50% of the CD8+ T cell compartment (4, 14). EBV-specific CD4+ T cells are also important for robust CD8+ cellular immunity and can also participate directly in cytotoxic killing of infected B cells (15, 16). During asymptomatic infection, NK cells help to restrict viral load by inhibiting their replication and can reduce the likelihood of EBV transformation of B cells (10, 17, 18). Furthermore, other innate effectors such as invariant natural killer T cells are also known to play a role in killing of infected B cells and can limit EBV transformation of B cells in vitro (19).
The advent of next-generation sequencing technology has enabled us to characterize primary immune deficiency (PID) states in humans caused by mutations in single immune-related genes that predispose them to certain pathogens. Indeed, several PIDs have now been recognized for their specific susceptibility to uncontrolled EBV infection and associated disease, sometimes referred to collectively as “EBV-opathies” (5, 20–22). In this review, we focus our attention on the incidence and severity of EBV infection in a recently characterized PID known as B-cell expansion with NF-κB and T-cell anergy (BENTA). Mechanistic insights into possible immunological shortcomings surrounding EBV infections in BENTA patients are provided below.
Benta Disease
Our group discovered a B-cell-specific lymphoproliferative congenital human disorder termed BENTA (23). BENTA disease is caused by heterozygous, germline-encoded gain-of-function mutations in the gene CARD11, which encodes a lymphocyte-specific scaffold protein (CARD11) also known as CARMA1. The CARD11 protein bridges the antigen receptor ligation in B or T cells with multiple downstream signaling pathways such as canonical NF-κB, c-Jun N-terminal kinase (JNK), and mechanistic target of rapamycin (mTOR) (24–26). Subsequent to antigen receptor ligation in lymphocytes, CARD11 is phosphorylated to facilitate BCL10 and MALT1 binding to form the CARD11–BCL10–MALT1 (CBM) complex, which further nucleates the dynamic signalosomes that activate inhibitor of κB kinase (IKK) and culminate in NF-κB translocation into the nucleus (27–30) to activate the canonical NF-κB pathway. The NF-κB family of transcription factors is critical for the induction of genes involved in cell survival, proliferation, and immune effector functions (31). GOF mutations in CARD11 render the protein in an open, active state irrespective of antigen receptor engagement, resulting in constitutive NF-κB activation (31–33).
To date, 16 different BENTA patients have been identified and definitively diagnosed, with five distinct CARD11 mutations. Polyclonal B cell lymphocytosis in early childhood is a hallmark of BENTA disease, often accompanied by splenomegaly and lymphadenopathy (23, 34–40). Immunologic phenotyping reveals the remarkable accumulation of both CD10+CD24hiCD38hi transitional and IgM+IgD+ mature naïve polyclonal B cells, even though T cell numbers frequently fall within the normal range (Table 1). Many BENTA patients also present with several signs of primary immunodeficiency despite the absence of any autoimmune disease symptoms. Recurrent ear and sinopulmonary infections are common in all patients, with other opportunistic viral infections such as molluscum contagiosum, BK virus, and EBV observed in some patients. In most patients, inadequate antibody responses against T-cell independent pneumococcal and meningococcal polysaccharide-based vaccines are noted. Some patients also show poor responses to T-cell-dependent vaccines such as Varicella Zoster virus and measles. Poor humoral immune responses in these patients are also reflected in very low frequencies of circulating class-switched and memory B cells, as well as low levels of IgM and IgA in the serum. Impaired humoral immunity in BENTA is evidenced by intrinsic defects in plasma cell differentiation and antibody secretion upon stimulation of naïve patient B cells in vitro, despite normal proliferation and enhanced survival (41). The hyporesponsiveness of BENTA patient T cells to in vitro stimulation, including poor proliferation and reduced IL-2 secretion, may also contribute to defective class-switched Ab responses (23, 35).
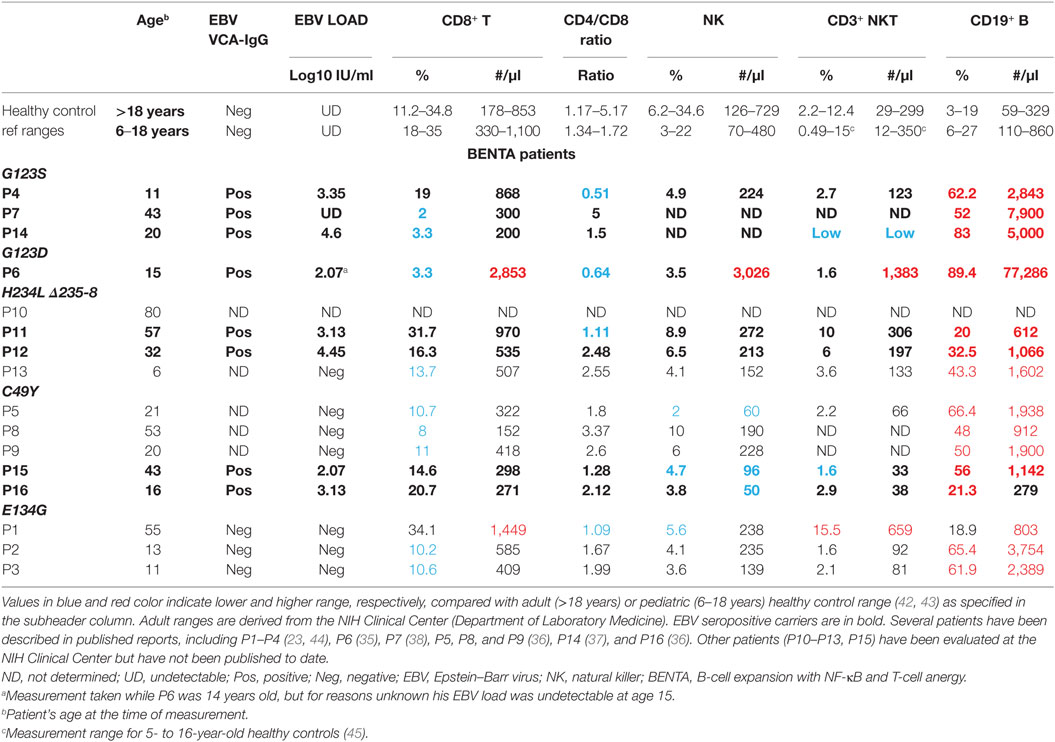
Table 1. Phenotypic analysis of BENTA patients.
Eight out of 16 patients are seropositive for EBV (Table 1). While EBV viral load is generally undetectable in healthy carriers, almost all BENTA patients (7/8) exposed to EBV are demonstrably viremic as measured by their DNA copy number (Table 1). However, EBV viral loads in BENTA patients are not nearly as high as seen in chronic active EBV (CA-EBV) patients and other PIDs (46). These data suggest that EBV-specific immunity is impaired in BENTA patients, but pales in comparison to other PIDs such as X-linked lymphoproliferative syndrome (XLP) or MAGT1/CD27/CD70/ITK or Coronin1A deficiency diseases featuring exquisite susceptibility to severe EBV infection and disease (5, 20–22). In the next section, we speculate on why gain-of-function CARD11 mutations might confer susceptibility to moderate EBV viremia in BENTA disease.
Mechanisms Underlying Benta Susceptibility to EBV
Too Many B Cells, Too Few T/NK Cells?
The consequences of distorted antigen receptor signaling in the presence of GOF CARD11 mutations reverberate throughout the lymphoid lineage in BENTA patients. Indeed, the size and makeup of lymphocyte compartments may influence EBV status in certain patients. Most notably, constitutive, canonical NF-κB activity induced by GOF CARD11 signaling in B cells drives excessive B cell accumulation in BENTA patients and may predispose them to malignant transformation as additional mutations are acquired over time. In fact, two patients in our cohort developed B-cell tumors in adulthood (P1 and P11), although neither was associated with EBV infection. Transgenic expression of a constitutively active form of IKKβ (caIKKβ) promotes the survival of mature murine B cells in vivo, though it is not sufficient to induce lymphomagenesis (47). Indeed, NF-κB-induced tumor suppressor genes such as A20 and IκB provide important negative feedback on NF-κB signaling, which must be overcome to promote lymphomagenesis (48). This negative feedback remains intact in primary BENTA B cells and may explain why only a fraction of BENTA B cells exhibit p65 nuclear localization at any given time (23).
Epstein–Barr virus itself is not likely a contributing factor for B-cell lymphocytosis in BENTA, as EBV-negative patients also have high B cell numbers. Although NF-κB actively represses lytic infection (49), the proliferation of latently infected EBV+ B cells relies on NF-κB signaling through viral proteins such as LMP1 and LMP2A, which mimic CD40 and BCR signaling, respectively (5). Hence, constitutive NF-κB activity in BENTA patient B cells could better enable EBV to expand the pool of latently infected B cells. Perhaps this could manifest in increased viral reactivation and viremia as CARD11-dependent NF-κB activity oscillates in infected BENTA B cells. Regardless, the presence of EBV may increase the risk of B cell malignancy later in life, simply given the increased size of target B cell compartment. An expanded pool of naïve B cells may simply support an increased level of lytic infection at any given time in EBV-infected BENTA patients, contributing to consistently higher viral loads. In support of this notion, there is an unconfirmed case of EBV-driven Hodgkin lymphoma in the maternal grandfather of patient P16.
Could relative reductions in the T and NK cell compartments also compromise immunity to EBV? In addition to the critical role served by CD8+ T cells, CD4+ T cells, NK cells, and NKT cells are also implicated in clearing EBV infection (16, 17, 19). As shown in Table 1, the absolute number of CD8+ T cells and the ratio of CD4+/CD8+ T cells are within normal range in most patients reported thus far. Nevertheless, the low number of NK/NKT cells observed in certain patients (e.g., P14, P15, and P16) could contribute to persistent EBV viremia in those individuals. Patient P5 also has a lower percentage and absolute number of NK cells, presenting a potential culprit for her frequent susceptibility to other viral infections.
Impaired T/NK Cell Function
A more likely explanation for uncontrolled EBV infection concerns impaired T cell function described in BENTA disease. In vitro, we observed poor proliferation and reduced IL-2 secretion from BENTA patient T cells stimulated with anti-CD3/anti-CD28 antibodies. This “anergic” response correlated with defects in TCR-mediated MAPK signaling and Ca++ flux (23, 35). Although the biochemical mechanisms remain nebulous, these defects are almost certainly linked to constitutive canonical NF-κB activation induced by GOF CARD11 signaling. Indeed, Krishna et al showed that restricting expression of constitutively active IKKβ (caIKKβ) to murine T cells also rendered them hyporesponsive to TCR/CD28 stimulation, marked by proximal TCR signaling defects and attenuated responses to bacterial infection (50). The authors connected some of these defects to enhanced expression of the transcriptional repressor Blimp-1, which has been shown to promote T cell exhaustion. Although we have not measured Blimp-1 in BENTA T cells, we recently characterized a profound, intrinsic defect in patient B cell differentiation linked to failed induction of Blimp-1 (41). Clearly much more work is required to understand how elevated NF-κB activation perturbs seemingly independent pathways downstream of TCR signaling. Regardless, it is tempting to speculate that any flaw in PLCγ1-mediated Ca++ flux underlies poor T cell-dependent control of EBV, as observed in more dramatic fashion in both ITK and MAGT1 deficiency (51–55).
Inactivation of CARD11 has also been shown to inhibit NK cell development and function (56). Although it is not clear how GOF CARD11 signaling may affect NK cells in BENTA, poor IL-2 secretion by anergic T cells upon stimulation could certainly weaken the NK cell response to viral infection. As mentioned earlier, some patients also display lower frequencies of NK and NKT cells, although EBV viremia is observed in several patients with normal NK/NKT counts. Thus, the increased susceptibility of BENTA patients to EBV could be linked to both excessive polyclonal B cell lymphocytosis and hyporesponsive T cells/NK cells that help in combating the viral infection.
Dysregulation of Key Receptor–Ligand Signals Required for EBV Control
The removal of latently infected B cells by cytotoxic T and NK cells requires several receptor–ligand interactions for recognition, cell–cell conjugation, and cytolysis, some of which may be weakened in BENTA patients (Figure 1). For example, cognate engagement of EBV-infected B cells by T cells requires SAP-dependent signaling through two receptors belonging to the SLAM family: 2B4 and NTB-A (57–60). Whereas most SLAM receptors participate in homotypic interactions in trans, 2B4 recognizes a distinct ligand on target cells known as CD48. The expression of both NTB-A and CD48 is dramatically upregulated on the B cell surface upon EBV infection, which promotes T cell and NK cell activation during asymptomatic infection and acute infection, respectively (5, 61). Recruitment of SAP, a small SH2 adaptor protein, to the cytoplasmic tails of NTB-A and 2B4 upon ligand binding is required for conveying downstream signals that ensure strong T:B cell conjugation and B cell killing (62). In this regard, SAP deficiency in XLP-1 patients makes them exquisitely susceptible to severe, uncontrolled EBV infection due to debilitated 2B4 and NTB-A signaling, presenting as FIM/HLH (63, 64). Based on our published RNA-Seq data, BENTA B cells activated in vitro with polyclonal stimuli display normal expression of CD48 and NTB-A compared with healthy human donors (41). Whether perturbed 2B4 and/or NTB-A signaling in BENTA patient T cells may influence EBV predisposition remains unclear, but warrants further investigation.
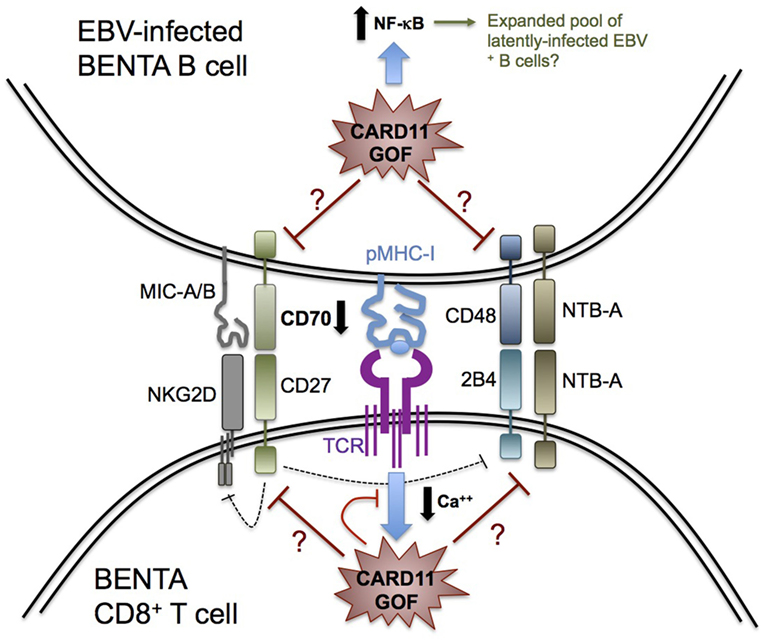
Figure 1. Possible determinants of impaired Epstein–Barr virus (EBV) control in B-cell expansion with NF-κB and T-cell anergy (BENTA) disease. Schematic diagram summarizing key receptor–ligand interactions that govern CD8+ T cell recognition and killing of EBV-infected B cells, based on our knowledge of primary immune deficiencies featuring enhanced susceptibility to EBV-driven disease. CARD11 GOF signaling could perturb several molecular signals required for optimal cytolysis of EBV-infected B cells, including signaling lymphocyte activation molecule receptors (2B4, NTB-A), NKG2D, and CD27. For example, decreased CD70 expression on BENTA B cells could impair CD27 signaling and contribute to reduced NKG2D or 2B4 expression on BENTA T cells. Alternatively, attenuated TCR signaling (e.g., reduced Ca++ flux) likely contributes to BENTA patient T cell hyporesponsiveness, which could disrupt generation of CD8+ effector T cells with optimal cytotoxic function. Finally, elevated NF-κB signaling in B cells could accelerate the expansion of latently infected EBV+ B cells, contributing to detectable viremia as the virus continuously reactivates.
Upon activation, the C-type lectin-like receptor NKG2D is also expressed on NK cells and CD8+ T cells and plays a major role in cytotoxic elimination of transformed and virally infected cells (65). The importance of NKG2D in EBV immunity was recently revealed by the discovery of X-linked immunodeficiency with magnesium defect, EBV infection, and neoplasia (XMEN) disease, caused by deficiency of the magnesium transporter MAGT1. Although NK cell and CD8+ T cell numbers are normal in XMEN patients, reduced intracellular Mg++ abrogates NKG2D receptor expression on activated NK cells and CD8+ T cells, which compromises cytolytic responses against EBV+ B cells (51, 66–68). Similarly, GOF CARD11 signaling may diminish NKG2D expression on BENTA NK cells or CD8+ T cells, perhaps linked to a Ca++ flux defect similar to that noted in XMEN T cells (54). On the other hand, there is no evidence for transcriptional upregulation of NKG2D receptor ligands MICA, MICB, and ULBPs on activated BENTA B cells compared with healthy control B cells. Simultaneous engagement of SAP-dependent 2B4 and SAP-independent NKG2D signaling is indispensable for CD8+ T cell-mediated killing of EBV-infected B cells (69), explaining why neither receptor (NKG2D or 2B4) can compensate for the absence of the other to maintain normal CTL activity in XMEN and XLP-1 patients. If constitutive CARD11 signaling indirectly impedes NKG2D or 2B4 signaling in BENTA CD8+ T cells/NK cells, this could jeopardize their ability to completely control EBV infection.
CD27, a costimulatory molecule belonging to the tumor necrosis factor receptor superfamily, is constitutively expressed on memory B cells and most T cells. CD27 engagement in B cells is known to play a key role in B cell activation and immunoglobulin synthesis (70). Our recent in vitro studies with BENTA B cells revealed an intrinsic defect in plasma cell differentiation and antibody production that correlated with poor induction of several genes related to plasma cell commitment, including CD27 (41), although CD27 expression is readily detected on patient T cells (data not shown). CD27 interacts with the ligand CD70, expressed transiently on activated B cells, T cells, and dendritic cells. EBV infection upregulates CD70 expression to greater levels on B cells (20). Recently described human patients with CD27 or CD70 deficiency present with similar disease phenotypes, including hypogammaglobulinemia, reduced memory B cells, increased viral infection, and EBV-induced lymphoproliferation and lymphoma. Heightened susceptibility to EBV-driven disease in these patients, despite normal numbers of T and NK cells, highlights a critical, non-redundant role for CD27–CD70 interactions in driving Ab responses and ensuring optimal cellular control of EBV (44, 71–74). Intriguingly, we recently discovered a significant reduction in CD70 expression on activated BENTA B cells in vitro compared with healthy control B cells (data not shown). Thereby, an impaired CD27–CD70 signaling axis in BENTA could significantly contribute to both specific Ab deficiency and impaired priming and function of EBV-specific CD8+ T cells. The latter could also be related to decreased NKG2D and 2B4 expression on memory CD8+ T cells, similar to CD70-deficient patients (44). Further exploration of a potential CD27-CD70 signaling deficit in BENTA patients is therefore warranted to elucidate a plausible mechanism to explain the inability of BENTA T and NK cells to fully contain EBV.
Clinical Management of EBV in Benta Patients
Assuming B cell lymphocytosis may predispose BENTA patients to greater risk of B cell malignancy later in life, patients are monitored closely for any evidence of B cell clonal outgrowth, using flow cytometry and Ig heavy chain rearrangement analysis. EBV viral load is also measured regularly, as increases in detectable viremia may reflect further debilitation of CD8+ T cell and NK cell function and could theoretically contribute to B cell transformation. However, viral loads in most EBV+ BENTA patients remain comparatively low relative to CA-EBV and other PIDs (46).
To the best of our knowledge, only one patient (P6) was actively treated for EBV-related complications (35). This patient was hospitalized at age 4 with acute EBV infection, featuring profound adenopathy and splenomegaly, as well as immune thrombocytic purpura. Lymph node biopsies revealed substantial polyclonal B cell accumulation in follicular and parafollicular areas, mixed with moderate numbers of CD8+ and CD4+ T cells. At this time, years before the causative CARD11 mutation was discovered, the patient was treated aggressively with intravenous immunoglobulin, rituximab, corticosteroids, and acyclovir. Symptoms resolved with treatment, and plasma EBV load was rendered undetectable by PCR. CD4+/CD8+ T cell ratio, which had dropped significantly during acute EBV infection, also recovered once infection was cleared. Following elective splenectomy 3 years later, his circulating B cell, T cell, and NK cell counts increased dramatically. This phenomenon has been observed in other patients following spleen removal (40) and likely reflects the loss of an important secondary lymphoid tissue niche for excess lymphocytes. Nevertheless, B cell counts in this patient remained 5–10 times higher than those noted in other BENTA patients. To control lymphocytosis, P6 was treated with methotrexate for 4 years until his lymphocyte count was reduced below 80 × 103/μl (35).
This distinctive case provides an illustrative example of successful treatment for acute EBV infection and may represent a blueprint for care if EBV viremia or lymphocytosis increases rapidly in any BENTA patient. Unlike CA-EBV patients, BENTA patients should not require more radical clinical interventions, such as hematopoietic stem cell transplantation or administration of autologous cytotoxic T cells to combat EBV infection (46). In the future, pharmacological inhibitors of NF-κB activation may be an attractive therapeutic tool for reducing B cell numbers in BENTA patients but must be approached with caution to avoid exacerbating underlying T/NK cell immunodeficiency. Inhibitors of MALT1 protease, which dampen canonical NF-κB activity without completely blocking it, may be a more attractive option and have recently yielded promising results for treatment of B cell lymphoma and autoimmune disease (75–78). Future clinical management will ultimately be guided by more basic research into possible aforementioned mechanisms that might explain impaired CTL and NK cell function and compromised EBV control in BENTA disease.
Conclusion
Although the current cohort of patients remains small, impaired control of EBV infection has emerged as a recurring problem in BENTA disease. In contrast to PIDs involving severe EBV-related complications (e.g., fulminant hepatitis and HLH) and complete deficiency of aforementioned receptors and signaling proteins required for EBV immunity, lower viremia in BENTA patients likely reflects attenuation, but not complete abrogation, of T and NK cell functions (Figure 1). Further research is required to connect CARD11 GOF signaling mechanistically to these moderate functional defects, which may involve aberrant signaling through NF-κB as well as other downstream signaling nodes, including JNK and mTORC1. Indeed, severe atopic disease observed in patients carrying CARD11 loss-of-function mutations can be attributed to defects in both NF-κB and mTORC1 activation in T cells (79), although none of these patients have presented with significant EBV infections. Continued identification and careful characterization of additional patients harboring novel CARD11 variants should yield further insights into how CARD11 signaling ultimately governs the immune response against EBV.
Author Contributions
SA wrote the manuscript. PA assembled data for Table 1. HS edited the manuscript and provided clinical perspective. AS supervised the project and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, although no other collaboration, with the authors PA and HS.
Acknowledgments
The authors thank all patients and their families for participating in research described in this review. They thank Dr. Thomas Fleisher (NIH Clinical Center) for supporting diagnostic flow cytometry on BENTA patients seen at NIH. They also thank all of our collaborating physicians for referring patients and providing helpful information, including Drs. Marylin Desjardins and Bruce Mazer at McGill University Health Centre and Dr. Ian Morison at the University of Otago, New Zealand.
Funding
This work was supported with funding from the National Institutes of Health (1R21AI109187 to AS) and the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases. This project has also been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
1. Cohen JI. Epstein-Barr virus infection. N Engl J Med (2000) 343(7):481–92. doi:10.1056/NEJM200008173430707
2. Cohen JI. Epstein-Barr virus vaccines. Clin Transl Immunol (2015) 4(1):e32. doi:10.1038/cti.2014.27
3. Hislop AD, Taylor GS, Sauce D, Rickinson AB. Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol (2007) 25:587–617. doi:10.1146/annurev.immunol.25.022106.141553
4. Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol (2015) 33:787–821. doi:10.1146/annurev-immunol-032414-112326
5. Tangye SG, Palendira U. Human immunity against EBV-lessons from the clinic. J Exp Med (2017) 214(2):269–83. doi:10.1084/jem.20161846
6. Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer (2004) 4(10):757–68. doi:10.1038/nrc1452
7. Chen MR. Epstein-Barr virus, the immune system, and associated diseases. Front Microbiol (2011) 2:5. doi:10.3389/fmicb.2011.00005
8. Odumade OA, Hogquist KA, Balfour HH Jr. Progress and problems in understanding and managing primary Epstein-Barr virus infections. Clin Microbiol Rev (2011) 24(1):193–209. doi:10.1128/CMR.00044-10
9. Ohga S, Nomura A, Takada H, Hara T. Immunological aspects of Epstein-Barr virus infection. Crit Rev Oncol Hematol (2002) 44(3):203–15. doi:10.1016/S1040-8428(02)00112-9
10. Azzi T, Lünemann A, Murer A, Ueda S, Béziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood (2014) 124(16):2533–43. doi:10.1182/blood-2014-01-553024
11. Thorley-Lawson DA. EBV persistence – introducing the virus. Curr Top Microbiol Immunol (2015) 390(Pt 1):151–209. doi:10.1007/978-3-319-22822-8_8
12. Kuppers R. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol (2003) 3(10):801–12. doi:10.1038/nri1201
13. Rickinson AB, Callan MF, Annels NE. T-cell memory: lessons from Epstein-Barr virus infection in man. Philos Trans R Soc Lond B Biol Sci (2000) 355(1395):391–400. doi:10.1098/rstb.2000.0579
14. Callan MF, Fazou C, Yang H, Rostron T, Poon K, Hatton C, et al. CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J Clin Invest (2000) 106(10):1251–61. doi:10.1172/JCI10590
15. Long HM, Haigh TA, Gudgeon NH, Leen AM, Tsang CW, Brooks J, et al. CD4+ T-cell responses to Epstein-Barr virus (EBV) latent-cycle antigens and the recognition of EBV-transformed lymphoblastoid cell lines. J Virol (2005) 79(8):4896–907. doi:10.1128/JVI.79.8.4896-4907.2005
16. Amyes E, Hatton C, Montamat-Sicotte D, Gudgeon N, Rickinson AB, McMichael AJ, et al. Characterization of the CD4+ T cell response to Epstein-Barr virus during primary and persistent infection. J Exp Med (2003) 198(6):903–11. doi:10.1084/jem.20022058
17. Chijioke O, Landtwing V, Munz C. NK cell influence on the outcome of primary Epstein-Barr virus infection. Front Immunol (2016) 7:323. doi:10.3389/fimmu.2016.00323
18. Strowig T, Brilot F, Arrey F, Bougras G, Thomas D, Muller WA, et al. Tonsilar NK cells restrict B cell transformation by the Epstein-Barr virus via IFN-gamma. PLoS Pathog (2008) 4(2):e27. doi:10.1371/journal.ppat.0040027
19. Chung BK, Tsai K, Allan LL, Zheng DJ, Nie JC, Biggs CM, et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood (2013) 122(15):2600–8. doi:10.1182/blood-2013-01-480665
20. Palendira U, Rickinson AB. Primary immunodeficiencies and the control of Epstein-Barr virus infection. Ann N Y Acad Sci (2015) 1356:22–44. doi:10.1111/nyas.12937
21. Parvaneh N, Filipovich AH, Borkhardt A. Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol (2013) 162(5):573–86. doi:10.1111/bjh.12422
22. Yang X, Nishida N, Zhao X, Kanegane H. Advances in understanding the pathogenesis of Epstein-Barr virus-associated lymphoproliferative disorders. Iran J Allergy Asthma Immunol (2015) 14(5):462–71.
23. Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med (2012) 209(12):2247–61. doi:10.1084/jem.20120831
24. Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev (2009) 228(1):199–211. doi:10.1111/j.1600-065X.2008.00749.x
25. Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal (2014) 7(329):ra55. doi:10.1126/scisignal.2005169
26. Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol (2010) 2(9):a003004. doi:10.1101/cshperspect.a003004
27. McCully RR, Pomerantz JL. The protein kinase C-responsive inhibitory domain of CARD11 functions in NF-kappaB activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol Cell Biol (2008) 28(18):5668–86. doi:10.1128/MCB.00418-08
28. Paul S, Traver MK, Kashyap AK, Washington MA, Latoche JR, Schaefer BC, et al. T cell receptor signals to NF-kappaB are transmitted by a cytosolic p62-Bcl10-Malt1-IKK signalosome. Sci Signal (2014) 7(325):ra45. doi:10.1126/scisignal.2004882
29. Qiao Q, Yang C, Zheng C, Fontán L, David L, Yu X, et al. Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell (2013) 51(6):766–79. doi:10.1016/j.molcel.2013.08.032
30. Sommer K, Guo B, Pomerantz JL, Bandaranayake AD, Moreno-García ME, Ovechkina YL, et al. Phosphorylation of the CARMA1 linker controls NF-kappaB activation. Immunity (2005) 23(6):561–74. doi:10.1016/j.immuni.2005.09.014
31. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol (2010) 2(5):a000182. doi:10.1101/cshperspect.a000182
32. Jattani RP, Tritapoe JM, Pomerantz JL. Cooperative control of caspase recruitment domain-containing protein 11 (CARD11) signaling by an unusual array of redundant repressive elements. J Biol Chem (2016) 291(16):8324–36. doi:10.1074/jbc.M115.683714
33. Jattani RP, Tritapoe JM, Pomerantz JL. Intramolecular interactions and regulation of cofactor binding by the four repressive elements in the caspase recruitment domain-containing protein 11 (CARD11) inhibitory domain. J Biol Chem (2016) 291(16):8338–48. doi:10.1074/jbc.M116.717322
34. Arjunaraja S, Snow AL. Gain-of-function mutations and immunodeficiency: at a loss for proper tuning of lymphocyte signaling. Curr Opin Allergy Clin Immunol (2015) 15(6):533–8. doi:10.1097/ACI.0000000000000217
35. Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J Clin Immunol (2014) 5(1):32–46. doi:10.1007/s10875-014-0106-4
36. Buchbinder D, Sassoon A. A case of bad Carma! Blood (2017) 129(12):1737. doi:10.1182/blood-2016-12-756007
37. Buchbinder D, Stinson JR, Nugent DJ, Heurtier L, Suarez F, Sukumar G, et al. Mild B-cell lymphocytosis in patients with a CARD11 C49Y mutation. J Allergy Clin Immunol (2015) 136(3):819–21.e1. doi:10.1016/j.jaci.2015.03.008
38. Casey TP. Chronic lymphocytic leukaemia in a child presenting at the age of two years and eight months. Australas Ann Med (1968) 17(1):70–4. doi:10.1111/imj.1968.17.1.70
39. Outinen T, Syrjänen J, Rounioja S, Saarela J, Kaustio M, Helminen M. Constant B cell lymphocytosis since early age in a patient with CARD11 mutation: a 20-year follow-up. Clin Immunol (2016) 165:19–20. doi:10.1016/j.clim.2016.02.002
40. Darte JM, McClure PD, Saunders EF, Weber JL, Donohue WL, et al. Congenital lymphoid hyperplasia with persistent hyperlymphocytosis. J Exp Med (1971) 284(8):431–2.
41. Arjunaraja S, Nosé BD, Sukumar G, Lott NM, Dalgard CL, Snow AL. Intrinsic plasma cell differentiation defects in B cell expansion with NF-kappaB and t cell anergy patient B cells. Front Immunol (2017) 8:913. doi:10.3389/fimmu.2017.00913
42. Huck K, Feyen O, Ghosh S, Beltz K, Bellert S, Niehues T. Memory B-cells in healthy and antibody-deficient children. Clin Immunol (2009) 131(1):50–9. doi:10.1016/j.clim.2008.11.008
43. Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, et al. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol (2003) 112(5):973–80. doi:10.1016/j.jaci.2003.07.003
44. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med (2017) 214(1):91–106. doi:10.1084/jem.20160849
45. Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Paediatric reference values for the peripheral T cell compartment. Scand J Immunol (2012) 75(4):436–44. doi:10.1111/j.1365-3083.2012.02671.x
46. Cohen JI, Jaffe ES, Dale JK, Pittaluga S, Heslop HE, Rooney CM, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood (2011) 117(22):5835–49. doi:10.1182/blood-2010-11-316745
47. Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, et al. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity (2006) 24(6):729–39. doi:10.1016/j.immuni.2006.04.005
48. Lim KH, Yang Y, Staudt LM. Pathogenetic importance and therapeutic implications of NF-kappaB in lymphoid malignancies. Immunol Rev (2012) 246(1):359–78. doi:10.1111/j.1600-065X.2012.01105.x
49. Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol (2003) 77(15):8532–40. doi:10.1128/JVI.77.15.8532-8540.2003
50. Krishna S, Xie D, Gorentla B, Shin J, Gao J, Zhong X-P. Chronic activation of the kinase IKKbeta impairs T cell function and survival. J Immunol (2012) 189(3):1209–19. doi:10.4049/jimmunol.1102429
51. Brigida I, Chiriaco M, Di Cesare S, Cittaro D, Di Matteo G, Giannelli S, et al. Large deletion of MAGT1 gene in a patient with classic Kaposi sarcoma, CD4 lymphopenia, and EBV infection. J Clin Immunol (2017) 37(1):32–5. doi:10.1007/s10875-016-0341-y
52. Ghosh S, Bienemann K, Boztug K, Borkhardt A. Interleukin-2-inducible T-cell kinase (ITK) deficiency – clinical and molecular aspects. J Clin Immunol (2014) 34(8):892–9. doi:10.1007/s10875-014-0110-8
53. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia (2012) 26(5):963–71. doi:10.1038/leu.2011.371
54. Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev (2009) 228(1):93–114. doi:10.1111/j.1600-065X.2008.00757.x
55. Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature (2011) 475(7357):471–6. doi:10.1038/nature10246
56. Hara H, Wada T, Bakal C, Kozieradzki I, Suzuki S, Suzuki N, et al. The MAGUK family protein CARD11 is essential for lymphocyte activation. Immunity (2003) 18(6):763–75. doi:10.1016/S1074-7613(03)00148-1
57. Bottino C, Falco M, Parolini S, Marcenaro E, Augugliaro R, Sivori S, et al. NTB-A [correction of GNTB-A], a novel SH2D1A-associated surface molecule contributing to the inability of natural killer cells to kill Epstein-Barr virus-infected B cells in X-linked lymphoproliferative disease. J Exp Med (2001) 194(3):235–46. doi:10.1084/jem.194.3.235
58. Nakajima H, Colonna M. 2B4: an NK cell activating receptor with unique specificity and signal transduction mechanism. Hum Immunol (2000) 61(1):39–43. doi:10.1016/S0198-8859(99)00170-6
59. Parolini S, Bottino C, Falco M, Augugliaro R, Giliani S, Franceschini R, et al. X-linked lymphoproliferative disease. 2B4 molecules displaying inhibitory rather than activating function are responsible for the inability of natural killer cells to kill Epstein-Barr virus-infected cells. J Exp Med (2000) 192(3):337–46. doi:10.1084/jem.192.3.337
60. Tangye SG, Cherwinski H, Lanier LL, Phillips JH. 2B4-mediated activation of human natural killer cells. Mol Immunol (2000) 37(9):493–501. doi:10.1016/S0161-5890(00)00076-6
61. Thorley-Lawson DA, Schooley RT, Bhan AK, Nadler LM. Epstein-Barr virus super induces a new human B cell differentiation antigen (B-LAST 1) expressed on transformed lymphoblasts. Cell (1982) 30(2):415–25. doi:10.1016/0092-8674(82)90239-2
62. Dupré L, Andolfi G, Tangye SG, Clementi R, Locatelli F, Aricò M, et al. SAP controls the cytolytic activity of CD8+ T cells against EBV-infected cells. Blood (2005) 105(11):4383–9. doi:10.1182/blood-2004-08-3269
63. Filipovich AH, Zhang K, Snow AL, Marsh RA. X-linked lymphoproliferative syndromes: brothers or distant cousins? Blood (2010) 116(18):3398–408. doi:10.1182/blood-2010-03-275909
64. Nakajima H, Cella M, Bouchon A, Grierson HL, Lewis J, Duckett CS, et al. Patients with X-linked lymphoproliferative disease have a defect in 2B4 receptor-mediated NK cell cytotoxicity. Eur J Immunol (2000) 30(11):3309–18. doi:10.1002/1521-4141(200011)30:11<3309::AID-IMMU3309>3.0.CO;2-3
65. Jamieson AM, Diefenbach A, McMahon CW, Xiong N, Carlyle JR, Raulet DH. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity (2002) 17(1):19–29. doi:10.1016/S1074-7613(02)00333-3
66. Chaigne-Delalande B, Li FY, O’Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science (2013) 341(6142):186–91. doi:10.1126/science.1240094
67. Dhalla F, Murray S, Sadler R, Chaigne-Delalande B, Sadaoka T, Soilleux E, et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol (2015) 35(2):112–8. doi:10.1007/s10875-014-0116-2
68. Patiroglu T, Haluk Akar H, Gilmour K, Unal E, Akif Ozdemir M, Bibi S, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol (2015) 159(1):58–62. doi:10.1016/j.clim.2015.04.015
69. Kwon HJ, Choi GE, Ryu S, Kwon SJ, Kim SC, Booth C, et al. Stepwise phosphorylation of p65 promotes NF-kappaB activation and NK cell responses during target cell recognition. Nat Commun (2016) 7:11686. doi:10.1038/ncomms11686
70. Borst J, Hendriks J, Xiao Y. CD27 and CD70 in T cell and B cell activation. Curr Opin Immunol (2005) 17(3):275–81. doi:10.1016/j.coi.2005.04.004
71. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol (2015) 136(3):703–12.e10. doi:10.1016/j.jaci.2015.02.022
72. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica (2013) 98(3):473–8. doi:10.3324/haematol.2012.068791
73. van Montfrans JM, Hoepelman AI, Otto S, van Gijn M, van de Corput L, de Weger RA, et al. CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. J Allergy Clin Immunol (2012) 129(3):787.e–93.e. doi:10.1016/j.jaci.2011.11.013
74. Izawa K, Martin E. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med (2017) 214(1):73–89. doi:10.1084/jem.20160784
75. Demeyer A, Staal J, Beyaert R. Targeting MALT1 proteolytic activity in immunity, inflammation and disease: good or bad? Trends Mol Med (2016) 22(2):135–50. doi:10.1016/j.molmed.2015.12.004
76. Lee CH, Bae SJ, Kim M. Mucosa-associated lymphoid tissue lymphoma translocation 1 as a novel therapeutic target for rheumatoid arthritis. Sci Rep (2017) 7(1):11889. doi:10.1038/s41598-017-12349-9
77. Saba NS, Wong DH, Tanios G, Iyer JR, Lobelle-Rich P, Dadashian EL, et al. MALT1 inhibition is efficacious in both naive and ibrutinib-resistant chronic lymphocytic leukemia. Cancer Res (2017) 77(24):7038–48. doi:10.1158/0008-5472.CAN-17-2485
78. Bardet M, Unterreiner A, Malinverni C, Lafossas F, Vedrine C, Boesch D, et al. The T-cell fingerprint of MALT1 paracaspase revealed by selective inhibition. Immunol Cell Biol (2017) 96(1):81–99. doi:10.1111/imcb.1018
Keywords: Epstein–Barr virus, B-cell expansion with NF-κB and T-cell anergy, CARD11, NF-κB, primary immune deficiency
Citation: Arjunaraja S, Angelus P, Su HC and Snow AL (2018) Impaired Control of Epstein–Barr Virus Infection in B-Cell Expansion with NF-κB and T-Cell Anergy Disease. Front. Immunol. 9:198. doi: 10.3389/fimmu.2018.00198
Received: 14 October 2017; Accepted: 23 January 2018;
Published: 08 February 2018
Edited by:
Jeffrey I. Cohen, National Institutes of Health (NIH), United StatesReviewed by:
Tri Giang Phan, Garvan Institute of Medical Research, AustraliaKaan Boztug, CeMM Research Center for Molecular Medicine (OAW), Austria
Copyright: © 2018 Arjunaraja, Angelus, Su and Snow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew L. Snow, YW5kcmV3LnNub3dAdXN1aHMuZWR1