 Alastair Copland1†
Alastair Copland1† Gil R. Diogo1†
Gil R. Diogo1† Peter Hart1†Shane Harris1Andy C. Tran1
Peter Hart1†Shane Harris1Andy C. Tran1 Mathew J. Paul1
Mathew J. Paul1 Mahavir Singh2
Mahavir Singh2 Simon M. Cutting3
Simon M. Cutting3 Rajko Reljic1*
Rajko Reljic1*
- 1St George’s Medical School, London, United Kingdom
- 2Lionex GmbH, Braunschweig, Germany
- 3School of Biological Sciences, Royal Holloway University of London, Egham, United Kingdom
Tuberculosis (TB) is the most deadly infectious disease in existence, and the only available vaccine, Bacillus Calmette-Guérin (BCG), is almost a century old and poorly protective. The immunological complexity of TB, coupled with rising resistance to antimicrobial therapies, necessitates a pipeline of diverse novel vaccines. Here, we show that Bacillus subtilis spores can be coated with a fusion protein 1 (“FP1”) consisting of Mycobacterium tuberculosis (Mtb) antigens Ag85B, ACR, and HBHA. The resultant vaccine, Spore-FP1, was tested in a murine low-dose Mtb aerosol challenge model. Mice were primed with subcutaneous BCG, followed by mucosal booster immunizations with Spore-FP1. We show that Spore-FP1 enhanced pulmonary control of Mtb, as evidenced by reduced bacterial burdens in the lungs. This was associated with elevated antigen-specific IgG and IgA titers in the serum and lung mucosal surface, respectively. Spore-FP1 immunization generated superior antigen-specific memory T-cell proliferation in both CD4+ and CD8+ compartments, alongside bolstered Th1-, Th17-, and Treg-type cytokine production, compared to BCG immunization alone. CD69+CD103+ tissue resident memory T-cells (Trm) were found within the lung parenchyma after mucosal immunization with Spore-FP1, confirming the advantages of mucosal delivery. Our data show that Spore-FP1 is a promising new TB vaccine that can successfully augment protection and immunogenicity in BCG-primed animals.
Introduction
In 2015, tuberculosis (TB) overtook HIV/AIDS as the leading cause of death due to infection (1). This statistic contrasts starkly with the fact that the only available TB vaccine, Mycobacterium bovis Bacillus Calmette-Guérin (BCG), is the most widely administered vaccine in history (2), having been developed almost a century ago. The proposed reasons for the failure of BCG to adequately protect against TB are many and varied. They include (i) BCG sub-strain heterogeneity (3), (ii) pre-exposure of the host to environmental non-tubercle mycobacteria (4), (iii) a failure to prevent pulmonary infection (5), and (iv) limited protection in adults compared to children (6). Despite these limitations, BCG is unlikely to be discontinued in clinical use. While its efficacy in many demographics is modest, there are accumulating data indicating that BCG may protect against non-TB diseases by training the innate immune system to respond non-specifically to diverse microbial threats (7, 8). A novel TB vaccine is therefore likely to supplement, rather than replace, BCG.
In 2011, the novel viral vector TB vaccine MVA85A, comprising Ag85A, was tested for safety and efficacy in a phase 2 clinical trial in South Africa, and it was found that parenteral administration of MVA85A in BCG-immunized infants offered no significant protection above that of BCG alone (9). The reasons for its failure are still unclear, since MVA85A protected against Mycobacterium tuberculosis (Mtb) in multiple animal models (10). But it is becoming increasingly apparent that the development pipeline for new TB vaccines will require technological diversity in order to maximize chances of success. In recent years, vaccines that are based upon particulate nano- or microscale delivery systems have made remarkable strides in both oncology and infectious diseases (11–13).
Bacillus subtilis is an environmental Gram-positive bacterium that is also found as a gut commensal in humans (14). Its spores have the desirable properties of being both safe and adjuvantic (15). But more importantly, they possess hydrophobic and electrostatic properties that allow them to readily bind protein antigens, making these spores pertinent to vaccine development as potential antigen delivery systems (16). The combination of intrinsic adjuvanticity and antigen-binding biophysical properties allows B. subtilis spores to act simultaneously as adjuvants and antigen carriers. Studies have shown that immunization of mice with B. subtilis spores coated with the influenza antigen M2e can induce strong antibody responses and protect against lethal challenge (17, 18). Similar findings have been observed in other immunization models, including immunogenicity against HIV and streptococci (19, 20). B. subtilis spores are thus an attractive platform for subunit vaccine enhancement.
We have previously shown that B. subtilis spores coated with TB antigens (21) or genetically engineered to express a TB antigen (22) can enhance protection against TB by BCG (prime-boost) in a mouse intranasal infection model. Although this provided a proof-of-principle framework for vaccine efficacy, the use of genetically modified components in a vaccine presents numerous regulatory barriers for clinical application (23). Here, we developed a novel TB vaccine—“Spore-FP1”—composed of B. subtilis spores non-covalently coated with a fusion protein (FP1) consisting of the antigens Ag85B, ACR and the epithelium-binding domain of HBHA (“FP1”). Ag85B and ACR were chosen to represent early and late stages of Mtb infection, respectively, while HBHA (heparin-binding domain only) was used for epithelial targeting in the lungs. Mucosal booster immunization with Spore-FP1 in BCG-primed mice enhanced protection in a low-dose aerosol Mtb challenge model, compared to BCG alone. The enhanced protection was concomitant with a wide array of boosted immunological parameters, including enhanced antigen-dependent T-cell proliferation and antibody production. Spore-FP1 is therefore a novel TB vaccine that has the potential to supplement pre-existing immunity conferred by BCG in human populations.
Materials and Methods
Ethics Statement
All animals were used with approval from St. George’s University of London Ethics Committee under an approved Home Office animal project license (70/7490) and used in accordance with the Animals (Scientific Procedures) Act 1986.
Mice and Immunizations
Female C57BL/6 mice were obtained from Charles River, UK, and were between 8 and 12 weeks of age before experimental use. For all bacterial challenge or immunogenicity experiments (except lung T-cell analysis), mice were immunized with 5 × 105 CFU BCG Pasteur (100 µL) subcutaneously or vehicle control. Intranasal booster immunizations consisted of 1 × 109 B. subtilis spores coated with 10 µg FP1 in 40 µL volumes per animal per dose, or vehicle control. These immunizations were performed under light anesthesia. For experiments involving dead spores, spores were autoclaved before protein adsorption. For some experiments, the adjuvant poly(I:C) (Sigma-Aldrich) was used intranasally at a dose of 20 µg.
Mice were infected with approximately 200 M. tuberculosis bacilli per animal delivered via low-dose aerosol, using a Biaera aerosol generator (Biaera Technologies). Infectious dose was routinely verified by standard plating techniques.
Bacteria and Colony Forming Unit Quantification
Bacillus Calmette-Guérin Pasteur and Mtb (strain H37Rv) were used for the in vivo experiments; these were kind gifts of Professor Juraj Ivanyi (King’s College, London). Both strains were grown to log phase at 37°C in 7H10 broth (Becton Dickinson) supplemented with ADC (Becton Dickinson), 0.05% Tween-80 and Selectab (Mast Diagnostics). Bacteria were then enumerated by the standard CFU method on 7H11 agar plates [supplemented with OADC (Becton Dickinson), glycerol and Selectab (Mast Diagnostics)] and cryopreserved in liquid nitrogen until use.
Bacterial burden from mouse organs was assessed by CFU enumeration. Lung and spleen homogenates were prepared in a stomacher containing 0.1% Triton X-100. Homogenates were plated in technical duplicates (lungs) or singlets (spleens) on 7H11 agar supplemented with OADC, glycerol and Selectatab. CFUs were counted after a 3–4-week incubation at 37°C.
Vaccines and Immunization
Amino acid sequences of the Mtb proteins ACR, Ag85B (pos. 23–25), and HBHA (pos. 160–199) were connected via linker peptides (GGGSGGGS), and six histidine residues were added to the C-terminus resulting in FP1. The amino acid sequence of FP1 was retranslated to DNA considering the codon usage of Escherichia coli DH5α, the host strain for protein production. In order to enable site-directed cloning, restriction sites for NcoI and HindIII were added to the 5′ and 3′ end, respectively. The synthetic gene was provided by GenScript (USA) inserted in pUC57. The gene of FP1 was excised from this plasmid using the abovementioned restriction endonucleases and ligated to expression vector pLEXWO481, an IPTG-inducible derivative of pMV261 (24), digested with the same enzymes as before. For production of FP1, the gene was expressed under control of lac-promoter while growing the host strain in APS medium at 30°C. Recombinant protein was isolated from inclusion bodies after denaturation in 8 M urea using metal chelate chromatography (Ni-NTA Superflow, Qiagen). Highly enriched FP1 was refolded by gel-filtration using sephadex G-200 material (GE Healthcare). Purity was assessed by fully automated SDS-PAGE with fluorimetric detection and densitometric purity (>97% purity). Western blots specific for component antigens were used to confirm the identity of the protein band. Endotoxin content was measured by LAL assay and determined to be <7 IU/mg. For formulation, FP1 was incubated with spores for 1 h at room temperature prior to the addition of polyI:C (if used). Vaccines were delivered immediately after formulation.
Antibody and Antigen Quantification
Antibody levels (IgA and IgG) in BAL and serum were quantified by ELISA. Antigens [Ag855, ACR; Lionex GmbH (Braunschweig, Germany)] were coated onto a plate at 2 µg/mL overnight, followed by blocking for 2 h with PBS containing 1% bovine serum albumin (BSA). BAL and serum were diluted 1:250 and 1:1,000, respectively, in PBS with 1% BSA and incubated on the plate in triplicate for 1 h at 37°C. Levels of IgA or IgG were detected using peroxidase-conjugated anti-mouse IgA or anti-mouse IgG (Sigma) and OPD substrate (Sigma). Plates were read on a Tecan200 plate-reader at 450 nm absorbance.
Assessment of protein loading onto spores and stability of final product was done by ELISA (quantifying unbound protein after adsorption) and by measuring charge and size using a ZetaSizer NanoZS (Malvern) according to manufacturer’s instructions and proprietary software. Significance was tested with a paired t-test. For ELISA measurements, FP1 was coated onto plates at varying concentrations as described for the antibody measurements, followed by detection by a peroxidase-conjugated anti-His antibody (Sigma) in conjunction with OPD substrate. Plates were read as described above.
General Flow Cytometry
For most experiments, cells were first stained with Fixable Viability Dye eFluor® 780 (1:1,000 dilution; eBioscience) in the presence of Fc receptor blockade (TruStain, 1:500 dilution; Biolegend). For surface staining, cells were then stained in flow cytometry buffer (PBS containing 0.5% BSA and 0.1% sodium azide—all from Sigma-Aldrich) for 30–45 min at 4°C. For some experiments, cells were subsequently fixed in the appropriate fixative for 30 min at 4°C, and then stained in a permeabilization buffer for 45 min, followed by acquisition on a BD FACSCanto II, unless otherwise specified. For compensation matrices, UltraComp beads were used according to the manufacturer’s instructions (eBioscience). Staining boundaries were determined by a combination of antibody titration, biological controls and fluorescence-minus-one samples.
Antigen-Presenting Cell (APC) Activation
Dendritic cells (DCs) were obtained according to a well-established protocol (25). Briefly, mouse femurs were aseptically flushed with complete RPMI (RPMI-1640 containing 100 U/mL penicillin/streptomycin, 2 mM l-glutamine, 10% fetal calf serum, and 50 µM 2-mercaptoethanol—all from Sigma-Aldrich) and the bone marrow cells were cultured in complete RPMI with 50 ng/mL GM-CSF (Peprotech) for 2 days, followed by complete removal of the liquid media containing non-adherent granulocytes, and replacement with fresh GM-CSF-supplemented media. Cells were then cultured for a further 3–4 days, and non-adherent and loosely adherent cells were gently detached. DCs were phenotyped by flow cytometry and were found to be>85% CD11c+ and expressing high levels of MHC Class II. DCs were cryopreserved in 10% DMSO until use. For experiments involving macrophages, the J774 cell line was used. Macrophages were cultured in complete DMEM (from Sigma, see RPMI), and sub-cultured every 3 days at ~80% confluency. Cells were>99% CD11b+ as assessed by flow cytometry.
To measure activation, APCs were stimulated for 48 h with B. subtilis spores at an MOI of 1, 10, or 100, or E. coli LPS (100 ng/mL; Sigma-Aldrich), and stained with a panel of antibodies: CCR7-PerCP/Cy5.5, CD80-APC, CD86-PE/Cy7, MHC Class I-FITC, MHC Class II-Brilliant Violet 510, PD-L1-Brilliant Violet 421, and PD-L2-PE—all from Biolegend. Supernatants were tested for IL-1β, IL-6, and TNF-α. IL-12p40 was detected by intracellular cytokine staining after 20 h stimulation of macrophages in the presence of 10 µg/mL brefeldin A (Sigma-Aldrich) using IL-12p40-PE (Biolegend) in flow cytometry buffer containing 0.5% saponin (Sigma-Aldrich). To detect transcription factor phosphorylation, macrophages were stimulated for 4 h and then fixed in 90% methanol as previously described (26), followed by staining with antibodies against phosphorylated forms of c-Jun (AP-1), NF-κB (p60), and IRF-3 (Cell Signaling).
T-Cell Proliferation
Splenocytes were obtained from mouse spleens that had been mechanically homogenized and treated with ACK lysis buffer (Sigma-Aldrich) to remove erythrocyte contamination. Splenocytes were cultured at 1 × 106/well in a 96-well plate. Cells were stimulated with 5 µg/mL recall antigen or 1 µg/mL α-CD3 (Biolegend) for 5–6 days, followed by surface staining with CD4-PerCP/Cy5.5, CD8-Brilliant Violet 510, CD44-FITC, CD62L-PE, and CD90.2-Brilliant Violet 421—all from Biolegend. Cells were then fixed and permeabilized using the eBioscience Foxp3/Transcription Factor Staining Buffer Set and stained with Ki67-APC.
Cytokine Quantification
Cytokines were measured by ELISA or Multiplex immunoassay. IL-1β, IL-6, and TNF-α in culture supernatant were measured by ELISA using eBioscience Ready-Set-Go kits according to the manufacturer’s instructions. For IL-4, IL-10, IL-17A, and IFN-γ, a 4-plex multiplex immunoassay (Biolegend) was used according to manufacturer’s instructions. Data was acquired on a BD FACSCanto, and analysis performed using Legendplex software (Biolegend).
Lung Cell Isolation and Analysis
For these experiments, mice were immunized with PBS or BCG (subcutaneously) or mucosally with the indicated vaccine component. Lungs were perfused of blood by flushing PBS through the right ventricle. Tissue was then dissected into 1 mm pieces using a scalpel, followed by digestion in 1 mg/mL collagenase and 0.5 mg/mL DNase I (Roche). Cells were then passed through a 70 µm strainer (Becton Dickinson), contaminating erythrocytes were lysed, and mononuclear cells were stained for CD3-APC, CD4-PerCP/Cy5.5, CD8-Brilliant Violet 510, CD44-FITC, CD62L-PE, CD69-PE/Cy7, and CD103-Brilliant Violet 421—all from Biolegend.
Statistical Analysis
Statistical tests are described in the relevant figure legends. All analysis was performed using FlowJo v10, Microsoft Excel 2010 and GraphPad Prism 7.
Results
Spores Can Effectively Bind to Mtb Fusion Proteins
We tested whether a biological carrier system, such as spores, could bind and facilitate carriage of the FP-1 fusion-protein. Spores were incubated with FP-1 prior to centrifugation to quantity the amount of free FP1 present in the supernatant after adsorption. Of 100 µg FP1, less than 2 µg free FP1 was detected in supernatants after adsorption indicating a high binding efficiency of over 98% (n = 3). To characterize the formulated Spore-FP1 vaccine, we assessed the size and charge of Spores either pre- or postadsorption using a ZetaSizer NanoZS. Size of the spores increased following FP-1 adsorption from 1,337 ± 13.8 to 1,389 ± 13.53 nm for naked and loaded spores, respectively (n = 3). Spores became marginally more negative when FP-1 was loaded, with charge decreasing from −47.13 ± 0.95 to −49.1 ± 0.44 mv (n = 3). While neither of the changes were significant, both trended toward significance (p = 0.064 and 0.068 for charge and size, respectively) and, in conjunction with the ELISA data, are suggestive of protein loading onto the spore surface. Importantly, FP1 loaded spores were moderately negatively charged and had a low polydispersity index (PDI = 0.237), indicating strong colloidal stability.
Spore-FP1 Can Enhance Bacterial Control Afforded by BCG
Mucosal vaccination against respiratory diseases offers distinct advantages over parenteral delivery routes, such as enhanced control of the pathogen, presumably due to localized immune effector cells (27). We therefore tested the ability of mucosally-delivered Spore-FP1 to enhance control of Mtb, compared to BCG immunization alone, in a “prime-boost” strategy. We hypothesized that a booster immunization with Spore-FP1 would lead to better protection than immunization with BCG alone. Mice were first primed with BCG or vehicle control, followed by two intranasal boosts with Spore-FP1. Mice were then challenged with ~100 CFU aerosolized Mtb, and bacterial burdens were quantified in the lungs and spleen. As shown in Figure 1A, mice immunized with BCG alone were better able to control Mtb compared to mock-immunized mice, as evidenced by reduced CFUs in the lungs (PBS CFUs: 5.54 ± 0.06; BCG CFUs: 4.84 ± 0.18, p < 0.05). When mice received the Spore-FP1 booster immunization, however, there was a significant improvement compared to BCG alone, with a near −1 log reduction in bacterial burden (Spore-FP1 CFUs: 4.10 ± 0.20, p < 0.0001 vs. PBS, p < 0.05 vs. BCG). In the spleens, BCG and Spore-FP1 offered comparable levels of protection, with both groups exhibiting significant protection compared to mock immunization (p < 0.0001). Next, Spore-FP1 was tested with codelivery of the adjuvant poly(I:C), a known inducer of the Th1 subset when used in the respiratory tract (28). As can be seen in the lungs (Figure 1B), Spore-FP1 was again able to induce significantly better protection than BCG (p < 0.01), with a trend—though not a statistically significant difference—for increased protection in the spleen (p = 0.06, BCG vs. Spore-FP1). These data collectively demonstrate that Spore-FP1 immunization could improve protection offered by BCG alone in multiple contexts, and therefore we sought to uncover any immunological phenomena that could be associated with efficacy.
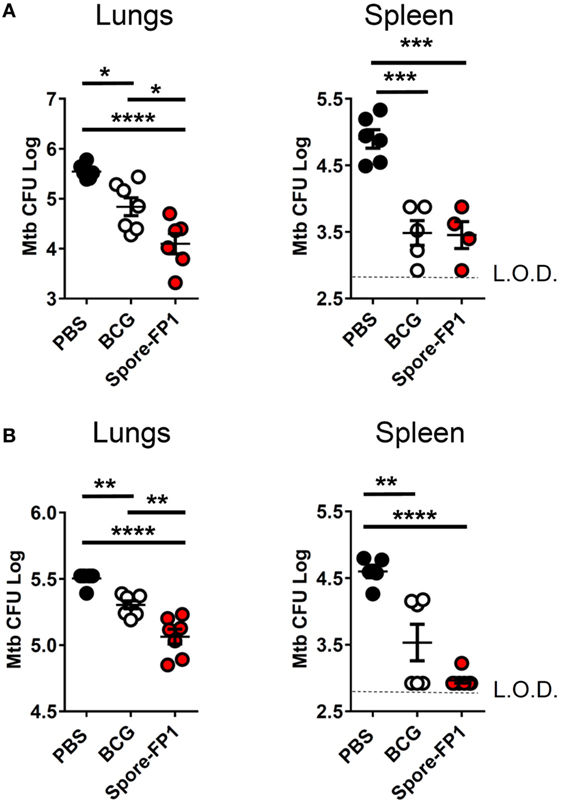
Figure 1. Spore-FP1 protects against aerosol Mycobacterium tuberculosis (Mtb) challenge. Mice received a Bacillus Calmette-Guérin subcutaneous prime (except the PBS control group) followed by two intranasal boosts with Spore-FP1. After 3 weeks, bacterial burdens in the lungs and spleens were quantified by CFU counting on 7H11 plates across three dilution ranges. (A) Mice were immunized with Spore-FP1 alone. (B) Mice were immunized with Spore-FP1 in combination with the adjuvant poly(I:C). Results are expressed as mean ± SEM. Data are derived from n = 4–7 individual mice. Significance was tested against the by one-way ANOVA with Tukey’s posttest, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Spore-FP1 Enhances Antigen-Specific Antibody Production
Evidence suggests that antibodies may play a role in protecting against TB, either directly (Fab-mediated) or indirectly (Fc-mediated): monoclonal IgA therapy can reduce pulmonary Mtb burden (29), and adoptive transfer of antibodies from hosts with latent TB can improve macrophage functionality (30). We therefore probed whether Spore-FP was generating antibodies against Ag85B and ACR. Spore-FP1 immunization significantly enhanced titers of Ag85B-specific IgA in the BAL compared to PBS (p < 0.05), whereas there was no difference with BCG (Figure 2A). There was also a trend for increased levels of Ag85B-specific IgG induced by Spore-FP1. With regards to ACR-specific antibodies (Figure 2B), Spore-FP1 was able to significantly enhance levels of α-ACR IgG in the serum, compared to PBS (p < 0.01). However, there were no changes in the α-ACR IgA within the BAL.
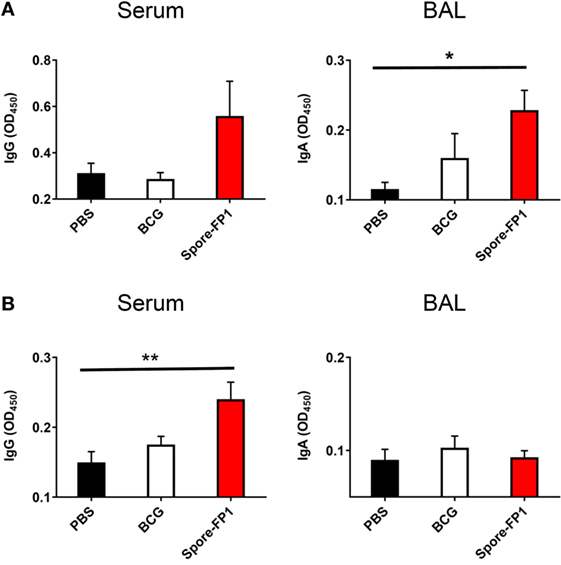
Figure 2. Enhanced humoral immunity caused by Spore-FP1. Immunized mice were tested for the presence of antigen-specific IgG in the serum (1:1,000 dilution) and IgA in the BAL (1 mL PBS flush; 1:10 dilution) by ELISA, with optical density read at 450 nm in duplicate. (A) Levels of IgG and IgA specific to Ag85B. (B) Levels of IgG and IgA specific to ACR. Results are expressed as mean ± SEM. Data shown are derived from n = 3 individual mice and are representative of two independent experiments. Significance was tested against the unstimulated control by one-way ANOVA with Tukey’s posttest, *p < 0.05 and **p < 0.01.
Spore-FP1 Generates Abundant Tcm and Tem Cells with High Proliferative Capacity
The observation that Spore-FP1 immunization led to higher Mtb-specific IgG and IgA titers suggested that T-cell immunity was also being modulated. We hypothesized that Spore-FP1 was inducing stronger T-cell immunity than BCG alone, leading to enhanced antibody levels. To test this, splenocytes from immunized mice were assessed for the expression of the cell cycle and proliferation marker Ki67 after exposure to the recall antigens Ag85B, ACR and FP1. The Ki67+ cells were then divided into naive (CD44loCD62Lhi), T central memory (Tcm; CD44hiCD62Lhi) or T effector memory (Tem; CD44hiCD62Llo) phenotypes. As shown in Figure 3, as expected, there was minimal proliferation in the PBS group in response to all antigens, with a background level of ~3% Ki67+ in memory cell subsets. There were modestly more proliferating cells in the BCG group, which is consistent with other studies showing that BCG induces a very small percentage of antigen-specific splenic T-cells (31, 32). For instance, there were 6.48% Ki67+ CD4+ Tem cells after ACR stimulation in this group, and a similar level in the CD8+ Tem cells. However, in the Spore-FP1 group, there was a sharp overall increase in the percentage of Ki67+ cells, with notable spikes (>20%) in proliferating CD8+ Tcm and Tem cells in response to Ag85B. Similarly, Spore-FP1 had the highest percentage of CD4+ Tem cells responding to Ag85B (>10%). Results for ACR in this group were more modest, but the trend remained consistent. These data support the ability of a mucosal vaccine to induce substantial T-cell responses at primary lymphoid sites.
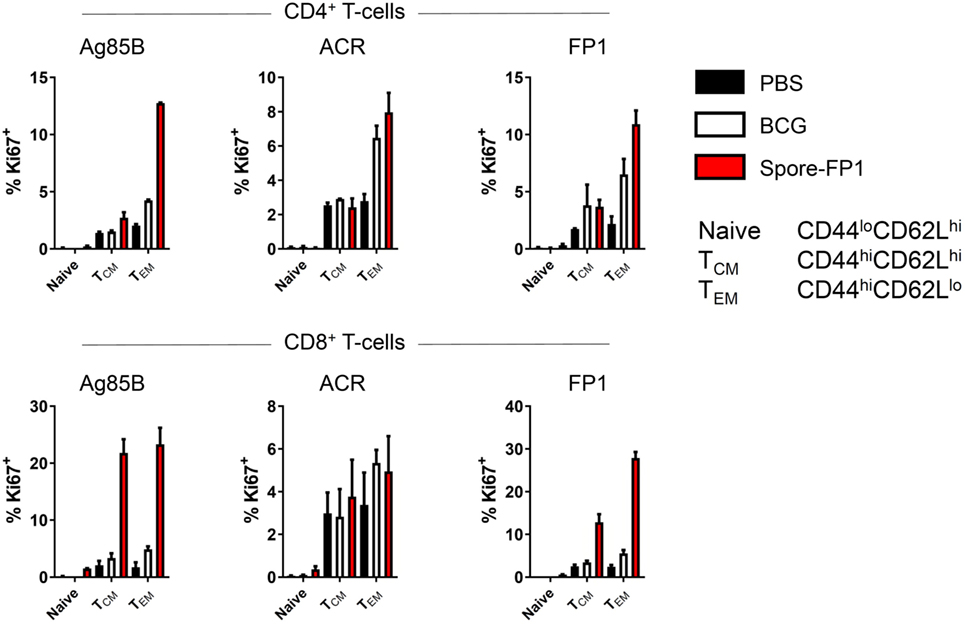
Figure 3. Enhanced T-cell proliferation due to Spore-FP1. Splenocytes were incubated in technical duplicates with 5 µg/mL recall antigen for 5–6 days and proliferation was measured by Ki67 staining. A gating strategy of live cells→single cells→CD3+→CD4+/CD8+ was used, followed by gating for Ki67+ cells and determination of memory cell phenotype by expression of CD44 and CD62L. Results are expressed as mean ± SEM. Data are derived from n = 3 pooled spleens per group.
Spore-FP1 Immunization Results in a Mixed T-Cell Cytokine Profile
The increase in T-cell proliferation in response to mucosal immunization with Spore-FP1 led us to question which subsets of T helper cells and cytotoxic T-cells were responding to antigen. Therefore, splenocytes from immunized animals were cultured with recall antigens (Ag85B, ACR and FP1) and assessed for the production of IFN-γ, IL-4, IL-10, and IL-17A, which are secreted from Th1/Tc1, Th2/Tc2, Treg, and Th17/Tc17 subsets, respectively. We found (Figure 4) that there was muted cytokine production across all analytes in the BCG group when cells were stimulated with recall antigens, with the exception of minor IFN-γ secretion (570.36 pg/mL) after ACR pulsing. In the Spore-FP1 group, however, there was profound cytokine release in response to all three antigens. After Ag85B pulsing, Spore-FP1 splenocytes produced copious amounts of IFN-γ (3519.6 pg/mL), IL-10 (86.26 pg/mL), and IL-17A (1837.5 pg/mL), suggesting that Spore-FP1 immunization generated mixed T-cell subsets that were specific for Mtb antigens. Similar results were observed for FP1 antigen recall, and there were modest levels of cytokines for ACR. No IL-4 was detected in any of the groups.
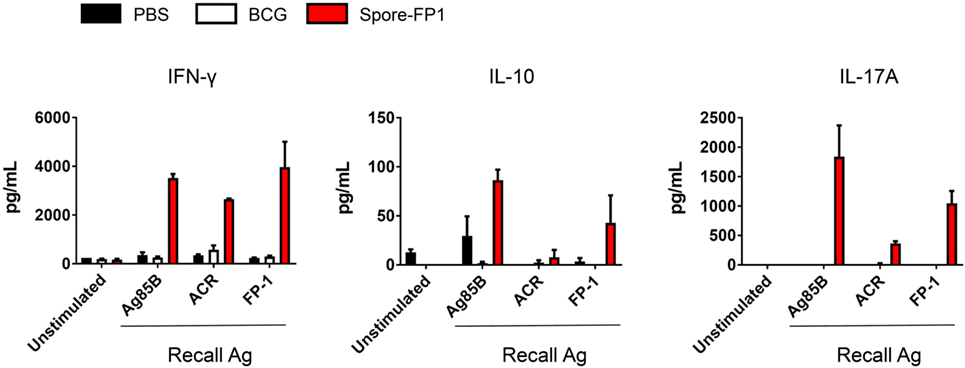
Figure 4. Cytokine profiles during splenocyte antigen recall. Splenocytes from immunized mice were stimulated in technical duplicates with 5 µg/mL recall antigen for 5–6 days and T-cell cytokines were measured by multiplex flow cytometry. Results are expressed as mean ± SEM. Data are derived from n = 3 pooled spleens per group.
Evidence of Tissue-Resident Memory T-Cells after Mucosal Immunization
Since Spore-FP1 was causing the development of antigen-specific T-cells in central lymphoid organs, we next interrogated the lungs for the presence of tissue-resident memory T-cells. Lungs were perfused and harvested from immunized animals, and then CD44hi (i.e., memory) T-cells were assessed for the expression of tissue retention markers CD69 and CD103. As shown in Figure 5, PBS and BCG immunization induced minimal levels of these cells (<4% in both CD4+ and CD8+ T-cells), with only a minor increase induced by FP1 alone. Notably, the mucosal delivery of B. subtilis spores alone did not lead to the generation of Trm, while the full vaccine construct, Spore-FP1, was able to induce 14.9% CD4+ and 12.5% CD8+ Trm, respectively.
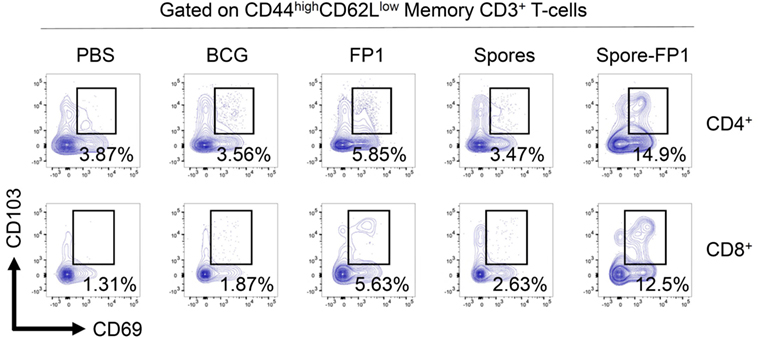
Figure 5. Spore-FP1 induces enrichment of tissue resident memory cells. Mice were first immunized with Bacillus Calmette-Guérin for 6 weeks (except the PBS group) and then received two intranasal doses of either spores alone, fusion protein 1 (FP1) alone, or Spore-FP1. Lung parenchymal cells were assessed by flow cytometry for T-cell markers. A gating strategy of live cells→single cells→CD3+→CD4+/CD8+→CD44hiCD62Llo was used to measure the frequency of double-positive CD69/CD103 Trm. Data are derived from n = 3 pooled mice per group showing a representative plot.
Bacillus Spores Activate Macrophages and DCs
Antigen-presenting cells are essential for the generation of T-cell immunity after immunization (33, 34). Empirical and systems biology approaches have revealed a correlation between APC activation by some antibody-inducing vaccines and protective immunity (35–37). Consequently, we next tested whether B. subtilis spores could activate DCs and macrophages (Figure 6). DCs and macrophages were pulsed with B. subtilis spores for 2 days at a range of MOIs and assessed for the upregulation of maturation markers. In DCs, spores significantly upregulated CD80, MHC Class I and CCR7 (CD80: p < 0.05, MHC Class I: p < 0.001, CCR7: p < 0.01), with strong trends for upregulation of CD86 and PD-L1 (Figure 6A). Interestingly, spores induced the downregulation of MHC Class II and PD-L2. This may reflect the time-point at which the markers were measured, since Class II is known to be upregulated initially, followed by late-phase downregulation in activated DCs (38). In macrophages, spores were able to induce the upregulation of PD-L1 (p < 0.05), PD-L2 (p < 0.001), and CCR7 (p < 0.001), with similar trends for CD80, CD86, and MHC Class II. With regards to the production of proinflammatory cytokines, there was evidence of secretion of IL-6 and TNF-α at an MOI of 100 in DCs and macrophages (Figure 6B), albeit at lower levels compared to the positive control LPS. There was modest IL-1β production at the highest dose of spores. Next, we used intracellular cytokine staining to detect IL-12p40 production after spore exposure (Figure 6C). Spores were found to induce comparable levels of IL-12p40+ APCs (23.3%) compared to LPS (18.3%). As expected, there was minimal production of IL-12p40 in unstimulated cells. Finally, we investigated three transcription factors downstream of Toll-like receptors (TLRs) to understand the cause of the phenotype. Intracellular flow cytometry at 4 h poststimulation (Figure 6D) revealed that B. subtilis spores were activating AP-1 (c-JUN), NF-κB and IRF-3 to a greater extent than LPS, suggesting engagement of both MyD88 and TRIF adaptors, in tandem with mitogen-activated protein kinase activity.
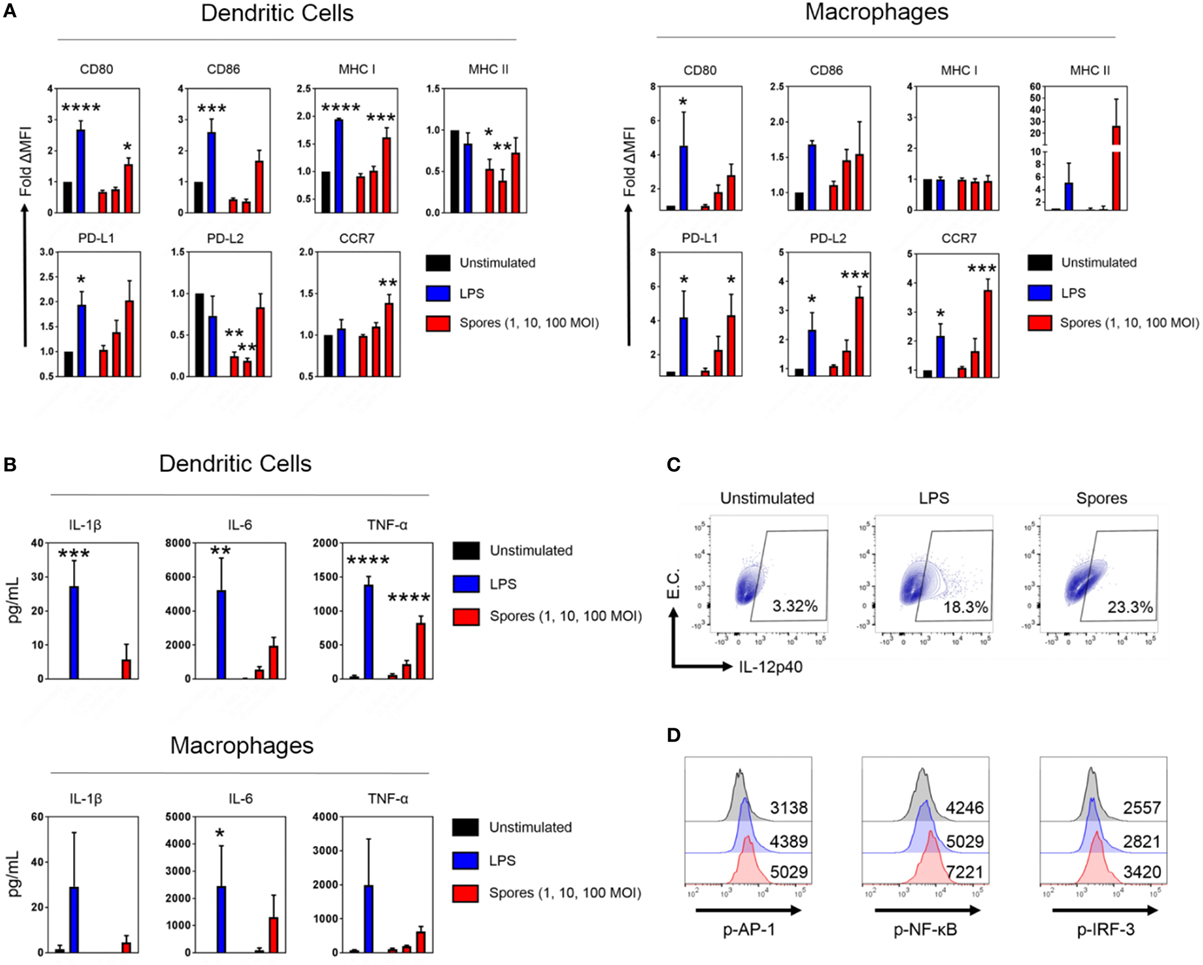
Figure 6. Bacillus subtilis spores activate antigen-presenting cells. (A) Dendritic cells (DCs) (left) and macrophages (right) were stimulated in duplicate for 48 h with LPS (100 ng/mL) or B. subtilis spores (1, 10, and 100 MOI) and surface molecule expression was measured by flow cytometry on gated viable cells. MFI was normalized to the unstimulated control. (B) Cytokines from the supernatants were tested for proinflammatory cytokine production by ELISA. (C) Macrophages were stimulated for 20 h with LPS (100 ng/mL) or B. subtilis spores (100 MOI) in the presence of brefeldin A (10 µg/mL), followed by intracellular detection of IL-12p40. EC, empty channel. A representative experiment is shown. (D) Transcription factor phosphorylation levels were determined by PhosphoFlow. Macrophages were stimulated with 100 ng/mL LPS (blue histograms) or 100 MOI spores (red histograms) for 4 h and then fixed and stained. Some cells were left untreated (black histograms). Representative MFI values are plotted on the relevant histogram. Data are from three (A–C) or one (D) independent experiments. Results are expressed as mean ± SEM. Significance was tested against the unstimulated control by one-way ANOVA with Fisher’s posttest, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Discussion
Tuberculosis is a disease defined by an immunological complexity that has hindered efforts toward vaccine development and allowed Mtb to persist perniciously across the globe. The cause of this complexity is a raft of exquisite immune evasion mechanisms that has evolved over thousands of years to manipulate host immunity. Therefore, at present, animal infection models are the most useful tool for predicting TB vaccine success (10).
We have previously reported that spores coated with Mtb antigens can provide good protection against Mtb in an intranasal infection model (21). A major limitation of this model, however, is that intranasal infection is not physiologically representative of natural Mtb infection. Furthermore, this study did not address whether such vaccine constructs could enhance pre-existing BCG-mediated immunity in the mouse model in a prime-boost immunization schedule. In the present study, we used a technologically superior low-dose aerosol Mtb challenge model to test the efficacy of a novel TB vaccine, Spore-FP1, on BCG-immunized mice, with the hypothesis that Spore-FP1 could boost the protection afforded by BCG. Spore-FP1 was designed to include antigens expressed during the early (Ag85B) and late (ACR) phases of Mtb infection, which could help to account for some of the observed dynamic changes in antigen expression during disease (39). Moreover, unlike previous studies, Spore-FP1 also contained a portion of the epithelium-binding domain of HBHA, which would encourage attachment to lung epithelium and dissemination of the vaccine (40).
Our most important finding was that intranasal immunization with Spore-FP1 in a BCG-primed mouse (i.e., the “prime-boost” strategy) could significantly enhance protection above that given by BCG alone. This effect was independent of any coadjuvants or whether the spores were viable or not. Bacterial burdens in the lungs were significantly reduced by the Spore-FP1 booster immunization compared to BCG alone. Interestingly, there was no statistically significant reduction in extra-pulmonary Mtb. This may reflect a limitation of detection in our assays, or at least an area of potential development for future utilization of spore vehicle systems. Distinct T-cell-derived cytokine patterns are known to exert dichotomous effects on bacterial burdens in the lungs and spleen (41), which may be the case here. Furthermore there was also a slight reduction in protective effect when Spore-FP1 was used with adjuvant. This may reflect ordinary biological variation between experiments, or perhaps non-optimal adjuvant choice, and will be the subject of future investigations.
To investigate immunological events associated with protection, we first evaluated antigen-dependent antibody production in serum and BAL. The role of antibodies in TB is contentious, although there have been recent reappraisals of the field generally in favor of their protective role (42). Mice immunized with Spore-FP1 were found to produce more Ag85B-specific IgG in the serum and IgA in the BAL than the BCG group; a similar trend was observed for ACR. These data strongly suggest that a Spore-FP1 boost immunization was better at inducing humoral immunity than a single BCG immunization.
T-cells are essential for protection against Mtb: Th1 cells prime macrophages for activation via IFN-γ (43), and Th17 cells can upregulate the production of antimicrobial peptides and lymphocyte chemoattractants (44, 45). Deficiency in either of these cytokines is extremely detrimental to the host during disease. It has been shown that during natural infection, Mtb can subvert the host immune system in order to restrict antigen presentation (46, 47). Hence a vaccine that enhances antigen presentation, and thus leads to a higher frequency of antigen-specific T-cells, is highly desirable. In our experiments, we observed a higher frequency of proliferating splenic T-cells in response to recall antigens in the Spore-FP1 group compared to mice that had only received BCG immunization. BCG is also able to restrict antigen presentation in vivo to a certain extent (47–49), and consistent with this fact, we observed minimal proliferative responses to Ag85B/ACR in BCG-immunized animals. Such small-magnitude responses in BCG-immunized mice are highly typical and described elsewhere in the literature, wherein cells specific for Ag85 typically represent ~0.1% of the total splenic polyclonal T-cell pool in cytokine capture assays (31, 32). In contrast to these constrained responses, Spore-FP1 was able to induce a dramatically larger percentage of proliferating T-cells, indicating either a higher frequency of memory cells, or at the very least cells with a higher proliferative capacity. Many of the proliferating CD8+ Ki67+ cells were of the Tcm phenotype, which act as a “reservoir” of cells in primary and secondary lymphoid organs with high potential for differentiation into effector cells in distal sites. For chronic diseases such as TB that include T-cell exhaustion as a definitive mechanism of immune evasion (i.e., terminal differentiation), the generation of proliferative Tcm by a prophylactic vaccine offers a distinct advantage.
In line with proliferative responses, Spore-FP1 was also a potent inducer of IFN-γ, IL-10, and IL-17A release after splenocyte exposure to recall antigens. Thus, the antigen-specific cells were fully functional by producing effector cytokines during proliferation. It could be surmised that Spore-FP1 therefore induced a mixed Th1-Th17-Treg response. The absence of IL-4 release is interesting, and suggests that Spore-FP1 induced a T-cell skewing away from the Th2 to a Th1/Th17 phenotype. IL-4 is largely believed to be detrimental during Mtb infection, since it antagonizes the biological effects of IFN-γ to promote alternatively activated macrophages (50). The role of IL-10 in TB is more contentious. While IL-10 can hamper antimycobacterial immunity during BCG immunization (51), recent evidence from Rhesus macaque infection models has suggested that CD4+ T-cells coexpressing a balance of pro- and anti-inflammatory cytokines are significantly associated with granuloma sterilization, possibly due to a reduction in “collateral damage” to the lung tissue (52). Furthermore, IL-10 is important for shielding CD8+ memory T-cells from apoptosis in inflammatory contexts (53), and IL-10 deficient mice are highly susceptible to reinfection by intracellular pathogens (54). We believe that the T-cell profile induced by Spore-FP1 is therefore beneficial in the context of immunization.
It is worth noting that for both humoral and cellular immunogenicity, there was generally a greater response to Ag85B than to ACR. This is perhaps due to the fact that Ag85B is a strong immunodominant antigen (55) that has formed the basis of many new TB vaccines. Notably, however, ACR was still able to elicit potent IFN-γ production in splenocytes from Spore-FP1-immunized mice.
Alongside conventional T-cell activation signatures, we also observed a striking accumulation of gross CD69+CD103+ Trm in lung tissue after immunization with Spore-FP1. These cells are likely to be directed toward epitopes found within FP1, since the vehicle control (spores alone) failed to induce any appreciable quantities of these cells. As to why no Trm were directed against B. subtilis spores themselves, it may be that B. subtilis, as a mammalian commensal (56) (in the absence of a “foreign” antigen such as those included in FP1), can suppress the mobilization of effector T-cells that would lead to its own clearance. In support of this hypothesis, B. subtilis secretory products can induce a Foxp3-dependent tolerogenic environment in the gut (57), and consistent with this fact, we observed modest IL-10 responses from splenocytes exposed to recall antigen (although much lower than IFN-γ and IL-17A). A recent study has elegantly demonstrated that mucosal immunization with BCG—as opposed to parenteral immunization—leads to the accumulation of Trm in the pulmonary tissue (27). These cells are sufficient for protection, since adoptive transfer of Trm into BCG-naive mice protects against Mtb challenge. We speculate that the enrichment of this cell type in the lungs, induced by Spore-FP1 in our experiments, is playing a major role in the protection afforded by our novel vaccine.
Turning our attention to the innate immune system, we detected potent activation signatures in macrophages and DCs pulsed with B. subtilis spores. While it is known that B. subtilis spores can activate TLR-2→MyD88 downstream pathways, these studies have largely restricted maturation marker analysis to CD40 and MHC Class I and II expression on DCs (19, 58). Here, we showed for the first time that spores can also simultaneously induce CCR7, PD-L1 and PD-L2 upregulation. Since minimal T-cell priming occurs in the lung (59, 60), CCR7 expression will be critical for DCs that have taken up Spore-FP1 to migrate to the lung-draining lymph nodes and present antigen to naive T-cells. The upregulation of PD-L1 and PD-L2, on the other hand, may mitigate the overall inflammatory response, which is an important boon for mucosal delivery. In justification of this notion, PD-L1 blockade during antigen delivery into the lungs leads to exacerbated irritation and inflammation via Treg depletion, which is ameliorated upon immune reconstitution (61). Underscoring all of these phenotypic characteristics was the observation that IRF-3 was phosphorylated alongside NF-κB upon APC stimulation with spores. These data allude to a novel activation pathway besides the TLR-2→MyD88 axis, which is driving APC activation by B. subtilis spores, and has hitherto remained unexplored. This proposition warrants further biochemical investigation.
To conclude, we have shown that Spore-FP1 can enhance protection offered by BCG and also activate multiple arms of the innate and adaptive immune systems. These data demonstrate the potential applicability of Spore-FP1 as a TB vaccine, but also offer fresh insights into the mechanisms of B. subtilis spores as a vaccine development platform.
Ethics Statement
The animal work was reviewed and approved by St George’s University of London Ethics Committee for animal experimentation and studies performed under a valid UK Home Office Project Licence.
Author Contributions
AC, PH, and GD performed most of the immunization and MTB challenge experiments. SH and ACT performed in vitro immunogenicity experiments. MS provided recombinant proteins. SC provided spores. MP performed immunological evaluations. RR conceived the study and wrote up the manuscript with AC.
Conflict of Interest Statement
Author MS was employed by company Lionex. All other authors declare no competing interests.
Funding
This study was funded by the European Commission H2020 grant no. 643558 awarded to the EMI-TB Consortium.
References
1. Churchyard G, Kim P, Shah NS, Rustomjee R, Gandhi N, Mathema B, et al. What we know about tuberculosis transmission: an overview. J Infect Dis (2017) 216:S629–35. doi:10.1093/infdis/jix362
2. Dye C. Making wider use of the world’s most widely used vaccine: bacille Calmette-Guerin revaccination reconsidered. J R Soc Interface (2013) 10:20130365. doi:10.1098/rsif.2013.0365
3. Castillo-Rodal AI, Castanon-Arreola M, Hernandez-Pando R, Calva JJ, Sada-Diaz E, Lopez-Vidal Y. Mycobacterium bovis BCG substrains confer different levels of protection against Mycobacterium tuberculosis infection in a BALB/c model of progressive pulmonary tuberculosis. Infect Immun (2006) 74:1718–24. doi:10.1128/IAI.74.3.1718-1724.2006
4. Poyntz HC, Stylianou E, Griffiths KL, Marsay L, Checkley AM, Mcshane H. Non-tuberculous mycobacteria have diverse effects on BCG efficacy against Mycobacterium tuberculosis. Tuberculosis (Edinb) (2014) 94:226–37. doi:10.1016/j.tube.2013.12.006
5. Moliva JI, Turner J, Torrelles JB. Prospects in Mycobacterium bovis bacille Calmette et Guerin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis? Vaccine (2015) 33:5035–41. doi:10.1016/j.vaccine.2015.08.033
6. Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PE, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis (2014) 58:470–80. doi:10.1093/cid/cit790
7. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci U S A (2012) 109:17537–42. doi:10.1073/pnas.1202870109
8. de Castro MJ, Pardo-Seco J, Martinon-Torres F. Nonspecific (heterologous) protection of neonatal BCG vaccination against hospitalization due to respiratory infection and sepsis. Clin Infect Dis (2015) 60:1611–9. doi:10.1093/cid/civ144
9. Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. doi:10.1016/S0140-6736(13)60177-4
10. McShane H, Williams A. A review of preclinical animal models utilised for TB vaccine evaluation in the context of recent human efficacy data. Tuberculosis (Edinb) (2014) 94:105–10. doi:10.1016/j.tube.2013.11.003
11. Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, et al. Co-delivery of cancer-associated antigen and toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine (2008) 26:5046–57. doi:10.1016/j.vaccine.2008.07.035
12. Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A (2012) 109:1080–5. doi:10.1073/pnas.1112648109
13. Kanekiyo M, Wei CJ, Yassine HM, Mctamney PM, Boyington JC, Whittle JR, et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature (2013) 499:102–6. doi:10.1038/nature12202
14. Hong HA, Khaneja R, Tam NM, Cazzato A, Tan S, Urdaci M, et al. Bacillus subtilis isolated from the human gastrointestinal tract. Res Microbiol (2009) 160:134–43. doi:10.1016/j.resmic.2008.11.002
15. Barnes AG, Cerovic V, Hobson PS, Klavinskis LS. Bacillus subtilis spores: a novel microparticle adjuvant which can instruct a balanced Th1 and Th2 immune response to specific antigen. Eur J Immunol (2007) 37:1538–47. doi:10.1002/eji.200636875
16. Wiencek KM, Klapes NA, Foegeding PM. Hydrophobicity of Bacillus and Clostridium spores. Appl Environ Microbiol (1990) 56:2600–5.
17. Zhao G, Miao Y, Guo Y, Qiu H, Sun S, Kou Z, et al. Development of a heat-stable and orally delivered recombinant M2e-expressing B. subtilis spore-based influenza vaccine. Hum Vaccin Immunother (2014) 10:3649–58. doi:10.4161/hv.36122
18. Lega T, Weiher P, Obuchowski M, Nidzworski D. Presenting influenza A M2e antigen on recombinant spores of Bacillus subtilis. PLoS One (2016) 11:e0167225. doi:10.1371/journal.pone.0167225
19. de Souza RD, Batista MT, Luiz WB, Cavalcante RC, Amorim JH, Bizerra RS, et al. Bacillus subtilis spores as vaccine adjuvants: further insights into the mechanisms of action. PLoS One (2014) 9:e87454. doi:10.1371/journal.pone.0087454
20. Tavares Batista M, Souza RD, Paccez JD, Luiz WB, Ferreira EL, Cavalcante RC, et al. Gut adhesive Bacillus subtilis spores as a platform for mucosal delivery of antigens. Infect Immun (2014) 82:1414–23. doi:10.1128/IAI.01255-13
21. Reljic R, Sibley L, Huang JM, Pepponi I, Hoppe A, Hong HA, et al. Mucosal vaccination against tuberculosis using inert bioparticles. Infect Immun (2013) 81:4071–80. doi:10.1128/IAI.00786-13
22. Sibley L, Reljic R, Radford DS, Huang JM, Hong HA, Cranenburgh RM, et al. Recombinant Bacillus subtilis spores expressing MPT64 evaluated as a vaccine against tuberculosis in the murine model. FEMS Microbiol Lett (2014) 358:170–9. doi:10.1111/1574-6968.12525
23. Frey J. Biological safety concepts of genetically modified live bacterial vaccines. Vaccine (2007) 25:5598–605. doi:10.1016/j.vaccine.2006.11.058
24. Stover CK, De La Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, et al. New use of BCG for recombinant vaccines. Nature (1991) 351:456–60. doi:10.1038/351456a0
25. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med (1992) 176:1693–702. doi:10.1084/jem.176.6.1693
26. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial regulatory T cells occupy a discrete TCR niche in human arthritis and require local signals to stabilize FOXP3 protein expression. J Immunol (2015) 195:5616–24. doi:10.4049/jimmunol.1500391
27. Perdomo C, Zedler U, Kuhl AA, Lozza L, Saikali P, Sander LE, et al. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. MBio (2016) 7:e1686–1616. doi:10.1128/mBio.01686-16
28. Trumpfheller C, Caskey M, Nchinda G, Longhi MP, Mizenina O, Huang Y, et al. The microbial mimic poly IC induces durable and protective CD4+ T cell immunity together with a dendritic cell targeted vaccine. Proc Natl Acad Sci U S A (2008) 105:2574–9. doi:10.1073/pnas.0711976105
29. Balu S, Reljic R, Lewis MJ, Pleass RJ, Mcintosh R, Van Kooten C, et al. A novel human IgA monoclonal antibody protects against tuberculosis. J Immunol (2011) 186:3113–9. doi:10.4049/jimmunol.1003189
30. Lu LL, Chung AW, Rosebrock TR, Ghebremichael M, Yu WH, Grace PS, et al. A functional role for antibodies in tuberculosis. Cell (2016) 167:433–43.e414. doi:10.1016/j.cell.2016.08.072
31. Goonetilleke NP, Mcshane H, Hannan CM, Anderson RJ, Brookes RH, Hill AV. Enhanced immunogenicity and protective efficacy against Mycobacterium tuberculosis of bacille Calmette-Guerin vaccine using mucosal administration and boosting with a recombinant modified vaccinia virus Ankara. J Immunol (2003) 171:1602–9. doi:10.4049/jimmunol.171.3.1602
32. Wozniak TM, Ryan AA, Britton WJ. Interleukin-23 restores immunity to Mycobacterium tuberculosis infection in IL-12p40-deficient mice and is not required for the development of IL-17-secreting T cell responses. J Immunol (2006) 177:8684–92. doi:10.4049/jimmunol.177.12.8684
33. Jung S, Unutmaz D, Wong P, Sano G, De Los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity (2002) 17:211–20. doi:10.1016/S1074-7613(02)00365-5
34. Fahlen-Yrlid L, Gustafsson T, Westlund J, Holmberg A, Strombeck A, Blomquist M, et al. CD11c(high)dendritic cells are essential for activation of CD4+ T cells and generation of specific antibodies following mucosal immunization. J Immunol (2009) 183:5032–41. doi:10.4049/jimmunol.0803992
35. Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med (2006) 203:413–24. doi:10.1084/jem.20051720
36. Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol (2014) 15:195–204. doi:10.1038/ni.2789
37. Kazmin D, Nakaya HI, Lee EK, Johnson MJ, Van Der Most R, Van Den Berg RA, et al. Systems analysis of protective immune responses to RTS,S malaria vaccination in humans. Proc Natl Acad Sci U S A (2017) 114:2425–30. doi:10.1073/pnas.1621489114
38. Villadangos JA, Cardoso M, Steptoe RJ, Van Berkel D, Pooley J, Carbone FR, et al. MHC class II expression is regulated in dendritic cells independently of invariant chain degradation. Immunity (2001) 14:739–49. doi:10.1016/S1074-7613(01)00148-0
39. Moguche AO, Musvosvi M, Penn-Nicholson A, Plumlee CR, Mearns H, Geldenhuys H, et al. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe (2017) 21:695–706.e695. doi:10.1016/j.chom.2017.05.012
40. Pethe K, Alonso S, Biet F, Delogu G, Brennan MJ, Locht C, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature (2001) 412:190–4. doi:10.1038/35084083
41. Sakai S, Kauffman KD, Sallin MA, Sharpe AH, Young HA, Ganusov VV, et al. CD4 T cell-derived IFN-gamma plays a minimal role in control of pulmonary Mycobacterium tuberculosis infection and must be actively repressed by PD-1 to prevent lethal disease. PLoS Pathog (2016) 12:e1005667. doi:10.1371/journal.ppat.1005667
42. Jacobs AJ, Mongkolsapaya J, Screaton GR, Mcshane H, Wilkinson RJ. Antibodies and tuberculosis. Tuberculosis (Edinb) (2016) 101:102–13. doi:10.1016/j.tube.2016.08.001
43. Herbst S, Schaible UE, Schneider BE. Interferon gamma activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS One (2011) 6:e19105. doi:10.1371/journal.pone.0019105
44. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med (2006) 203:2271–9. doi:10.1084/jem.20061308
45. Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog (2014) 10:e1004099. doi:10.1371/journal.ppat.1004099
46. Bold TD, Banaei N, Wolf AJ, Ernst JD. Suboptimal activation of antigen-specific CD4+ effector cells enables persistence of M. tuberculosis in vivo. PLoS Pathog (2011) 7:e1002063. doi:10.1371/journal.ppat.1002063
47. Grace PS, Ernst JD. Suboptimal antigen presentation contributes to virulence of Mycobacterium tuberculosis in vivo. J Immunol (2016) 196:357–64. doi:10.4049/jimmunol.1501494
48. Pecora ND, Fulton SA, Reba SM, Drage MG, Simmons DP, Urankar-Nagy NJ, et al. Mycobacterium bovis BCG decreases MHC-II expression in vivo on murine lung macrophages and dendritic cells during aerosol infection. Cell Immunol (2009) 254:94–104. doi:10.1016/j.cellimm.2008.07.002
49. Sakai S, Kawamura I, Okazaki T, Tsuchiya K, Uchiyama R, Mitsuyama M. PD-1-PD-L1 pathway impairs T(h)1 immune response in the late stage of infection with Mycobacterium bovis bacillus Calmette-Guerin. Int Immunol (2010) 22:915–25. doi:10.1093/intimm/dxq446
50. Buccheri S, Reljic R, Caccamo N, Ivanyi J, Singh M, Salerno A, et al. IL-4 depletion enhances host resistance and passive IgA protection against tuberculosis infection in BALB/c mice. Eur J Immunol (2007) 37:729–37. doi:10.1002/eji.200636764
51. Pitt JM, Stavropoulos E, Redford PS, Beebe AM, Bancroft GJ, Young DB, et al. Blockade of IL-10 signaling during Bacillus Calmette-Guerin vaccination enhances and sustains Th1, Th17, and innate lymphoid IFN-gamma and IL-17 responses and increases protection to Mycobacterium tuberculosis infection. J Immunol (2012) 189:4079–87. doi:10.4049/jimmunol.1201061
52. Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog (2015) 11:e1004603. doi:10.1371/journal.ppat.1004603
53. Laidlaw BJ, Cui W, Amezquita RA, Gray SM, Guan T, Lu Y, et al. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat Immunol (2015) 16:871–9. doi:10.1038/ni.3224
54. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature (2002) 420:502–7. doi:10.1038/nature01152
55. Huygen K. The immunodominant T-cell epitopes of the mycolyl-transferases of the antigen 85 complex of M. tuberculosis. Front Immunol (2014) 5:321. doi:10.3389/fimmu.2014.00321
56. Salzman NH, De Jong H, Paterson Y, Harmsen HJ, Welling GW, Bos NA. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology (2002) 148:3651–60. doi:10.1099/00221287-148-11-3651
57. Paynich ML, Jones-Burrage SE, Knight KL. Exopolysaccharide from Bacillus subtilis induces anti-inflammatory M2 macrophages that prevent T cell-mediated disease. J Immunol (2017) 198:2689–98. doi:10.4049/jimmunol.1601641
58. Aps LR, Diniz MO, Porchia BF, Sales NS, Moreno AC, Ferreira LC. Bacillus subtilis spores as adjuvants for DNA vaccines. Vaccine (2015) 33:2328–34. doi:10.1016/j.vaccine.2015.03.043
59. Belz GT, Bedoui S, Kupresanin F, Carbone FR, Heath WR. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat Immunol (2007) 8:1060–6. doi:10.1038/ni1505
60. Pastva AM, Mukherjee S, Giamberardino C, Hsia B, Lo B, Sempowski GD, et al. Lung effector memory and activated CD4+ T cells display enhanced proliferation in surfactant protein A-deficient mice during allergen-mediated inflammation. J Immunol (2011) 186:2842–9. doi:10.4049/jimmunol.0904190
Keywords: tuberculosis, spores, vaccine, immunity, adjuvants
Citation: Copland A, Diogo GR, Hart P, Harris S, Tran AC, Paul MJ, Singh M, Cutting SM and Reljic R (2018) Mucosal Delivery of Fusion Proteins with Bacillus subtilis Spores Enhances Protection against Tuberculosis by Bacillus Calmette-Guérin. Front. Immunol. 9:346. doi: 10.3389/fimmu.2018.00346
Received: 13 November 2017; Accepted: 07 February 2018;
Published: 12 March 2018
Edited by:
Jeffrey K. Actor, University of Texas Health Science Center at Houston, United StatesCopyright: © 2018 Copland, Diogo, Hart, Harris, Tran, Paul, Singh, Cutting and Reljic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajko Reljic, cnJlbGppY0BzZ3VsLmFjLnVr
†These authors have contributed equally to this work.