 Jeffrey I. Cohen
Jeffrey I. Cohen- Medical Virology Section, Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
The phosphatidylinositol-3-kinase (PI3K)/Akt pathway is important for multiple stages of herpesvirus replication including virus entry, replication, latency, and reactivation. Recently, patients with gain-of-function mutations in the p110δ-catalytic subunit of PI3K or in the p85-regulatory subunit of PI3K have been reported. These patients have constitutively active PI3K with hyperactivation of Akt. They present with lymphoproliferation and often have infections, particularly recurrent respiratory infections and/or severe virus infections. The most frequent virus infections are due to Epstein–Barr virus (EBV) and cytomegalovirus (CMV); patients often present with persistent EBV and/or CMV viremia, EBV lymphoproliferative disease, or CMV lymphadenitis. No patients have been reported with CMV pneumonia, colitis, or retinitis. Other herpesvirus infections have included herpes simplex pneumonia, recurrent zoster, and varicella after vaccination with the varicella vaccine. Additional viral infections have included adenovirus viremia, severe warts, and extensive Molluscum contagiosum virus infection. The increased susceptibility to virus infections in these patients is likely due to a reduced number of long-lived memory CD8 T cells and an increased number of terminally differentiated effector CD8 T cells.
Introduction
Viruses often exploit intracellular signaling pathways to facilitate entry, replication, latency, and reactivation. Among the many pathways that viruses manipulate is the phosphatidylinositol-3-kinase (PI3K)/Akt pathway. This pathway has several important activities including inhibiting apoptosis, regulating the cell cycle, and enhancing protein synthesis, resulting in increased cell survival and control of cell growth (1). Activation of the pathway by the binding of viruses, growth factors, or cytokines to receptors on the plasma membrane results in the movement of the PI3K complex from the cytoplasm to the plasma membrane. Class I PI3K complexes are important for virus infection and consist of a regulatory subunit (p50, p55, or p85) and a catalytic subunit (p110α, β, γ, or δ). The interaction of phosphorylated tyrosines on receptors with the p85 subunit of PI3K reduces its inhibitory effect on the p110 subunit, resulting in the phosphorylation of phosphatidylinositol 4, 5-triphosphate (PIP2) and the activation of downstream signaling molecules including PDK1, Akt, and mTOR. Mutations in the p85-regulatory subunit or the p110δ-catalytic subunit have been associated with immunodeficiencies often presenting with lymphoproliferative disease, recurrent respiratory infections, and severe herpesvirus infections (see below).
Herpesviruses Modulate the PI3K Pathway
Eight herpesviruses infect humans: herpes simplex viruses (HSV) 1 and 2, varicella–zoster virus (VZV), cytomegalovirus (CMV), Epstein–Barr virus (EBV), human herpesviruses (HHV) 6 and 7, and Kaposi’s sarcoma-associated herpesvirus (KSHV). All herpesviruses infect and are shed from the epithelial cells, and all undergo latency and reactivation. HSV-1, HSV-2, and VZV establish latency in sensory neurons, CMV and HHV-6 in monocytes and CD34 cells, HHV-7 in CD4 cells, and EBV and KSHV in B cells. In healthy persons, HSV causes orolabial and genital herpes, VZV results in varicella and zoster, CMV, EBV, and HHV-6 cause mononucleosis, and HHV-6 and HHV-7 cause roseola. Each of the herpesviruses can result in a severe disease involving multiple organs in immunocompromised persons; CMV and EBV are frequently detected in the blood of immunocompromised persons. EBV is associated with lymphoproliferative disease in immunocompromised persons and B cell lymphoma, while KSHV is associated with Kaposi’s sarcoma and primary effusion lymphoma. While antibody contributes to protection from initial infection with herpesviruses, T cells are critical for reducing the severity of infection and disease associated with reactivation. Thus, mutations in genes important for the function of T cells can impair the control of herpesviruses by the host.
The PI3K pathway has a critical role for herpesvirus infection as well as for the control of herpesviruses by the immune system (2–4). Accordingly, herpesviruses manipulate this pathway to enhance virus entry, replication, latency, and reactivation. The binding of HSV (5), CMV (6), EBV (7), and KSHV (8) to the cell results in the activation of PI3K/Akt. Several viral glycoproteins including HSV gD and gB (9), CMV gB (10), EBV gp350 (7), and KSHV gB (11), each of which is required for initial infection of cells, activate the PI3K/Akt pathway. Viral proteins expressed during infection of cells by HSV (12), VZV (13), CMV (14), EBV (15), and KSHV (16) activate PI3K/Akt. These include the first proteins expressed in infected cells, the immediate-early proteins, including CMV IE1 and IE2 (14), EBV Rta (15) and KSHV Rta (17) which activate the PI3K/Akt pathway.
The PI3K/Akt pathway is also important for maintaining latency in HSV (18), EBV (19), and KSHV (20). Proteins and RNAs expressed during latency including HSV LAT (21) and EBV LMP1 (22), LMP2 (23), and EBNA2 (24) all activate the PI3K/Akt pathway. In addition, this pathway is critical for the reactivation of HSV (18), EBV (25), and KSHV (26) from latency.
Several herpesvirus proteins including HSV VP11 (27), VZV ORF12 (13), EBV LMP1 (22), and KSHV K1 (28) directly interact with the p85-regulatory subunit of PI3K to activate the PI3K/Akt pathway. Additional herpesvirus proteins interact with other proteins in the PI3K/Akt pathway.
Immunodeficiencies Associated with Mutations in PIK3CD or PIK3R1
Two laboratories (29, 30) independently reported a new immunodeficiency due to heterozygous gain-of-function mutations in the p110δ-catalytic subunit of PI3K, which is encoded by PIK3CD. Angulo et al. (29) reported a series of 17 patients with activated PI3K-δ syndrome (APDS) due to an E1012K mutation in PIK3CD. Lymphocytes from the patients had increased levels of activated Akt and phosphatidylinositol 3, 4, 5-triphosphate (PIP3), and increased activation-induced cell death. The patients had lymphopenia, elevated levels of IgM and transitional B cells, and reduced levels of antibodies to Streptococcus pneumoniae and Haemophilus influenzae type B with a reduced number of circulating B cells and class-switched memory B cells. They also had repeated respiratory infections with damage to the lungs; some had severe virus infections. Lucas et al. (30) reported nine patients with p110δ-activating mutations causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) with N334K, E525K, or E1012K mutations in PIK3CD. The patients’ lymphocytes had an increased phosphorylation of Akt and mTOR, an increased number of senescent effector T cells and transitional B cells, and a reduced number of naïve T cells, CD4 cells, and class-switched memory B cells. The patients had lymphoid hyperplasia often with obstructive lymphoid nodules and recurrent sinopulmonary infections; several patients had autoimmune cytopenias. Two had EBV lymphomas and all had EBV and/or CMV viremia.
Two laboratories (31, 32) reported a new immunodeficiency due to heterozygous gain-of-function mutations in the p85-regulatory subunit of PI3K, which is encoded by PIK3R1. These patients’ immune system and clinical phenotype were similar to those with PIK3CD mutations. Deau et al. (31) reported four patients with two different heterozygous splice mutations in PIK3R1, whose lymphocytes showed low numbers of memory B cells and naïve T cells, and increased levels of activated Akt, IgM, transitional B cells, senescent CD8 cells, and activation-induced cell death. One of the patients had CMV and EBV viremia and enterovirus enteritis. Lucas et al. (32) described four patients with heterozygous splice-site mutations in PIK3R1. Lymphocytes from the patients had an increased phosphorylation of Akt and an increased number of senescent effector T cells and CD8 cells; they had low numbers of CD4 cells and low levels of IgG. The patients had lymphoproliferative disease and frequent sinopulmonary infections.
With the recognition of similar phenotypes in patients with gain-of-function mutations in PIK3CD and PIK3R1, APDS and PASLI have now been divided into APDS1 and PASLI-CD when reporting patients with mutations in PIK3CD, and APDS2 and PASLI-R1 when reporting patients with mutations in PIK3R1. A different type of mutation was reported in PIK3R1. Conley et al. (33) reported a patient with a homozygous stop codon in the p85-regulatory subunit of PI3K. The patient had no B cells, normal numbers and activity of T cells, and no history of severe virus infection.
Herpesvirus Infections in Patients with Gain-of-Function PIK3CD Mutations
The most frequent viral infectious complications associated with PIK3CD mutations have been EBV and CMV viremia and EBV lymphoproliferative disease or CMV lymphadenitis. In the first report of gain-of-function PIK3CD mutations (29), patients were screened for frequent respiratory infections and family histories of increased susceptibility to infection; accordingly, all 17 had recurrent upper or lower respiratory tract infections. Four of the 17 patients had infections caused by EBV, CMV, VZV, or HSV, including one patient with HSV pneumonia. In the next report of PIK3CD gain-of-function mutations (30), patients were screened based on the persistence of CMV and EBV in the peripheral blood; all nine patients had EBV viremia, with peak viral loads ranging from <250 to 63,350 copies/µl in the blood (30). Two patients had EBV-positive B cell lymphomas; one had an EBV-diffuse B cell lymphoma and one had EBV-nodular sclerosis Hodgkin disease. In one patient without lymphoma, two other family members had EBV lymphoma. Interestingly, while patients had high EBV DNA levels in the blood and some developed EBV lymphomas, in the patients who had EBV-specific CD8 T cells quantified by staining with HLA tetramers specific for EBV lytic and latency proteins, normal or elevated numbers of EBV-specific CD8 T cells were noted. Two patients had CMV viremia and three had CMV lymphadenitis. While it is possible that chronic infection with CMV or EBV could have resulted in the observed senescence of CD8 T cells, there was no correlation with the EBV and CMV load in the blood and the number of senescent CD8 T cells.
Since these two reports were published, additional papers have reported persistent herpesvirus viremia or severe herpesvirus infections in patients with PIK3CD gain-of-function mutations (Table 1). Mutations at seven sites in PIK3CD—E81K, G124D, N334K, C416R, E525K, E1021K, and E1025G—have been reported in persons with herpesvirus viremia or severe virus infections. In the largest review to date, a total of 53 patients with PIK3CD gain-of-function mutations were reported, and 49% had persistent or recurrent herpesvirus infections (40). EBV viremia was detected in 26% (14/53) of the patients, and 6% were reported to have a disseminated infection. EBV was detected in multiple biopsies including lymph nodes, tonsils, and the gastrointestinal tract as well as in the cerebrospinal fluid. Seven patients had diffuse lymphadenopathy; EBV and/or CMV was detected by PCR in the blood of six of these patients. One case of EBV encephalitis was reported. Two patients had EBV lymphomas, one had Hodgkin lymphoma and one had diffuse large B cell lymphoma; both patients died. CMV viremia was reported in 15% (8/53) of patients, 4 of whom responded to ganciclovir. EBV and CMV coinfection was reported in four patients, one of whom had a lymph node biopsy that was positive for EBV, CMV, and HHV-6 by PCR. Severe or persistent HSV or VZV infections were detected in 21% (11/53) of patients. One patient had HSV pneumonitis and one had recurrent HSV keratitis. Varicella infections resulted in hospitalization of two patients, and two had recurrent zoster.
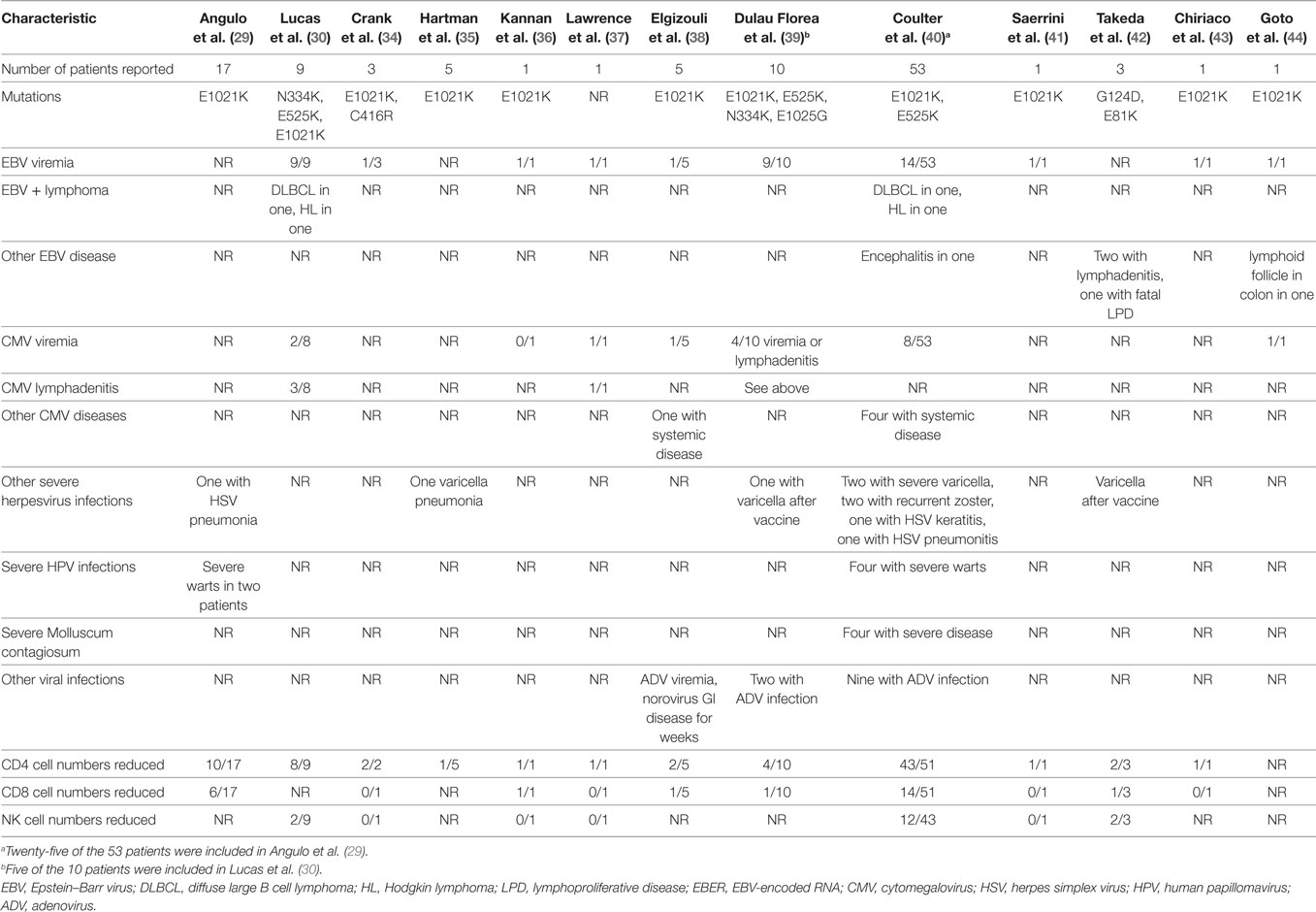
Table 1. Viral infections in patients with germline gain-of-function mutations in PIK3CD.
Two other patients have been reported with PIK3CD gain-of-function mutations and EBV lymphomas (30). One patient had fatal EBV lymphoproliferative disease (42), two had EBV lymphadenitis (42), and one had EBV encephalitis (40). Two developed varicella after receiving the varicella vaccine (39, 42) and one had varicella pneumonia (35). Interestingly, despite frequent reports of CMV viremia and lymphadenitis, severe complications of CMV including pneumonia, colitis, or retinitis have not been reported. The treatment of patients with CMV lymphadenitis is often unsatisfactory; while the disease responds to antiviral therapy, it often recurs when treatment is stopped.
Herpesvirus Infections in Patients with Gain-of-Function PIK3R1 Mutations
In the first report of patients with gain-of-function PIK3R1 mutations, Deau et al. (31) described four patients with recurrent respiratory bacterial infections, two of whom had EBV viremia and one of whom had both CMV and EBV viremia (9,300 and 1,500 copies/ml, respectively). In the next report of PIK3R1 gain-of-function mutations, Lucas et al. (32) reported four patients, one of whom had CMV lymphadenitis. Since these two papers were published, additional papers have reported persistent EBV or CMV viremia or severe herpesvirus infections in patients with gain-of-function mutations in PIK3R1 (Table 2). All patients with severe virus infections have had splice donor-site mutations resulting in loss of exon 11. In the largest series to date, Elkaim et al. (45) reported 36 patients with mutations in PIK3R1. EBV viremia was detected in 22% (8/36) of patients, 4 of whom were asymptomatic and 4 of whom had EBV lymphoproliferative disease. One of these patients had two episodes of EBV Hodgkin lymphoma. Asymptomatic CMV viremia was present in 17% (6/36) of patients, and two had CMV lymphadenitis. Two patients were hospitalized for severe VZV infections. In another report (47), a 15-year-old boy had CMV viremia and CMV lymphadenitis that was refractory to therapy. A lymph node biopsy showed 240,000 copies of CMV/mg of tissue and follicular hyperplasia. While he initially responded to ganciclovir and valganciclovir, his lymphadenopathy recurred associated with the obstruction of the upper airway. A repeat lymph node biopsy was CMV-positive, and he received additional valganciclovir and corticosteroids and had a good response, though he had intermittent low-grade CMV viremia. He later presented with recurrent massive lymphadenopathy, and a repeat lymph node biopsy was CMV-positive, and he was treated again with ganciclovir and valganciclovir. He relapsed once valganciclovir was stopped and when he became refractory to antivirals, hematopoietic stem cell transplantation was performed, and he responded well. Like patients with mutations in PIK3CD, no cases of severe CMV involving the lungs, colon, liver, or retina have been reported in patients with mutations in PIK3R1.
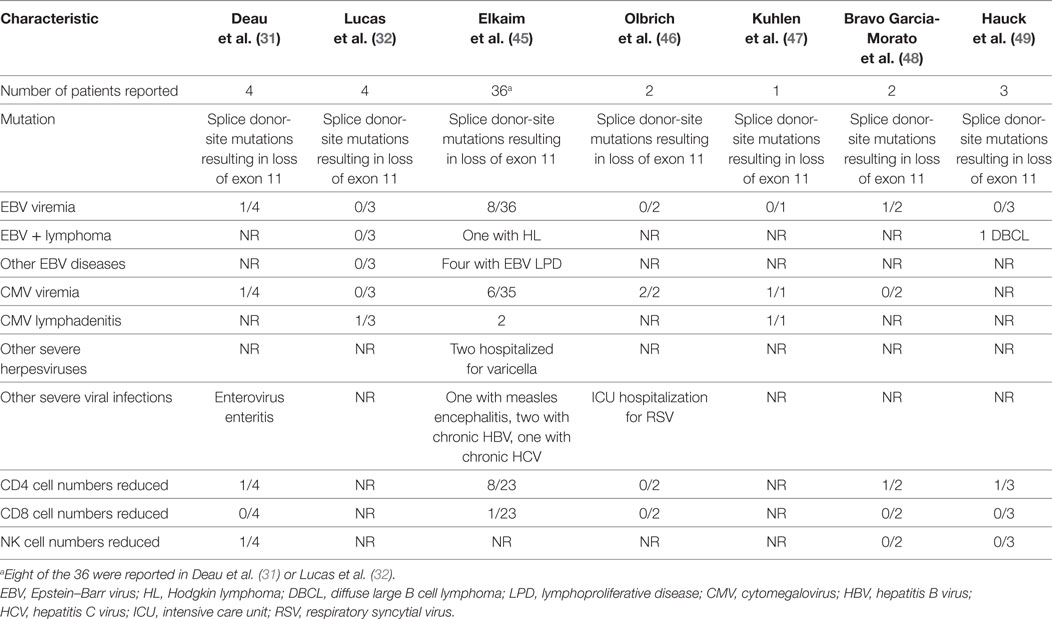
Table 2. Viral infections in patients with germline gain-of-function mutations in PIK3R1.
While most case reports of patients with EBV or CMV viremia did not indicate the level of viral DNA, in 13 patients with EBV viremia and 5 with CMV viremia and mutations in either PIK3CD or PI3KR1, the levels were quantified and expressed as copies of viral DNA/ml. In these cases, the mean and median EBV loads were 9,146 and 2,250 copies/ml, respectively, and the mean and median CMV loads were 2,749 and 1,211 copies/ml, respectively. Thus, the levels of EBV and CMV in the blood generally were not markedly elevated. EBV and CMV viremia were not reported as initiating with symptomatic primary infection; instead, they were associated with virus reactivation.
Other Virus Infections in Patients with Gain-of-Function PIK3CD Mutations
In the first report of gain-of-function PIK3CD mutations, two patients were described with severe warts (29); subsequently, two additional patients with severe warts have been reported (40). Four patients have been reported with severe Molluscum contagiosum infections (40), one with adenovirus viremia (38), and 11 others reported with adenovirus infections with virus isolated from the blood, bronchoalveolar lavage fluid, and/or stool (39, 40). One patient was reported with norovirus infection that lasted for several weeks and was associated with persistent diarrhea (38). CD4 T cell numbers were reduced in 72% of patients, and CD8 T cell and NK cell numbers were reduced in 27% of patients with severe virus infections and PIK3CD mutations (Table 1).
Other Virus Infections in Patients with Gain-of-Function PIK3R1 Mutations
In the first report of gain-of-function PIK3R1 mutations, one patient was reported with enterovirus gastroenteritis (31). In the largest report of PIK3R1 mutations to date, Elkaim et al. (45) reported one patient with measles encephalitis and hydrocephalus and other patients with chronic hepatitis B and hepatitis C virus infections. In another report, one patient was hospitalized in the intensive care unit for bronchiolitis due to respiratory syncytial virus infection (46). CD4 T cell numbers were reduced in 32% of patients and NK cell numbers were reduced in 11% of patients with severe virus infections and PIK3R1 mutations (Table 2).
Mechanism for Impaired Control of Herpesvirus Infections in Patients with PI3K Mutations
Lucas et al. (30) found that patients with PIK3CD gain-of-function mutations had normal or high levels of EBV-specific CD8 T cells in the blood by tetramer staining, but that EBV-specific CD8 T cells were predominantly CCR7−CD45RA− indicative of terminal effector memory cells and had more CD38 than control cells indicative of an increased activity. They postulated that the persistent hyperactivation of Akt results in an increase in the proliferation of CD8 T cells and an increase in the number of terminal differentiated effector CD8 T cells with a corresponding increase in senescent CD8 T cells and a decrease in long-lived memory CD8 T cells. Together, this may result in impaired control of EBV- and CMV-infected cells. The increased proliferation of EBV-infected B cells could also increase the risk for additional chromosomal mutations and result in an increased risk of EBV lymphomas. Interestingly, while older persons have a similar number of T cells than younger persons, they have a higher frequency of senescent T cells (50, 51). Older persons also have higher levels of EBV and CMV in the blood than younger persons (52, 53), and older persons may develop EBV lymphoproliferative disorders in the absence of an immune deficiency disease (54). Thus, an increased number of senescent T cells has been associated with impaired control of EBV and CMV infections. Coulter et al. (40) also noted that a reduced number of CD4, CD8, or NK cells was not associated with herpesvirus infections in patients with PIK3CD mutations, indicating that a functional rather than a quantitative abnormality in lymphocytes was responsible for the infections.
Cytomegalovirus and EBV persist predominantly in the blood, lymph nodes, and spleen, and the disease is often associated with lymphadenopathy, lymphadenitis, or lymphomas involving the lymphoid tissues. By contrast, HSV and VZV are latent in the nervous system and most often result in disease involving the skin. CD8-naïve and central or effector memory T cells are the predominant T cell types in the blood, spleen, and lymph nodes, while tissue-resident memory T cells and terminally differentiated effector CD8 T cells are the predominant CD8 T cell subsets in the peripheral tissues including the skin (55, 56). In addition, the persistence of EBV and CMV in the blood allows for clonal expansions of T cells, with the persistence of memory T cells after initial infection. Thus, the increase in terminal differentiated effector CD8 T cells and the reduction in memory CD8 T cells in patients with mutations in PIK3CD or PIK3R1 may allow EBV and CMV to proliferate in the blood and lymphoid organs while having less of an effect on HSV and VZV in the skin. Furthermore, the reduction in memory CD8 T cells in the blood and lymphoid tissues may allow EBV and CMV to proliferate to higher levels resulting in viremia, lymphadenitis, and EBV lymphoma. In addition, the increased numbers of terminal differentiated effector CD8 T cells in patients with PIK3CD or PIK3R1, which are generally more often present in the peripheral tissues than in the blood, may have a protective effect from CMV involvement of the lungs, colon, and liver in these patients.
Screening and Treatment
Since EBV viremia was detected in 46 and 22% of patients with mutations in PIK3CD and PIK3R1, respectively, and CMV viremia was detected in 20 and 21% of patients with mutations in PIK3CD and PIK3R1, respectively, testing for mutations in these two genes should be considered in patients with unexplained EBV or CMV viremia. The frequency of other virus infections in these patients is much lower, and therefore screening for mutations in PIK3CD or PIK3R1 would be less likely to be useful.
The treatment of patients with PIK3CD mutations with a PI3K inhibitor, leniolisib (57), or with an mTOR inhibitor, rapamycin (30, 40) reduced lymphoproliferation and the number of senescent T cells and increased the number of naïve T cells. Despite its immunosuppressive effects, patients with PIK3CD mutations treated with leniolisib did not have an increase in EBV or CMV viremia while on therapy (57). Similarly, complications associated with EBV or CMV have not been reported in patients treated with rapamycin (30, 40). Rapamycin has been shown to inhibit the development of EBV-positive B cell lymphomas in a mouse model (58) and has been associated with the resolution of EBV-positive lymphoproliferative disease in patients (59). Similarly, rapamycin modestly reduces CMV replication in vitro (60), and the use of rapamycin instead of other immunosuppressant drugs has resulted in the reduction in CMV infection and disease (61). Thus, despite its immunosuppressive activities, rapamycin or PI3K inhibitors do not appear to increase the risk of EBV or CMV disease in patients with PIK3CD mutations.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases.
References
1. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell (2017) 170:605–35. doi:10.1016/j.cell.2017.07.029
2. Liu X, Cohen JI. The role of PI3K/Akt in human herpesvirus infection: from the bench to the bedside. Virology (2015) 479-480:568–77. doi:10.1016/j.virol.2015.02.040
3. Diehl N, Schaal H. Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses (2013) 5:3192–212. doi:10.3390/v5123192
4. Dunn EF, Connor JH. HijAkt: the PI3K/Akt pathway in virus replication and pathogenesis. Prog Mol Biol Transl Sci (2012) 106:223–50. doi:10.1016/B978-0-12-396456-4.00002-X
5. MacLeod IJ, Minson T. Binding of herpes simplex virus type-1 virions leads to the induction of intracellular signalling in the absence of virus entry. PLoS One (2010) 5:e9560. doi:10.1371/journal.pone.0009560
6. Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol (2001) 75:6022–32. doi:10.1128/JVI.75.13.6022-6032.2001
7. Barel M, Balbo M, Le Romancer M, Frade R. Activation of Epstein–Barr virus/C3d receptor (gp140, CR2, CD21) on human cell surface triggers pp60src and Akt–GSK3 activities upstream and downstream to PI 3-kinase, respectively. Eur J Immunol (2003) 33:2557–66. doi:10.1002/eji.200324059
8. Naranatt PP, Akula SM, Zien CA, Krishnan HH, Chandran B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase–PKC-zeta–MEK–ERK signaling pathway in target cells early during infection: implications for infectivity. J Virol (2003) 77:1524–39. doi:10.1128/JVI.77.2.1524-1539.2003
9. Cheshenko N, Trepanier JB, Stefanidou M, Buckley N, Gonzalez P, Jacobs W, et al. HSV activates Akt to trigger calcium release and promote viral entry: novel candidate target for treatment and suppression. FASEB J (2013) 27:2584–99. doi:10.1096/fj.12-220285
10. Cobbs C, Khan S, Matlaf L, McAllister S, Zider A, Yount G, et al. HCMV glycoprotein B is expressed in primary glioblastomas and enhances growth and invasiveness via PDGFR-alpha activation. Oncotarget (2014) 5:1091–100. doi:10.18632/oncotarget.1787
11. Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase–Src–phosphatidylinositol 3-kinase–rho GTPase signal pathways and cytoskeletal rearrangements. J Virol (2004) 78:4207–23. doi:10.1128/JVI.78.8.4207-4223.2004
12. Laing JM, Smith CC, Aurelian L. Multi-targeted neuroprotection by the HSV-2 gene ICP10PK includes robust bystander activity through PI3-K/Akt and/or MEK/ERK-dependent neuronal release of vascular endothelial growth factor and fractalkine. J Neurochem (2010) 112:662–76. doi:10.1111/j.1471-4159.2009.06475.x
13. Liu X, Cohen JI. Varicella–zoster virus ORF12 protein activates the phosphatidylinositol 3-kinase/Akt pathway to regulate cell cycle progression. J Virol (2013) 87:1842–8. doi:10.1128/JVI.02395-12
14. Yu Y, Alwine JC. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3'-OH kinase pathway and the cellular kinase Akt. J Virol (2002) 76:3731–8. doi:10.1128/JVI.76.8.3731-3738.2002
15. Darr CD, Mauser A, Kenney S. Epstein–Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J Virol (2001) 75:6135–42. doi:10.1128/JVI.75.13.6135-6142.2001
16. Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res (2001) 61:2641–8.
17. Li X, Chen S, Sun R. Cdk1 inhibition induces mutually inhibitory apoptosis and reactivation of Kaposi’s sarcoma-associated herpesvirus. J Virol (2012) 86:6668–76. doi:10.1128/JVI.06240-11
18. Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, et al. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe (2010) 8:320–30. doi:10.1016/j.chom.2010.09.007
19. Lam N, Sugden B. CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal (2003) 15:9–16. doi:10.1016/S0898-6568(02)00083-9
20. Sharma-Walia N, Patel K, Chandran K, Marginean A, Bottero V, Kerur N, et al. COX-2/PGE2: molecular ambassadors of Kaposi’s sarcoma-associated herpes virus oncoprotein-v-FLIP. Oncogenesis (2012) 1:e5. doi:10.1038/oncsis.2012.5
21. Li S, Carpenter D, Hsiang C, Wechsler SL, Jones C. Herpes simplex virus type 1 latency-associated transcript inhibits apoptosis and promotes neurite sprouting in neuroblastoma cells following serum starvation by maintaining protein kinase B (Akt) levels. J Gen Virol (2010) 91:858–66. doi:10.1099/vir.0.015719-0
22. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein–Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem (2003) 278:3694–704. doi:10.1074/jbc.M209840200
23. Fukuda M, Longnecker R. Epstein–Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt pathway. J Virol (2007) 81:9299–306. doi:10.1128/JVI.00537-07
24. Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, et al. Cell target genes of Epstein–Barr virus transcription factor EBNA-2: induction of the p55alpha regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J Gen Virol (2006) 87:2859–67. doi:10.1099/vir.0.82128-0
25. Goswami R, Gershburg S, Satorius A, Gershburg E. Protein kinase inhibitors that inhibit induction of lytic program and replication of Epstein–Barr virus. Antiviral Res (2012) 96:296–304. doi:10.1016/j.antiviral.2012.09.021
26. Peng L, Wu TT, Tchieu JH, Feng J, Brown HJ, Feng J, et al. Inhibition of the phosphatidylinositol 3-kinase–Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J Gen Virol (2010) 91:463–9. doi:10.1099/vir.0.015073-0
27. Wagner MJ, Smiley JR. Herpes simplex virus requires VP11/12 to activate Src family kinase phosphoinositide 3-kinase–Akt signaling. J Virol (2011) 85:2803–12. doi:10.1128/JVI.01877-10
28. Lee BS, Lee SH, Feng P, Chang H, Cho NH, Jung JU. Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J Virol (2005) 79:12173–84. doi:10.1128/JVI.79.19.12173-12184.2005
29. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi:10.1126/science.1243292
30. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15:88–97. doi:10.1038/ni.2771
31. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest (2014) 124:3923–8. doi:10.1172/JCI75746
32. Lucas CL, Zhang Y, Venida A, Wang Y, Hughes J, McElwee J, et al. Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. J Exp Med (2014) 211:2537–47. doi:10.1084/jem.20141759
33. Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med (2012) 209:463–70. doi:10.1084/jem.20112533
34. Crank MC, Grossman JK, Moir S, Pittaluga S, Buckner CM, Kardava L, et al. Mutations in PIK3CD can cause hyper IgM syndrome (HIGM) associated with increased cancer susceptibility. J Clin Immunol (2014) 34:272–6. doi:10.1007/s10875-014-0012-9
35. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of function mutations of PIK3CD as a cause of primary sclerosing cholangitis. J Clin Immunol (2015) 35:11–4. doi:10.1007/s10875-014-0109-1
36. Kannan JA, Dávila-Saldaña BJ, Zhang K, Filipovich AH, Kucuk ZY. Activated phosphoinositide 3-kinase δ syndrome in a patient with a former diagnosis of common variable immune deficiency, bronchiectasis, and lymphoproliferative disease. Ann Allergy Asthma Immunol (2015) 115:452–4. doi:10.1016/j.anai.2015.08.009
37. Lawrence MG, Uzel G. 6-year-old boy with recurrent sinopulmonary infections and lymphadenopathy. J Allergy Clin Immunol Pract (2015) 3:461.e–3.e. doi:10.1016/j.jaip.2014.10.017
38. Elgizouli M, Lowe DM, Speckmann C, Schubert D, Hülsdünker J, Eskandarian Z, et al. Activating PI3Kδ mutations in a cohort of 669 patients with primary immunodeficiency. Clin Exp Immunol (2016) 183:221–9. doi:10.1111/cei.12706
39. Dulau Florea AE, Braylan RC, Schafernak KT, Williams KW, Duab J, Goval RK, et al. Abnormal B-cell maturation in the bone marrow of patients with germline mutations in PIK3CD. J Allergy Clin Immunol (2016) 139:1032–5. doi:10.1016/j.jaci.2016.08.028
40. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol (2017) 139:597–606. doi:10.1016/j.jaci.2016.06.021
41. Saettini F, Pelagatti MA, Sala D, Moratto D, Giliani S, Badolato R, et al. Early diagnosis of PI3Kδ syndrome in a 2 years old girl with recurrent otitis and enlarged spleen. Immunol Lett (2017) 190:279–81. doi:10.1016/j.imlet.2017.08.021
42. Takeda AJ, Zhang Y, Dornan GL, Siempelkamp BD, Jenkins ML, Matthews HF, et al. Novel PIK3CD mutations affecting N-terminal residues of p110δ cause activated PI3Kδ syndrome (APDS) in humans. J Allergy Clin Immunol (2017) 140:1152.e10. doi:10.1016/j.jaci.2017.03.026
43. Chiriaco M, Brigida I, Ariganello P, Di Cesare S, Di Matteo G, Taus F, et al. The case of an APDS patient: defects in maturation and function and decreased in vitro anti-mycobacterial activity in the myeloid compartment. Clin Immunol (2017) 178:20–8. doi:10.1016/j.clim.2015.12.008
44. Goto F, Uchiyama T, Nakazawa Y, Imai K, Kawai T, Onodera M. Persistent impairment of T-cell regeneration in a patient with activated PI3K δ syndrome. J Clin Immunol (2017) 37:347–50. doi:10.1007/s10875-017-0393-7
45. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol (2016) 138:210.e–8.e. doi:10.1016/j.jaci.2016.03.022
46. Olbrich P, Lorenz M, Cura Daball P, Lucena JM, Rensing-Ehl A, Sanchez B, et al. Activated PI3Kδ syndrome type 2: two patients, a novel mutation, and review of the literature. Pediatr Allergy Immunol (2016) 27:640–4. doi:10.1111/pai.12585
47. Kuhlen M, Hönscheid A, Loizou L, Nabhani S, Fischer U, Stepensky P, et al. De novo PIK3R1 gain-of-function with recurrent sinopulmonary infections, long-lasting chronic CMV-lymphadenitis and microcephaly. Clin Immunol (2016) 162:27–30. doi:10.1016/j.clim.2015.10.008
48. Bravo García-Morato M, García-Miñaúr S, Molina Garicano J, Santos Simarro F, Del Pino Molina L, López-Granados E, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol (2017) 179:77–80. doi:10.1016/j.clim.2017.03.004
49. Hauck F, Magg T, Krolo A, Bilic I, Hirschmugl T, Laass M, et al. Variant PIK3R1 hypermorphic mutation and clinical phenotypes in a family with short statures, mild immunodeficiency and lymphoma. Klin Padiatr (2017) 229:113–7. doi:10.1055/s-0043-104218
50. Johnstone J, Millar J, Lelic A, Verschoor CP, Walter SD, Devereaux PJ, et al. Immunosenescence in the nursing home elderly. BMC Geriatr (2014) 17:50. doi:10.1186/1471-2318-14-50
51. Wikby A, Johansson B, Olsson J, Löfgren S, Nilsson BO, Ferguson F. Expansions of peripheral blood CD8 T-lymphocyte subpopulations and an association with cytomegalovirus seropositivity in the elderly: the Swedish NONA immune study. Exp Gerontol (2002) 37:445–53. doi:10.1016/S0531-5565(01)00212-1
52. Thomasini RL, Pereira DS, Pereira FSM, Mateo EC, Mota TN, Guimarães GG, et al. Aged-associated cytomegalovirus and Epstein–Barr virus reactivation and cytomegalovirus relationship with the frailty syndrome in older women. PLoS One (2017) 12:e0180841. doi:10.1371/journal.pone.0180841
53. Parry HM, Zuo J, Frumento G, Mirajkar N, Inman C, Edwards E, et al. Cytomegalovirus viral load within blood increases markedly in healthy people over the age of 70 years. Immun Ageing (2016) 13:1. doi:10.1186/s12979-015-0056-6
54. Asano N, Yamamoto K, Tamaru J, Oyama T, Ishida F, Ohshima K, et al. Age-related Epstein–Barr virus (EBV)-associated B-cell lymphoproliferative disorders: comparison with EBV-positive classic Hodgkin lymphoma in elderly patients. Blood (2009) 113:2629–36. doi:10.1182/blood-2008-06-164806
55. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38:187–97. doi:10.1016/j.immuni.2012.09.020
56. Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol (2014) 14:24–35. doi:10.1038/nri3567
57. Rao VK, Webster S, Dalm VASH, Šedivá A, van Hagen PM, Holland S, et al. Effective ‘activated PI3Kδ syndrome’-targeted therapy with the PI3Kδ inhibitor leniolisib. Blood (2017) 130:2307–16. doi:10.1182/blood-2017-08-801191
58. Nepomuceno RR, Balatoni CE, Natkunam Y, Snow AL, Krams SM, Martinez OM. Rapamycin inhibits the interleukin 10 signal transduction pathway and the growth of Epstein–Barr virus B-cell lymphomas. Cancer Res (2003) 63:4472–80.
59. Boratynska M, Smolska D. Inhibition of mTOR by sirolimus induces remission of post-transplant lymphoproliferative disorders. Transpl Int (2008) 21:605–8. doi:10.1111/j.1432-2277.2008.00655.x
60. Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol (2004) 78:11030–9. doi:10.1128/JVI.78.20.11030-11039.2004
61. Nashan B, Gaston R, Emery V, Säemann MD, Mueller NJ, Couzi L, et al. Review of cytomegalovirus infection findings with mammalian target of rapamycin inhibitor-based immunosuppressive therapy in de novo renal transplant recipients. Transplantation (2012) 93:1075–85. doi:10.1097/TP.0b013e31824810e6
Keywords: phosphatidylinositol-3-kinase, Akt, PIK3CD, PIK3R1, APDS, PASLI, Epstein–Barr virus, cytomegalovirus
Citation: Cohen JI (2018) Herpesviruses in the Activated Phosphatidylinositol-3-Kinase-δ Syndrome. Front. Immunol. 9:237. doi: 10.3389/fimmu.2018.00237
Received: 24 November 2017; Accepted: 26 January 2018;
Published: 23 February 2018
Edited by:
Carrie L. Lucas, Yale University, United StatesReviewed by:
Silvia Clara Giliani, University of Brescia, ItalyFlore Rozenberg, Université Paris Descartes, France
Copyright: © 2018 This work is authored by Jeffrey I. Cohen on behalf of the U.S. Government and, as regards Dr. Cohen and the US government, is not subject to copyright protection in the United States. Foreign and other copyrights may apply. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey I. Cohen, amNvaGVuQG5pYWlkLm5paC5nb3Y=