Abstract
Blockade of programmed death 1 (PD-1) protein and its ligand programmed death ligand 1 (PD-L1) has been used as cancer immunotherapy in recent years, with the blockade of PD-1 being more widely used than blockade of PD-L1. PD-1 and PD-L1 blockade therapy showed benefits in patients with various types of cancer; however, such beneficial effects were seen only in a subgroup of patients. Improving the efficacy of PD-1 and PD-L1 blockade therapy is clearly needed. In this review, we summarize the recent studies on the effects of gut microbiota on PD-1 and PD-L1 blockade and discuss the new perspectives on improving efficacy of PD-1 and PD-L1 blockade therapy in cancer treatment through modulating gut microbiota. We also discuss the possibility that chronic infections or inflammation may impact on PD-1 and PD-L1 blockade therapy.
Introduction
The immune system uses various effector cells and molecules to control and eradicate infectious agents and cancer cells. Cytotoxic T cells (CTL) are the major effector cells in anti-tumor immune responses (1, 2). However, the functions of these effector cells are inhibited in the tumor microenvironment, which contributes to cancer cell immune evasion (3). In recent years, the blockade of immune checkpoint proteins and molecules that deliver inhibitory signals to activated T cells, have shown great promise in cancer treatment. However, the beneficial effects of these treatment strategies were seen only in a subgroup of patients (4). In this review, we summarize the emerging evidence of improving immune checkpoint protein blockade therapy efficacy by modulating gut microbiota and discuss the possibility that chronic infections or inflammation may impact on programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1) blockade therapy.
PD-1 and Its Ligands
Programmed death 1, also known as cluster of differentiation 279 (CD279), is a cell surface receptor that was discovered in 1992 (5). PD-L1 and PD-L2, the two molecules that interact with PD-1, were identified in the following years (6, 7). PD-L1 is also known as CD274 or B7 homolog 1 (B7-H1) and PD-L2 known as CD273 or B7-DC.
Programmed death 1 is expressed on T, B cells, and myeloid cells (8). PD-L1 is expressed by a variety of cells in the immune system and non-immune cells. However, the expression level of PD-L1 in normal human tissues is low; despite the presence of PD-L1 mRNA, PD-L1 protein is rarely detected on the cell surfaces in most of normal human tissues except for a subset of human tissue macrophages (6, 9). PD-L2 is predominately expressed by antigen-presenting cells, such as dendritic cells (DCs) and macrophages (10–13). The expression of both PD-L1 and PD-L2 is regulated by cytokines, for example interferon (IFN)-γ greatly increases the expression of PD-L1 and several cytokines are able to induce the expression of PD-L2 in other immune cells and non-immune cells in addition to the DCs and macrophages (9–11, 14–16).
Programmed death 1 and its ligands are members of the immune checkpoint proteins delivering inhibitory signals to activated T cells. The interaction of PD-1 with PD-L1 or PD-L2 leads to suppression of T cells, a regulatory mechanism to prevent overstimulation of immune responses and autoimmunity (6, 7, 9, 16–21). However, such a mechanism is hijacked in the tumor microenvironment, providing opportunities for tumor cells to evade the attack from the immune system.
PD-1 and PD-L1 Blockade in Cancer Immunotherapy
In anti-tumor immune responses, the tumor antigens generated by gene mutations, are recognized by the immune system and specific CD8+ CTLs targeting tumor antigens are generated (22). These specific effector CTLs recognize the target tumor cells and induce tumor cell apoptosis.
However, tumor cells employ various strategies to escape the attack from the immune system, one of which is to resist the killing effects from the anti-tumor CTLs by increasing PD-L1 expression in tumor tissues (9, 23, 24). Most normal human tissues do not express detectable PD-L1 on their cell surface, in contrast PD-L1 is abundantly expressed by tumor cells, the immune and non-immune cells in various tumor tissues (6, 9, 25–30). IFN-γ released by the anti-tumor CTLs infiltrating into tumor tissues plays a major role in inducing the expression of PD-L1 (9–11, 14–16). Other cytokines, such as tumor necrosis factor (TNF) -α, interleukin (IL)-4, and IL-10 can also increase PD-L1 expression (31, 32).
The interaction of PD-L1 with PD-1 in the tumor microenvir-onment enables the tumor cells to resist the endogenous anti-tumor activities from the immune system. PD-L1 expressed in tumor tissues interacting with PD-1 expressed on the activated T cells leads to the dysfunction of the effector T cells, via multiple mechanisms, such as promoting T cell apoptosis, anergy, and exhaustion (6, 7, 9, 16–21). More recently, it was found that interaction of PD-L1 with PD-1 expressed on tumor-associated macrophages inhibits the phagocytic potency of macrophages against tumor cells (33). The importance of PD-L1 and PD-1 interaction in tumor cell evasion has led scientists to explore the use of these molecules as therapeutic targets in cancer immunotherapy (Figure 1).
Figure 1
Dong et al. showed that PD-L1 positive human tumor cells induced apoptosis of co-cultured activated effector T cells and this effect was blocked by an anti-human PD-L1 monoclonal antibody (mAb). They also showed that the growth of PD-L1 positive murine tumors in syngeneic mice was suppressed by an anti-murine PD-L1 mAb (9). Other researchers later reported similar findings in examination of different types of cancer cells using mice models (24, 34–36). These important laboratory observations led to numerous clinical trials of using monoclonal antibodies targeting PD-1 or PD-L1 in cancer immunotherapy for a variety of cancers. In addition to affecting the immunological pathways, PD-L1 and PD-1 blockade may also work in part by disrupting autologous PD-1 and PD-L1 signaling within tumors (37, 38).
To date, the U.S. Food and Drug Administration (FDA) has approved the use of five monoclonal antibodies targeting PD-L1 or PD-1 in cancer treatment. The details of the clinical trials of these five monoclonal antibodies are summarized in Table 1. Despite the clear benefits of PD-L1 and PD-1 blockade in treating some cancer patients, not all cases responded to treatment (Table 1). Given this, strategies to improve the efficacy of cancer immunotherapy are needed. Emerging evidence suggests that modulation of the gut microbiota is a promising approach.
Table 1
| Commercial name (active ingredient) | Target | Treatment of disease | Targeting patients | Clinical cases | Clinical phase | Overall response rate (95% CI) | Objective response rate (95% CI) | Clinical study (clinical trial ID) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Bavencio (Avelumab) | PD-L1 | Metastatic MCC | Metastatic MCC patients whose disease had progressed on or after chemotherapy administered | 88 | Phase 2 | 33% (23.3%, 43.8%) | Not applicable | JAVELIN Merkel 200 Trial (NCT02155647) | (39, 40) |
| Tecentriq (Atezolizumab) | PD-L1 | Advanced or metastatic urothelial carcinoma | Cisplatin-ineligible patients with locally advanced or metastatic urothelial carcinoma | 119 | Phase 2 | 23.5% (16.2%, 32.2%) | Not applicable | IMvigor210 (NCT02951767) | (41) |
| Previously treated patients with locally advanced or metastatic urothelial carcinoma | 310 | Phase 2 | 14.8% (11.1%, 19.3%) | Not applicable | IMvigor210 (NCT02951767) | (41) | |||
| Metastatic NSCLC | Previously treated patients with metastatic non-small cell lung cancer | 22 | Phase 2 | Not applicable | 15% (10%, 22%) | POPLAR (NCT01903993) | (42) | ||
| Imfinzi (Durvalumab) | PD-L1 | Locally advanced or metastatic urothelial carcinoma | Patients with locally advanced or metastatic urothelial carcinoma in total | 182 | Phase 1 and 2 | Not applicable | 17.0% (11.9%, 23.3%) | Study 1108 (NCT01693562) | (43–45) |
| Patients with locally advanced or metastatic urothelial carcinoma who showed high PD-L1 expression on tumor cells | 95 | Phase 1 and 2 | Not applicable | 26.3% (17.8%, 36.4%) | Study 1108 (NCT01693562) | (43–45) | |||
| Patients with locally advanced or metastatic urothelial carcinoma who showed low or non-PD-L1 expression on tumor cells | 73 | Phase 1 and 2 | Not applicable | 4.1% (0.9%, 11.5%) | Study 1108 (NCT01693562) | (43–45) | |||
| Keytruda (Pembrolizumab) | PD-1 | Melanoma | Patients with Ipilimumab-Naïve melanoma (receive KEYTRUDA at a dose of 10 mg/Kg every 3 weeks) | 277 | Phase 3 | 33% (27%, 39%) | Not applicable | KEYNOTE-006 (NCT01866319) | (46, 47) |
| Patients with Ipilimumab-Naïve melanoma (receive KEYTRUDA at a dose of 10 mg/Kg every 2 weeks) | 279 | Phase 3 | 34% (28%, 40%) | Not applicable | KEYNOTE-006 (NCT01866319) | (46, 47) | |||
| Patients with Ipilimumab-refractory melanoma (receive KEYTRUDA at a dose of 2 mg/Kg every 3 weeks) | 180 | Phase 2 | Not applicable | 21% (15%, 28%) | KEYNOTE-002 (NCT01704287) | (48) | |||
| Patients with Ipilimumab-refractory melanoma (receive KEYTRUDA at a dose of 10 mg/Kg every 3 weeks) | 181 | Phase 2 | Not applicable | 25% (19%, 32%) | KEYNOTE-002 (NCT01704287) | (48) | |||
| NSCLC | Metastatic NSCLC patients with first-line treatment with a single agent | 154 | Phase 3 | Not applicable | 45% (37%, 53%) | KEYNOTE-024 (NCT02142738) | (49) | ||
| Metastatic NSCLC patients with first-line treatment in combination with pemetrexed and carboplatin | 60 | Phase 1 and 2 | 55% (42%, 68%) | Not applicable | KEYNOTE-021 (NCT02039674) | (50) | |||
| Previously treated NSCLC patients (all randomized patients who receive KEYTRUDA at a dose of 2 mg/Kg every 3 weeks) | 344 | Phase 2 and 3 | Not applicable | 18% (14%, 23%) | KEYNOTE-010 (NCT01905657) | (51) | |||
| Previously treated NSCLC patients (all randomized patients who receive KEYTRUDA at a dose of 10 mg/Kg every 3 weeks) | 346 | Phase 2 and 3 | Not applicable | 19% (15%, 23%) | KEYNOTE-010 (NCT01905657) | (51) | |||
| HNSCC | HNSCC patients whose disease had progressed on or after chemotherapy administered | 174 | Phase 1 | 16% (11%, 22%) | Not applicable | KEYNOTE-012 (NCT01848834) | (52) | ||
| Urothelial Carcinoma | Cisplatin-ineligible patients with urothelial carcinoma | 370 | Phase 2 | Not applicable | 29% (24%, 34%) | KEYNOTE-052 (NCT02335424) | (53) | ||
| Previously treated urothelial carcinoma patients | 270 | Phase 3 | Not applicable | 21% (16%, 27%) | KEYNOTE-045 (NCT02256436) | (54) | |||
| cHL | Patients with cHL | 210 | Phase 2 | 69% (62%, 75%) | Not applicable | KEYNOTE-087 (NCT02453594) | (55, 56) | ||
| MSI-H | Patients with MSI-H or mismatch repair deficient (dMMR) | 149 | Phase 1 Phase 2 Phase 1 Phase 2 Phase 2 | Not applicable | 39.6% (31.7%, 47.9%) | KEYNOTE-012 (NCT01848834) KEYNOTE-016 (NCT01876511) KEYNOTE-028 (NCT02054806) KEYNOTE-158 (NCT02628067) KEYNOTE-164 (NCT02460198) | (52, 57–59) | ||
| Opdivo (Nivolumab) | PD-1 | Unresectable or metastatic melanoma | Previously treated patients with unresectable or metastatic melanoma in the treatment of OPDIVO | 316 | Phase 3 | Not applicable | 40% (34%, 46%) | CheckMate-067 (NCT01844505) | (60, 61) |
| Previously treated patients with unresectable or metastatic melanoma in the treatment of OPDIVO plus Ipilimumab (anti-CTLA4 antibody) | 314 | Phase 3 | Not applicable | 50% (44%, 55%) | CheckMate-067 (NCT01844505) | (60, 61) | |||
| Metastatic NSCLC | NSCLC patients who had experienced disease progressed during or after one prior platinum doublet-based chemotherapy regimen | 272 | Phase 3 | Not applicable | 20% (14%, 28%) | CheckMate-017 (NCT01642004) | (62) | ||
| Patients with metastatic non-squamous NSCLC who had experienced disease progressed during or after one prior platinum doublet-based chemotherapy regimen | 292 | Phase 3 | Not applicable | 19% (15%, 24%) | CheckMate-057 (NCT01673867) | (63) | |||
| Renal cell carcinoma | Patients with advanced RCC who had experienced disease progressed during or after one or two prior anti-angiogenic therapy regimes | 410 | Phase 3 | Not applicable | 21.5% (17.6%, 25.8%) | CheckMate-025 (NCT01668784) | (64, 65) | ||
| cHL | Adult patients with cHL | 258 | Phase 2 Phase 1 | Not applicable | 69% (63%, 75%) | CheckMate-205 (NCT02181738) CA209-039 (NCT01592370) | (66, 67) | ||
| Recurrent or metastatic SCCHN | Patients with metastatic or recurrent SCCHN | 240 | Phase 3 | Not applicable | 13.3% (9.3%, 18.3%) | CheckMate-141 (NCT02105636) | (68, 69) |
Five monoclonal antibodies targeting programmed death ligand-1 (PD-L1) or programmed death 1 (PD-1) were approved by the U.S. Food and Drug Association to treat cancer.
Five monoclonal PD-L1 or PD-1 antibodies granted after May 2017 by FDA for cancer treatments were not included in the table.
MCC, metastatic Merkel cell carcinoma; NSCLC, non-small cell lung cancer; HNSCC, head and neck squamous cell cancer; cHL, classical Hodgkin lymphoma; MSI-H, microsatellite instability-high cancer; dMMR, mismatch repair deficient; SCCHN, recurrent or metastatic squamous cell carcinoma of the head and neck.
Modulation of Gut Microbiota Enhances the Anti-Tumor Efficacy of PD-1 and PD-L1 Blockade Therapy
A very interesting study by Sivan et al. provided strong evidence that the efficacy of PD-L1 blockage therapy can be improved by the modulation of gut microbiota (70). In this study, Sivan et al. examined the subcutaneous growth of B16.SIY melanoma in genetically similar C57BL/6 mice raised in the Jackson Laboratory (JAX) and Taconic Farms (TAC), and found that the tumor growth was more aggressive in TAC mice as compared to that in JAX mice and that TAC mice had a significantly lower intratumoral CD8+ T cell accumulation. They then conducted various experiments, which demonstrated that gut microbiota contributed to this difference.
They first showed that prophylactic transfer of fecal material from JAX mice to TAC mice was sufficient to delay tumor growth. To examine whether microbial community alone was effective as a therapy, they administered fecal material from JAX mice alone or in combination with anti-PD-L1 mAbs to TAC mice. These experiments showed that fecal material alone was sufficient to significantly inhibit tumor growth and that the combination treatment further improved tumor control. To identify the responsible bacterial species, they used 16S ribosomal RNA (16S rRNA) sequencing and identified Bifidobacterium species, particularly Bifidobacterium breve, Bifidobacterium longum, and Bifidobacterium adolescentis as the candidate species. The role of these Bifidobacterium species in enhancing protective immunity against tumors were further investigated by administering TAC mice bearing established tumors with a cocktail of Bifidobacterium species containing B. breve and B. longum by oral gavage. This experiment resulted in Bifidobacterium-treated mice having significantly improved tumor control as compared to mice that did not receive Bifidobacterium. Sivan et al. also showed that the possible mechanisms by which Bifidobacterium species inhibited tumor growth were through activating DCs, which in turn, improves the effector function of tumor-specific CD8+ T cells. Given that the enhanced anti-melanoma effect from Bifidobacterium species had occurred at the innate immunity level, the authors anticipated that Bifidobacterium species also provide anti-tumor beneficial effects to other types of tumors. However, the mechanisms by which Bifidobacterium species activated DCs improved the effects of anti-tumor CD8+ cells still need to be clarified.
The findings by Sivan et al. using mice models suggest that it is possible to enhance the anti-tumor efficacy of PD-L1 blockade therapy in treating cancer patients by modulating their gut microbiota and their findings are summarized in Figure 2. Interestingly, a very recent study by Matson et al. examining the stool samples collected from patients with metastatic melanoma before anti-PD-1 immunotherapy found that B. longum, Collinsella aerofaciens, and Enterococcus faecium were more abundant in the anti-PD-1 immunotherapy responders, supporting the anti-tumor effects of Bifidobacterium species (71).
Figure 2
Several additional studies also compared the gut microbiota in patients with metastatic melanoma receiving anti-PD-1 therapy. A recent study by Frankel et al. using metagenomic shotgun sequencing method showed that melanoma patients who responded to immune checkpoint inhibitors were enriched with Bacteroides caccae (72). Furthermore, they showed that the bacteria that are enriched within responders are most likely to be antibody dependent. Patients who responded to nivolumab (PD-1 antibody) were enriched with Fecalibacterium prausnitzii, Bacteroides thetaiotamicron, and Holdemania filiformis, whereas patients who responded to pembrolizumab (another PD-1 antibody), their gut microbiota enriched with Dorea formicogenerans. However, the mechanisms responsible for these changes are not clear. Studies comparing the gut microbiota changes prior to and following anti-PD-1 therapy of individual patients are required, which will provide information regarding whether anti-PD-1 antibodies directly affect gut bacterial species.
A study by Wargo et al. examined the human gut microbiota and metabolites of metastatic melanoma patients who received anti-PD-1 therapy using 16S rRNA and whole genome shotgun sequencing (73). They found that bacterial diversity and composition in patients that responded to the therapy were significantly different from that in patients who did not respond to the therapy. The responding patients had a higher diversity of bacteria and a higher abundance of Clostridiales, and the non-responders had a higher abundance of Bacteroidales. In a very recent study with multiple first authors and J. A. Wargo being the responding author, they further compared the gut microbiota of patients with metastatic melanoma receiving anti-PD-1 therapy (74). They found that patients who responded to anti-PD-1 therapy were associated with a significantly higher bacterial diversity and abundance of bacteria from the Ruminococcaceae family, which belongs to the Clostridiales order, as compared to patients who did not respond to the therapy. Furthermore, they performed fecal microbiota transplantation experiments in germ-free mice, in which they showed that germ-free mice transplanted with stool samples from patients who responded to anti-PD-1 and anti-PD-L1 therapy had a significantly reduced tumor growth and improved responses to anti-PD-1 and anti-PD-L1 therapy, coupled with a higher density of CD8+ T cells. However, it is not clear which bacterial species in the Ruminococcaceae family has played the role in enhancing the PD-1 blockade therapy.
Another recent study by Routy et al. investigated the effects of gut microbiota in PD-1 blockade therapy (75). In their study, data from 140 patients with advanced non-small-cell-lung cancer, 67 patients with renal cell carcinoma, and 42 patients with urothelial carcinoma were collected, and they found that 69 patients who took antibiotics before or soon after starting the PD-1 blockade therapy had shorter progression-free survival and overall survival. They then explored the composition of the gut microbiota using shotgun sequencing, which showed that Akkermansia muciniphila was enriched in patients who responded to anti-PD-1 therapy. This suggests that A. muciniphila may enhance patient response to PD-1 blockade therapy. They verified this observation by transplanting the patients stool samples in specific pathogen-free mice or germ-free mice and observed tumor growth in these mice. They also found that A. muciniphila alone was able to restore the anti-tumor effects of PD-1 blockade that was inhibited by antibiotics. However, the mechanism by which A. muciniphila enhancing PD-1 blockade therapy is not known.
Bacterial species that are positively associated with PD-1 and PD-L1 blockade therapy are summarized in Table 2. Some bacterial species have also been demonstrated to affect CTLA-4 blockade immunotherapy, which were not reviewed here (76, 77).
Table 2
| Bacteria | Model | Methods | Main findings | Reference |
|---|---|---|---|---|
| Bifidobacterium breve, Bifidobacterium longum, Bifidobacterium adolescentis | Mouse | Fecal transplantationMicrobial DNA analysisBacterial administrationCell sortingGene expression profiling |
| (70) |
| Fecalibacterium prausnitzii, Bacteroides thetaiotamicron, Holdemania filiformis, Dorea formicogenerans | Human | Metagenomic shotgun sequencingGut metabolomic profiling |
| (72) |
| Clostridiales | Human | 16S rRNA gene sequencingWhole genome shotgun sequencingImmunohistochemistryFlow cytometryCytokines assayGene expression profiling |
| (73) |
| Ruminococcaceaea | MouseHuman | 16S rRNA gene sequencingWhole genome shotgun sequencingImmunohistochemistryFlow cytometryCytokines assayGene expression profilingFecal microbiota transplantation |
| (74) |
| Akkermansia muciniphila | MouseHuman | Metagenomic shotgun sequencingFecal microbiota transplantationImmunohistochemistryFlow cytometryCytokines assay |
| (75) |
| B. longum, Collinsella aerofaciens, Enterococcus faecium | MouseHuman | 16S rRNA gene sequencingMetagenomic shotgun sequencingSpecies-specific quantitative PCRImmunohistochemistryFecal transplantation |
| (71) |
Bacterial species that are positively associated with programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1) blockade therapy.
aBacteria of Ruminococcaceae family belongs to the Clostridiales order.
bPatients here include patients with advanced non-small-cell-lung cancer, renal cell carcinoma, and urothelial carcinoma.
Potential Mechanisms of Gut Microbes on Improving the Efficacy of PD-1 and PD-L1 Blockade Therapy
Despite the exciting findings in this research field, the underlying molecular mechanisms by which the identified gut bacterial species in the above studies enhance PD-1 and PD-L1 blockade therapy remain largely unknown.
Recently, unmethylated CpG oligodeoxynucleotides, which are abundant in bacterial DNA, were found to enhance CD8+ T cell anti-tumor immunity by downregulating PD-1 expression via the IL-12 pathway, suggesting that gut bacterial species that are positively associated with PD-1 and PD-L1 blockade therapy may release components that directly downregulate PD-1 or PD-L1 expression (78, 79).
It is also possible that the gut bacterial species indirectly affect PD-1 and PD-L1 expression through locally or systematically regulating immune responses, thereby affecting the efficacy of PD-1 and PD-L1 blockade therapy. Gut microbiota has been shown to impact on both innate and adaptive immune cells. Germ-free animals had a reduced number of intestinal DCs and administration of Escherichia coli in these animals was able to recruit sufficient DCs to the intestines (80, 81). In Germ-free pigs, systemic circulating macrophages were also reduced and their functions were compromised (82). Germ-free mice had markedly decreased presence of lamina propria CD4+ T cells and absence of lymphocyte zones in spleens and mesenteric lymph nodes (83, 84). Polysaccharide A from Bacteroides fragilis was found to induce the Th1 response (83). Reduction of commensal microbiota in mice by using broad-spectrum antibiotics resulted in defective T and B cell responses against influenza infection (85). The findings that gut microbes can affect the immune functions, both locally and systematically suggest that bacterial species positively associated with PD-1 and PD-L1 blockade may enhance PD-1 and PD-L1 immunotherapy through regulation of the immune response. The previous study by Sivan et al. showed that Bifidobacterium species that inhibited tumor growth activated DCs, further supporting this view (70).
The Possible Impact of Chronic Infections and Inflammation on PD-1 and PD-L1 Blockade Therapy
Several microbes cause chronic infections in humans, some of which are known to increase host PD-1 and PD-L1 expression (86–94). However, studies have not examined whether existing chronic infections in patients with cancer affect the efficacy of PD-1 and PD-L1 blockade therapy.
An example of a chronic infection is Helicobacter pylori infection. H. pylori are a Gram-negative bacterium that colonizes the stomach of more than 50% of the world population. While most of the individuals colonized with H. pylori are asymptomatic, some may develop chronic gastritis and peptic ulcers, and H. pylori colonization is also a risk factor for gastric cancer (95). Previous studies have shown that patients with H. pylori infection have a significantly higher level of pro-inflammatory cytokines, such as TNF-α (96–98). Das et al. showed that H. pylori increased the gastric epithelial expression of PD-L1 using a gastric epithelial cell line model (86). Furthermore, they showed that gastric epithelial cells exposed to H. pylori inhibited the proliferation of CD4+ T cells isolated from blood and the inhibitory effect can be blocked using antibodies PD-L1. Similarly, Wu et al. observed increased PD-L1 expression in gastric biopsies of individuals infected with H. pylori, and co-culture of H. pylori infected primary gastric epithelial cells with T cells isolated from blood induced T cell apoptosis (87). These results suggest that H. pylori infection may cause the non-specific inhibition of circulating T cells, including tumor-specific T cells. In addition to H. pylori, several viruses, such as the hepatitis B virus, hepatitis C virus, human papillomavirus, and Epstein–Barr virus are also able to establish chronic infections in humans and increase host PD-1 or PD-L1 expression (88–94). Future studies are needed to examine whether chronic infections or inflammation impact on the efficacy of PD-1 and PD-L1 blockade. A recent study by Kottke et al. using a mouse model showed that pro-inflammatory cytokine TNF-α promoted tumor recurrence, while TNF-α blockade prevented tumor recurrence (99–102). Some bacterial species that are known to reduce chronic inflammation after administration orally may be examined to see whether they can improve cancer treatment (103–108). If chronic infections or inflammation reduce the efficacy of PD-1 and PD-L1 blockade, it would be through mechanisms other than the induction of the PD-1 and PD-L1 expression in the tumor tissues, as previous studies observed better responses to PD-1 blockade in patients with higher expression of PD-L1 in tumor tissues (51).
Future Directions
As discussed, despite the clear benefits of PD-1 and PD-L1 blockade in treating some cancer patients, the efficacy and the recurrence of tumor are issues that remain to be tackled. Emerging evidence suggests that modulation of the gut microbiota is a promising approach for improving PD-L1 and PD-1 blockade therapy. However, future studies are needed to further develop this research area.
The Bifidobacterium species, particularly B. longum, increased anti-PD-L1 efficacy in mice models and was positively associated with anti-PD-1 efficacy in metastatic melanoma patients. Future studies are needed to understand the molecular mechanisms of these Bifidobacterium species in enhancing PD-1 and PD-L1 blockade therapy. In addition to the Bifidobacterium species, various studies reported positive associations of gut microbes with PD-1 and PD-L1 blockade therapy at genus level. These microbes need to be identified at species and strain level and their potential anti-tumor mechanisms require further investigation.
Several bacterial and viral pathogens are known to cause chronic human infections and the pro-inflammatory cytokines are known to induce host PD-1 and PD-L1 expression. In addition, some of these pathogens are known to directly attach immune cells. Whether chronic infections caused by different pathogens impact on PD-1 and PD-L1 blockade therapy should be investigated, and appropriate strategies to enhance PD-1 and PD-L1 blockade therapy in these patients can then be developed accordingly. A suggested course of action is outlined in Table 3.
Table 3
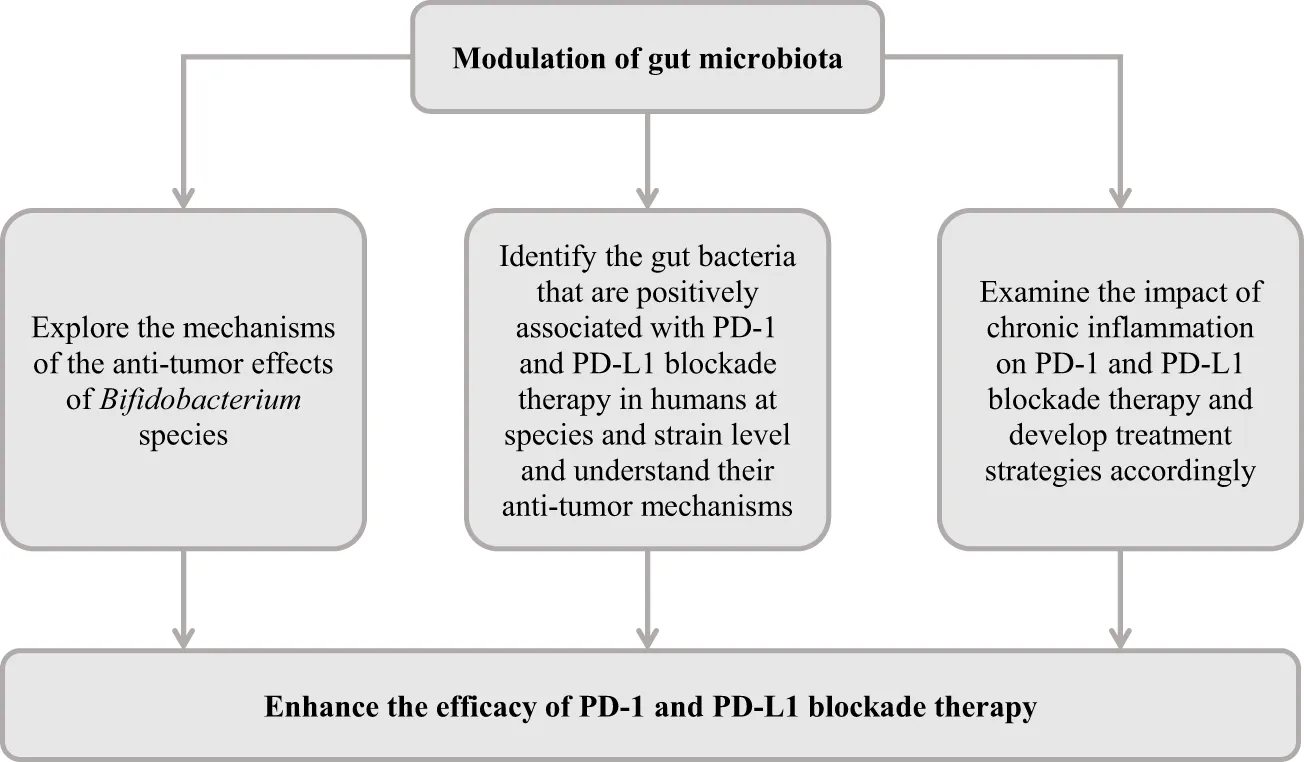 |
Suggested future directions.
Statements
Author contributions
Wrote the paper: YW, LZ. Figures and tables: YW. Revised the paper: YW, LZ, RM, FL, SL. All authors have approved the final version of the manuscript.
Funding
This work is supported by a Faculty Research Grant awarded to LZ from the University of New South Wales (Grant No.: PS46772).
Acknowledgments
The authors would like to thank Dr. Jiezhong Chen (School of Biomedical Sciences, University of Queensland) for reading the manuscript and providing feedback on this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
ErcoliniAMLadleBHManningEAPfannenstielLWArmstrongTDMachielsJ-PHet alRecruitment of latent pools of high-avidity CD8+ T cells to the antitumor immune response. J Exp Med (2005) 201(10):1591–602.10.1084/jem.20042167
2
MeliefCJ. Tumor eradication by adoptive transfer of cytototic T lymphocytes. Adv Cancer Res (1992) 58:143–75.10.1016/S0065-230X(08)60294-8
3
ZouW. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer (2005) 5(4):263–74.10.1038/nrc1586
4
PostowMACallahanMKWolchokJD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33(17):1974–82.10.1200/JCO.2014.59.4358
5
IshidaYAgataYShibaharaKHonjoT. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887.
6
DongHZhuGTamadaKChenL. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5(12):1365–9.10.1038/70932
7
FreemanGJLongAJIwaiYBourqueKChernovaTNishimuraHet alEngagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34.10.1084/jem.192.7.1027
8
KeirMEButteMJFreemanGJSharpeAH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704.10.1146/annurev.immunol.26.021607.090331
9
DongHStromeSESalomaoDRTamuraHHiranoFFliesDBet alTumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800.10.1038/nm730
10
YamazakiTAkibaHIwaiHMatsudaHAokiMTannoYet alExpression of programmed death 1 ligands by murine T cells and APC. J Immunol (2002) 169(10):5538–45.10.4049/jimmunol.169.10.5538
11
IshidaMIwaiYTanakaYOkazakiTFreemanGJMinatoNet alDifferential expression of PD-L1 and PD-L2, ligands for an inhibitory receptor PD-1, in the cells of lymphohematopoietic tissues. Immunol Lett (2002) 84(1):57–62.10.1016/S0165-2478(02)00142-6
12
TsengS-YOtsujiMGorskiKHuangXSlanskyJEPaiSIet alB7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193(7):839–46.10.1084/jem.193.7.839
13
ShinTKennedyGGorskiKTsuchiyaHKosekiHAzumaMet alCooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med (2003) 198(1):31–8.10.1084/jem.20030242
14
TamuraHDongHZhuGSicaGLFliesDBTamadaKet alB7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood (2001) 97(6):1809–16.10.1182/blood.V97.6.1809
15
MazanetMMHughesCC. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol (2002) 169(7):3581–8.10.4049/jimmunol.169.7.3581
16
Selenko-GebauerNMajdicOSzekeresAHöflerGGuthannEKorthäuerUet alB7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol (2003) 170(7):3637–44.10.4049/jimmunol.170.7.3637
17
GhiottoMGauthierLSerriariNPastorSTrunehANunèsJAet alPD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol (2010) 22(8):651–60.10.1093/intimm/dxq049
18
ChenL. Co-inhibitory molecules of the B7–CD28 family in the control of T-cell immunity. Nat Rev Immunol (2004) 4(5):336–47.10.1038/nri1349
19
ZouWChenL. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol (2008) 8(6):467–77.10.1038/nri2326
20
TsushimaFYaoSShinTFliesAFliesSXuHet alInteraction between B7-H1 and PD-1 determines initiation and reversal of T-cell anergy. Blood (2007) 110(1):180–5.10.1182/blood-2006-11-060087
21
GoldbergMVMarisCHHipkissELFliesASZhenLTuderRMet alRole of PD-1 and its ligand, B7-H1, in early fate decisions of CD8 T cells. Blood (2007) 110(1):186–92.10.1182/blood-2006-12-062422
22
EngelsBEngelhardVHSidneyJSetteABinderDCLiuRBet alRelapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell (2013) 23(4):516–26.10.1016/j.ccr.2013.03.018
23
MühlbauerMFleckMSchützCWeissTFrohMBlankCet alPD-L1 is induced in hepatocytes by viral infection and by interferon-α and-γ and mediates T cell apoptosis. J Hepatol (2006) 45(4):520–8.10.1016/j.jhep.2006.05.007
24
IwaiYIshidaMTanakaYOkazakiTHonjoTMinatoN. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99(19):12293–7.10.1073/pnas.192461099
25
KonishiJYamazakiKAzumaMKinoshitaIDosaka-AkitaHNishimuraM. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res (2004) 10(15):5094–100.10.1158/1078-0432.CCR-04-0428
26
BrownJADorfmanDMMaF-RSullivanELMunozOWoodCRet alBlockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol (2003) 170(3):1257–66.10.4049/jimmunol.170.3.1257
27
CurielTJWeiSDongHAlvarezXChengPMottramPet alBlockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat Med (2003) 9(5):562–7.10.1038/nm863
28
KuangD-MZhaoQPengCXuJZhangJ-PWuCet alActivated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med (2009) 206(6):1327–37.10.1084/jem.20082173
29
LiuYZengBZhangZZhangYYangR. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol (2008) 129(3):471–81.10.1016/j.clim.2008.07.030
30
PardollDM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64.10.1038/nrc3239
31
SznolMChenL. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res (2013) 19(5):1021–34.10.1158/1078-0432.CCR-12-2063
32
KondoAYamashitaTTamuraHZhaoWTsujiTShimizuMet alInterferon-γ and tumor necrosis factor-α induce an immunoinhibitory molecule, B7-H1, via nuclear factor-κB activation in blasts in myelodysplastic syndromes. Blood (2010) 116(7):1124–31.10.1182/blood-2009-12-255125
33
GordonSRMauteRLDulkenBWHutterGGeorgeBMMcCrackenMNet alPD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545(7655):495–9.10.1038/nature22396
34
BlankCBrownIPetersonACSpiottoMIwaiYHonjoTet alPD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res (2004) 64(3):1140–5.10.1158/0008-5472.CAN-03-3259
35
PetroffMChenLPhillipsTHuntJ. B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta (2002) 23:S95–101.10.1053/plac.2002.0813
36
HiranoFKanekoKTamuraHDongHWangSIchikawaMet alBlockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65(3):1089–96.
37
ClarkCAGuptaHBSareddyGPandeswaraSLaoSYuanBet alTumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res (2016) 76(23):6964–74.10.1158/0008-5472.CAN-16-0258
38
OrzechowskiADongH. Abstract 5026: B7-H1 confers tumor chemoresistance by regulating MAPK/ERK activation. Cancer Res (2014) 74(19 Suppl):5026.10.1158/1538-7445.AM2014-5026
39
KaufmanHRussellJSHamidOBhatiaSTerheydenPD’AngeloSPet alAvelumab (MSB0010718C; anti-PD-L1) in patients with metastatic Merkel cell carcinoma previously treated with chemotherapy: results of the phase 2 JAVELIN Merkel 200 trial. J Clin Oncol (2016) 34(15):9508.10.1200/JCO.2016.34.15_supll.9508
40
KaufmanHLRussellJHamidOBhatiaSTerheydenPD’AngeloSPet alAvelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol (2016) 17(10):1374–85.10.1016/S1470-2045(16)30364-3
41
BalarAVGalskyMDRosenbergJEPowlesTPetrylakDPBellmuntJet alAtezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet (2017) 389(10064):67–76.10.1016/S0140-6736(16)32455-2
42
SpiraAIParkKMazièresJVansteenkisteJFRittmeyerABallingerMet alEfficacy, safety and predictive biomarker results from a randomized phase II study comparing MPDL3280A vs docetaxel in 2L/3L NSCLC (POPLAR). J Clin Oncol (2015) 33(15):8010.10.1200/jco.2015.33.15_suppl.8010
43
JinCZhengYJinXMukhopadhyayPGuptaAKDennisPAet alExposure-efficacy and safety analysis of durvalumab in patients with urothelial carcinoma (UC) and other solid tumors. J Clin Oncol (2017) 35(15):2568.10.1200/JCO.2017.35.15_suppl.2568
44
IbrahimRStewartRShalabiA. PD-L1 blockade for cancer treatment: MEDI4736. Semin Oncol (2015) 42:474–83.10.1053/j.seminoncol.2015.02.007
45
SyedYY. Durvalumab: first global approval. Drugs (2017) 77(12):1369–76.10.1007/s40265-017-0782-5
46
RibasAHamidODaudAHodiFSWolchokJDKeffordRet alAssociation of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA (2016) 315(15):1600–9.10.1001/jama.2016.4059
47
SchachterJRibasALongGVAranceAGrobJJMortierLet alPembrolizumab versus ipilimumab for advanced melanoma: final overall survival analysis of KEYNOTE-006. J Clin Oncol (2016) 34(15):9504.10.1200/JCO.2016.34.15_suppl.9504
48
RibasAPuzanovIDummerRSchadendorfDHamidORobertCet alPembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol (2015) 16(8):908–18.10.1016/S1470-2045(15)00083-2
49
ReckMRodríguez-AbreuDRobinsonAGHuiRCsősziTFülöpAet alPembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med (2016) 2016(375):1823–33.10.1056/NEJMoa1606774
50
LangerCJGadgeelSMBorghaeiHPapadimitrakopoulouVAPatnaikAPowellSFet alCarboplatin and pemetrexed with or without pembroli-zumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol (2016) 17(11):1497–508.10.1016/S1470-2045(16)30498-3
51
HerbstRSBaasPKimD-WFelipEPérez-GraciaJLHanJ-Yet alPembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387(10027):1540–50.10.1016/S0140-6736(15)01281-7
52
SeiwertTYBurtnessBMehraRWeissJBergerREderJPet alSafety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol (2016) 17(7):956–65.10.1016/S1470-2045(16)30066-3
53
BalarAVCastellanoDEO’DonnellPHGrivasPVukyJPowlesTet alPembrolizumab as first-line therapy in cisplatin-ineligible advanced urothelial cancer: results from the total KEYNOTE-052 study population. J Clin Oncol (2017) 35(6):284.10.1200/JCO.2017.35.6_suppl.284
54
BellmuntJde WitRVaughnDJFradetYLeeJ-LFongLet alPembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med (2017) 376(11):1015–26.10.1056/NEJMoa1613683
55
MoskowitzCHZinzaniPLFanaleMAArmandPJohnsonNARadfordJAet alPembrolizumab in relapsed/refractory classical Hodgkin lymphoma: primary end point analysis of the phase 2 Keynote-087 study. Blood (2016) 128(22):1107.
56
ChenRZinzaniPLFanaleMAArmandPJohnsonNABricePet alPhase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol (2017) 35:2125.10.1200/JCO.2016.72.1316
57
LeDTYoshinoTJägerDAndreTBendellJCWangRet alKEYNOTE-164: phase II study of pembrolizumab (MK-3475) for patients with previously treated, microsatellite instability-high advanced colorectal carcinoma. J Clin Oncol (2016) 34(4).10.1200/jco.2016.34.4_suppl.tps787
58
OttPAFernandezMEEHiretSKimD-WMossRAWinserTet alPembrolizumab (MK-3475) in patients (pts) with extensive-stage small cell lung cancer (SCLC): preliminary safety and efficacy results from KEYNOTE-028. J Clin Oncol (2015) 33(15):7502.10.1200/jco.2015.33.15_suppl.7502
59
OttPABangY-JBerton-RigaudDElezEPishvaianMJRugoHSet alPembrolizumab in advanced endometrial cancer: preliminary results from the phase Ib KEYNOTE-028 study. J Clin Oncol (2016) 34(15):5581.10.1200/JCO.2016.34.15_suppl.5581
60
WolchokJDChiarion-SileniVGonzalezRRutkowskiPGrobJJCoweyCLet alUpdated results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naive patients (pts) with advanced melanoma (MEL)(CheckMate 067). J Clin Oncol (2016) 34(15):9505.10.1200/JCO.2016.34.15_suppl.9505
61
WolchokJDChiarion-SileniVGonzalezRRutkowskiPGrobJJCoweyCLet alEfficacy and safety results from a phase III trial of nivolumab (NIVO) alone or combined with ipilimumab (IPI) versus IPI alone in treatment-naive patients (pts) with advanced melanoma (MEL)(CheckMate 067). J Clin Oncol (2015) 33(18).10.1200/jco.2015.33.18_suppl.lba1
62
SpigelDRReckampKLRizviNAPoddubskayaEWestHJEberhardtWEEet alA phase III study (CheckMate 017) of nivolumab (NIVO; anti-programmed death-1 [PD-1]) vs docetaxel (DOC) in previously treated advanced or metastatic squamous (SQ) cell non-small cell lung cancer (NSCLC). J Clin Oncol (2015) 33(15):8009.10.1200/jco.2015.33.15_suppl.8009
63
Paz-AresLHornLBorghaeiHSpigelDRSteinsMReadyNet alPhase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J Clin Oncol (2015) 33(18).10.1200/jco.2015.33.18_suppl.lba109
64
MotzerRJEscudierBMcDermottDFGeorgeSHammersHJSrinivasSet alNivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) 373(19):1803–13.10.1056/NEJMoa1510665
65
CellaDGrünwaldVNathanPDoanJDastaniHTaylorFet alQuality of life in patients with advanced renal cell carcinoma given nivolumab versus everolimus in CheckMate 025: a randomised, open-label, phase 3 trial. Lancet Oncol (2016) 17(7):994–1003.10.1016/S1470-2045(16)30125-5
66
YounesASantoroAZinzaniPLTimmermanJAnsellSMArmandPet alCheckmate 205: nivolumab (nivo) in classical Hodgkin lymphoma (cHL) after autologous stem cell transplant (ASCT) and brentuximab vedotin (BV)—A phase 2 study. J Clin Oncol (2016) 34(15):7535.10.1200/JCO.2016.34.15_suppl.7535
67
AnsellSArmandPTimmermanJMShippMAGarelikMBBZhuLet alNivolumab in patients (pts) with relapsed or refractory classical Hodgkin lymphoma (R/R cHL): clinical outcomes from extended follow-up of a phase 1 study (CA209-039). Blood (2015) 126:583.
68
FerrisRLBlumenscheinGJrFayetteJGuigayJColevasADLicitraLet alNivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med (2016) 2016(375):1856–67.10.1056/NEJMoa1602252
69
FerrisRLBlumenscheinGRFayetteJGuigayJColevasADLicitraLFet alFurther evaluations of nivolumab (nivo) versus investigator’s choice (IC) chemotherapy for recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CheckMate 141. J Clin Oncol (2016) 34(15):6009.10.1200/JCO.2016.34.15_suppl.6009
70
SivanACorralesLHubertNWilliamsJBAquino-MichaelsKEarleyZMet alCommensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science (2015) 350(6264):1084–9.10.1126/science.aac4255
71
MatsonVFesslerJBaoRChongsuwatTZhaYAlegreM-Let alThe commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science (2018) 359(6371):104–8.10.1126/science.aao3290
72
FrankelAECoughlinLAKimJFroehlichTWXieYFrenkelEPet alMetagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia (2017) 19(10):848–55.10.1016/j.neo.2017.08.004
73
WargoJAGopalakrishnanVSpencerCKarpinetsTReubenAAndrewsMCet alAssociation of the diversity and composition of the gut microbiome with responses and survival (PFS) in metastatic melanoma (MM) patients (pts) on anti-PD-1 therapy. J Clin Oncol (2017) 35(15):3008.10.1200/JCO.2017.35.15_suppl.3008
74
GopalakrishnanVSpencerCNeziLReubenAAndrewsMKarpinetsTet alGut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103.10.1126/science.aan4236
75
RoutyBLe ChatelierEDerosaLDuongCPAlouMTDaillèreRet alGut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7.10.1126/science.aan3706
76
VétizouMPittJMDaillèreRLepagePWaldschmittNFlamentCet alAnticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84.10.1126/science.aad1329
77
PittJMVétizouMGomperts BonecaILepagePChamaillardMZitvogelL. Enhancing the clinical coverage and anticancer efficacy of immune checkpoint blockade through manipulation of the gut microbiota. Oncoimmunology (2017) 6(1):e1132137.10.1080/2162402X.2015.1132137
78
YinPLiuXMansfieldASHarringtonSMLiYYanYet alCpG-induced antitumor immunity requires IL-12 in expansion of effector cells and down-regulation of PD-1. Oncotarget (2016) 7(43):70223.10.18632/oncotarget.11833
79
WangSCamposJGallottaMGongMCrainCNaikEet alIntratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A (2016) 113(46):E7240–9.10.1073/pnas.1608555113
80
HaversonKRehakovaZSinkoraJSverLBaileyM. Immune development in jejunal mucosa after colonization with selected commensal gut bacteria: a study in germ-free pigs. Vet Immunol Immunopathol (2007) 119(3):243–53.10.1016/j.vetimm.2007.05.022
81
WilliamsAMProbertCSStepankovaRTlaskalova-HogenovaHPhillipsABlandPW. Effects of microflora on the neonatal development of gut mucosal T cells and myeloid cells in the mouse. Immunology (2006) 119(4):470–8.10.1111/j.1365-2567.2006.02458.x
82
ZhangWWenKAzevedoMSGonzalezASaifLJLiGet alLactic acid bacterial colonization and human rotavirus infection influence distribution and frequencies of monocytes/macrophages and dendritic cells in neonatal gnotobiotic pigs. Vet Immunol Immunopathol (2008) 121(3):222–31.10.1016/j.vetimm.2007.10.001
83
MazmanianSKLiuCHTzianabosAOKasperDL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell (2005) 122(1):107–18.10.1016/j.cell.2005.05.007
84
MacphersonAMartinicMHarrisN. The functions of mucosal T cells in containing the indigenous commensal flora of the intestine. Cell Mol Life Sci (2002) 59(12):2088–96.10.1007/s000180200009
85
IchinoheTPangIKKumamotoYPeaperDRHoJHMurrayTSet alMicrobiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci U S A (2011) 108(13):5354–9.10.1073/pnas.1019378108
86
DasSSuarezGBeswickEJSierraJCGrahamDYReyesVE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol (2006) 176(5):3000–9.10.4049/jimmunol.176.5.3000
87
WuYYLinCWChengKSLinCWangYMLinITet alIncreased programmed death-ligand-1 expression in human gastric epithelial cells in Helicobacter pylori infection. Clin Exp Immunol (2010) 161(3):551–9.10.1111/j.1365-2249.2010.04217.x
88
XieZChenYZhaoSYangZYaoXGuoSet alIntrahepatic PD-1/PD-L1 up-regulation closely correlates with inflammation and virus replication in patients with chronic HBV infection. Immunol Invest (2009) 38(7):624–38.10.1080/08820130903062210
89
PengGLiSWuWTanXChenYChenZ. PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol Immunol (2008) 45(4):963–70.10.1016/j.molimm.2007.07.038
90
Golden-MasonLPalmerBKlarquistJMengsholJACastelblancoNRosenHR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol (2007) 81(17):9249–58.10.1128/JVI.00409-07
91
ShenTChenXChenYXuQLuFLiuS. Increased PD-L1 expression and PD-L1/CD86 ratio on dendritic cells were associated with impaired dendritic cells function in HCV infection. J Med Virol (2010) 82(7):1152–9.10.1002/jmv.21809
92
YangWSongYLuYLSunJZWangHW. Increased expression of programmed death (PD)-1 and its ligand PD-L1 correlates with impaired cell-mediated immunity in high-risk human papillomavirus-related cervical intraepithelial neoplasia. Immunology (2013) 139(4):513–22.10.1111/imm.12101
93
FangWZhangJHongSZhanJChenNQinTet alEBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: implications for oncotargeted therapy. Oncotarget (2014) 5(23):12189.10.18632/oncotarget.2608
94
DerksSLiaoXChiaravalliAMXuXCamargoMCSolciaEet alAbundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget (2016) 7(22):32925.10.18632/oncotarget.9076
95
ParsonnetJFriedmanGDVandersteenDPChangYVogelmanJHOrentreichNet alHelicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med (1991) 325(16):1127–31.10.1056/NEJM199110173251603
96
CrabtreeJShallcrossTHeatleyRWyattJ. Mucosal tumour necrosis factor alpha and interleukin-6 in patients with Helicobacter pylori associated gastritis. Gut (1991) 32(12):1473–7.10.1136/gut.32.12.1473
97
BodgerKWyattJHeatleyR. Gastric mucosal secretion of interleukin-10: relations to histopathology, Helicobacter pylori status, and tumour necrosis factor-alpha secretion. Gut (1997) 40(6):739–44.10.1136/gut.40.6.739
98
PerriFClementeRFestaVDe AmbrosioCQuitadamoMFusilloMet alSerum tumour necrosis factor-alpha is increased in patients with Helico-bacter pylori infection and CagA antibodies. Ital J Gastroenterol Hepatol (1999) 31(4):290–4.
99
KottkeTEvginLShimKGRommelfangerDBoisgeraultNZaidiSet alSubversion of NK-cell and TNFα immune surveillance drives tumor recurrence. Cancer Immunol Res (2017) 5(11):1029–45.10.1158/2326-6066.CIR-17-0175
100
ChenJJiangCJinLZhangX. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol (2015) 27(3):409–16.10.1093/annonc/mdv615
101
GowrishankarKGunatilakeDGallagherSJTiffenJRizosHHerseyP. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-κB. PLoS One (2015) 10(4):e0123410.10.1371/journal.pone.0123410
102
WangXYangLHuangFZhangQLiuSMaLet alInflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett (2017) 184:7–14.10.1016/j.imlet.2017.02.006
103
MatsumotoSHaraTHoriTMitsuyamaKNagaokaMTomiyasuNet alProbiotic Lactobacillus-induced improvement in murine chronic inflammatory bowel disease is associated with the down-regulation of pro-inflammatory cytokines in lamina propria mononuclear cells. Clin Exp Immunol (2005) 140(3):417–26.10.1111/j.1365-2249.2005.02790.x
104
AmdekarSSinghVSinghRSharmaPKeshavPKumarA. Lactobacillus casei reduces the inflammatory joint damage associated with collagen-induced arthritis (CIA) by reducing the pro-inflammatory cytokines: Lactobacillus casei: COX-2 inhibitor. J Clin Immunol (2011) 31(2):147–54.10.1007/s10875-010-9457-7
105
LiuYFathereeNYMangalatNRhoadsJM. Human-derived probiotic Lactobacillus reuteri strains differentially reduce intestinal inflammation. Am J Physiol Gastrointest Liver Physiol (2010) 299(5):G1087–96.10.1152/ajpgi.00124.2010
106
TienMTGirardinSERegnaultBLe BourhisLDilliesMACoppeeJYet alAnti-inflammatory effect of Lactobacillus casei on Shigella-infected human intestinal epithelial cells. J Immunol (2006) 176(2):1228–37.10.4049/jimmunol.176.2.1228
107
VieiraATGalvaoIAmaralFATeixeiraMMNicoliJRMartinsFS. Oral treatment with Bifidobacterium longum 51A reduced inflammation in a murine experimental model of gout. Benef Microbes (2015) 6(6):799–806.10.3920/BM2015.0015
108
ZhangLSuPHenrikssonAO’RourkeJMitchellH. Investigation of the immunomodulatory effects of Lactobacillus casei and Bifidobacterium lactis on Helicobacter pylori infection. Helicobacter (2008) 13(3):183–90.10.1111/j.1523-5378.2008.00595.x
Summary
Keywords
gut microbiota, programmed death 1, programmed death ligand 1, cancer immunotherapy, efficacy
Citation
Wang Y, Ma R, Liu F, Lee SA and Zhang L (2018) Modulation of Gut Microbiota: A Novel Paradigm of Enhancing the Efficacy of Programmed Death-1 and Programmed Death Ligand-1 Blockade Therapy. Front. Immunol. 9:374. doi: 10.3389/fimmu.2018.00374
Received
27 November 2017
Accepted
09 February 2018
Published
05 March 2018
Volume
9 - 2018
Edited by
Amedeo Amedei, Università degli Studi di Firenze, Italy
Reviewed by
Dr Kawaljit Kaur, University of California, Los Angeles, United States; Haidong Dong, Mayo Clinic Minnesota, United States; Sungjune Kim, Moffitt Cancer Center, United States
Updates
Copyright
© 2018 Wang, Ma, Liu, Lee and Zhang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhang, l.zhang@unsw.edu.au
Specialty section: This article was submitted to Cancer Immunity and Immunotherapy, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.