 Yang Zhao1†
Yang Zhao1† Xiao-Fei Shen2†Ke Cao3†Jie Ding2Xing Kang2Wen-xian Guan2*Yi-tao Ding2*Bao-rui Liu1*
Xiao-Fei Shen2†Ke Cao3†Jie Ding2Xing Kang2Wen-xian Guan2*Yi-tao Ding2*Bao-rui Liu1* Jun-Feng Du4*
Jun-Feng Du4*
- 1The Comprehensive Cancer Center of Drum Tower Hospital, Medical School of Nanjing University, Clinical Cancer Institute of Nanjing University, Nanjing, China
- 2Department of General Surgery, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, China
- 3Department of Critical Care Medicine, Nanjing Drum Tower Hospital, The Affiliated Hospital of Nanjing University Medical School, Nanjing, China
- 4Department of General Surgery, PLA Army General Hospital, Beijing, China
How to induce immune tolerance without long-term need for immunosuppressive drugs has always been a central problem in solid organ transplantation. Modulating immunoregulatory cells represents a potential target to resolve this problem. Myeloid-derived suppressor cells (MDSCs) are novel key immunoregulatory cells in the context of tumor development or transplantation, and can be generated in vitro. However, none of current systems for in vitro differentiation of MDSCs have successfully achieved long-term immune tolerance. Herein, we combined dexamethasone (Dex), which is a classic immune regulatory drug in the clinic, with common MDSCs inducing cytokine granulocyte macrophage colony stimulating factor (GM-CSF) to generate MDSCs in vitro. Addition of Dex into GM-CSF system specifically increased the number of CD11b+ Gr-1int/low MDSCs with an enhanced immunosuppressive function in vitro. Adoptive transfer of these MDSCs significantly prolonged heart allograft survival and also favored the expansion of regulatory T cells in vivo. Mechanistic studies showed that inducible nitric oxide sythase (iNOS) signaling was required for MDSCs in the control of T-cell response and glucocorticoid receptor (GR) signaling played a critical role in the recruitment of transferred MDSCs into allograft through upregulating CXCR2 expression on MDSCs. Blockade of GR signaling with its specific inhibitor or genetic deletion of iNOS reversed the protective effect of Dex-induced MDSCs on allograft rejection. Together, our results indicated that co-application of Dex and GM-CSF may be a new and important strategy for the induction of potent MDSCs to achieve immune tolerance in organ transplantation.
Introduction
The ultimate goal for organ transplantation is to achieve long-term acceptance without the need for immunosuppressive drugs (1). Cellular immunotherapy is a promising alternative for preventing allograft rejection and adoptive transfer of immune cells has been tried to establish immune tolerance in animal models (2–4). Our previous results showed that adoptive transfer of adaptive immune cells, regulatory T cells (Treg), can help to establish mixed bone marrow chimeras and lead to the prolongation of intestinal allograft survival (5). However, the possibility of adoptive transfer of other immune cells such as innate immune cells to achieve immune tolerance has not been fully elucidated.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of innate immune cells generated from immature myeloid cells in the bone marrow and were first characterized in tumor-bearing mice or in patients with cancer (6). With the increased understanding of MDSC development, it is now clear that pathological situations such as inflammation and tumor development can lead to a partial block in the differentiation of immature myeloid cells into mature myeloid cells, resulting in the expansion of MDSCs (6). In mice, the phenotype of MDSCs was CD11b (also known as integrin-αM) and granulocyte differentiation antigen-1 (Gr-1), and in humans, their phenotype is Lin−HLA−DR−CD33+ or CD11b+CD14−CD33+ (6). Once generated in a pathological context, MDSCs were able to control T-cell immune response through the upregulated expression of immune suppressive factors such as arginase, inducible nitric oxide synthase (iNOS), nitric oxide, and reactive oxygen species. Thus, despite early evidence of MDSCs in the control of tumor development, accumulating results have also suggested a critical role of MDSCs in the amelioration of allogenic transplantation based on their immune suppressive function (4, 7). Manipulating both the development and function of MDSCs has been suggested to be a potential therapeutic target to achieve immune tolerance (8). However, although numerous studies have identified multiple signaling pathways to contribute to the induction and function of MDSCs in vivo and in vitro, none of them have been applied to clinical trials possibly due to the instability of MDSCs in vivo.
Myeloid-derived suppressor cells can be induced from bone marrow progenitors in vitro, and the most common way to induce MDSCs in vitro is the use of granulocyte macrophage colony stimulating factor (GM-CSF) at high concentrations for 4 days (9). In addition, in vitro-generated MDSCs by GM-CSF have also been shown to prolong the survival of pancreatic islet allograft upon adoptive transfer (7), which provides promising prospects of cellular immunotherapy based on in vitro-generated MDSCs in the context of transplantation. Dexamethasone (Dex) belongs to the family of glucocorticoid and has been widely used in treating inflammatory disorders (10). Dex can not only inhibit the production of inflammatory cytokines in vivo but also modulate the phenotype and/or function of both classic innate and adaptive immune cells such as dendritic cells (11), macrophages (12), and Treg (13). However, the role of Dex in the modulation of MDSCs has not been fully addressed. In the present study, we sought to determine whether the use of Dex together with GM-CSF can promote the differentiation of MDSCs in vitro with an enhanced immunosuppressive ability, and also explore whether adoptive transfer of these in vitro-induced MDSC can help to achieve immune tolerance in vivo.
Materials and Methods
Mice
BALB/c, C57BL/6 (B6), and CD45.1+ B6 mice were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). All the mice were bred and maintained in the condition of specific pathogen-free. Male 8- to 10-week-old littermate mice were used for experiments. iNOS knockout (KO) mice (B6 background, CD45.2) were purchased from Jackson Lab. All the animal experiments were carried out following the Guidelines for the Care and Use of Laboratory Animals of Nanjing University and were approved by the ethical review committee of Nanjing University.
Cell Culture
Bone marrow cells were acquired from tibias and femurs of B6 mice (CD45.1 or CD45.2) as described previously (14). Tibias and femurs were removed using sterile techniques, and bone marrow was flushed with PBS. Red blood cells were lysed with ammonium chloride. Total bone marrow cells were planted into 100-mm dishes (Corning, USA) for 2 h. The non-adherent cells were collected and were further cultured in the presence of 50 ng/ml GM-CSF, with or without Dex at different concentration as indicated in 60-mm-diameter dish (Corning, USA) in 4 ml of complete RPMI 1640 medium (containing 10% heat-inactivated FBS, 2mM L-glutamine, 10 mM HEPES, 20 µM 2-ME, 150 U/ml streptomycin and 200 U/ml penicillin) at 37°C, 5% CO2 for 4 days.
T Cell Proliferation Assay
Splenic cells were labeled with 2 µM CFSE for 5 min in PBS at 37°C and were then washed with RPMI 1640. The labeled cells were further cultured with ConA (2 µg/ml) (15) in the presence of different doses of MDSCs as indicated for 72 h. Cell proliferation was assessed by flow cytometry after stained with anti-CD4 or anti-CD8 mAb.
Flow Cytometry Antibodies and Reagents
Anti-mCD11b-FITC, anti-mCD11b-PE, anti-mGr-1-PE-Cy5, anti-mCD31-PE, anti-mF4/80-PE, anti-mCD11c-PE, anti-mI-Ab-PE, anti-mCD115-PE, anti-mCD274-PE, anti-mCD80-PE, anti-mCD86-FITC, anti-mCD115-PE, anti-mCD62L-PE, anti-mIgG-PE, anti-mIgG-FITC, and anti-mFoxp3-PE were purchased from BD Biosciences Pharmingen (San Diego, CA, USA). Anti-mCD8-PE-Cy5, anti-mCD4-FITC, and anti-mCD4-PE were purchased from eBioscience (San Diego, CA, USA). Anti-mCD45.2-PE-Cy5, anti-mCD45.1-APC, anti-mLy6G-PE, and anti-mCD11b-FITC were purchased from biolegend. Recombinant mouse GM-CSF was purchased from PeproTech (Rock Hill, NJ, USA). Dex, RU486 (16), bacterial lipopolysaccharide (LPS; E. coli 055:B5), and NG-monomethyl-l-arginine (L-NMMA) (17) were purchased from Sigma-Aldrich. Anti-CXCR2 mAb (16) and IgG isotype were purchased from R&D Systems (Minneapolis, MN, USA).
NO Production Assay
The Griess reaction was used to measure nitrite production. Equal volumes of culture supernatants (100 µl) were incubated in 96-well plate with Griess reagent at room temperature for 10 min, and absorbance was determined at 550 nm in a microplate reader (Bio-Rad). Nitrite concentrations were calculated by comparing the absorbance values for the test samples to a standard curve generated by a serial dilution of sodium nitrite.
Cardiac Allograft Transplantation
Hearts from Balb/c donors were transplanted heterotopically into the abdominal cavity of B6 recipients. Anastomoses were performed between the donor ascending aorta/pulmonary arties and the recipient descending aorta/inferior vena cava. The function of the cardiac graft was monitored daily by palpation through the abdominal wall. Graft loss was defined as no palpable beat. In some experiments, grafts were excised, fixed in 10% buffered formaldehyde, paraffin embedded, sectioned (3 µm), and stained with hematoxylin and eosin (H&E) as we described previously (5).
Cell Isolation, Flow Cytometry, and Adoptive Transfer
Splenic cells and peripheral white blood cells were prepared as we described previously (5). For isolation of infiltrated cells in cardiac allografts, allografts were cut into small pieces and then digested for 40 min at 37°C with 400 U/ml collagenase IV (Sigma-Aldrich), 10 mM HEPES, and 0.01% DNase I (MP Biomedicals) in HBSS. Digested suspensions were passed through a nylon mesh. Then cells were collected after centrifugation and re-suspended in FACS staining buffer for cell surface marker staining. Samples were analyzed on a Beckman Coulter Epics XL benchtop FCM (Beckman Coulter) with FCS express software (De Novo Software, Thornhill, ON, Canada). CD11b+ Gr-1int/low cells were sorted after in vitro culture using a FACSAria II (BD Biosciences) and were adoptively transferred into B6 recipient immediately after cardiac allograft transplantation (3 × 106 per mouse, i.v.).
Quantitative Real-time PCR
Total RNA of the indicated cells was extracted using RNAeasy mini kit (Qiagen) and reverse transcribed with Superscript II (Qiagen) according to the manufacturer’s instructions. The cDNA served as template for the amplification of target genes and the housekeeping gene (HPRT) by real-time PCR. Target gene expression was calculated using the comparative method for relative quantification upon normalization to HPRT gene expression. Primer sequences:
HPRT Forward: AGTACAGCCCCAAAATGGTTAAG,
Reverse: CTTAGGCTTTGTATTTGGCTTTTC;
iNOS Forward: CACCAAGCTGAACTTGAGCG,
Reverse: CGTGGCTTTGGGCTCCTC;
GRα Forward: ACTGCCCCAAGTGAAAACAGA,
Reverse: GCCCAGTTTCTCCTGCTTAATTAC;
CXCR2 Forward: TCACAAACAGCGTCGTAGA,
Reverse: GACAGCATCTGGCAGAATAG.
Statistical Analysis
Data were reported as Mean ± SEM. Student’s t-test was used to compare the differences between two groups, and one-way or two-way ANOVA analysis was used for comparison among multiple groups. Survival studies were analyzed with the log-rank test. P < 0.05 was considered statistically significant.
Results
Dex Enhances the Differentiation and Suppressive Capacity of MDSCs Induced by GM-CSF In Vitro
To address the role of Dex in the induction of MDSCs, MDSCs were generated from bone marrow cells in the presence of GM-CSF as previously described, with or without different concentrations of Dex for 4 days. As shown in Figure 1A, GM-CSF or GM-CSF with different concentrations of Dex all induced CD11b+ Gr-1+ cells. Interestingly, we found Dex could increase the percentage and cell number of CD11b+Gr-1int/low cells but not CD11b+Gr-1hi cells in a dose-dependent manner (Figures 1A,B). Based on the increased percentage and cell number of CD11b+Gr-1int/low cells induced by GM-CSF and Dex, CD11b+Gr-1int/low cells were gated to further analyze cell surface markers. The expression of CD115, F4/80, and CD11c were identical between the cells treated with GM-CSF or GM-CSF + Dex (Figure 1C), indicating Dex treatment does not alter cell phenotype. Other surface markers indicative for MDSCs suppressive action, including CD80 and CD86 were significantly decreased in cells induced by Dex. CD62L, which enables MDSCs homing into lymphoid organs, was slightly increased on the surface of MDSCs induced by Dex and GM-CSF (Figure 1C).
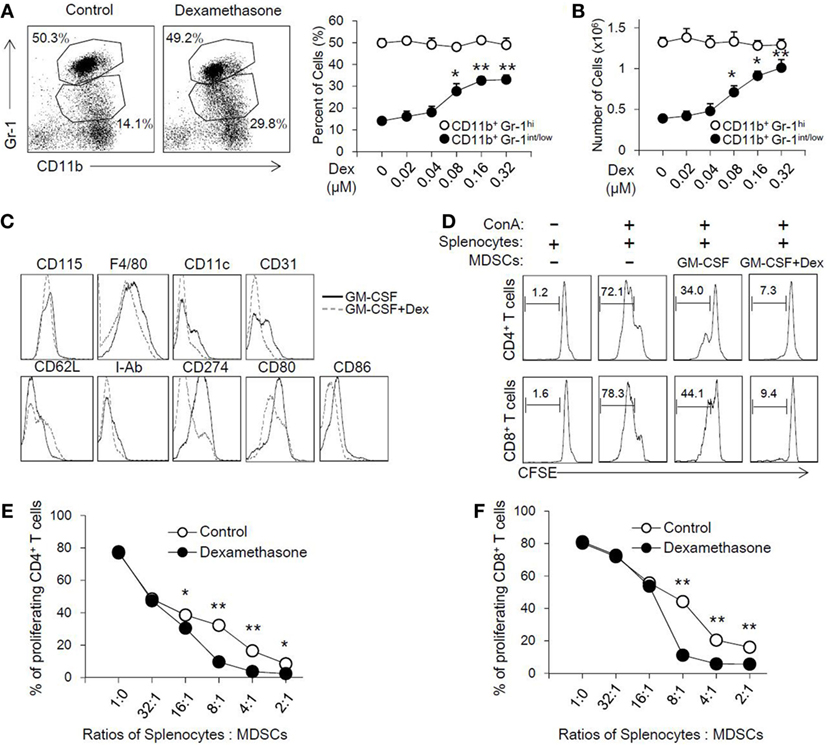
Figure 1. Dexamethasone (Dex) increases the cell number of CD11b+ Gr-1int/low Myeloid-derived suppressor cells (MDSCs) with an enhanced suppressive function in vitro. MDSCs were induced from bone marrow cells in the presence of granulocyte macrophage colony stimulating factor (GM-CSF), with or without Dex for 4 days. The percentage (A) and cell number (B) of MDSCs induced by GM-CSF or GM-CSF + Dex were shown. The phenotype markers (C) of MDSCs induced in vitro were also summarized by flow cytometry. (D) CFSE-labeled splenic cells (2 × 105 per well) were stimulated by ConA (2 µg/ml), and MDSCs induced by GM-CSF or GM-CSF + Dex were added at different ratios between splenic cells and MDSCs for 72 h. The pattern of proliferation was examined by flow cytometric analysis of CFSE dilution in the gated CD4+ and CD8+ T cells. The percentages indicated CFSE dilution in the gated T cell population (number of splenic cells and MDSCs: 2 × 105 and 0.25 × 105). The inhibitory rates in CD4+ T cells (E) and CD8+ T cells (F) were calculated based on the proliferation rates determined by flow cytometry analysis. The data (Mean ± SEM) are representative from three independent experiments (*P < 0.05, **P < 0.01).
To directly assess the suppressive function of MDSCs induced by GM-CSF or GM-CSF + Dex, cell proliferation of T cells stimulated by ConA was determined in the presence of the induced MDCs at different ratios. GM-CSF-induced MDSCs can inhibit the proliferation of CD8+ and CD4+ T cells in a dose-dependent manner, while GM-CSF + Dex-induced MDSCs displayed stronger suppressive capacity (Figure 1D). MDSCs induced by GM-CSF + Dex can inhibit T cell proliferation even in lower ratios (Figures 1E,F). Therefore, these results suggested Dex can enhance the immunosuppressive function of GM-CSF-induced MDSCs in vitro.
The Enhanced Immunosuppressive Function of MDSCs Induced by Dex Is Mediated by iNOS In Vitro
To further explore the mechanisms accounting for the enhanced immunosuppressive function seen in GM-CSF + Dex-induced MDSCs, immunosuppressive molecules were assessed in MDSCs induced by GM-CSF or GM-CSF + Dex in the presence or absence of splenocytes. There were no significant differences in potential relevant genes, including Arg1, Nox2, COX2, HO-1, TGF-β, and IDO between MDSCs induced by GM-CSF and by GM-CSF + Dex (data not shown), while the expression of iNOS in GM-CSF + Dex-induced MDSCs was significantly higher than that in GM-CSF-induced MDSCs (Figure 2A). Next, we assessed the level of NO production in MDSCs co-cultured with T cells activated by ConA. The NO production was also significantly higher in the culture system with MDSCs induced by GM-CSF + Dex (Figure 2B), suggesting the critical role of iNOS pathway in the immunosuppressive function of Dex-induced MDSCs. Moreover, addition of a nitric oxide synthase inhibitor, L-NMMA, which can specifically block NO production, significantly reversed the inhibitory effects of MDSCs induced by GM-CSF or GM-CSF + Dex on T cell proliferation in a dose-dependent manner (Figures 2C,D). In addition, there was no significant difference in the immunosuppressive function between iNOS-deficient MDSCs induced by GM-CSF or GM-CSF + Dex, both of which displayed significantly decreased inhibitory function (Figures 2E,F). Inhibition of IDO, Cox2, and Arg1 with their corresponding inhibitors also failed to recover T cell proliferation in the culture system with MDSCs induced by GM-CSF alone or GM-CSF + Dex (data not shown). Taken together, all these results clearly showed that iNOS pathway is responsible for the enhanced immunosuppressive function of MDSCs induced by GM-CSF + Dex in vitro.
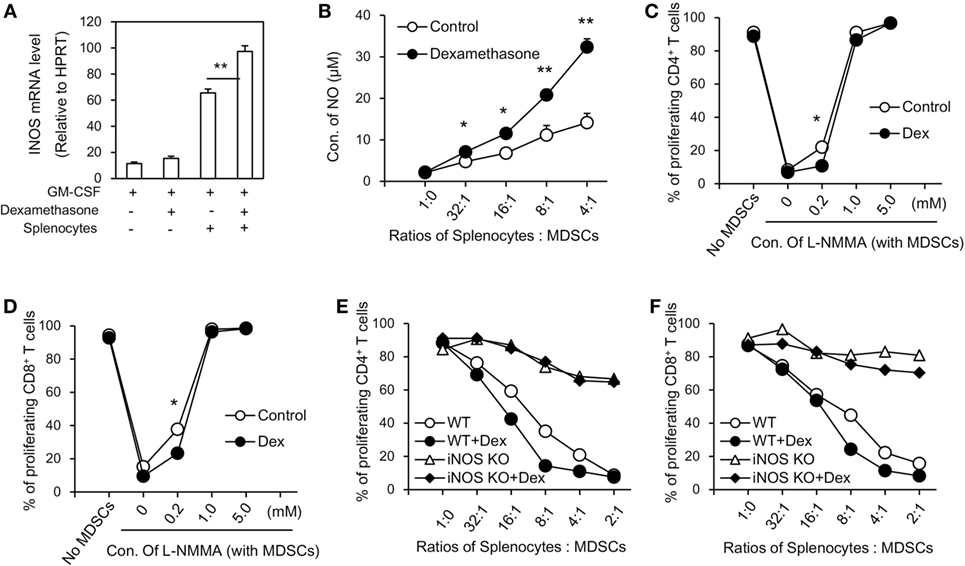
Figure 2. Dexamethasone (Dex) enhances the inhibitory function of myeloid-derived suppressor cells (MDSCs) through iNOS pathway. (A) MDSCs (C57BL/6, CD45.1) induced by granulocyte macrophage colony stimulating factor (GM-CSF) or GM-CSF + Dex were cocultured with splenic cells (C57BL/6, CD45.2) in the presence of conA (2 µg/ml) at a ratio of 1:1 for 3 days, and MDSCs were harvested by flow cytometry based on the surface staining of CD45.1. The mRNA expression of iNOS in harvested MDSCs was determined by RT-PCR. (B) MDSCs induced in vitro were cocultured with CFSE-labeled splenic cells (2 × 105 per well) stimulated by ConA (2 µg/ml) at different ratios for 72 h and the NO levels in the coculture medium were assessed. iNOS inhibitor L-NMMA at different concentrations were added into T cells and MDSCs coculture system for 72 h. Percentage of proliferating CD4+ T cells (C) and CD8+ T cells (D) were shown. MDSCs induced from iNOS knockout (KO) mice by GM-CSF or GM-CSF + Dex were added to CFSE-labeled splenic cell proliferation assay. iNOS deficiency significantly reversed the inhibitory effect of MDSCs induced by GM-CSF + Dex on CD4+ T cells (E) and CD8+ T cells (F). Data are shown as Mean ± SEM. All the data are composite of three independent experiments (*P < 0.05, **P < 0.01).
Adoptive Transfer of MDSC-Dex Prolongs Cardiac Allograft Survival in an iNOS-Dependent Manner
To examine the role of MDSCs in allograft rejection in vivo, GM-CSF-induced MDSCs (MDSC-control) or GM-CSF + Dex-induced MDSCs (MDSC-Dex) were transferred to allogeneic cardiac transplant mice. Adoptive transfer of MDSC-Dex significantly prolonged cardiac allograft survival when compared to that of MDSC-control (Figures 3A,B). Previous results showed iNOS pathway accounted for the enhanced immunosuppressive function of MDSC-Dex in vitro. To explore whether iNOS pathway is also required for MDSC-Dex mediated suppressive function in vivo, MDSC isolated from iNOS KO mice were induced by GM-CSF + Dex in vitro and were adoptively transferred to allogeneic cardiac transplant mice. Absent of iNOS pathway in MDSC induced by GM-CSF + Dex significantly reversed the effect of MDSC-Dex on allograft survival (Figure 3C). The infiltration of immune cells including CD8+ T, CD4+ T, and macrophages were also significantly increased in iNOS KO mice transferred with MDSCs induced by GM-CSF + Dex (Figure 3D). Therefore, NO is required for Dex-induced MDSC amelioration of allograft immune response both in vivo and in vitro.
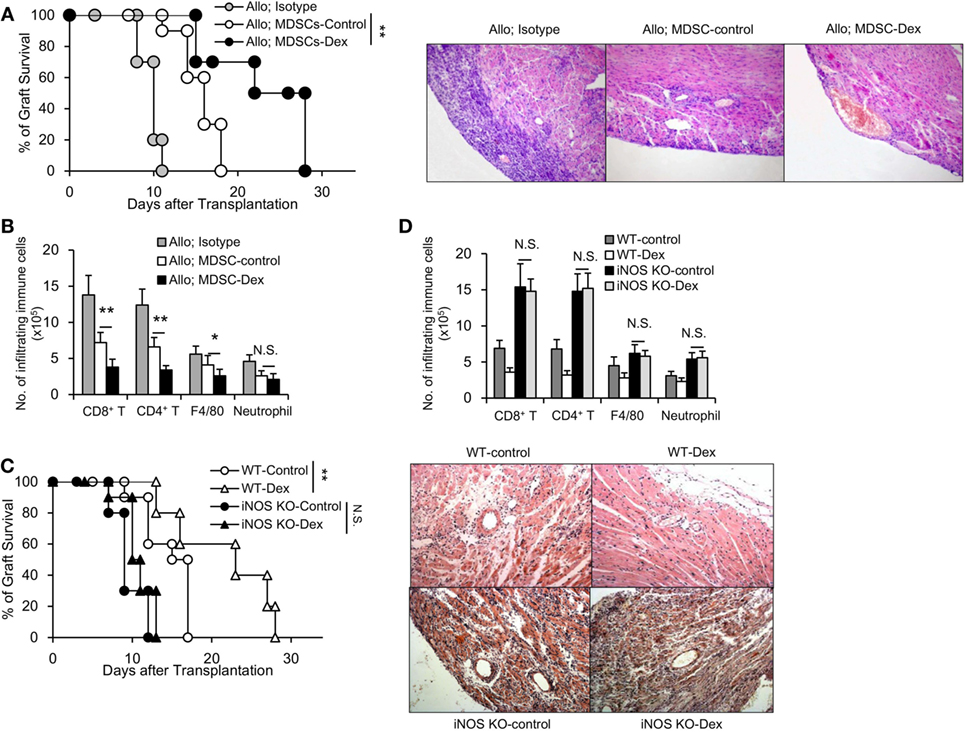
Figure 3. Adoptive transfer of granulocyte macrophage colony stimulating factor (GM-CSF) + dexamethasone (Dex)-induced Myeloid-derived suppressor cells (MDSCs) prolongs cardiac allograft survival in an iNOS-dependent manner. (A) Cardiac allograft survival (BALB/c donor hearts to C57BL/6J recipient mice) from mice transferred with sorted CD11b+Gr-1int/low MDSCs (3 × 106 per mouse, i.v.) induced by GM-CSF or GM-CSF + Dex immediately after cardiac allograft transplantation was monitored (n = 8 per group), and H&E staining of representative sections at postoperative day 7 from mice transferred with MDSCs were shown. (B) Numbers of infiltrating immune cells in cardiac allografts were assessed by flow cytometry. (C) Deficiency of iNOS reversed the protective function of Dex-induced MDSCs on cardiac allograft in recipient mice (n = 8 per group). (D) The infiltration of immune cells in cardiac allografts were analyzed in WT or iNOS knockout (KO) mice transferred with MDSCs induced by GM-CSF or GM-CSF + Dex. *P < 0.05, **P < 0.01, N.S., not significant.
MDSCs Induced by Dex Displayed an Enhanced Migration into Allograft through Upregulating the Expression of Glucocorticoid Receptor (GR) and CXCR2
The migration of MDSCs into allograft is critical for its immunosuppressive function, as well as for the prolongation of allograft survival (18). To explore whether the prolongation of allograft seen in Dex-MDSCs treated mice was due to an enhanced migration of MDSCs, we first assessed the expression of CXCR2, which was implicated in mediating MDSCs recruitment into local inflammatory and tumor sites (19, 20). We found the expression of CXCR2 was significantly upregulated in MDSCs induced by GM-CSF + Dex (Figure 4A), and MDSCs induced by GM-CSF + Dex also displayed a significantly increased infiltration into allograft (Figure 4B). In addition, anti-CXCR2 treatment significantly reduced the number of MDSCs in allograft, resulting in a decreased allograft survival in both MDSCs-Dex group and MDSCs-control group (Figures 4B,C). Thus, Dex promotes the migration of MDSCs into allograft through upregulating the expression of CXCR2.
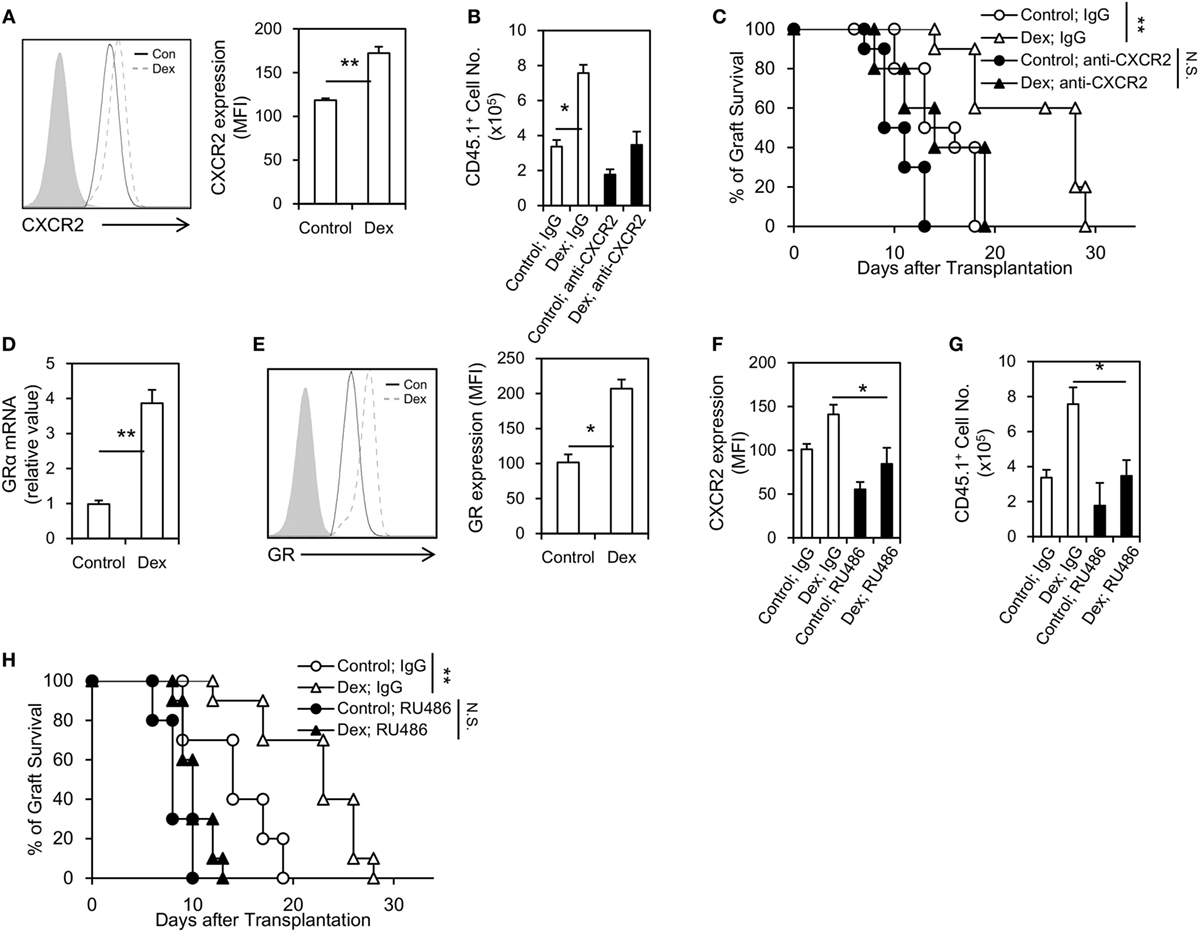
Figure 4. Dexamethasone (Dex) treatment promotes Myeloid-derived suppressor cells (MDSCs) migration into cardiac allograft through upregulating the expression of glucocorticoid receptor (GR) and CXCR2. CXCR2 expression (A) in MDSCs induced by granulocyte macrophage colony stimulating factor (GM-CSF) or GM-CSF + Dex was determined by flow cytometry, and the mean fluorescent intensity (MFI) of CXCR2 was summarized. Allograft recipient mice (CD45.2, C57BL6/J) were administered 50 µg neutralizing anti-CXCR2 mAb or IgG isotype control via i.v. injection 1 h before transplantation, and then MDSCs induced by GM-CSF or GM-CSF + Dex (3 × 106 per mice, i.v.) from CD45.1 mice (C57BL6/J) were adoptively transferred into these recipients. The infiltrating donor CD45.1+ CD11b+ Gr-1+ cell number (B) and graft survival (C) were determined (n = 8). Dex treatment upregulated the expression of GR in MDSCs both at the protein (D) and mRNA levels (E). The GR antagonist RU486 efficiently diminished the CXCR2 expression on MDSCs (F) and decreased the infiltration of donor CD45.1+ CD11b+ Gr-1+ MDSCs in cardiac allograft (G). RU486 treatment (10 mg/kg, i.p., 1 h before transplantation) significantly diminished graft survival in recipient received MDSCs induced by GM-CSF + Dex (H). Data (Mean ± SEM) are representative of two independent experiments (n = 5–10 in each group). *P < 0.05, **P < 0.01, N.S., not significant.
Next, we explored how Dex modulates the expression of CXCR2 on MDSCs. GR signaling is regarded to be essential for GC effects, and we found Dex treatment significantly upregulated the expression of GR in MDSCs both at the mRNA and protein levels (Figures 4D,E). Moreover, the use of a GR specific inhibitor, RU486, decreased the expression of CXCR2 on MDSCs (Figure 4F). RU486 treatment also reduced the infiltration of MDSCs in allograft (Figure 4G), leading to a diminished allograft survival (Figure 4H). Taken together, these results suggest Dex promotes the migration of MDSCs into allograft through GR-dependent pathway.
Dex-Induced MDSCs Promote CD4+ Foxp3+ Treg Expansion
Previously, MDSCs have been shown to mediate CD4+ Foxp3+ Treg development in mouse tumor models in vivo (21, 22). Recent studies further suggested that MDSCs can induce Treg development in animal transplantation model (23) and renal transplant patients (24). To address whether MDSCs in our model can also promote the expansion of Treg in vivo, the percentages of Treg in the allografts, the peripheral blood and the spleens of mice receiving MDSC-control or MDSC-Dex were analyzed at post transplantation day 7. As shown in Figure 5A, the percentage of Treg in the allografts of mice receiving MDSC-Dex was significantly higher than that receiving MDSC-control. Similar results were also found in the peripheral blood and the spleen (Figures 5B,C). To direct assess the effect of MDSCs on the expansion of Treg, we cocultured MDSCs induced by GM-CSF or GM-CSF + Dex with purified CD4+ T cells from the spleen for 3 days. As shown in Figures 5D,E, MDSCs induced by Dex + GM-CSF significantly enhanced the pool of Treg. Taken together, all these results suggested MDSCs induced by GM-CSF + Dex favors the expansion of Treg.
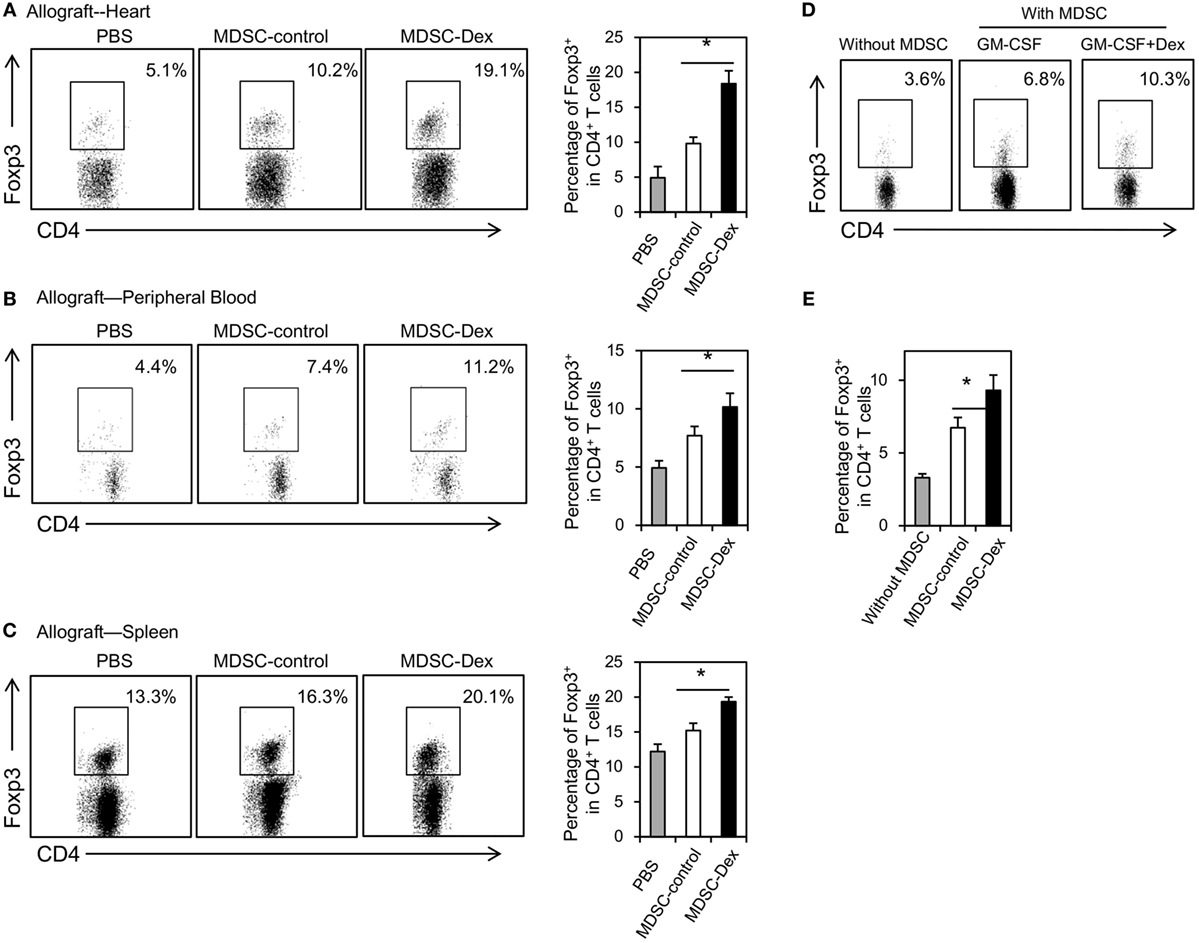
Figure 5. Adoptive transfer of myeloid-derived suppressor cells (MDSCs) induced by granulocyte macrophage colony stimulating factor (GM-CSF) and dexamethasone (Dex) favors the expansion of CD4+ Foxp3+ regulatory T cells (Treg) in vivo. MDSCs induced by GM-CSF or GM-CSF + Dex (3 × 106 per mice) were adoptively transferred into recipient mice via the tail vein immediately after cardiac allograft transplantation. On post transplantation day 7, recipient mice were euthanized and the percentages of CD4+ Foxp3+ Treg in allograft (A), peripheral blood (B), and spleen (C) were assessed. (D,E) MDSCs (CD45.2, C57BL6/J) induced by GM-CSF or GM-CSF + Dex mice were cocultured with CD4+ T cells (CD45.2, C57BL6/J) purified by flow cytometry from the spleen at a ratio of 1:4 in the presence of anti-CD3 (5 μg/ml)/CD28(2 µg/ml) for 3 days. The percentages of Foxp3+ cells in CD4+ T cells were assessed. Data are Mean ± SEM. All the data are composite of three independent experiments (n = 3–5 in each group). P values were determined by Student’s t-test. *P < 0.05, N.S., not significant.
Discussion
Historically, developing therapeutic strategies of transplantation tolerance mainly focuses on targeting adaptive immune response, based on the findings that T cells are both necessary and sufficient to induce allograft rejection (25–28). Deletion of activated T cells and development of Treg are the two main mechanisms. However, deletion of activated T cells is necessary but not sufficient for the induction of immune tolerance and Treg also do not seem to contribute to immune tolerance during the early phase (29, 30). Modulation of innate immune cells may be a candidate to resolve this problem. Previous studies identified that classic antigen-presenting cell dendritic cells can serve as a potent innate immune cell population which promotes transplantation tolerance through inducing antigen-specific Treg (31–35). Recently, accumulating evidence from animal models also suggest a great potential of MDSCs for preventing allograft rejection (6, 36, 37), which may be a new potential therapy target on innate immune cells to control graft rejection in the clinic.
To develop a clinically acceptable method through manipulating MDSCs, in vitro induction of potent and stable MDSCs has been tried (6, 7). Dex is a classic drug used in clinical trials to ameliorate immune-mediated allograft rejection associated with inflammation, and can modulate immune cell phenotype and function (11–13). Recently, systemic administration of Dex has been shown to modulate MDSCs expansion in vivo both in skin transplantation (16) and renal disease model (38). Dex can also favor the migration of MDSCs into inflammatory sites to control immune response (16). In addition, Dex modulates MDSCs immunosuppressive function through GR signaling in an immunological hepatic injury model (39). Therefore, Dex is an important regulator of MDSCs expansion and/or function. However, since Dex can also modulate GR signaling in other cells (40, 41), systemic injection of Dex may not be an ideal way to manipulate MDSCs in future clinical trials. In our study, we used Dex combined with classic MDSC induction cytokine GM-CSF to generate the differentiation of MDSCs in vitro. We found Dex can specifically increase the cell number of CD11b+ Gr-1int/low MDSCs in vitro with more powerful immune suppressive function on T cell proliferation. Mechanistic study showed that iNOS pathway is required for the immune regulatory function of Dex-induced MDSCs as significantly increased production of NO was found in Dex-induced MDSCs system. In addition, inhibition of NO production with genetic deletion of iNOS or chemical inhibitor blocked the immunosuppressive function of Dex-induced MDSCs in vitro. Consistent with these in vitro results, adoptive transfer of Dex-induced MDSCs significantly prolongs the survival of cardiac allograft in vivo.
Glucocorticoid receptor is one of the key mechanisms for the effect of Dex on MDSCs. Modulation of GR by Dex can suppress HIF1α and HIF1α-dependent glycolysis in MDSCs which promotes the immune suppressive activity of MDSCs (39). On the other hand, pharmacological inhibition of GR by RU486 can also suppress the function of MDSCs, leading to the diminished allo skin graft survival (16). To explore whether GR signaling is also involved in our study, we first assessed the GR level in MDSCs. We found GR expression was significantly increased in Dex-induced MDSCs, indicating the importance of GR signaling in Dex-induced MDSCs. The migration of MDSCs into inflammation site, i.e., the allograft in transplantation model, mainly depends on surface expression of CXCR2, which also plays a critical for immune tolerance. We found Dex treatment increased the expression of CXCR2 on MDSCs. Importantly, blockade of CXCR2 with a CXCR2 mAb or blockade of GR with its specific inhibitor RU486 significantly decreased the expression of CXCR2 on MDSCs and also recruitment of MDSCs into allograft, leading to a diminished survival of allograft survival. Taken together, these results demonstrated that GR signaling is required for the immune regulation of Dex-induced MDSCs.
Myeloid-derived suppressor cells and Treg are both important immune regulator cells in the context of organ transplantation (42). Since Treg or MDSCs alone was not sufficient to induce long-term tolerance, it was attractive to investigate interaction between MDSCs and Treg. Indeed, several studies have shown MDSCs promote the development and homeostasis of Treg in tumor (21, 22, 43) and organ transplantation (1, 18). Moreover, the interaction of Dex, MDSCs, and Treg has been identified in a focal segmental glomerulosclerosis mice mode (38). Both systemic administration of Dex or adoptive transfer of MDSCs can increase the percentages of Treg in the spleen and the kidney, and pharmacological depletion of MDSCs resulted in decreased levels of Treg (38). However, this study did not assess the direct effect of MDSCs on Treg expansion in vitro, and the renal injury itself can trigger strong expansion of Treg expansion (38). Thus, it is possible that depletion of MDSCs may lead to the changes of other cells which have an indirect impact on Treg expansion. We also investigated whether adoptive transfer of Dex-induced MDSCs can have an impact on Tregs in vivo. Consistent with other studies, adoptive transfer of Dex-induced MDSCs can favor the expansion of Treg both systemically and in allograft in vivo. Detailed mechanisms for the expansion of Treg upon adoptive transfer of Dex-induced MDSCs are not clear. Several mechanisms have been suggested to promote the expansion of Treg by MDSCs, including chemokine driven attraction of Treg (43, 44), and induced proliferation of natural Treg (45, 46). We identified a systemic expansion of Treg after adoptive transfer of MDSCs with an increased number of total Treg pool (data not shown), and in vitro culture of MDSCs with CD4+ T cells can promote the expansion of Treg. Therefore, in the context of our model, MDSCs may directly expand the population of Treg through one or combined mechanisms mentioned above. Molecules responsible for the expansion of Treg by MDSCs includes iNOS signaling (1), CD274 (46), arginase (45), and IL-10 (22). In our study, no significant differences in CD274, Arg1 and TGF-β levels were found between MDSCs induced by GM-CSF or GM-CSF + Dex (Figure 1C, data not shown). Blockade of iNOS signaling in MDSCs with the use of iNOS KO mice also did not have an impact on the expansion of CD4+ Foxp3+ T cells in vivo (data not shown). The production of IL-10, which was shown to maintain Foxp3 expression in Treg (47) and also potentiated differentiation of human induced Treg (48), was significantly increased in Dex-induced MDSCs (data not shown). Future studies on the molecules responsible for the expansion of Treg are urged to fully address these questions.
In summary, we have shown that Dex treatment can enhance the expansion and immunosuppressive function of in vitro-generated MDSCs. Adoptive transfer of these Dex-induced MDSCs in vitro can also prolong cardiac allograft survival and favor the expansion of Treg in vivo, which may be a new approach of cellular therapy for organ transplantation.
Ethics Statement
All the animal experiments were carried out following the Guidelines for the Care and Use of Laboratory Animals of Nanjing University, and were approved by the ethical review committee of Nanjing University.
Author Contributions
J-FD and Y-tD conceived and designed the experiments; X-FS, YZ, and KC performed the experiments; JD did the allogenic cardiac transplantation; X-FS and XK analyzed the data; J-FD and W-xG contributed reagents/materials/analysis tools; B-rL designed the experiments of Treg induction in vitro, X-FS wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by National Natural Science Foundation of China (No. 81500432, X-FS, No. 81571563, J-FD), and National Key Clinical Specialist Construction Program of China (No. 2012-649, Y-tD).
References
1. Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest (2010) 120(7):2486–96. doi:10.1172/JCI41628
2. Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol (2010) 185(10):5828–34. doi:10.4049/jimmunol.0903636
3. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116(25):5738–47. doi:10.1182/blood-2010-06-287839
4. Messmann JJ, Reisser T, Leithauser F, Lutz MB, Debatin KM, Strauss G. In vitro-generated MDSCs prevent murine GVHD by inducing type 2 T cells without disabling antitumor cytotoxicity. Blood (2015) 126(9):1138–48. doi:10.1182/blood-2015-01-624163
5. Shen XF, Jiang JP, Yang JJ, Wang WZ, Guan WX, Du JF. Donor-specific regulatory T cells acquired from tolerant mice bearing cardiac allograft promote mixed chimerism and prolong intestinal allograft survival. Front Immunol (2016) 7:511. doi:10.3389/fimmu.2016.00511
6. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
7. Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity (2010) 32(6):790–802. doi:10.1016/j.immuni.2010.05.010
8. Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells (2010) 28(3):620–32. doi:10.1002/stem.301
9. Rossner S, Voigtlander C, Wiethe C, Hanig J, Seifarth C, Lutz MB. Myeloid dendritic cell precursors generated from bone marrow suppress T cell responses via cell contact and nitric oxide production in vitro. Eur J Immunol (2005) 35(12):3533–44. doi:10.1002/eji.200526172
10. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med (2006) 203(8):1883–9. doi:10.1084/jem.20060336
11. Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol (2004) 4(1):24–34. doi:10.1038/nri1256
12. Zheng G, Zhong S, Geng Y, Munirathinam G, Cha I, Reardon C, et al. Dexamethasone promotes tolerance in vivo by enriching CD11clo CD40lo tolerogenic macrophages. Eur J Immunol (2013) 43(1):219–27. doi:10.1002/eji.201242468
13. Kang Y, Xu L, Wang B, Chen A, Zheng G. Cutting edge: immunosuppressant as adjuvant for tolerogenic immunization. J Immunol (2008) 180(8):5172–6. doi:10.4049/jimmunol.180.8.5172
14. Du J, Shen X, Zhao Y, Hu X, Sun B, Guan W, et al. Wip1-deficient neutrophils significantly promote intestinal ischemia/reperfusion injury in mice. Curr Mol Med (2015) 15(1):100–8. doi:10.2174/1566524015666150114122929
15. Han C, Wu T, Na N, Zhao Y, Li W, Zhao Y. The effect of immunosuppressive drug cyclosporine A on myeloid-derived suppressor cells in transplanted mice. Inflamm Res (2016) 65(9):679–88. doi:10.1007/s00011-016-0949-7
16. Liao J, Wang X, Bi Y, Shen B, Shao K, Yang H, et al. Dexamethasone potentiates myeloid-derived suppressor cell function in prolonging allograft survival through nitric oxide. J Leukoc Biol (2014) 96(5):675–84. doi:10.1189/jlb.2HI1113-611RR
17. Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep (2016) 6:20250. doi:10.1038/srep20250
18. Nakamura T, Nakao T, Yoshimura N, Ashihara E. Rapamycin prolongs cardiac allograft survival in a mouse model by inducing myeloid-derived suppressor cells. Am J Transplant (2015) 15(9):2364–77. doi:10.1111/ajt.13276
19. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi:10.1126/scitranslmed.3007974
20. David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R axis: a double agent in tumor immune resistance. Vaccines (Basel) (2016) 4(3):E22. doi:10.3390/vaccines4030022
21. Pan PY, Ma G, Weber KJ, Ozao-Choy J, Wang G, Yin B, et al. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res (2010) 70(1):99–108. doi:10.1158/0008-5472.CAN-09-1882
22. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66(2):1123–31. doi:10.1158/0008-5472.CAN-05-1299
23. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol (2011) 187(9):4598–610. doi:10.4049/jimmunol.1100519
24. Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, et al. Monocytic myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4(+) Foxp3(+) Treg expansion. Am J Transplant (2013) 13(12):3123–31. doi:10.1111/ajt.12461
25. Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, et al. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med (1999) 5(11):1303–7. doi:10.1038/15260
26. Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol (2003) 3(3):199–210. doi:10.1038/nri1027
27. Sakaguchi S, Sakaguchi N. Thymus and autoimmunity: capacity of the normal thymus to produce pathogenic self-reactive T cells and conditions required for their induction of autoimmune disease. J Exp Med (1990) 172(2):537–45. doi:10.1084/jem.172.2.537
28. Chai JG, Xue SA, Coe D, Addey C, Bartok I, Scott D, et al. Regulatory T cells, derived from naive CD4+CD25- T cells by in vitro Foxp3 gene transfer, can induce transplantation tolerance. Transplantation (2005) 79(10):1310–6. doi:10.1097/01.TP.0000159147.56408.9C
29. Wu Z, Bensinger SJ, Zhang J, Chen C, Yuan X, Huang X, et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med (2004) 10(1):87–92. doi:10.1038/nm965
31. Thomson AW, Turnquist HR, Zahorchak AF, Raimondi G. Tolerogenic dendritic cell-regulatory T-cell interaction and the promotion of transplant tolerance. Transplantation (2009) 87(9 Suppl):S86–90. doi:10.1097/TP.0b013e3181a2dcec
32. Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol (2007) 7(8):610–21. doi:10.1038/nri2132
33. Ezzelarab M, Thomson AW. Tolerogenic dendritic cells and their role in transplantation. Semin Immunol (2011) 23(4):252–63. doi:10.1016/j.smim.2011.06.007
34. Castellaneta A, Mazariegos GV, Nayyar N, Zeevi A, Thomson AW. HLA-G level on monocytoid dendritic cells correlates with regulatory T-cell Foxp3 expression in liver transplant tolerance. Transplantation (2011) 91(10):1132–40. doi:10.1097/TP.0b013e31821414c9
35. Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol (2007) 178(11):7018–31. doi:10.4049/jimmunol.178.11.7018
36. Thomson AW, Turnquist HR. Regulators with potential: substantiating myeloid-derived suppressor cells in human organ transplantation. Am J Transplant (2013) 13(12):3061–2. doi:10.1111/ajt.12464
37. Boros P, Ochando JC, Chen SH, Bromberg JS. Myeloid-derived suppressor cells: natural regulators for transplant tolerance. Hum Immunol (2010) 71(11):1061–6. doi:10.1016/j.humimm.2010.08.001
38. Li L, Zhang T, Diao W, Jin F, Shi L, Meng J, et al. Role of myeloid-derived suppressor cells in glucocorticoid-mediated amelioration of FSGS. J Am Soc Nephrol (2015) 26(9):2183–97. doi:10.1681/ASN.2014050468
39. Lu Y, Liu H, Bi Y, Yang H, Li Y, Wang J, et al. Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1alpha-dependent glycolysis. Cell Mol Immunol (2017) 14:1–12. doi:10.1038/cmi.2017.5
40. Engler JB, Kursawe N, Solano ME, Patas K, Wehrmann S, Heckmann N, et al. Glucocorticoid receptor in T cells mediates protection from autoimmunity in pregnancy. Proc Natl Acad Sci U S A (2017) 114(2):E181–90. doi:10.1073/pnas.1617115114
41. Baschant U, Frappart L, Rauchhaus U, Bruns L, Reichardt HM, Kamradt T, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci U S A (2011) 108(48):19317–22. doi:10.1073/pnas.1105857108
42. Nakamura T, Nakao T, Ashihara E, Yoshimura N. Myeloid-derived suppressor cells recruit CD4(+)/Foxp3(+) regulatory T cells in a murine cardiac allograft. Transplant Proc (2016) 48(4):1275–8. doi:10.1016/j.transproceed.2015.10.060
43. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189(12):5602–11. doi:10.4049/jimmunol.1201018
44. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, et al. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One (2010) 5(1):e8922. doi:10.1371/journal.pone.0008922
45. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68(13):5439–49. doi:10.1158/0008-5472.CAN-07-6621
46. van der Merwe M, Abdelsamed HA, Seth A, Ong T, Vogel P, Pillai AB. Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplantation. J Immunol (2013) 191(11):5764–76. doi:10.4049/jimmunol.1302191
47. Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, et al. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol (2009) 10(11):1178–84. doi:10.1038/ni.1791
Keywords: myeloid-derived suppressor cells, immune tolerance, dexamethasone, iNOS, glucocorticoid receptor
Citation: Zhao Y, Shen X-F, Cao K, Ding J, Kang X, Guan W-x, Ding Y-t, Liu B-r and Du J-F (2018) Dexamethasone-Induced Myeloid-Derived Suppressor Cells Prolong Allo Cardiac Graft Survival through iNOS- and Glucocorticoid Receptor-Dependent Mechanism. Front. Immunol. 9:282. doi: 10.3389/fimmu.2018.00282
Received: 05 September 2017; Accepted: 31 January 2018;
Published: 15 February 2018
Edited by:
Luis Graca, Universidade de Lisboa, PortugalReviewed by:
Huanfa Yi, Jilin University, ChinaBruce Milne Hall, University of New South Wales, Australia
Copyright: © 2018 Zhao, Shen, Cao, Ding, Kang, Guan, Ding, Liu and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-xian Guan, Z3Vhbnd4LW5qdUBob3RtYWlsLmNvbQ==;
Yi-tao Ding, ZGluZ3l0LW5qdUBob3RtYWlsLmNvbQ==;
Bao-rui Liu, YmFvcnVpbGl1MDdAMTYzLmNvbQ==;
Jun-Feng Du, ZHVqZjY2QDEyNi5jb20=
†These authors have contributed equally to this work.