 Wook-Jin Chae
Wook-Jin Chae Alfred L. M. Bothwell
Alfred L. M. Bothwell- Department of Immunobiology, Yale University School of Medicine, New Haven, CT, United States
Regulatory T cells (Tregs) are an important subset of adaptive immune cells and control immune reactions for maintaining homeostasis. Tregs are generated upon their encounter with self or non-self-antigen and mediate tolerance or suppress aberrant immune responses. A high level of specificity of Tregs to recognize antigen(s) suggested their instrumental potential to treat various inflammatory diseases. This review will first introduce seminal basic research findings in the field of Tregs over the last two decades pertinent to therapeutic approaches in progress. We will then discuss the previous approaches to use Tregs for therapeutic purposes and the more recent development of gene-modification approaches. The suppressive function of Tregs has been studied intensively in clinical settings, including cancer, autoimmunity, and allotransplantation. In cancer, Tregs are often aberrantly increased in their number, and their suppressor function inhibits mounting of effective antitumor immune responses. We will examine potential approaches of using gene-modified Tregs to treat cancer. In autoimmunity and allotransplantation, chronic inflammation due to inherent genetic defects in the immune system or mismatch between organ donor and recipient results in dysfunction of Tregs, leading to inflammatory diseases or rejection, respectively. Since the recognition of antigen is a central part in Treg function and their therapeutic use, the modulation of T cell receptor specificity will be discussed. Finally, we will focus on future novel strategies employing the therapeutic potential of Tregs using gene modification to broaden our perspective.
Introduction
Regulatory T cells (Tregs) are an important T cell subpopulation that maintains immunological homeostasis. In the early 1970s, a few papers provided evidence for an inhibitory role of thymus-derived T cells. Seminal research by Gershon and Kondo introduced the concept of infectious tolerance (1, 2). Over the next decade, these thymus-derived T cells were termed “suppressor T cells.” Later, suppressor T cells were renamed as “regulatory T cells,” based on their capability to regulate autoimmunity. Since the 1990s, Tregs have been one of the most intense research fields in immunology. The negative regulation of immune responses by Tregs is vital in autoimmune and auto-inflammatory disorders, acute and chronic infection, allergy, metabolic inflammation, transplantation, and cancer. Accordingly, modulation of Tregs holds the therapeutic potential to treat numerous disease classes. The unique history of Tregs has been well-reviewed previously (3).
Identifying Treg Markers and Mechanistic Studies of Treg Function in Mice
Initial attempts to isolate and to identify Tregs for immunological studies used IL-2 receptor α-chain (CD25), which is highly expressed on Tregs. Adoptive transfer of CD25+ Tregs prevented various experimental mouse models of autoimmunity (4–6). Other Treg markers were subsequently identified including lymphocyte activation gene (LAG)-3, CTLA-4, folate receptor-4, latency-associated peptide, and IL-35 (7–10). Often these markers were also expressed in activated effector CD4 T cells, necessitating the identification of more definitive markers of Tregs.
In this regard, one of the most important findings in Treg biology was the discovery of Treg lineage transcriptional factor FOXP3. Mutations in the FOXP3 gene were identified in Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) human patients. The murine counterpart of IPEX patients, scurfy mice, lacks functional FOXP3 and showed similar phenotypes to IPEX patients (11–13). Two studies demonstrated the importance of FOXP3 in Treg development and function (14, 15). Development of biological tools including Red Fluorescent Protein (RFP) FOXP3 reporter mice, Foxp3-Diphtheria Toxin Receptor mice which permitted Treg depletion, and the development of Treg-specific Foxp3YFP–Cre mice which allowed the conditional deletion of a gene in Tregs, facilitated the understanding of Treg biology in mice (16–19).
CD4+FOXP3+ Tregs can be induced from peripheral CD4+ naïve T cells in the periphery by many factors such as tolerogenic dendritic cells expressing indoleamine 2,3-dioxygenase (IDO), commensal bacteria, retinoic acid, or transforming growth factor (TGF)-β and are designated peripheral Tregs (pTregs) to distinguish them from the thymic-derived Tregs (tTregs) (20–24). Similar to their tTregs, pTregs regulate immune responses in various types of inflammatory disease environments including spontaneous intestinal tumorigenesis, inflammatory bowel disease, asthma, and experimental autoimmune encephalomyelitis (EAE) (25–28). It has been reported that FOXP3+ Tregs express the immunosuppressive cytokine IL-10. Later, IL-10-expressing Tregs were further dissected into IL-10+FOXP3+ Tregs and Foxp3-negative type 1 Tregs (Tr1) that are induced by dexamethasone and Vitamin D (29–31).
Using genetic, biochemical, and molecular biological approaches, functional modules of Foxp3 such as dimerization/oligomerization of the transcriptional factor were identified, and the regulatory mechanism of Foxp3-mediated gene expression in Tregs was extensively studied (32–40). The molecular mechanism of stable FOXP3 expression has been under intense investigation by measuring DNA demethylation at the Treg-specific demethylated region (TSDR), a conserved CpG-rich region within the Foxp3 locus where methylation maintains stable lineage commitment of Tregs (41, 42). In parallel to the regulation of FOXP3 expression, posttranslational modification by acetylation, ubiquitination, or phosphorylation has an important role in modulating the Foxp3-mediated transcriptional repression that is required for suppressor function (43–48).
Human Tregs in Basic and Clinical Studies
In the past two decades, there has been significant progress in the understanding of regulatory mechanisms of tolerance in humans. Various markers for the identification of human Tregs were found including CD25, FOXP3, and CD127 (IL-7Rα chain) (49–52). Further studies revealed that human conventional T cells transiently express FOXP3 without acquiring suppressive activity (53). Human Tregs are functionally and phenotypically distinguished by their activation status. Suppressive Treg cells are CD45RA+FOXP3lo in resting state and CD45RA−FOXP3hi in activated state while CD45RA−FOXP3lo T cells are non-suppressive. The proportion of the three subpopulations was markedly different between aged individuals, cord blood and patients with immunological diseases (54, 55).
Expansion of Tregs using rapamycin or induction of Tr1 cells has been utilized to induce polyclonal Tregs for clinical intervention (56, 57). Tr1 cells express similar markers to FOXP3+ Tregs such as CTLA-4, PD-1, CD39, and ICOS. Tr1 cells do not express FOXP3 constitutively, but they do express IL-10 and TGF-β once they are activated via T cell Receptor (TCR). Tr1 cells show bystander suppressor activity (58). IL-10 and TGF-β from Tregs inhibit effector CD4 T cells proliferation and production of effector cytokines, such as IL-2 and IFN-γ (59). Other than cytokine-mediated suppression, it is known that granzyme B-mediated cell death of myeloid APCs is mediated by the stable adhesion between HLA-class I molecules on Tr1 cells and CD112/CD115 on myeloid APCs (60).
In clinical settings, modification of TCR has been utilized to modulate Treg activity to intervene in various types of inflammatory diseases in an antigen-specific manner (61, 62). Treg-based therapies with freshly isolated or expanded Tregs have been implemented in clinical practice for patients undergoing allogeneic hemopoietic stem cell transplantation to prevent graft-versus-host disease (GVHD) (63), inhibiting rejection in solid organ transplantation and controlling autoimmunity in patients [e.g., Type 1 Diabetes (T1D)] (64). Since Tregs have multiple roles in a variety of clinical settings, the generation of gene-modified Tregs and administration of those Tregs via adoptive transfer is a promising approach to treat chronic inflammatory diseases, cancer, or rejection in transplantation medicine.
Gene-Modified Tregs in Cancer Immunotherapy
Regulatory T cells are found at high frequencies in the tumor microenvironment in a variety of cancers (65). Analysis in a variety of human carcinomas suggested that the accumulation of Tregs in the tumor microenvironment is associated with a poor prognosis (65).
Generating Tumor Antigen-Specific CAR+ Tregs
Over a decade ago, a seminal study proposed the therapeutic potential of genetically engineered T cells bearing a tumor antigen-specific TCR in cancer immunotherapy (66). Overexpression of the α and β chains of a specific TCR has been used as a traditional approach to engineer T cell specificity. The antigen-specific suppressor function of Tregs on effector T cells was demonstrated by the tumor antigen NY-ESO-1. Depletion of Tregs enabled the activation of NY-ESO-1-specific naïve CD4 T cells in healthy subjects and melanoma patients with NY-ESO-1-expressing tumors (67, 68). TCRs recognizing melanoma antigens have been successfully transduced in human Tregs in vitro (69). Interestingly, the affinity of the TCR did not affect the antigen-specific suppressive function. This indicated that tumor antigen-mediated TCR signals do not affect the function of fully differentiated Tregs ex vivo.
An alternative strategy is to transduce a chimeric antigen receptor (CAR) into Tregs to generate antigen-specific Tregs. CARs are synthetic proteins generated by fusing an extracellular domain for antigen recognition with transmembrane and signaling domains from the TCR and co-stimulatory receptors (70). The antigen-recognizing domain of a CAR is generated by a single-chain variable fragment (scFv) fusion protein of the complementarity determining regions of the heavy and light chains of a monoclonal antibody. A major advantage of generating scFvs is to avoid the limitation of MHC restriction. This expands the pool of treatable patients compared to the TCR overexpression approach. Expression and engineering of CARs that are specific to tumor antigens is now a primary interest in cancer immunotherapy employing CAR Tregs (71). Further studies for a more diverse set of tumor antigens are warranted to broaden the therapeutic potential of this approach.
Modulating Foxp3 Expression in Tregs
Another approach to inhibit the suppressor function of Tregs is to downregulate FOXP3 expression. Use of lentiviral FOXP3 shRNA delivery inhibited Treg-like leukemia in mice (72). This lentiviral strategy was used to knockdown FOXP3 mRNA in human Tregs, and this approach demonstrated the loss of suppressor function, indicating that it has potential to be used in cancer immunotherapy (73). However, Tregs that are transduced with the lentivirus have not been tested for safety, and thus further research is needed. Stat3 has been reported to play a crucial role to maintain FOXP3 expression in human. Delivery of small interfering RNA (siRNA) for Stat3 into Tregs demonstrated the loss of the suppressor function (74). Recently, it has been reported that siRNA can be delivered in gold nanoparticles, circumventing the issue with a lentiviral system in human patients (75). A stable FOXP3 expression is dependent on posttranslational modification. Genetic or pharmacologic modulation of FOXP3 acetylation via the histone/protein acetyltransferases (HATs), p300, and CBP downregulated suppressive function of Treg and promoted antitumor immunity (76).
A recent study demonstrated that the pharmacologic inhibition of a single de-ubiquitination enzyme, Usp7, determines the fate of FOXP3 and Tip60 in Tregs, thus providing a target for therapeutic modulation of Treg function in antitumor immunity (77). It has been shown that selective small molecule inhibitors for the bromodomains of CREBBP/EP300 reduced FOXP3 expression, as well as expression of functional markers in Tregs (e.g., LAG-3, CTLA-4, and TIM-3) (78). It has also been reported that intranuclear interactomic inhibition of FOXP3 could abrogate suppressor function via nuclear delivery of FOXP3 (79). These approaches are promising at a preclinical stage, yet assessment of target-specific delivery of siRNA or Protein Transduction Domain-FOXP3 protein, and their side effects have not been assessed. Potential autoimmune responses should be considered when Treg dysfunction is implemented as a therapeutic approach in cancer immunotherapy. An additional concern is that a series of surprising reports found that a high incidence of tumor-infiltrating Tregs is associated with improved prognosis in cancer patients (80–83). Thus, the inhibition of FOXP3 expression needs further study and careful consideration regarding the role of Tregs in a given tumor microenvironment.
Gene-Modified Tregs in Autoimmunity
Past successes using genetically enhanced T-cells in the cancer arena have prompted interest in the development of related approaches to suppress unwanted autoimmune responses. Refractory autoimmune disease is associated with a markedly decreased life expectancy urging consideration of intensive therapeutic approaches. Tregs provide an attractive tool for genetic targeting against autoantigens present in the organ(s) of interest.
Modulating Antigen Specificity and CAR Approach in Tregs to Treat Autoimmunity
Therapeutic effect of purified Tregs have been demonstrated in preclinical studies in a range of autoimmune disease models in mice, including Systemic lupus erythematosus (84), T1D, autoimmune hepatitis, inflammatory bowel diseases, and autoimmune encephalomyelitis (85–88). Subsequently, studies in several disease model systems have demonstrated that antigen-specific Tregs were present in diseased animals and more potent in suppressing pathogenic immune responses compared to polyclonal Tregs (9, 89).
Among autoimmune diseases, T1D has been an intense area of development for gene-modified Treg-mediated therapy with islet-specific Tregs. Most recently, it has been demonstrated that lentiviral TCR gene transfer to polyclonal human Tregs achieved human islet-specific and viral-specific CD4 T cell clones. This enabled antigen-specific suppression at increased potency compared to polyclonal Tregs, increasing optimism for the success of this approach (90). However, T cells transduced with islet-specific TCRs were less responsive to cognate antigen than T cells with virus-specific TCRs, suggesting further work in this area is needed. The animal model of multiple sclerosis, EAE, has been instrumental in testing gene-modified Tregs for therapeutic intervention in neurological autoimmune diseases. For example, a lentiviral gene delivery system was used to express a CAR targeting myelin oligodendrocyte glycoprotein with the murine FOXP3 in CD4 T cells. Intranasal administration of these cells diminished ongoing neuronal inflammation in vivo (61).
Several other attempts to utilize CAR+ Tregs to treat autoimmunity have revealed the important fact that activation of Tregs needs to be antigen-specific; this was found in murine colitis and arthritis models as well as in human Treg activation (91–93). This appears a critical point since in many autoimmune disorders autoantigen(s) that trigger autoimmune responses are unknown. In the case of murine arthritis, naïve CD4+ T-cells were engineered to co-express FOXP3 with HLA-DR1, covalently linked to an immunodominant peptide capable of driving collagen-induced arthritis. HLA-DR1 is associated with human rheumatoid arthritis. By this approach, T-cells were equipped with a bait molecule that allowed them to engage collagen autoreactive CD4+ T-cells in a TCR-dependent manner. In DR1 humanized mice, the engineered T-cells could inhibit the development of autoimmune arthritis more effectively than cells engineered to express FOXP3 alone (94). However, this approach warrants further studies, among other reasons because of the distinct subset of CD4+CD25+ Tregs expressing HLA-class II in humans (95).
Inducing FOXP3 Expression to Treat Autoimmunity
In addition to TCR modulation, modulation of FOXP3 expression itself is a promising strategy to treat autoimmune diseases. IPEX syndrome is a hereditary immunodeficiency characterized by the loss of function of FOXP3-expressing Tregs (11). A recent study demonstrated the lentiviral delivery of the FOXP3 gene into IPEX-derived CD4 T cells produced a stable Treg population. In this study, CD4 T cells from IPEX patients were converted into FOXP3-expressing Tregs, and they acquired Treg-like phenotypes in vivo. When FOXP3 is expressed by lentiviral gene transduction, T-cells express several Treg markers such as CD25, CTLA-4, and GITR. Functionally, the cells resembled Tregs with decreased proliferation, hypo-responsiveness, reduced cytokine release, and suppressive activity similar to purified Tregs (96). This approach for FOXP3 gene transfer with adoptive cell therapy may potentially be a promising approach to treat IPEX patients as well as other autoimmune patients with dysfunctional human Tregs. Further studies regarding the stability of FOXP3 expression in these CD4 T cells and further assessment of the efficacy of this approach in clinical settings are warranted.
Retroviral delivery of the FOXP3 gene into purified CD4+CD25−CD45RO− human T cells showed unstable levels of FOXP3 and Treg-associated phenotypic markers while lentiviral delivery using elongation factor-1α showed reliable expression of CD25 and GITR (97). An alternative approach may be to enforce Treg differentiation using a cell-permeable form of FOXP3 protein with a transduction domain. The introduction of FOXP3 in protein form induced a Treg phenotype in human and mouse T cells, respectively (98, 99). Repeated infusion of FOXP3 with a transduction domain showed amelioration of the scurfy phenotype, and inflammatory bowel disease and rheumatoid arthritis mouse models (100, 101). The cost of infusion for protein delivery in a clinical setting for human patients, the stability of a functional Foxp3 protein in vivo, and lack of specificity in immunosuppression due to Foxp3 protein delivery to the nucleus awaits further optimization of this approach.
Manipulation of Tregs in Allotransplantation and Other Diseases
Clinical evaluation of adoptive immunotherapy using Tregs is attracting increasing interest. Most experience has been gained using donor-derived Tregs, which have been infused safely in patients treated with allogeneic stem cells (102). These studies have also provided encouraging evidence of efficacy in prevention of GVHD, even in the context of haploidentical stem cell transplantation (102, 103).
Approaches to Generate Alloantigen-Specific Tregs
Similar to improved ability of autoantigen-specific Treg to control autoimmune inflammation, alloantigen-specific Tregs are more effective than polyclonal Tregs at preventing organ or tissue graft rejection. These alloantigen-specific Tregs were enriched by in vitro alloantigen-stimulated expansion or the expression of a TCR transgene (104–106). A humanized mouse model of skin graft rejection has also shown the potency of suppressor function of alloantigen-expanded human Tregs (107). Tregs expressing CARs could also be used in the context of transplantation. For example, a CAR approach targeting HLA-A2 has been used to produce alloantigen-specific Tregs (108). CAR-stimulated Tregs showed minimal cytotoxicity. In vitro, HLA-A2-CAR Tregs maintained high levels of FOXP3 expression and other Treg markers, and stable demethylation of the TSDR ensured suppressor function. The HLA-A2 approach may have significant advantages in the clinical setting where a sufficient number of APCs are required (107), and the potential loss of FOXP3 after repeated stimulation has been reported (109). With improved stability alloantigen-specific Tregs will have more versatile uses in future transplantation trials.
Other Gene-Modification Approaches for Generating Suppressor Lymphocytes
In vitro generation of Tr1 cells has been developed for clinical purposes. However, a major caveat of clinical use of Tr1 cell therapy is lack of purity. Andolfi et al. showed lentiviral delivery of IL-10 and GFP could generate a homogeneous Tr1 cell population to circumvent this issue (110). These “pure” Tr1 cells showed an anergic phenotype and TGF-β/IL-10 -dependent suppression of allogeneic T-cell responses and successfully controlled GVHD (110). Tr1 cells were generated in vitro using genetically modified B cells in an allergy model in an antigen-specific manner. Retroviral transduction of the fusion protein, Derp 2, a major house dust mite allergen, with an endosomal targeting sequence (gp75) was performed in B cells for efficient MHC class II presentation. The engineered B cells were adoptively transferred to the host (BALB/c mice) before or after peptide immunization. The production of IL-10 from these retrogenic B cells and the induction of IL-10 expressing Tr1 cells achieved allergen-specific immune tolerance against asthma (111). Although the result is encouraging, more studies with different types of allergens, or the use of humanized mouse models, could be considered to assess the potential scope of this approach.
Corneal allograft failure is mediated by CD4 T cells (112). CD25 in CD4 T cells plays an important role in the induction of corneal graft rejection. CD25-mediated signaling is associated not only with the expression of Treg cytokines (IL-10, TGF-β) but also T helper 1 type cytokines (IFN-γ, IL-1β, and TNF-α). A recent study showed that the use of CD25 siRNA in a corneal transplantation model significantly prolonged graft survival time on Sprague-Dawley rat recipients with Wistar rat donors (113). In this study, neovascularization and maintenance of transparency of the cornea were significantly improved. However, similar studies have not been extended to human patients, and the safety of this approach remains to be tested.
Future Perspective in Gene-Modified Treg Therapy
Recent technical advances and developments in the field of gene-modified Treg therapy have evolved into a new era. It is clear that the approach is very promising, yet several hurdles need to be overcome before broad clinical implementation. One of the biggest concerns is to ensure the purity of clinical products using GMP-based protocols. There is some concern about the stability of engineered Tregs and the fact that some Tregs might be converted into effector T cells, particularly into Th17 type cells (114, 115). Approaches that may be helpful to maintain FOXP3 expression have been discussed including all-trans-retinoic acid, IL-2, vitamin C or ex vivo treatment with rapamycin (116–119). For lentiviral gene transfer approaches, studies in the past showed long-term safety in human immunodeficiency virus patients who received gene-modified T-cells without genotoxic effects such as clonal expansion (120). Development in vector engineering has also achieved enhanced genetic stability and greater stability of transgene expression, providing greater safety (121). In TCR engineering, there is a concern about TCR cross-reactivity which is caused by recognition of low-affinity antigen by a TCR. There were two cases in which T cells were engrafted with an affinity-enhanced TCR selected for the tumor antigen, MAGE A3, and this TCR was found to have cross-reactivity and cardiovascular toxicity (122, 123).
Conclusion
The regulatory functions of Tregs and specificity to various types of stimuli triggered intense research efforts to develop these cells for various clinical treatments. For example, CAR-T cells, lentiviral gene transfer, small molecule compounds that regulate FOXP3 expression, and infusion of cell-permeable FOXP3 proteins were developed (Figure 1). Potential uncertainties of gene-modified Treg therapy remain, as well as the challenges of the manipulation of Tregs under GMP conditions, and concerns of effector-mediated toxicity due to lack of purity, unstable Treg phenotypes and TCR cross-reactivity. However, alternate approaches are being sought and tested and as the clinical data emerge, these challenges shift to the further evolution of innovative therapeutic approaches.
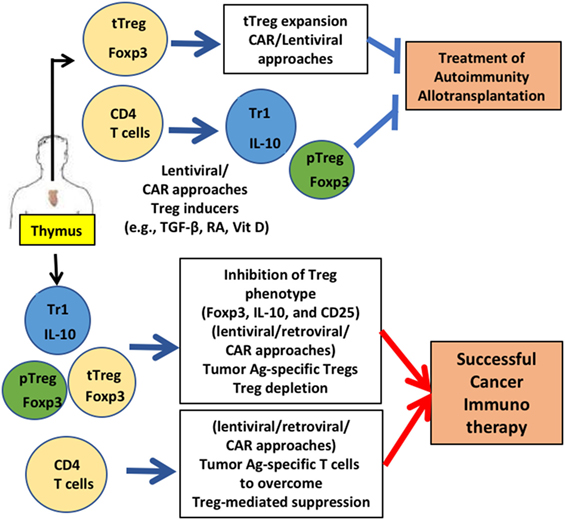
Figure 1. Gene-modified regulatory T cell (Treg)-mediated therapy. Engineering of Tregs aims to generate Tregs that are functionally similar to various types of Tregs that were identified in human: thymic-derived thymic-derived Treg (tTreg), transforming growth factor (TGF)-β-induced peripheral Treg (pTreg), and IL-10 expressing Tr1 cells. CD4 T cells and Tregs are generated from thymus. For treating autoimmunity or allotransplantation, gene-modification approaches aim to acquire a stable Treg phenotype and sufficient numbers via in vitro expansion to obtain enough cells for treatment. Generation of antigen-specific Tregs reduces the number of Tregs for therapy significantly. For successful cancer immunotherapy, Treg function needs to be downregulated. Destabilizing Treg functions by inhibiting FOXP3 and other functional Treg proteins (e.g., CD25) by gene modification is under development. Applications for these gene-modified Tregs are currently being expanded in mice (e.g., humanized mice) and men (human Tregs for clinical trials).
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. Diane McMahon-Pratt and Allison Ehrlich for their critical reading of this manuscript.
Funding
This work is supported by NIH R01CA168670, R21AI23858, R56AI27794, R21AI107957, and P30CA016359 from the National Cancer Institute (awarded to AB).
References
1. Gershon RK, Lance EM, Kondo K. Immuno-regulatory role of spleen localizing thymocytes. J Immunol (1974) 112(2):546–54.
3. Germain RN. Special regulatory T-cell review: a rose by any other name: from suppressor T cells to Tregs, approbation to unbridled enthusiasm. Immunology (2008) 123(1):20–7. doi:10.1111/j.1365-2567.2007.02779.x
4. Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol (2000) 164(1):183–90. doi:10.4049/jimmunol.164.1.183
5. Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol (2000) 18:423–49. doi:10.1146/annurev.immunol.18.1.423
6. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Pillars article: immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995. J Immunol (2011) 186(7):3808–21.
7. Tran DQ, Andersson J, Hardwick D, Bebris L, Illei GG, Shevach EM. Selective expression of latency-associated peptide (LAP) and IL-1 receptor type I/II (CD121a/CD121b) on activated human FOXP3+ regulatory T cells allows for their purification from expansion cultures. Blood (2009) 113(21):5125–33. doi:10.1182/blood-2009-01-199950
8. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi:10.1016/j.immuni.2004.08.010
9. Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol (2004) 34(11):2996–3005. doi:10.1002/eji.200425143
10. Yamaguchi T, Hirota K, Nagahama K, Ohkawa K, Takahashi T, Nomura T, et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity (2007) 27(1):145–59. doi:10.1016/j.immuni.2007.04.017
11. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
12. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27(1):68–73. doi:10.1038/83784
13. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest (2000) 106(12):R75–81. doi:10.1172/JCI11679
14. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi:10.1038/ni904
15. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi:10.1126/science.1079490
16. Wan YY, Flavell RA. Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A (2005) 102(14):5126–31. doi:10.1073/pnas.0501701102
17. Kim J, Lahl K, Hori S, Loddenkemper C, Chaudhry A, deRoos P, et al. Cutting edge: depletion of Foxp3+ cells leads to induction of autoimmunity by specific ablation of regulatory T cells in genetically targeted mice. J Immunol (2009) 183(12):7631–4. doi:10.4049/jimmunol.0804308
18. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science (2010) 329(5999):1667–71. doi:10.1126/science.1191996
19. Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, et al. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med (2007) 204(1):57–63. doi:10.1084/jem.20061852
20. Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature (2011) 474(7351):298–306. doi:10.1038/nature10208
21. Munn DH, Sharma MD, Lee JR, Jhaver KG, Johnson TS, Keskin DB, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science (2002) 297(5588):1867–70. doi:10.1126/science.1073514
22. Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science (2007) 317(5835):256–60. doi:10.1126/science.1145697
23. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol (2005) 6(12):1219–27. doi:10.1038/ni1265
24. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol (2013) 14(4):307–8. doi:10.1038/ni.2554
25. Chae WJ, Bothwell AL. Spontaneous intestinal tumorigenesis in Apc (/Min+) mice requires altered T cell development with IL-17A. J Immunol Res (2015) 2015:860106. doi:10.1155/2015/860106
26. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
27. Klunker S, Chong MM, Mantel PY, Palomares O, Bassin C, Ziegler M, et al. Transcription factors RUNX1 and RUNX3 in the induction and suppressive function of Foxp3+ inducible regulatory T cells. J Exp Med (2009) 206(12):2701–15. doi:10.1084/jem.20090596
28. Selvaraj RK, Geiger TL. Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3+ regulatory T lymphocytes through the induction of anergy and infectious tolerance. J Immunol (2008) 180(5):2830–8. doi:10.4049/jimmunol.180.5.2830
29. Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med (2013) 19(6):739–46. doi:10.1038/nm.3179
30. Gregori S, Goudy KS, Roncarolo MG. The cellular and molecular mechanisms of immuno-suppression by human type 1 regulatory T cells. Front Immunol (2012) 3:30. doi:10.3389/fimmu.2012.00030
31. Battaglia M, Stabilini A, Draghici E, Migliavacca B, Gregori S, Bonifacio E, et al. Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes (2006) 55(6):1571–80. doi:10.2337/db05-1576
32. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A (2006) 103(17):6659–64. doi:10.1073/pnas.0509484103
33. Chae WJ, Henegariu O, Lee SK, Bothwell AL. The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci U S A (2006) 103(25):9631–6. doi:10.1073/pnas.0600225103
34. Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, et al. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol (2006) 177(5):3133–42. doi:10.4049/jimmunol.177.5.3133
35. Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol (2007) 19(7):825–35. doi:10.1093/intimm/dxm043
36. Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, et al. Structure of the forkhead domain of FOXP2 bound to DNA. Structure (2006) 14(1):159–66. doi:10.1016/j.str.2005.10.005
37. Bandukwala HS, Wu Y, Feuerer M, Chen Y, Barboza B, Ghosh S, et al. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity (2011) 34(4):479–91. doi:10.1016/j.immuni.2011.02.017
38. Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep (2012) 1(6):665–75. doi:10.1016/j.celrep.2012.04.012
39. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature (2007) 445(7130):931–5. doi:10.1038/nature05478
40. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell (2012) 151(1):153–66. doi:10.1016/j.cell.2012.06.053
41. Toker A, Engelbert D, Garg G, Polansky JK, Floess S, Miyao T, et al. Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. J Immunol (2013) 190(7):3180–8. doi:10.4049/jimmunol.1203473
42. Wang L, Liu Y, Han R, Beier UH, Thomas RM, Wells AD, et al. Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell Biol (2013) 33(20):4106–15. doi:10.1128/MCB.00144-13
43. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A (2007) 104(11):4571–6. doi:10.1073/pnas.0700298104
44. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi:10.1182/blood-2009-02-207118
45. Chen Z, Barbi J, Bu S, Yang HY, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity (2013) 39(2):272–85. doi:10.1016/j.immuni.2013.08.006
46. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity (2013) 39(2):259–71. doi:10.1016/j.immuni.2013.05.018
47. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem (2014) 289(39):26872–81. doi:10.1074/jbc.M114.586651
48. Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem (2013) 288(34):24494–502. doi:10.1074/jbc.M113.467704
49. Rallon NI, Lopez M, Soriano V, Garcia-Samaniego J, Romero M, Labarga P, et al. Level, phenotype and activation status of CD4+FoxP3+ regulatory T cells in patients chronically infected with human immunodeficiency virus and/or hepatitis C virus. Clin Exp Immunol (2009) 155(1):35–43. doi:10.1111/j.1365-2249.2008.03797.x
50. Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) t regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med (2001) 193(11):1295–302. doi:10.1084/jem.193.11.1295
51. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med (2006) 203(7):1701–11. doi:10.1084/jem.20060772
52. Rodriguez-Perea AL, Arcia ED, Rueda CM, Velilla PA. Phenotypical characterization of regulatory T cells in humans and rodents. Clin Exp Immunol (2016) 185(3):281–91. doi:10.1111/cei.12804
53. Battaglia M, Roncarolo MG. The fate of human Treg cells. Immunity (2009) 30(6):763–5. doi:10.1016/j.immuni.2009.06.006
54. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity (2009) 30(6):899–911. doi:10.1016/j.immuni.2009.03.019
55. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3(+)CD4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22(6):679–84. doi:10.1038/nm.4086
56. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58(3):652–62. doi:10.2337/db08-1168
57. Gregori S, Roncarolo MG, Bacchetta R. Methods for in vitro generation of human type 1 regulatory T cells. Methods Mol Biol (2011) 677:31–46. doi:10.1007/978-1-60761-869-0_3
58. Gregori S, Passerini L, Roncarolo MG. Clinical outlook for type-1 and FOXP3(+) T regulatory cell-based therapy. Front Immunol (2015) 6:593. doi:10.3389/fimmu.2015.00593
59. Roncarolo MG, Gregori S, Bacchetta R, Battaglia M. Tr1 cells and the counter-regulation of immunity: natural mechanisms and therapeutic applications. Curr Top Microbiol Immunol (2014) 380:39–68. doi:10.1007/978-3-662-43492-5_3
60. Magnani CF, Alberigo G, Bacchetta R, Serafini G, Andreani M, Roncarolo MG, et al. Killing of myeloid APCs via HLA class I, CD2 and CD226 defines a novel mechanism of suppression by human Tr1 cells. Eur J Immunol (2011) 41(6):1652–62. doi:10.1002/eji.201041120
61. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation (2012) 9:112. doi:10.1186/1742-2094-9-112
62. Hoeppli RE, MacDonald KG, Levings MK, Cook L. How antigen specificity directs regulatory T-cell function: self, foreign and engineered specificity. HLA (2016) 88(1–2):3–13. doi:10.1111/tan.12822
63. Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol (2011) 23(5):679–84. doi:10.1016/j.coi.2011.06.006
64. Leslie M. Immunology. regulatory T cells get their chance to shine. Science (2011) 332(6033):1020–1. doi:10.1126/science.332.6033.1020
65. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi:10.1038/nri1806
66. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi:10.1126/science.1129003
67. Danke NA, Koelle DM, Yee C, Beheray S, Kwok WW. Autoreactive T cells in healthy individuals. J Immunol (2004) 172(10):5967–72. doi:10.4049/jimmunol.172.10.5967
68. Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A (1997) 94(5):1914–8. doi:10.1073/pnas.94.5.1914
69. Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood (2012) 119(15):3420–30. doi:10.1182/blood-2011-09-377051
70. Riviere I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther (2017) 25(5):1117–24. doi:10.1016/j.ymthe.2017.03.034
71. Curran KJ, Brentjens RJ. Chimeric antigen receptor T cells for cancer immunotherapy. J Clin Oncol (2015) 33(15):1703–6. doi:10.1200/JCO.2014.60.3449
72. Tsai BY, Suen JL, Chiang BL. Lentiviral-mediated Foxp3 RNAi suppresses tumor growth of regulatory T cell-like leukemia in a murine tumor model. Gene Ther (2010) 17(8):972–9. doi:10.1038/gt.2010.38
73. Amendola M, Passerini L, Pucci F, Gentner B, Bacchetta R, Naldini L. Regulated and multiple miRNA and siRNA delivery into primary cells by a lentiviral platform. Mol Ther (2009) 17(6):1039–52. doi:10.1038/mt.2009.48
74. Pallandre JR, Brillard E, Crehange G, Radlovic A, Remy-Martin JP, Saas P, et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: implications in graft-versus-host disease and antitumor immunity. J Immunol (2007) 179(11):7593–604. doi:10.4049/jimmunol.179.11.7593
75. Gamrad L, Rehbock C, Westendorf AM, Buer J, Barcikowski S, Hansen W. Efficient nucleic acid delivery to murine regulatory T cells by gold nanoparticle conjugates. Sci Rep (2016) 6:28709. doi:10.1038/srep28709
76. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang LC, et al. Inhibition of p300 impairs Foxp3(+) T regulatory cell function and promotes antitumor immunity. Nat Med (2013) 19(9):1173–7. doi:10.1038/nm.3286
77. Wang L, Kumar S, Dahiya S, Wang F, Wu J, Newick K, et al. Ubiquitin-specific protease-7 inhibition impairs Tip60-dependent Foxp3+ T-regulatory cell function and promotes antitumor immunity. EBioMedicine (2016) 13:99–112. doi:10.1016/j.ebiom.2016.10.018
78. Ghosh S, Taylor A, Chin M, Huang HR, Conery AR, Mertz JA, et al. Regulatory T cell modulation by CBP/EP300 bromodomain inhibition. J Biol Chem (2016) 291(25):13014–27. doi:10.1074/jbc.M115.708560
79. Park JH, Ko JS, Shin Y, Cho JY, Oh HA, Bothwell AL, et al. Intranuclear interactomic inhibition of FoxP3 suppresses functions of Treg cells. Biochem Biophys Res Commun (2014) 451(1):1–7. doi:10.1016/j.bbrc.2014.06.141
80. Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother (2011) 60(7):909–18. doi:10.1007/s00262-011-1046-y
81. Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood (2006) 108(9):2957–64. doi:10.1182/blood-2006-04-018218
82. Alvaro T, Lejeune M, Salvado MT, Bosch R, Garcia JF, Jaen J, et al. Outcome in Hodgkin’s lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res (2005) 11(4):1467–73. doi:10.1158/1078-0432.CCR-04-1869
83. Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay Nel H, et al. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res (2006) 12(2):465–72. doi:10.1158/1078-0432.CCR-05-1886
84. Scalapino KJ, Tang Q, Bluestone JA, Bonyhadi ML, Daikh DI. Suppression of disease in New Zealand Black/New Zealand White lupus-prone mice by adoptive transfer of ex vivo expanded regulatory T cells. J Immunol (2006) 177(3):1451–9. doi:10.4049/jimmunol.177.3.1451
85. Mukherjee R, Chaturvedi P, Qin HY, Singh B. CD4+CD25+ regulatory T cells generated in response to insulin B:9-23 peptide prevent adoptive transfer of diabetes by diabetogenic T cells. J Autoimmun (2003) 21(3):221–37. doi:10.1016/S0896-8411(03)00114-8
86. Lapierre P, Beland K, Yang R, Alvarez F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology (2013) 57(1):217–27. doi:10.1002/hep.26023
87. Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol (2003) 170(8):3939–43. doi:10.4049/jimmunol.170.8.3939
88. Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol (2002) 169(9):4712–6. doi:10.4049/jimmunol.169.9.4712
89. Masteller EL, Tang Q, Bluestone JA. Antigen-specific regulatory T cells – ex vivo expansion and therapeutic potential. Semin Immunol (2006) 18(2):103–10. doi:10.1016/j.smim.2006.01.004
90. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73. doi:10.1016/j.jaut.2017.01.001
91. Wright GP, Notley CA, Xue SA, Bendle GM, Holler A, Schumacher TN, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci U S A (2009) 106(45):19078–83. doi:10.1073/pnas.0907396106
92. Hombach AA, Kofler D, Rappl G, Abken H. Redirecting human CD4+CD25+ regulatory T cells from the peripheral blood with pre-defined target specificity. Gene Ther (2009) 16(9):1088–96. doi:10.1038/gt.2009.75
93. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology (2009) 136(5):1721–31. doi:10.1053/j.gastro.2009.01.049
94. Qian Z, Latham KA, Whittington KB, Miller DC, Brand DD, Rosloniec EF. Engineered regulatory T cells coexpressing MHC class II:peptide complexes are efficient inhibitors of autoimmune T cell function and prevent the development of autoimmune arthritis. J Immunol (2013) 190(11):5382–91. doi:10.4049/jimmunol.1300024
95. Baecher-Allan C, Wolf E, Hafler DA. MHC class II expression identifies functionally distinct human regulatory T cells. J Immunol (2006) 176(8):4622–31. doi:10.4049/jimmunol.176.8.4622
96. Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4(+) T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med (2013) 5(215):215ra174. doi:10.1126/scitranslmed.3007320
97. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16(1):194–202. doi:10.1038/sj.mt.6300341
98. Shen W, Liu J, Peng X, Zong Y, Xu X, Shao Q. [Effect of PTD-deltaPRD Foxp3 fusion protein on the mixed lymphocyte reaction in mice]. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2009) 34(12):1224–30.
99. Liu X, Xu X, Lin X, Tian Y, Ji B, Xia S, et al. PTD-hFOXP3 protein acts as an immune regulator to convert human CD4(+)CD25(-) T cells to regulatory T-like cells. J Cell Biochem (2012) 113(12):3797–809. doi:10.1002/jcb.24255
100. Choi JM, Shin JH, Sohn MH, Harding MJ, Park JH, Tobiasova Z, et al. Cell-permeable Foxp3 protein alleviates autoimmune disease associated with inflammatory bowel disease and allergic airway inflammation. Proc Natl Acad Sci U S A (2010) 107(43):18575–80. doi:10.1073/pnas.1000400107
101. Liu X, Ji B, Sun M, Wu W, Huang L, Sun A, et al. Cell-penetrable mouse forkhead box protein 3 alleviates experimental arthritis in mice by up-regulating regulatory T cells. Clin Exp Immunol (2015) 181(1):87–99. doi:10.1111/cei.12630
102. Reisner Y, Hagin D, Martelli MF. Haploidentical hematopoietic transplantation: current status and future perspectives. Blood (2011) 118(23):6006–17. doi:10.1182/blood-2011-07-338822
103. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi:10.1182/blood-2010-07-293795
104. Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest (2008) 118(11):3619–28. doi:10.1172/JCI33185
105. Golshayan D, Jiang S, Tsang J, Garin MI, Mottet C, Lechler RI. In vitro-expanded donor alloantigen-specific CD4+CD25+ regulatory T cells promote experimental transplantation tolerance. Blood (2007) 109(2):827–35. doi:10.1182/blood-2006-05-025460
106. Joffre O, Santolaria T, Calise D, Al Saati T, Hudrisier D, Romagnoli P, et al. Prevention of acute and chronic allograft rejection with CD4+CD25+Foxp3+ regulatory T lymphocytes. Nat Med (2008) 14(1):88–92. doi:10.1038/nm1688
107. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Transl Med (2011) 3(83):83ra42. doi:10.1126/scitranslmed.3002076
108. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi:10.1172/JCI82771
109. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol (2009) 39(4):1088–97. doi:10.1002/eji.200838904
110. Andolfi G, Fousteri G, Rossetti M, Magnani CF, Jofra T, Locafaro G, et al. Enforced IL-10 expression confers type 1 regulatory T cell (Tr1) phenotype and function to human CD4(+) T cells. Mol Ther (2012) 20(9):1778–90. doi:10.1038/mt.2012.71
111. Ahangarani RR, Janssens W, VanderElst L, Carlier V, VandenDriessche T, Chuah M, et al. In vivo induction of type 1-like regulatory T cells using genetically modified B cells confers long-term IL-10-dependent antigen-specific unresponsiveness. J Immunol (2009) 183(12):8232–43. doi:10.4049/jimmunol.0901777
112. Jia Z, Jiao C, Zhao S, Li X, Ren X, Zhang L, et al. Immunomodulatory effects of mesenchymal stem cells in a rat corneal allograft rejection model. Exp Eye Res (2012) 102:44–9. doi:10.1016/j.exer.2012.06.008
113. Qin Q, Luo D, Shi Y, Zhao Q, Chen Y, Wu J, et al. CD25 siRNA induces Treg/Th1 cytokine expression in rat corneal transplantation models. Exp Eye Res (2016) 151:134–41. doi:10.1016/j.exer.2016.08.010
114. Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol (2007) 178(11):6725–9. doi:10.4049/jimmunol.178.11.6725
115. Afzali B, Mitchell P, Lechler RI, John S, Lombardi G. Translational mini-review series on Th17 cells: induction of interleukin-17 production by regulatory T cells. Clin Exp Immunol (2010) 159(2):120–30. doi:10.1111/j.1365-2249.2009.04038.x
116. Zhou X, Kong N, Wang J, Fan H, Zou H, Horwitz D, et al. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol (2010) 185(5):2675–9. doi:10.4049/jimmunol.1000598
117. Tresoldi E, Dell’Albani I, Stabilini A, Jofra T, Valle A, Gagliani N, et al. Stability of human rapamycin-expanded CD4+CD25+ T regulatory cells. Haematologica (2011) 96(9):1357–65. doi:10.3324/haematol.2011.041483
118. Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med (2016) 213(3):377–97. doi:10.1084/jem.20151438
119. Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol (2011) 186(11):6329–37. doi:10.4049/jimmunol.1100061
120. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med (2012) 4(132):132ra53. doi:10.1126/scitranslmed.3003761
121. Knight S, Zhang F, Mueller-Kuller U, Bokhoven M, Gupta A, Broughton T, et al. Safer, silencing-resistant lentiviral vectors: optimization of the ubiquitous chromatin-opening element through elimination of aberrant splicing. J Virol (2012) 86(17):9088–95. doi:10.1128/JVI.00485-12
122. Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med (2013) 5(197):197ra103. doi:10.1126/scitranslmed.3006034
Keywords: regulatory T cell, autoimmunity, cancer, gene modification, therapy
Citation: Chae W-J and Bothwell ALM (2018) Therapeutic Potential of Gene-Modified Regulatory T Cells: From Bench to Bedside. Front. Immunol. 9:303. doi: 10.3389/fimmu.2018.00303
Received: 01 June 2017; Accepted: 02 February 2018;
Published: 16 February 2018
Edited by:
Christine Happle, Hannover Medical School, GermanyReviewed by:
Bin Li, Shanghai Jiao Tong University School of Medicine, ChinaBruno LUCAS, Centre national de la recherche scientifique (CNRS), France
Copyright: © 2018 Chae and Bothwell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wook-Jin Chae, d29va2ppbi5jaGFlQHlhbGUuZWR1;
Alfred L. M. Bothwell, YWxmcmVkLmJvdGh3ZWxsQHlhbGUuZWR1