 Zhe-Kun Jia
Zhe-Kun Jia Hai-Yun Li
Hai-Yun Li Yu-Lin Liang1
Yu-Lin Liang1 Lawrence Albert Potempa
Lawrence Albert Potempa Yi Wu
Yi Wu- 1MOE Key Laboratory of Cell Activities and Stress Adaptations, School of Life Sciences, Lanzhou University, Lanzhou, China
- 2MOE Key Laboratory of Environment and Genes Related to Diseases, School of Basic Medical Sciences, Xi’an Jiaotong University, Xi’an, China
- 3Roosevelt University College of Pharmacy, Schaumburg, IL, United States
- 4The Affiliated Children’s Hospital of Xi’an Jiaotong University, Xi’an, China
C-reactive protein (CRP) is an established marker of rheumatoid arthritis (RA) but with ill-defined actions in the pathogenesis. Here, we show that CRP regulates the differentiation of osteoclasts, a central mediator of joint inflammation and bone erosion in RA, in a conformation- and receptor activator of NF-κB ligand (RANKL)-dependent manner. CRP in the native conformation is ineffective, whereas the monomeric conformation (mCRP) actively modulates osteoclast differentiation through NF-κB and phospholipase C signaling. Moreover, mCRP can bind RANKL, the major driver of osteoclast differentiation, and abrogate its activities. The binding and inhibition of RANKL are mediated by the cholesterol binding sequence (CBS) of mCRP. Corroborating the in vitro results, CRP knockout exacerbates LPS-induced bone resorption in mice. These results suggest that mCRP may be protective in joint inflammation by inhibiting pathological osteoclast differentiation and that the CBS peptide could be exploited as a potential RANKL inhibitor.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic joint inflammation and bone erosion (1). Autoimmunity in RA is likely triggered first at mucosal sites in response to environmental insults but needs to specifically target joints to initiate the disease. Osteoclasts play a major part in homing systemic autoimmunity to joints (2). These cells locate in the bone compartments, and are differentiated from myeloid precursors driven primarily by receptor activator of NF-κB ligand (RANKL) (3). They respond to autoantibodies frequently found in RA by secreting cytokines that evoke and amplify joint inflammation. Moreover, osteoclasts mediate bone resorption directly causing joint damage (4). Therefore, modulating the differentiation and actions of osteoclasts represents a promising strategy for treatment of RA (2, 5, 6).
C-reactive protein (CRP) is an established biomarker of RA (7, 8) with its blood levels closely associated with disease severity and progression (9). However, it remains elusive how CRP acts in the underlying pathological process. CRP appears to be protective in mice with collagen-induced arthritis (10, 11), but fails to influence autoantibody responses (11). This suggests that the protection by CRP might be exerted at joint level. Therefore, recent studies have examined the effects of CRP on the differentiation of osteoclasts, but yielding opposite conclusions (12, 13). Here, we address this controversy by demonstrating the conformation- and RANKL-dependent actions of CRP. Only with the monomeric conformation (mCRP) that is converted specifically at inflammatory loci (14–18), can this protein regulate osteoclast differentiation and neutralize the activities of RANKL. Therefore, exploiting mCRP-RANKL interactions might provide a novel osteoclast-targeting strategy in RA.
Materials and Methods
Reagents
Native CRP (nCRP; purity > 97%) purified from human ascites was purchased from the BindingSite (Birmingham, UK; catalog number: BP300.X; lot number: 361639 and 404353) and repurified with p-aminophenyl phosphoryl choline agarose (Thermo Fisher Scientific, Rockford, IL, USA; catalog number: 20307). Wide-type and mutant mCRP were prepared as described (19, 20). Proteins were dialyzed to remove NaN3, and passed through Detoxi-Gel Columns (Thermo Fisher Scientific, Rockford, IL, USA; catalog number: 20344) to remove endotoxin when necessary. CRP peptides (purity > 98%) were synthesized by Science Peptide Biological Technology (Shanghai, China). Lyophilized peptides were reconstituted aseptically with DMSO at 40 mg/ml and stored at −20°C in aliquots or kept at 4°C for a maximum of 1 week. Mouse antihuman mCRP mAbs were generated as described (21). 2.5 µg/ml polymyxin B (PMB, Inalco Pharmaceuticals, San Luis Obispo, CA, USA; catalog number: 1758-9325; lot number: R1/51/121) was included in all cell response experiments to neutralize residual endotoxin.
Osteoclast Differentiation
RAW 264.7 murine macrophage cells (13) and bone marrow-derived macrophages (BMDMs) (22) were prepared and cultured as described. Briefly, bone marrow cells were isolated from femurs and tibias of 6-week-old male C57BL/6 mice. Following removal of red blood cells, the remaining cells were cultured in α-MEM (Hyclone, Logan, UT, USA; catalog number: SH30265.01; lot number: AC10207087, AB10164750) containing 10% FBS and 5 ng/ml M-CSF (R&D Systems, Minneapolis, MN, USA; catalog number: 416-ML; lot number: ME2915062 and ME3919011) for 12 h. Nonadherent cells were cultured for another 3 days with 10 ng/ml M-CSF. After vigorous washing, adherent cells were harvested and seeded in 24-well plates for 1–2 days and used as BMDMs. 10 ng/ml M-CSF was copresent in all treatments of BMDMs.
Raw cells or BMDMs were treated with nCRP, mCRP, RANKL (R&D Systems, Minneapolis, MN, USA; catalog number: 462-TEC; lot number: CWA1815111), or their combinations to induce osteoclast differentiation. Culture media were changed every two days. In some experiments, BMDM were pretreated with signaling inhibitors and then stimulated with mCRP or RANKL for 24 h. The used inhibitors were: Bay11-7082 (10 µM, 1 h pretreatment; NF-κB inh), SB20358 (20 µM, 15 min pretreatment; p38 MAPK inh), SP600125 (10 µM, 1 h pretreatment; JNK inh), U0126 (10 µM, 1 h pretreatment; ERK inh), U73122 [10 µM, 0.5 h pretreatment; phospholipase C (PLC) inh], LY294002 (50 µM, 1 h pretreatment; PI3K inh), MK-2206 2HCl (5 µM, 1 h pretreatment; Akt inh), Piceatannol (10 µM, 1 h pretreatment; Syk inh), and GW5074 (20 µM, 0.5 h pretreatment; Raf inh).
Total RNA was extracted with RNAiso Plus reagent (Takara, Shiga, Japan; catalog number: 9109; lot number: AKA3402, AKA5802). cDNA was synthesized from 2 µg total RNA using PrimeScript RT Master Mix system (Takara; catalog number: RR036A; lot number: AK4102, AK4403). The expression of osteoclast marker genes was determined with quantitative PCR using RealStar Green Power Mixture (Genestar, Beijing, China; catalog number: A311; lot number: 7AB01) in a StepOne Plus real-time PCR system (Thermo Fisher Scientific). The gene expression levels were normalized to that of GAPDH. The primer sequences used were: TRAP (forward: 5′-GCAACATCCCCTGGTATGTG-3′; reverse: 5′-GCAAACGGTAGTAAGGGCTG-3′); Cathepsin K (forward: 5′-GCATTACCAACATGGCCAGC-3′; reverse: 5′-CTCCCTTCCAAAGCCACCAA-3′); RANKL (forward: 5′-CAGCATCGCTCTGTTCCTGTA-3′; reverse: 5′-CTGCGTTTTCATGGAGTCTCA-3′); NFKB2 (forward: 5′-GGCCGGAAGACCTATCCTACT-3′; reverse: 5′-CTACAGACACAGCGCACACT-3′); SOCS1 (forward: 5′-CTGCGGCTTCTATTGGGGAC-3′; reverse: 5′-AAAAGGCAGTCGAAGGTCTCG-3′); SOCS3 (forward: 5′-ATGGTCACCCACAGCAAGTTT-3′; reverse: 5′-TCCAGTAGAATCCGCTCTCCT-3′); PPARG (forward: 5′-GGAAGACCACTCGCATTCCTT-3′; reverse: 5′-GTAATCAGCAACCATTGGGTCA-3′); GAPDH (forward: 5′-GGGCTACACTGAGGACCAGGTT-3′; reverse: 5′-TGCTGTAGCCGTATTCATTGTCA-3′).
Gene expression profiles were determined with SurePrint G3 Mouse Gene Expression 8*60K Microarray (Agilent Technologies, Santa Clara, CA, USA) by CapitalBio Technology (Beijing, China). Expression ratios were calculated, first normalized to the 75th percentile per chip, and finally normalized to medians per gene.
Following differentiation for 6 days, cells were fixed with formaldehyde and the number of osteoclasts was determined with a TRAP staining kit (Sigma-Aldrich, St. Louis, MO, USA; catalog number: 387A-1KT; lot number: SLBP7795V) according to the manufacture’s instruction. TRAP-positive multinucleated cells (>3 nuclei/cell) were counted by a light microcopy as osteoclasts.
Bone Resorption
RAW cells or BMDMs were seeded on fresh bovine femur slices of 20-µm thick in 24-well plates. Osteoclast differentiation was induced by treatment of nCRP, mCRP, RANKL or their combinations with culture media changed every 2 days. Six days later, the slices were washed with 1 M ammonia for 3 min to break the osteoclasts, then fixed with 50% glutaric acid for 3 min, and finally stained with 1% toluidine blue for 5–10 min. Bone resorption was measured by pit area.
RANKL Binding
The interaction of mCRP and RANKL was determined with ELISA. Briefly, microtiter wells were coated with 1 µg/ml RANKL overnight at 4°C. All the following steps were performed at 37°C, and after each incubation step wells were washed 3 times with TBS (10 mM Tris, 140 mM NaCl, pH 7.4) containing 0.02% NP-40. Wells were washed and blocked with 1% BSA in TBS. nCRP, mCRP, or mCRP mutant was added to immobilized RANKL for 1 h followed by washing. Binding were then detected with a sheep antihuman CRP polyclonal antibody (BindingSite; catalog number: PC044; lot number: 352325) and a donkey antisheep IgG (H + L) secondary antibody (Abbkine, Wuhan, China; catalog number: A21060; lot number: ATQMA0601).
Inflammatory Osteolysis
CRP knockout (KO) mice of C57BL/6 background were generated by insertion of a floxed STOP cassette at the translation start site of CRP gene via homologous recombination using CRISPR/Cas9 technique (Shanghai Biomodel Organism Science & Technology Development, Shanghai, China). CRP KO mice are fertile and grossly healthy. Inflammatory osteolysis was induced in wild-type or CRP KO mice (25 ± 2 g) of 7–8 weeks age as described (23). Briefly, 5 mg/kg LPS (Sigma-Aldrich; catalog number: L2880, lot number: 25M4040V), 2.5 mg/kg mCRP or vehicle was injected into the subcutaneous tissues overlying calvaria. The injections were performed every other day for 7 days. The calvaria were harvested and fixed in 4% paraformaldehyde for 2 days, followed by decalcification with 10% neutral buffered EDTA and embedding in paraffin. Samples were sectioned and TRAP and hematoxylin and eosin (HE) staining were performed to evaluate osteoclastogenesis and bone damage. The experiments conformed to the Guide for the Care and Use of Laboratory Animals published by NIH and were conducted according to the protocols approved by the Ethics Committee of Animal Experiments of Xi’an Jiaotong University.
Fluorescence Imaging
Raw264.7 and BMDM cells cultured on coverslips were rinsed twice with sterile PBS and incubated with FITC-labeled nCRP or mCRP for 30 min at 4°C. After gently rising, cell membrane was marked with FM 4-64 (Invitrogen, Carlsbad, CA, USA; catalog number: F34653; lot number: 1814727) at 4°C for 1 min. Nuclei were counterstained with DAPI (SouthernBiotech, Birmingham, AL, USA; catalog number: 0100-20; lot number: F0617-S327). Samples were examined by a LSM 710 confocal microscopy (Zeiss, Jena, Germany).
Statistical Analysis
Data were presented as mean ± SEM. Statistical analysis was performed by two-tailed Student’s t-test, one-way ANOVA with Tukey post hoc or Kolmogorov–Smironv tests as appropriate. Values of p < 0.05 were considered significant.
Results
mCRP Induces Osteoclast Differentiation
Circulating CRP is composed of five identical subunits, but dissociates into the monomeric conformation, i.e., mCRP, upon entering local lesions (14–18). Indeed, mCRP has been identified as the major conformation present in synovium tissues of RA patients (24). CRP in different conformations exhibit distinct or even contrasting activities (14–18), which may account for the controversies on its role in osteoclast differentiation (12, 13). We thus first examined this issue using Raw 264.7 macrophage cell line. Treating Raw cells with nCRP for 2 days did not alter the expression of osteoclast maker genes TRAP (also called ACP5) and Cathepsin K (Figure 1A). By contrast, these genes were markedly upregulated by mCRP treatment.
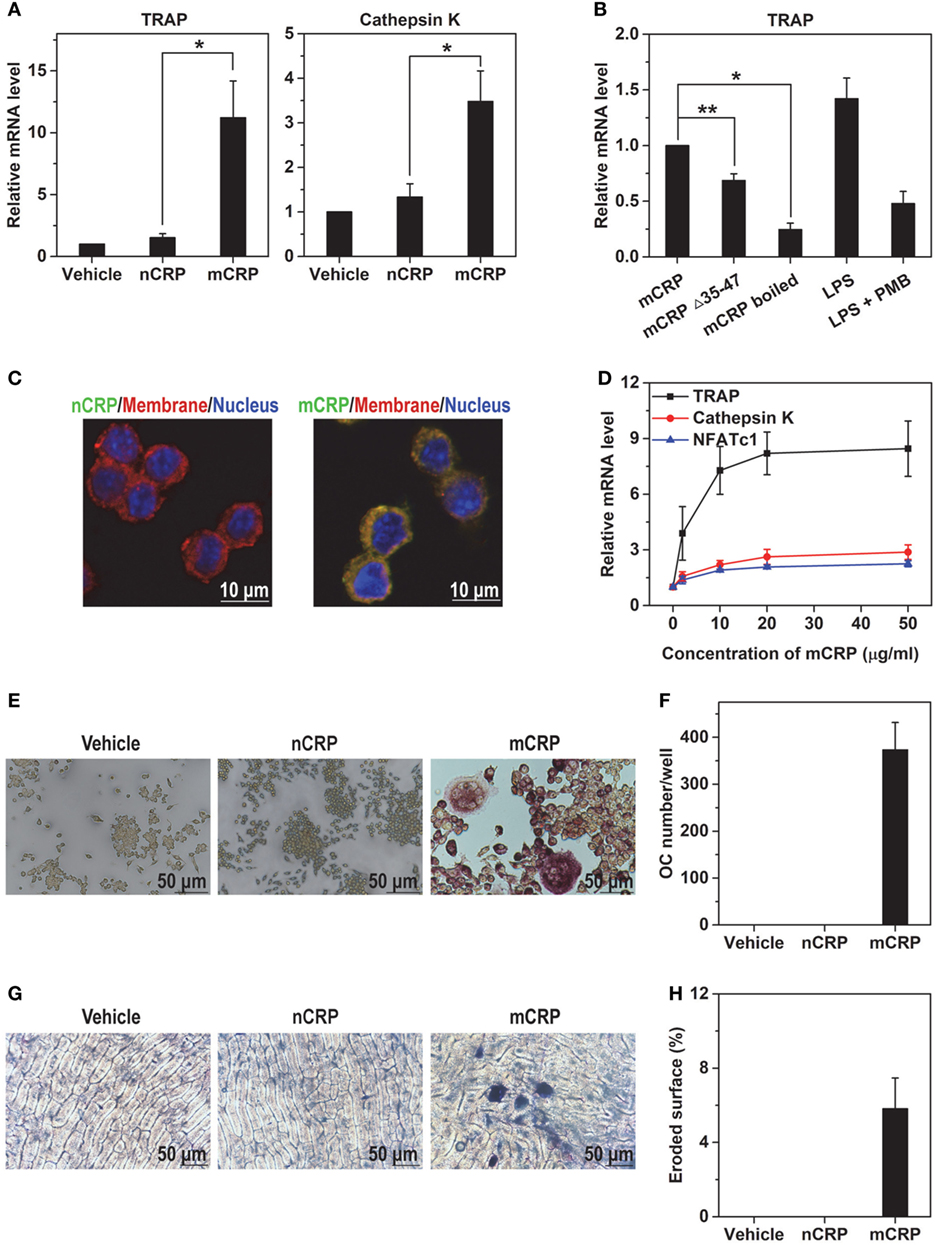
Figure 1. mCRP but not native C-reactive protein (nCRP) induces osteoclast differentiation of Raw 264.7 cell line. Raw cells were treated with the indicated reagents for 2 days and the expression of osteoclast marker genes was determined with q-PCR. (A) 100 µg/ml mCRP upregulated the expression of TRAP and Cathepsin K, while nCRP at the same concentration was ineffective. (B) mCRP mutant lacking cholesterol-binding sequence motif (Δ35–47) or boiled wild-type mCRP showed impaired capacity to upregulate the expression of TRAP. The effects of LPS at 100 ng/ml, which is 50-fold higher than the residual level of endotoxin in our mCRP preparation, could be abrogated by the copresence of 2.5 µg/ml polymyxin B (PMB) that was included in all differentiation experiments. (C) FITC-labeled nCRP and mCRP were incubated with Raw cells at 4°C and visualized by confocal microscopy. Cell membranes and nuclei were counterstained with FM-4-64 and DAPI, respectively. mCRP showed intense binding to Raw cells, but nCRP did not. (D) Dose-dependent induction of TRAP expression by mCRP. Following treatment with 100 µg/ml nCRP or mCRP for 6 days, Raw cells were stained for TRAP (E) and osteoclast number was counted as TRAP-positive multinucleated cells (F). Raw cells were plated on bone slices and treated with nCRP or mCRP for 6 days. The slices were then stained by toluidine blue (G) to measure eroded surface (H). mCRP but not nCRP induced the differentiation of Raw cells to mature osteoclasts with bone resorption activities.
The effects of mCRP were not due to endotoxin contaminant because experiments were performed in the presence of PMB. Moreover, boiling or deleting the key recognition motif, i.e., cholesterol-binding sequence (CBS; a.a. 35–47) (20) that interacts with the lipid raft receptor (25, 26), impaired the actions of mCRP (Figure 1B). mCRP also showed much stronger binding to Raw cells than nCRP (Figure 1C), and evoked substantial responses at a concentration of 2 µg/ml (Figure 1D). The capability of mCRP to drive osteoclast differentiation was further confirmed by functional assays of TRAP staining (Figures 1E,F) and bone resorption (Figures 1G,H), in which nCRP was ineffective.
We next validated the conformation-dependent actions of CRP using mouse BMDMs. nCRP showed only weak binding to BMDMs and was unable to drive osteoclast differentiation; while mCRP bound BMDMs intensely (Figure 2A) and induced strong expression of TRAP (Figure 2B), leading to the formation of multinucleated osteoclasts (Figures 2C,D) with bone resorption activities (Figures 2E,F). Consistent with the in vitro results, subcutaneous injection of mCRP on calvaria of healthy mice resulted in increased number of osteoclasts (Figures 2G,H) and obvious trabecular damage (Figures 2I,J). We thus conclude that the induction of osteoclast differentiation by CRP depends on the monomeric conformation.
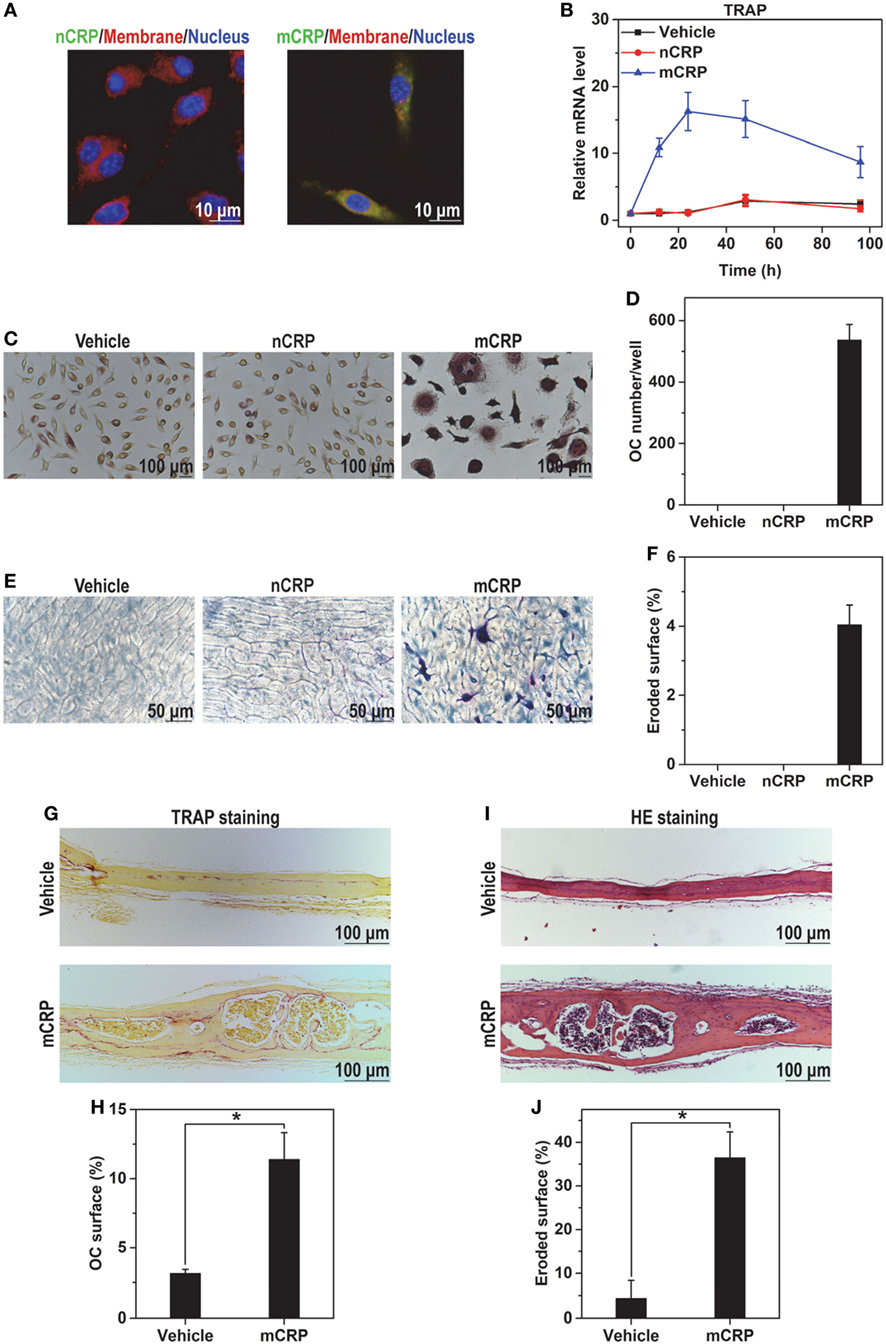
Figure 2. mCRP but not native C-reactive protein (nCRP) induces osteoclast differentiation of bone marrow-derived macrophages (BMDMs) and in mice. (A) FITC-labeled nCRP or mCRP was incubated with BMDMs at 4°C and visualized by confocal microscopy. Cell membranes and nuclei were counterstained with FM-4-64 and DAPI, respectively. mCRP bound BMDMs strongly, while nCRP did not. (B) BMDMs were treated with 100 µg/ml nCRP or mCRP for the indicated times. TRAP expression was induced by mCRP but not nCRP. BMDMs were treated with 100 µg/ml nCRP or mCRP for 6 days, and then stained for TRAP (C) to count the number of TRAP-positive multinucleated cells as osteoclasts (D). BMDMs were plated on bone slices and treated with nCRP or mCRP for 6 days. The slices were then stained by toluidine blue (E) to measure eroded surface (F). mCRP but not nCRP induced the differentiation of BMDMs to mature osteoclasts with bone resorption activities. mCRP (2.5 mg/kg) or saline buffer (Vehicle) was s.c. injected on calvaria of healthy wild-type mice every 2 days for 1 week. TRAP (G,H) and hematoxylin and eosin staining (I,J) were conducted to evaluate osteoclastogenesis and bone damage, respectively. Osteolysis was actively induced by mCRP injection.
mCRP Does Not Act via Induction of RANKL
RANKL is considered as the major inducer of osteoclast differentiation (3) and has been reported to be upregulated by CRP (12). To clarify whether the effects of mCRP are mediated via downstream RANKL, we performed expression profiling on mCRP-treated BMDMs by DNA microarray. Osteoclast differentiation emerged as one of the top-ranked pathways activated by mCRP. Of the 35 relevant genes, 11 were differentially expressed in response to mCRP following a 4-h treatment (Figures 3A,B). Importantly, mCRP did not induce the expression of RANKL, its activating receptor RANK, or the inhibitory receptor LGR4 (22), but downregulated the expression of the decoy receptor OPG, suggesting little involvement of the canonical RANKL pathway in mediating the downstream effects of mCRP.
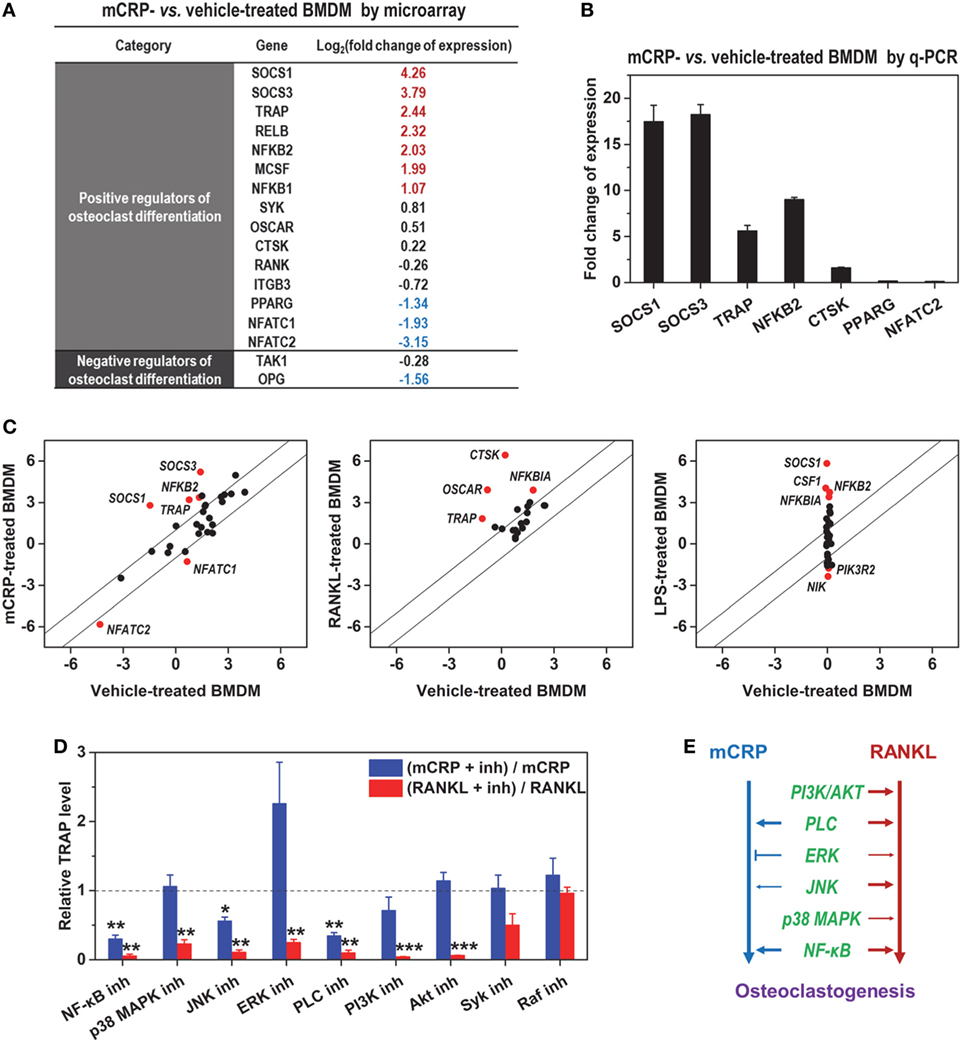
Figure 3. mCRP acts via receptor activator of NF-κB ligand (RANKL)-independent pathway. (A) The expression profiles of mCRP- and vehicle-treated bone marrow-derived macrophages (BMDMs) were determined with DNA microarray. The expression changes of confidently determined genes relevant to osteoclast differentiation were listed. mCRP did not induce the expression of RANKL or its activating receptor RANK. (B) q-PCR validation of gene expression induced by mCRP. (C) Comparison of the effects of mCRP, RANKL (GSE57468), and LPS (GSE21895) on the expression of osteoclastogenesis-relevant genes. Remarkable differences in their effects were evident. (D) BMDMs were treated with mCRP or RANKL in the presence or absence of the indicated signaling inhibitors: Bay11-7082 (10 µM, NF-κB inh), SB20358 (20 µM, p38 MAPK inh), SP600125 (10 µM, JNK inh), U0126 (10 µM, ERK inh), U73122 (10 µM, Phospholipase C (PLC) inh), LY294002 (50 µM, PI3K inh), MK-2206 2HCl (5 µM, Akt inh), Piceatannol (10 µM, Syk inh), and GW5074 (20 µM, Raf inh). The mRNA levels of TRAP were determined by q-PCR and normalized to that of controls treated only with inhibitor. Results are shown as (TRAP level of cells treated with mCRP or RANKL plus the indicated inhibitor)/(TRAP level of cells treated with mCRP or RANKL). (E) The signaling pathways evoked by the two inducers differed significantly.
Additional analysis revealed remarkable differences between the gene expression profiles induced by mCRP and RANKL (Figure 3C). Moreover, screening with signaling inhibitors demonstrated that they activated distinct pathways (Figure 3D). mCRP acted primarily through NF-κB and phospholipase C, while a more extensive network was involved in RANKL signaling (Figure 3E). Of particular interest, ERK inhibition with U0126 markedly enhanced the effects of mCRP but suppressed that of RANKL. These results together demonstrate that the induction of osteoclast differentiation by mCRP does not depend on RANKL.
mCRP Inhibits RANKL-Induced Osteoclast Differentiation
As mCRP appeared to act differently from RANKL, we asked whether these two inducers have any synergy in osteoclast differentiation. When applied alone, RANKL was 3–10-fold more potent than mCRP in inducing the expression of osteoclast maker genes in BMDMs (Figure 4A). When applied together with mCRP, however, the effects of RANKL was completely absent with the net responses comparable to that of mCRP acting alone. Similar findings were also obtained in TRAP staining (Figures 4B,C) and bone resorption assays (Figures 4D,E). These results suggest that mCRP might instead inhibit the activities of RANKL on osteoclast differentiation.
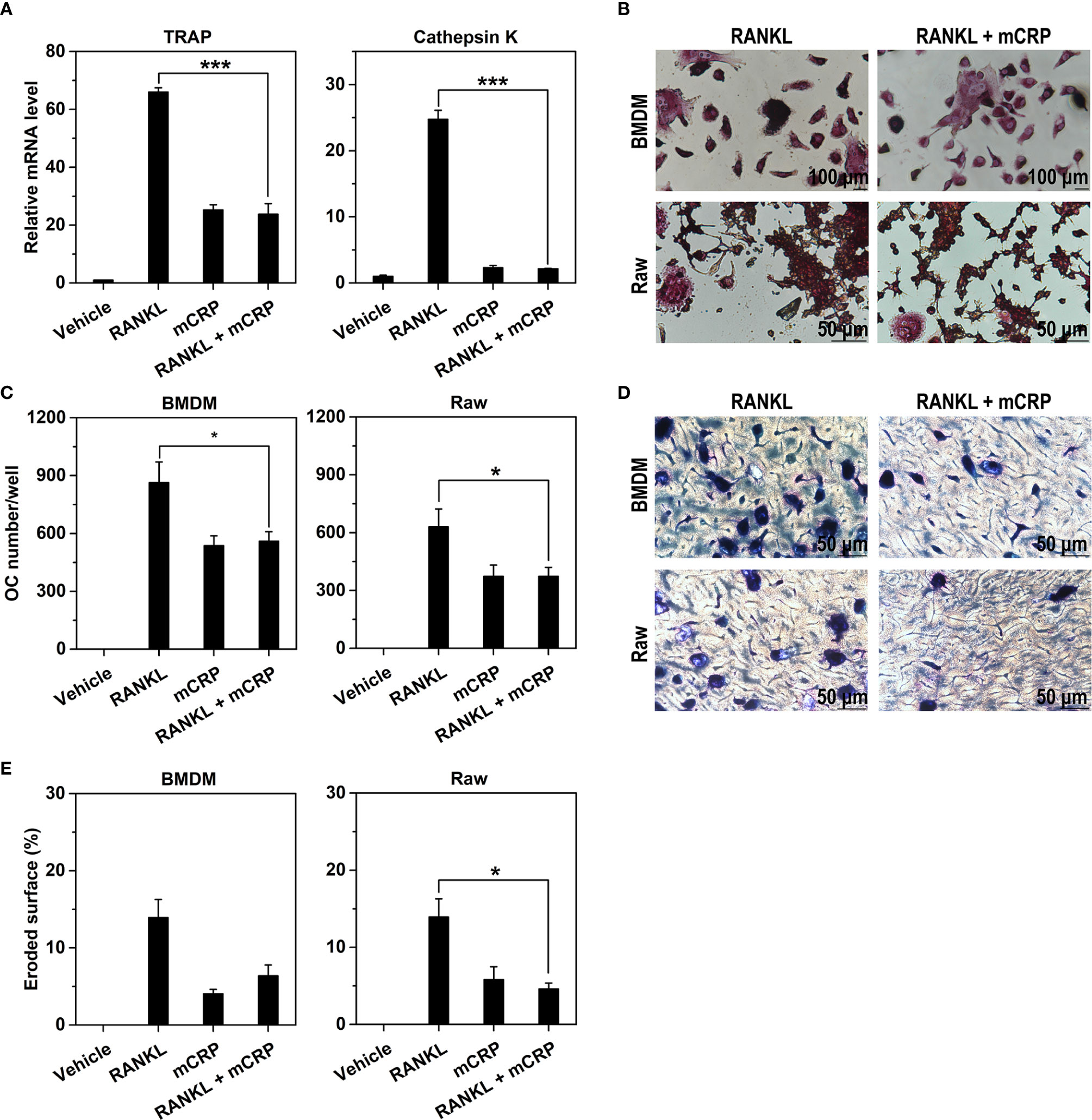
Figure 4. mCRP neutralizes the effects of receptor activator of NF-κB ligand (RANKL). Bone marrow-derived macrophages (BMDMs) were treated with 10 ng/mL of M-CSF and 50 ng/ml RANKL in the presence or absence of 100 µg/mL of mCRP. After treatment for 2 days, the expression of TRAP and Cathepsin K were determined by q-PCR (A). After treatment for 6 days, cells were stained for TRAP (B) and counted for the number of osteoclasts (C). Their bone resorption activities were evaluated by toluidine blue staining (D) and the quantification of eroded surface (E). The potent effects of RANKL on BMDMs were absent when treated together with mCRP. Comparable results were also obtained with Raw cells (B–E). *p < 0.05; ***p < 0.001.
mCRP is specifically converted in inflamed tissues, but does not exist in normal tissues (14–18). Therefore, it is plausible that the actions of mCRP are confined to pathological osteoclast differentiation where overproduced RANKL also plays a predominant role (4–6). The overall effects of mCRP in such a scenario, however, are likely suppressive due to inhibition of RANKL. In line with this speculation, the bone resorption phenotype of CRP KO mice (Figures 5A,B) is indistinguishable from that of wild-type mice in normal calvaria, but manifested increased number of osteoclasts (Figures 5C,D) and enhanced damage of trabeculae (Figures 5E,F) following subcutaneous injection of LPS.
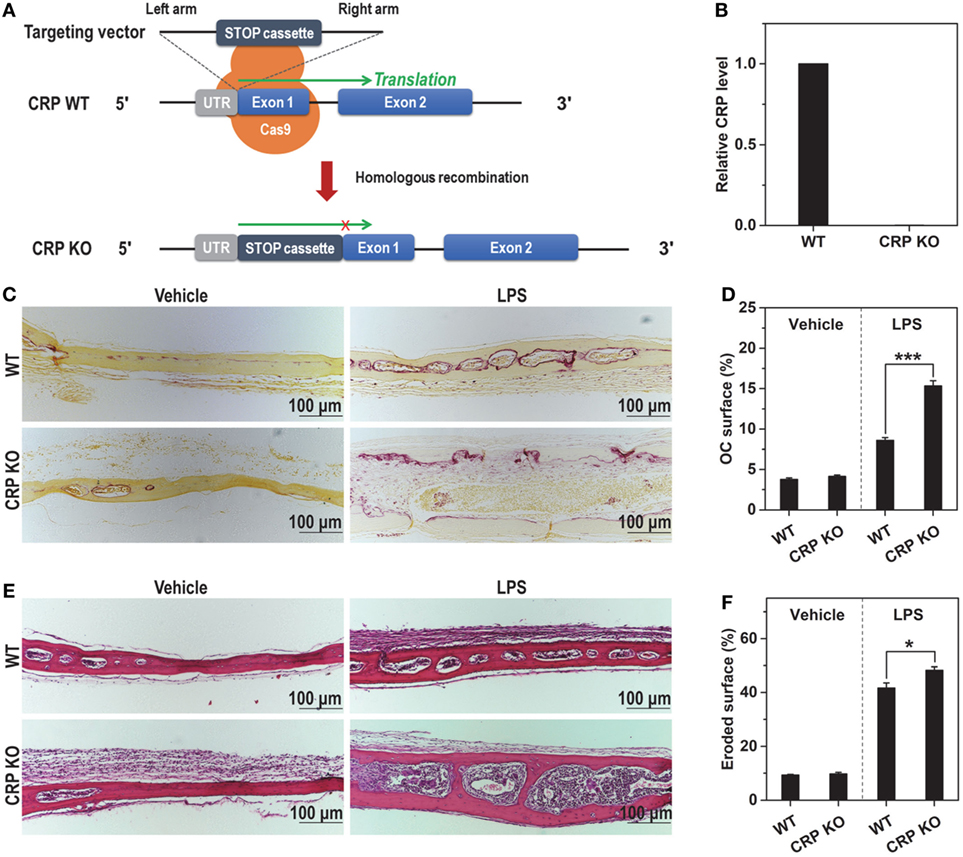
Figure 5. C-reactive protein (CRP) knockout (KO) aggravates proinflammatory bone damage in mice. (A) The design of CRP KO mice. (B) The mRNA level of CRP in liver tissues of wild-type (WT) and CRP KO mice. No CRP expression could be detected in CRP KO mice. LPS (5 mg/kg) or saline buffer (Vehicle) was s.c. injected on calvaria of healthy wild-type and CRP KO mice every 2 days for 1 week. TRAP (C,D) and hematoxylin and eosin staining (E,F) were conducted to evaluate osteoclastogenesis and bone damage, respectively. These pathological indices were aggravated in CRP KO versus wide type mice.
CBS Mediates the Binding and Inhibition of RANKL
The inhibition of RANKL by mCRP could be due to interference between signaling pathways evoked by them. However, the complete and persistent inhibition observed in mouse BMDMs (Figure 6A) and peripheral blood mononuclear cells (PBMCs) (Figure 6B) made this possibility unlikely. Rather, inhibition due to their direct physical interactions appeared more likely. Indeed, mCRP bound RANKL with high affinity (Figure 6C). Deleting CBS (a.a. 35–47), the major recognition motif of mCRP (20), or competing with the synthetic CBS peptide markedly impaired the binding (Figure 6D). Moreover, the CBS peptide efficiently inhibited RANKL-induced osteoclastogenesis (Figures 6E–H). Therefore, the physical interactions between mCRP and RANKL are mediated by CBS.
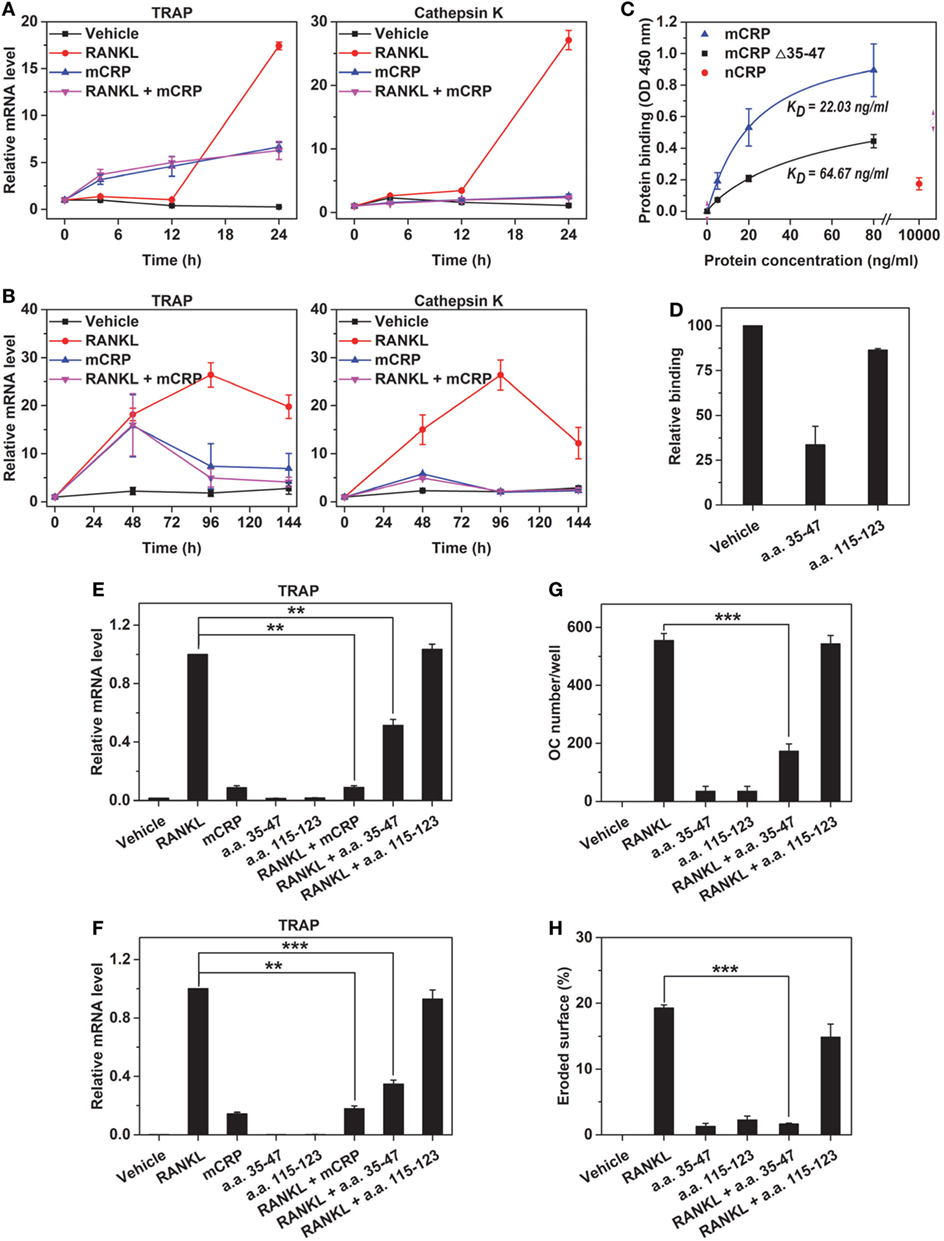
Figure 6. mCRP binds receptor activator of NF-κB ligand (RANKL) via cholesterol-binding sequence. Bone marrow-derived macrophages (BMDMs) (A) or peripheral blood mononuclear cells (B) were treated with RANKL, mCRP, or their combination for the indicated times. In both cell types, the effects of RANKL were abrogated by cotreatment of mCRP at all time points tested. (C) mCRP, mCRP Δ35–47, or native C-reactive protein (nCRP) at the indicated concentrations were added to RANKL immobilized on microtiter wells. mCRP avidly bound immobilized RANKL, but this binding was impaired by deleting a.a. 35–47. Weak binding of nCRP was only detected at very high concentration. a.a. 35–47 peptide (20 µg/ml) nearly abrogated the binding of mCRP to RANKL (D), halved RANKL-induced TRAP expression in Raw (E) and BMDM (F), and greatly suppressed RANKL-induced differentiation of osteoclasts (G) and their bone resorption activities (H). By contrast, another CRP peptide, i.e., a.a. 115–123 (20 µg/ml), showed no effect in these assays.
Discussion
The effects of CRP on osteoclast differentiation have been controversial. One study reports that CRP inhibits RANKL-induced osteoclast differentiation of Raw 264.7 cells via TLR signaling (13). However, residual endotoxin is a notorious contaminant that confuses the interpretation of CRP’s activity (27, 28). Another study reports that CRP promotes osteoclast differentiation of human PBMCs via induction of RANKL (12). However, the prolonged incubation time (3–21 days) and the prominent death of primary cells cultured ex vivo would favor the conversion of nCRP to mCRP (29–34). Interestingly, the authors claim that the actions of CRP are mediated at least partly by CD16 (12), an established receptor for mCRP (35).
In the present study, with tight control of endotoxin contaminant and protein conformation, we show that CRP in its native conformation, i.e., nCRP, has no effect on osteoclast differentiation of Raw 264.7 cells, mouse BMDMs or PBMCs; while mCRP actively regulates their differentiation. The remarkable difference between gene expression profiles induced by mCRP and LPS further exclude possible confounding of endotoxin. Our findings thus establish that the effects of CRP on osteoclast differentiation are conformation-dependent. This may account for the aforementioned controversy and argues that conformation control is critical for interpreting the actions of CRP.
The actions of mCRP in osteoclastogenesis appear to depend on crosstalk among NF-κB, phospholipase C and ERK signaling pathways. Indeed, NF-κB, phospholipase C, and p38 MAPK have been shown to be responsible for mCRP-induced cytokine induction in endothelial cells (36, 37), while ERK and PI3K/Akt are more important in mediating the effects of mCRP on angiogenesis (38–41) and survival of neutrophils (42). These findings reveal a cell type- and biological process-dependent signaling evoked by mCRP. Such a versatility in activating various signaling pathways is likely duo to the capacity of mCRP to interact with lipid rafts (25, 26), signaling platforms on cell surfaces. Therefore, adaptor(s) in lipid rafts that mediates the context-specific actions of mCRP warrants further investigation.
It has been increasingly recognized that the actions of CRP also depend on localization (14–18). nCRP circulates in the blood as a pentamer, but undergoes irreversible conformation changes, forming mCRP in inflamed tissues due to interaction with damaged membranes (29–34, 43–46), amyloid aggregates (47), neutrophil extracellular traps (48), or acidic pH (49). mCRP exhibits markedly enhanced activities, and is consequently considered to be the major conformation acting in local lesions (14–18), such as synovium tissues of RA patients (24). Therefore, the interaction of mCRP with osteoclast precursors would be expected to occur in inflamed but not normal joints.
In inflamed joints, dysregulated RANKL signaling likely plays a major role in driving pathological osteoclast differentiation (4–6), and its neutralizing mAb, Denosumab, has become an approved therapeutic (50) that prevents bone loss in RA (51). As such, the overall effects of mCRP in pathological osteoclast differentiation may be protective through antagonizing RANKL. This speculation is supported by exacerbated inflammatory bone resorption in CRP KO versus wild-type mice, and is also consistent with the beneficial role of CRP in collagen-induced arthritis (10, 11). In this regard, CBS that mediates the interaction of mCRP with RANKL and exhibits anti-inflammatory activities as a synthetic peptide (20, 52), might emerge as a potential RANKL inhibitor to target pathological osteoclast differentiation.
Though mCRP is a potent proinflammatory molecule, it also possesses anti-inflammatory actions (53, 54). Therefore, the net contribution of mCRP in diseases might be context-dependent. Indeed, a protective role of mCRP in early atherogenesis has been reported (55). Recently, we have demonstrated that mCRP may be protective in lupus nephritis by recruiting CFH, and autoantibodies against CBS abrogating this effect predicts worse prognosis (52). Of note, dysregulated complement activation is critically involved in both atherosclerosis and lupus nephritis, which may explain the beneficial role of mCRP. In RA, however, dysregulated osteoclast differentiation is a central pathogenic mechanism (2) and mCRP may thus exert protective effects by neutralizing RANKL-induced osteoclastogenesis.
Ethics Statement
The experiments conformed to the Guide for the Care and Use of Laboratory Animals published by NIH and were conducted according to the protocols approved by the Ethics Committee of Animal Experiments of Xi’an Jiaotong University.
Author Contributions
YW and S-RJ designed the research. Z-KJ, H-YL, and Y-LL performed the research. YW, S-RJ, Z-KJ, H-YL, and LP analyzed the data and wrote the article. All authors reviewed the results and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JA and handling editor declared their shared affiliation.
Acknowledgments
We thank the Core Facility of School of Life Sciences, Lanzhou University for technical and instrumental support. This work was supported by grants from the National Natural Science Foundation of China (grant numbers, 31671339, 31570749, and 31470718) and grants from the Fundamental Research Funds for the Central Universities (grant numbers, lzujbky-2016-k11).
References
1. Malmstrom V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol (2017) 17:60–75. doi:10.1038/nri.2016.124
2. Catrina AI, Svensson CI, Malmstrom V, Schett G, Klareskog L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat Rev Rheumatol (2017) 13:79–86. doi:10.1038/nrrheum.2016.200
3. Nakashima T, Takayanagi H. New regulation mechanisms of osteoclast differentiation. Ann N Y Acad Sci (2011) 1240:E13–8. doi:10.1111/j.1749-6632.2011.06373.x
4. Novack DV, Teitelbaum SL. The osteoclast: friend or foe? Annu Rev Pathol (2008) 3:457–84. doi:10.1146/annurev.pathmechdis.3.121806.151431
5. Szentpétery Á, Horváth Á, Gulyás K, Pethö Z, Bhattoa HP, Szántó S, et al. Effects of targeted therapies on the bone in arthritides. Autoimmun Rev (2017) 16:313–20. doi:10.1016/j.autrev.2017.01.014
6. Okamoto K, Nakashima T, Shinohara M, Negishi-Koga T, Komatsu N, Terashima A, et al. Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev (2017) 97:1295–349. doi:10.1152/physrev.00036.2016
7. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, et al. 2010 Rheumatoid arthritis classification criteria: an American college of rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum (2010) 62:2569–81. doi:10.1002/art.27584
8. Felson DT, Smolen JS, Wells G, Zhang B, van Tuyl LH, Funovits J, et al. American College of Rheumatology/European League Against Rheumatism provisional definition of remission in rheumatoid arthritis for clinical trials. Arthritis Rheum (2011) 63:573–86. doi:10.1002/art.30129
9. Mallya RK, de Beer FC, Berry H, Hamilton ED, Mace BE, Pepys MB. Correlation of clinical parameters of disease activity in rheumatoid arthritis with serum concentration of C-reactive protein and erythrocyte sedimentation rate. J Rheumatol (1982) 9:224–8.
10. Jiang S, Xia D, Samols D. Expression of rabbit C-reactive protein in transgenic mice inhibits development of antigen-induced arthritis. Scand J Rheumatol (2006) 35:351–5. doi:10.1080/03009740600757963
11. Jones NR, Pegues MA, McCrory MA, Kerr SW, Jiang H, Sellati R, et al. Collagen-induced arthritis is exacerbated in C-reactive protein-deficient mice. Arthritis Rheum (2011) 63:2641–50. doi:10.1002/art.30444
12. Kim KW, Kim BM, Moon HW, Lee SH, Kim HR. Role of C-reactive protein in osteoclastogenesis in rheumatoid arthritis. Arthritis Res Ther (2015) 17:41. doi:10.1186/s13075-015-0563-z
13. Cho IJ, Choi KH, Oh CH, Hwang YC, Jeong IK, Ahn KJ, et al. Effects of C-reactive protein on bone cells. Life Sci (2016) 145:1–8. doi:10.1016/j.lfs.2015.12.021
14. Ma X, Ji SR, Wu Y. Regulated conformation changes in C-reactive protein orchestrate its role in atherogenesis. Chinese Sci Bull (2013) 58:1642–9. doi:10.1007/s11434-012-5591-3
15. Wu Y, Potempa LA, El Kebir D, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem (2015) 396:1181–97. doi:10.1515/hsz-2015-0149
16. Thiele JR, Zeller J, Bannasch H, Stark GB, Peter K, Eisenhardt SU. Targeting C-reactive protein in inflammatory disease by preventing conformational changes. Mediators Inflamm (2015) 2015:372432. doi:10.1155/2015/372432
17. Slevin M, Krupinski J. A role for monomeric C-reactive protein in regulation of angiogenesis, endothelial cell inflammation and thrombus formation in cardiovascular/cerebrovascular disease? Histol Histopathol (2009) 24:1473–8. doi:10.14670/HH-24.1473
18. Singh SK, Suresh MV, Voleti B, Agrawal A. The connection between C-reactive protein and atherosclerosis. Ann Med (2008) 40:110–20. doi:10.1080/07853890701749225
19. Potempa LA, Yao Z-Y, Ji S-R, Filep JG, Wu Y. Solubilization and purification of recombinant modified C-reactive protein from inclusion bodies using reversible anhydride modification. Biophys Rep (2015) 1:18–33. doi:10.1007/s41048-015-0003-2
20. Li HY, Wang J, Meng F, Jia ZK, Su Y, Bai QF, et al. An intrinsically disordered motif mediates diverse actions of monomeric C-reactive protein. J Biol Chem (2016) 291:8795–804. doi:10.1074/jbc.M115.695023
21. Ying SC, Gewurz H, Kinoshita CM, Potempa LA, Siegel JN. Identification and partial characterization of multiple native and neoantigenic epitopes of human C-reactive protein by using monoclonal antibodies. J Immunol (1989) 143:221–8.
22. Luo J, Yang Z, Ma Y, Yue Z, Lin H, Qu G, et al. LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med (2016) 22:539–46. doi:10.1038/nm.4076
23. Li L, Khansari A, Shapira L, Graves DT, Amar S. Contribution of interleukin-11 and prostaglandin(s) in lipopolysaccharide-induced bone resorption in vivo. Infect Immun (2002) 70:3915–22. doi:10.1128/IAI.70.7.3915-3922.2002
24. Shi P, Li XX, Zhu W, Yang H, Dong C, Li XM. Immunohistochemical staining reveals C-reactive protein existing predominantly as altered conformation forms in inflammatory lesions. Acta Biol Hung (2014) 65:265–73. doi:10.1556/ABiol.65.2014.3.3
25. Wang MY, Ji SR, Bai CJ, El Kebir D, Li HY, Shi JM, et al. A redox switch in C-reactive protein modulates activation of endothelial cells. FASEB J (2011) 25:3186–96. doi:10.1096/fj.11-182741
26. Ji SR, Ma L, Bai CJ, Shi JM, Li HY, Potempa LA, et al. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J (2009) 23:1806–16. doi:10.1096/fj.08-116962
27. Nerurkar SS, McDevitt PJ, Scott GF, Johanson KO, Willette RN, Yue TL. Lipopolysaccharide (LPS) contamination plays the real role in C-reactive protein-induced IL-6 secretion from human endothelial cells in vitro. Arterioscler Thromb Vasc Biol (2005) 25:e136. doi:10.1161/01.ATV.0000175753.82842.cc
28. Taylor KE, Giddings JC, van den Berg CW. C-reactive protein-induced in vitro endothelial cell activation is an artefact caused by azide and lipopolysaccharide. Arterioscler Thromb Vasc Biol (2005) 25:1225–30. doi:10.1161/01.ATV.0000164623.41250.28
29. Ji SR, Wu Y, Zhu L, Potempa LA, Sheng FL, Lu W, et al. Cell membranes and liposomes dissociate C-reactive protein (CRP) to form a new, biologically active structural intermediate: mCRP(m). FASEB J (2007) 21:284–94. doi:10.1096/fj.06-6722com
30. Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105:128–37. doi:10.1161/CIRCRESAHA.108.190611
31. Mihlan M, Blom AM, Kupreishvili K, Lauer N, Stelzner K, Bergström F, et al. Monomeric C-reactive protein modulates classic complement activation on necrotic cells. FASEB J (2011) 25:4198–210. doi:10.1096/fj.11-186460
32. Lauer N, Mihlan M, Hartmann A, Schlötzer-Schrehardt U, Keilhauer C, Scholl HP, et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J Immunol (2011) 187:4374–83. doi:10.4049/jimmunol.1002488
33. Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation (2014) 130:35–50. doi:10.1161/CIRCULATIONAHA.113.007124
34. Braig D, Kaiser B, Thiele JR, Bannasch H, Peter K, Stark GB, et al. A conformational change of C-reactive protein in burn wounds unmasks its proinflammatory properties. Int Immunol (2014) 26:467–78. doi:10.1093/intimm/dxu056
35. Heuertz RM, Schneider GP, Potempa LA, Webster RO. Native and modified C-reactive protein bind different receptors on human neutrophils. Int J Biochem Cell Biol (2005) 37:320–35. doi:10.1016/j.biocel.2004.07.002
36. Khreiss T, Jozsef L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation (2004) 109:2016–22. doi:10.1161/01.CIR.0000125527.41598.68
37. Li HY, Wang J, Wu YX, Zhang L, Liu ZP, Filep JG, et al. Topological localization of monomeric C-reactive protein determines proinflammatory endothelial cell responses. J Biol Chem (2014) 289:14283–90. doi:10.1074/jbc.M114.555318
38. Turu MM, Slevin M, Matou S, West D, Rodríguez C, Luque A, et al. C-reactive protein exerts angiogenic effects on vascular endothelial cells and modulates associated signalling pathways and gene expression. BMC Cell Biol (2008) 9:47. doi:10.1186/1471-2121-9-47
39. Slevin M, Matou-Nasri S, Turu M, Luque A, Rovira N, Badimon L, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol (2010) 20:151–65. doi:10.1111/j.1750-3639.2008.00256.x
40. Slevin M, Matou S, Zeinolabediny Y, Corpas R, Weston R, Liu D, et al. Monomeric C-reactive protein – a key molecule driving development of Alzheimer’s disease associated with brain ischaemia? Sci Rep (2015) 5:13281. doi:10.1038/srep13281
41. Boras E, Slevin M, Alexander MY, Aljohi A, Gilmore W, Ashworth J, et al. Monomeric C-reactive protein and Notch-3 co-operatively increase angiogenesis through PI3K signalling pathway. Cytokine (2014) 69:165–79. doi:10.1016/j.cyto.2014.05.027
42. Khreiss T, József L, Hossain S, Chan JS, Potempa LA, Filep JG. Loss of pentameric symmetry of C-reactive protein is associated with delayed apoptosis of human neutrophils. J Biol Chem (2002) 277:40775–81. doi:10.1074/jbc.M205378200
43. Molins B, Pena E, de la Torre R, Badimon L. Monomeric C-reactive protein is prothrombotic and dissociates from circulating pentameric C-reactive protein on adhered activated platelets under flow. Cardiovasc Res (2011) 92:328–37. doi:10.1093/cvr/cvr226
44. de la Torre R, Pena E, Vilahur G, Slevin M, Badimon L. Monomerization of C-reactive protein requires glycoprotein IIb-IIIa activation: pentraxins and platelet deposition. J Thromb Haemost (2013) 11:2048–58. doi:10.1111/jth.12415
45. Habersberger J, Strang F, Scheichl A, Htun N, Bassler N, Merivirta RM, et al. Circulating microparticles generate and transport monomeric C-reactive protein in patients with myocardial infarction. Cardiovasc Res (2012) 96:64–72. doi:10.1093/cvr/cvs237
46. Wang MS, Messersmith RE, Reed SM. Membrane curvature recognition by C-reactive protein using lipoprotein mimics. Soft Matter (2012) 8:7909–18. doi:10.1039/C2SM25779C
47. Strang F, Scheichl A, Chen YC, Wang X, Htun NM, Bassler N, et al. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: a novel pathomechanism driving cortical inflammation in Alzheimer’s disease? Brain Pathol (2012) 22:337–46. doi:10.1111/j.1750-3639.2011.00539.x
48. Xu PC, Lin S, Yang XW, Gu DM, Yan TK, Wei L, et al. C-reactive protein enhances activation of coagulation system and inflammatory response through dissociating into monomeric form in antineutrophil cytoplasmic antibody-associated vasculitis. BMC Immunol (2015) 16:10. doi:10.1186/s12865-015-0077-0
49. Hammond DJ Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, Pangburn MK, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem (2010) 285:36235–44. doi:10.1074/jbc.M110.142026
50. Lacey DL, Boyle WJ, Simonet WS, Kostenuik PJ, Dougall WC, Sullivan JK, et al. Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov (2012) 11:401–19. doi:10.1038/nrd3705
51. Chiu YG, Ritchlin CT. Denosumab: targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin Biol Ther (2017) 17:119–28. doi:10.1080/14712598.2017.1263614
52. Li QY, Li HY, Fu G, Yu F, Wu Y, Zhao MH. Autoantibodies against C-reactive protein influence complement activation and clinical course in lupus nephritis. J Am Soc Nephrol (2017) 28:3044–54. doi:10.1681/ASN.2016070735
53. Ji SR, Wu Y, Potempa LA, Qiu Q, Zhao J. Interactions of C-reactive protein with low-density lipoproteins: implications for an active role of modified C-reactive protein in atherosclerosis. Int J Biochem Cell Biol (2006) 38:648–61. doi:10.1016/j.biocel.2005.11.004
54. Ji SR, Wu Y, Potempa LA, Liang YH, Zhao J. Effect of modified C-reactive protein on complement activation: a possible complement regulatory role of modified or monomeric C-reactive protein in atherosclerotic lesions. Arterioscler Thromb Vasc Biol (2006) 26:935–41. doi:10.1161/01.ATV.0000206211.21895.73
Keywords: inflammation, rheumatoid arthritis, osteoclast, receptor activator of NF-κB ligand, C-reactive protein
Citation: Jia Z-K, Li H-Y, Liang Y-L, Potempa LA, Ji S-R and Wu Y (2018) Monomeric C-Reactive Protein Binds and Neutralizes Receptor Activator of NF-κB Ligand-Induced Osteoclast Differentiation. Front. Immunol. 9:234. doi: 10.3389/fimmu.2018.00234
Received: 23 October 2017; Accepted: 26 January 2018;
Published: 19 February 2018
Edited by:
Mark Slevin, Manchester Metropolitan University, United KingdomReviewed by:
Jerzy Krupinski, Hospital Universitari Mutua de Terrassa, SpainJason Jonathan Ashworth, Manchester Metropolitan University, United Kingdom
Copyright: © 2018 Jia, Li, Liang, Potempa, Ji and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shang-Rong Ji, anNyQGx6dS5lZHUuY24=;
Yi Wu, d3V5QGx6dS5lZHUuY24=