 Victoria Sundblad1†
Victoria Sundblad1† Amado A. Quintar2,3†
Amado A. Quintar2,3† Luciano G. Morosi1,4
Luciano G. Morosi1,4 Sonia I. Niveloni5
Sonia I. Niveloni5 Ana Cabanne6
Ana Cabanne6 Edgardo Smecuol5
Edgardo Smecuol5 Eduardo Mauriño5
Eduardo Mauriño5 Karina V. Mariño4
Karina V. Mariño4 Julio C. Bai5,7‡
Julio C. Bai5,7‡ Cristina A. Maldonado2,3‡
Cristina A. Maldonado2,3‡ Gabriel A. Rabinovich1,8*‡
Gabriel A. Rabinovich1,8*‡
- 1Laboratorio de Inmunopatología, Instituto de Biología y Medicina Experimental (IBYME), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
- 2Centro de Microscopía Electrónica, Facultad de Ciencias Médicas, Universidad Nacional de Córdoba, Córdoba, Argentina
- 3Instituto de Investigaciones en Ciencias de la Salud (INICSA), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Córdoba, Argentina
- 4Laboratorio de Glicómica Funcional y Molecular, Instituto de Biología y Medicina Experimental (IBYME), Consejo de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
- 5Sección Intestino Delgado, Departamento de Medicina, Hospital de Gastroenterología Dr. C. Bonorino Udaondo, Buenos Aires, Argentina
- 6Unidad de Patología, Hospital de Gastroenterología, Bonorino Udaondo, Buenos Aires, Argentina
- 7Instituto de Investigaciones, Universidad del Salvador, Buenos Aires, Argentina
- 8Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Buenos Aires, Argentina
Galectins, a family of animal lectins characterized by their affinity for N-acetyllactosamine-enriched glycoconjugates, modulate several immune cell processes shaping the course of innate and adaptive immune responses. Through interaction with a wide range of glycosylated receptors bearing complex branched N-glycans and core 2-O-glycans, these endogenous lectins trigger distinct signaling programs thereby controling immune cell activation, differentiation, recruitment and survival. Given the unique features of mucosal inflammation and the differential expression of galectins throughout the gastrointestinal tract, we discuss here key findings on the role of galectins in intestinal inflammation, particularly Crohn’s disease, ulcerative colitis, and celiac disease (CeD) patients, as well as in murine models resembling these inflammatory conditions. In addition, we present new data highlighting the regulated expression of galectin-1 (Gal-1), a proto-type member of the galectin family, during intestinal inflammation in untreated and treated CeD patients. Our results unveil a substantial upregulation of Gal-1 accompanying the anti-inflammatory and tolerogenic response associated with gluten-free diet in CeD patients, suggesting a major role of this lectin in favoring resolution of inflammation and restoration of mucosal homeostasis. Thus, a coordinated network of galectins and their glycosylated ligands, exerting either anti-inflammatory or proinflammatory responses, may influence the interplay between intestinal epithelial cells and the highly specialized gut immune system in physiologic and pathologic settings.
Introduction: Deciphering Glycocodes in Immunity
Complex sugar structures play essential roles as hardware for storage of biological information, which can be deciphered by endogenous glycan-binding proteins or lectins (1). The singular role of lectins in translating glycan-containing information into a myriad of cellular responses invigorated further studies aimed at understanding their expression patterns and molecular mechanisms of action.
Galectins, a family of lectins with affinity for N-acetyllactosamine (LacNac) residues, have diverse roles in shaping the course of innate and adaptive immunity and tailoring inflammatory responses, thereby modulating tumor immunity and autoimmune reactions (2, 3). In this perspective article, we review current knowledge on the role of galectins in inflammatory intestinal disorders, and present new findings on the regulated expression of galectin (Gal)-1 in intestinal tissue of celiac disease (CeD) patients.
Galectins
Galectins, evolutionarily conserved glycan-binding proteins, play key roles in multiple immune cell processes. Either through protein-glycan or protein–protein interactions, these lectins function within the extracellular milieu by interacting with various glycosylated receptors, or work inside the cells by controlling distinct signaling pathways and modulating intracellular processes (3, 4).
To date, 15 members of the galectin family have been identified in vertebrates, which were classified into three groups based on their molecular architecture: (a) “proto-type” galectins, comprising a single polypeptide chain with one carbohydrate recognition domain (CRD) that is able to dimerize (Gal-1, -2, -5, -7, -10, -11, -13, -14, and -15); (b) “chimera-type” Gal-3, which consists of a C-terminal CRD linked to an N-terminal peptide, and (c) “tandem repeat-type” galectins composed of a single polypeptide chain exhibiting two CRDs in tandem connected by a linker peptide (Gal-4, -6, -8, -9, and -12) (4, 5). While some members of the family (e.g., Gal-1 and Gal-3) are widely distributed among different tissues and species (6–8), others have more restricted tissue localization. For example, Gal-7 is preferentially found in the skin (9, 10), Gal-12 is mostly expressed in adipose tissue (11, 12), Gal-5 is restricted to rat reticulocytes (13, 14) and Gal-10 is found in human but not mouse eosinophils (15).
Once synthesized, galectins may remain within the intracellular compartment and participate in protein-protein interactions to regulate intracellular events (16, 17). For example, both Gal-1 and -3 participate in pre-mRNA splicing (18) whereas Gal-10 modulates functionality of human CD25+ Treg cells (19). However, despite the lack of a classical secretory signal peptide, most galectins are released through an unconventional route to the extracellular compartment (20). Secreted galectins can specifically decipher biological information encoded in complex saccharide structures (particularly LacNac-enriched complex branched N-glycans and core 2 O-glycans), and convey this biochemical information into functional cellular responses (3, 17). Although saccharide structures are widely distributed in a range of glycoconjugates, individual galectins may co-opt a particular set of glycosylated receptors, generated by the coordinated action of glycosyltransferases and glycosidases which are differentially regulated in distinct target cells (4, 21, 22). Notably, one-CRD galectins can dimerize via the back sides of their CRDs, whereas chimera-type Gal-3 can pentamerize via its non-lectin N-terminal domain, and tandem-repeat galectins can oligomerize (17). Thus, through formation of multivalent galectin–glycan complexes, galectins can promote cross-linking, reorganization, and clustering of glycosylated receptors thereafter regulating their activation and signaling (23, 24). Within the immune compartment, galectin–glycan complexes may control signaling thresholds of relevant receptors such as the T-cell receptor (25), pre-B cell receptor (26), and cytokine receptors (27) among others, thereby modulating lymphoid and myeloid regulatory programs.
Galectins: Key Players in the Inflammatory Response
Compelling evidence highlights major roles for galectins in controlling innate and adaptive immune responses. These lectins may influence the capacity of innate immune cells [e.g., neutrophils, dendritic cells (DCs), monocytes/macrophages, eosinophils, and mast cells] to respond to chemotactic gradients, migrate across endothelial cell surfaces, synthesize and release pro- or anti-inflammatory cytokines, and recognize, engulf, and kill microbes and damaged cells (28). In this regard, some galectins trigger innate immune responses, while others influence the resolution of acute inflammation (28). Galectins can also tailor adaptive immunity by influencing T-cell signaling and activation, modulating T-cell survival, controlling the suppressive function of regulatory T cells (Tregs), altering the cytokine balance and regulating B-cell maturation and differentiation (3). Both the specificity of the CRD as well as glycan presentation in the corresponding receptors make distinct contributions to the specific effects of individual galectins, selectively mediating different biological processes. The final balance of their synchronized actions contributes to activation, polarization, and resolution of adaptive immune responses (29). Although the specific immunoregulatory activities of each individual galectin is beyond the scope of the present work, and are described elsewhere (3, 29), some of the most relevant activities displayed by Gal-1, the central core of the present article, are summarized herein. This endogenous lectin, composed of two subunits of 14.5 kDa, functions as a regulatory signal which undermines acute inflammatory responses by controlling neutrophil adhesion, function and turnover (30, 31) and modulating monocyte and macrophage activation and polarization (32–35). Moreover, Gal-1 influences DC maturation, immunogenicity, and migration (36–40). Interestingly upon exposure to this lectin, DCs acquire an IL-27-dependent regulatory function leading to IL-10-mediated T-cell tolerance, suppression of T-helper (Th)1 and Th17 responses, promotion of tumor-immune escape and suppression of autoimmune neuroinflammation (40).
Regarding the T-cell compartment, Gal-1 controls T-cell viability, blunts Th1- and Th17-mediated responses and skews the balance of the immune response toward a Th2 cytokine profile (17, 41–43). Interestingly, we found that Th1- and Th17-differentiated cells express the repertoire of cell surface glycans that are critical for Gal-1 binding and induction of apoptosis; whereas Th2 cells are protected from this lectin through α2,6-sialylation of surface glycoproteins (43). Remarkably, Gal-1 also controls the immunosuppressive activity of Tregs and promotes their differentiation (44–46). Finally, by influencing B-cell development, differentiation, signaling and survival, Gal-1 also controls B-cell function (47–50).
The essential role of Gal-1 in the control of inflammation has been widely demonstrated in experimental models of autoimmunity, allergy and cancer (29, 51–53). In cancer settings, Gal-1 contributes to create immunosuppressive microenvironments, allowing tumor cell evasion of immune responses (46, 54–63). On the other hand, in experimental models of autoimmune disease including collagen-induced arthritis (64), myelin-oligodendrocyte glycoprotein35–55-induced encephalomyelitis (43, 65), diabetes (66), uveitis (67), and orchitis (68), Gal-1 elicits a broad spectrum of immunoregulatory activities leading to the resolution of chronic inflammation. The mechanisms underlying these immunosuppressive effects recapitulate those observed in vitro and in vivo including T-cell dysfunction and inhibition of proinflammatory cytokines (43, 58, 64, 69, 70), induction of tolerogenic DCs (40), expansion of Foxp3+ and Foxp3− Tregs (60, 67) and generation of alternatively activated “M2-type” macrophages (71).
Galectins in the Gut: A Sweet Path at the Cross-Roads of Tolerance and Inflammation
Despite the broad immunoregulatory activities of galectins, only few studies have uncovered the role of these lectins in gut immune homeostasis and the implications of these findings in intestinal inflammation. Interestingly, Gal-1, -2, -3, -4, and -9 are typically expressed in particular gut areas: whereas Gal-1 is mainly present in the lamina propria (LP), Gal-2, -3, -4, -7, and -9 are constitutively expressed within the epithelial compartment of the mouse intestine (72, 73). Epithelial cells (ECs) of small and large intestine express high levels of Gal-3 and Gal-4, although Gal-2 is only found in the large intestine (72). Interestingly, while Gal-3 may interact with commensal bacteria possibly influencing their colonization capacity (74), Gal-4 and Gal-8 mediate bacterial recognition and killing (75). Notably, Gal-1 is broadly expressed in small bowel enterocytes and may influence their viability (76). Moreover, studies reporting the galectin signature of human intestinal cells were mainly focused on pathologic conditions. Thus far, Gal-1, -3, -4, and -9 have shown to be homogeneously expressed across different sections of the large intestine (77).
Galectins in Intestinal Inflammatory Diseases
Inflammatory Bowel Diseases (IBD)
Crohn’s disease (CD) and ulcerative colitis (UC) represent the two main forms of IBD, chronic relapsing inflammatory conditions that affect the gastrointestinal tract. Despite some shared clinical features, these diseases can be distinguished by differences in risk factors, and clinical, anatomical, histological, and immunological features (78–80). Both conditions may involve an aberrant activation of mucosal T-cells against the commensal microbiota and deregulation of the EC compartment, thus compromising normal intestinal function and promoting an exuberant inflammatory response (81, 82). Whereas CD is characterized by an overactivation of mucosal Th1 and/or Th17 cells (with the concomitant secretion of IFN-γ, IL-17, and IL-22), UC patients exhibit a marked Th2 bias (with higher levels of IL-5 and IL-13) (79, 80, 83).
In a murine model of acute and chronic 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis, treatment with recombinant Gal-1 (rGal-1) resulted in improvement of the clinical, histopathological, and immunological manifestations of the disease. Further analysis revealed increased apoptosis of TNBS-specific CD4+ T-cells in the LP, decreased percentage of activated T-cells and diminished levels of proinflammatory and Th1-type cytokines, effects that were accompanied by normalization of the mucosal architecture (69). Accordingly, Gal-1 was found to be upregulated in inflamed areas of IBD patients when compared with non-inflamed areas of the same patient or with control subjects. Indeed, expression of common mucosal-associated galectins (Gal-1, -3, -4, -9) was found dysregulated in these inflamed tissues, suggesting that alteration in galectin expression pattern may represent an endogenous compensatory mechanisms likely aimed at limiting the inflammatory process and restoring mucosal homeostasis (77). Notably, the viability of human and mouse enterocytes was also controlled by Gal-1 in human IBD biopsies and in murine models of intestinal inflammation. Interestingly, proinflammatory stimuli promoted Gal-1 binding to EC which in turn influenced their survival and secretion of proresolving cytokines, thereby protecting the intestinal epithelium from inflammatory responses (76, 84). Thus, through elimination of antigen-experienced T-cells, modulation of proinflammatory cytokines or direct stimulation of epithelial-derived anti-inflammatory factors, Gal-1 contributes to the resolution of gut inflammation (Figure 1).
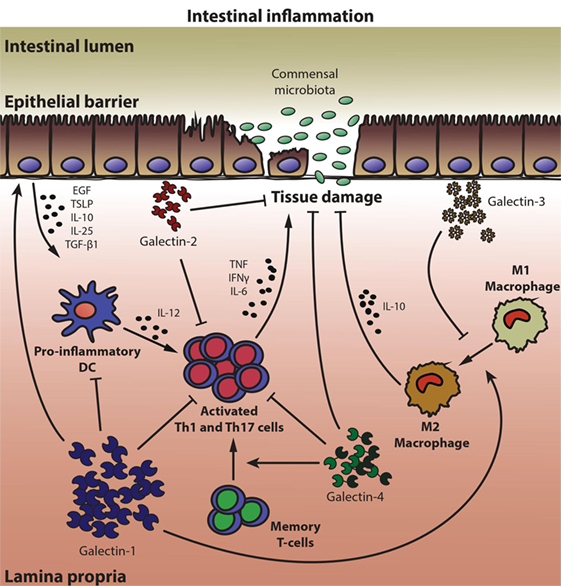
Figure 1. Multifunctional roles of galectins in mucosal intestinal inflammation. A coordinated network of galectin family members, which exerts either anti-inflammatory or proinflammatory responses, conditions epithelial barrier maintenance and immune gut homeostasis. Galectin-1 (Gal-1) promotes the secretion of growth factors and anti-inflammatory cytokines by epithelial cells (ECs), induces apoptosis of activated Th1 and Th17 cells, inhibits secretion of proinflammatory cytokines by dendritic cells (DCs) and T lymphocytes, and favors an anti-inflammatory (M2) macrophage phenotype. Similar to Gal-1, Gal-2 displays several anti-inflammatory properties, but also promotes wound healing and tissue regeneration in ECs. In animal models of colitis, Gal-3 shows mostly proinflammatory functions, inhibiting the polarization of macrophages toward an M2 phenotype, whereas Gal-4 exhibits both anti- and proinflammatory properties within the intestinal inflamed mucosa, depending on the experimental setting analyzed. By blocking production of proinflammatory cytokines, Gal-4 prevented inflammation and favored epithelial regeneration. However, through binding to memory T-cells, Gal-4 led to T-cell activation and perpetuated intestinal inflammation.
Notably, other members of the galectin family could also be involved in controlling intestinal inflammation (Figure 1). Gal-3 may function as a proinflammatory mediator that aggravates dextran sulfate sodium (DSS)-induced colitis through promotion of an M1 macrophage phenotype (85). Deletion of Gal-3 gene in mice or pharmacological inhibition of this lectin promoted macrophage polarization toward a M2 phenotype in colonic tissue (85). In line with these observations, peritoneal macrophages lacking Gal-3 are more prone to undergo apoptosis than their wild-type counterparts, strongly suggesting a role for Gal-3 as a proinflammatory mediator in the peritoneal cavity (86). Notably, in IBD patients Gal-3 levels are reduced in active inflamed areas, probably aimed at limiting the inflammatory process and restoring mucosal homeostasis (77, 87–89). In contrast, a protective role for this lectin was suggested in both the DSS-induced and the T-cell transfer colitis models, through suppression of IL-6 production by colonic LP fibroblasts or by induction of Foxp3+ Tregs (90, 91). These discrepancies could be explained not only by differences in experimental models (92), but also by dissimilar roles of endogenous versus exogenous Gal-3 during different stages of the inflammatory response (16, 29).
Similarly, Gal-4 has been shown to act either as an anti-inflammatory or as a proinflammatory factor in IBD. An anti-inflammatory function for both Gal-4 and Gal-2 was described, which contributed to ameliorate mucosal inflammation in the DSS colitis model through mechanisms involving apoptosis of activated mucosal LP T-cells and diminished proinflammatory cytokine secretion (93, 94) (Figure 1). Within the EC compartment, Gal-2 and Gal-4 (but not Gal-1) promoted wound-healing (95). Gal-4 may also function as a glycoprotein trafficking carrier, which generates an apical endocytic-recycling pathway via complex-type N-glycans (96, 97). Notably, during IBD progression, local inflammation was also associated with dysregulated expression of glycosyltransferases, leading to exposure of altered glycan structures on memory CD4+ T-cells (98). In fact, downregulation of core 2 β1,6-N-acetylglucosaminyltransferase 1 (C2GnT1) allowed Gal-4-O-glycan interactions resulting in expansion of memory CD4+ T-cells, enhanced IL-6 production and perpetuation of intestinal inflammation (98, 99). Notably, inflamed IBD mucosa could be distinguished from control tissue and from other types of intestinal inflammatory conditions by a specific galectin signature, as revealed by a multivariate-linear discriminant analysis of Gal-1, -3, -4, and -9 in IBD patient biopsies (77).
Celiac Disease
Oral tolerance to dietary antigens is a key active process in which immune responses to innocuous antigens, commensal bacteria, and pathogens are suppressed (100). In CeD, intolerance to indigestible wheat gluten peptides results in chronic intestinal inflammation associated with an extensive Th1 and Th17 responses (101). Similar to most chronic inflammatory diseases, CeD has a multifactorial etiology involving environmental factors as well as genetic components. Among them, HLA-DQ2 and HLA-DQ8 have been identified to confer susceptibility to CeD development (102–104). In genetically susceptible individuals, intestinal inflammation is triggered when ingested gliadin (proline-rich and glutamine-rich gluten proteins) found in wheat, rye, barley, and oats (105, 106) is partially processed and presented to CD4+ T-cells that infiltrate the LP of the small intestine. Thus HLA-DQ2/8 molecules may orchestrate a gluten-specific CD4+ T-cell response (107).
Celiac disease patients on a gluten-containing diet show increased levels of serum antibodies specific for gliadin and tissue transglutaminase, an enzyme that plays a key role in disruption of tolerance to gluten, among other antigens (108, 109). To date, the only known effective treatment for CeD is a lifelong gluten-free diet (GFD) (109), which allows the complete recovery of intestinal structure and function, and normalization of serum antibodies (110). In spite of considerable progress in our understanding of the mechanisms underlying CeD development and progression, there is no clear answer to how breaking mucosal tolerance to gluten turns a controlled local immune response into chronic inflammation and epithelial destruction (111).
Although the involvement of galectins in IBD has been well documented, their relevance in CeD development and progression is poorly understood. In this regard, a significant increase in Gal-10 expression has been correlated with mucosal damage and number of eosinophils in duodenal lesions of CeD patients (112). In addition, despite some discrepancies, evidence suggest a role for Gal-9 in human and mouse food allergy, a broad entity with some common features with CeD (113–115).
Galectin-1 Expression Delineates Response to GFD in CeD Patients
Since several immunoregulatory mechanisms are dysregulated in mucosal tissue of CeD patients (108) and Gal-1 displays broad tolerogenic and anti-inflammatory activities in mucosal tissues (29), we evaluated the expression of this lectin in biopsies of CeD patients with or without gluten withdrawal (Table 1).
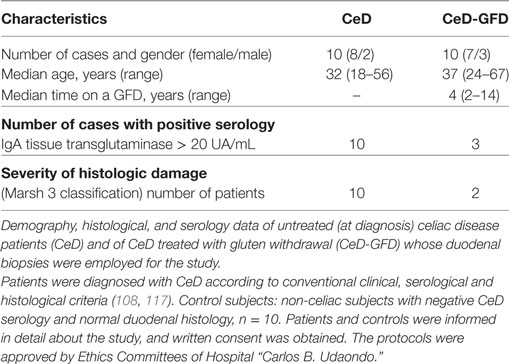
Table 1. Analysis of duodenal biopsies from control subjects, untreated CeD patients, and CeD patients subjected to gluten withdrawal.
Hematoxylin/eosin staining of duodenal biopsies showed that, unlike the conserved LP structures observed in control subjects (Figure 2A), CeD patients exhibited atrophic villi with enlarged hyperplastic crypts and increased intraepithelial lymphocytes infiltration (Figure 2B). Mucosa from CeD patients after GFD (CeD-GFD patients) presented considerably recovered villi (Figure 2C). In control biopsies, Gal-1 labeling (Table 2) was mainly localized in stromal cells, while most ECs exhibited weak positive staining (Figure 2D). Biopsies from CeD patients exhibited a poorly labeled stromal fibrillar network, while atrophic epithelia showed no considerable staining. Subepithelial and periglandular infiltrating cells appeared negative for Gal-1 (Figure 2E). Duodenal biopsies from CeD-GFD patients exhibited a substantial increase in Gal-1 immunoreactivity, especially in the interstitium of the recovered villi. Numerous subepithelial fibroblast-like cells, as well as round nucleus-containing cells scattered in the LP compatible with macrophages, and a few lymphocytes were Gal-1-positive. Notably, ECs recovered their Gal-1 weak positive staining (Figure 2F). Moreover, no significant differences were observed in the expression of Gal-4 (Table 2)—a galectin family member mostly expressed in ECs of the intestinal tract—in biopsies from CeD patients before or after gluten withdrawal (Figures 2G–I).
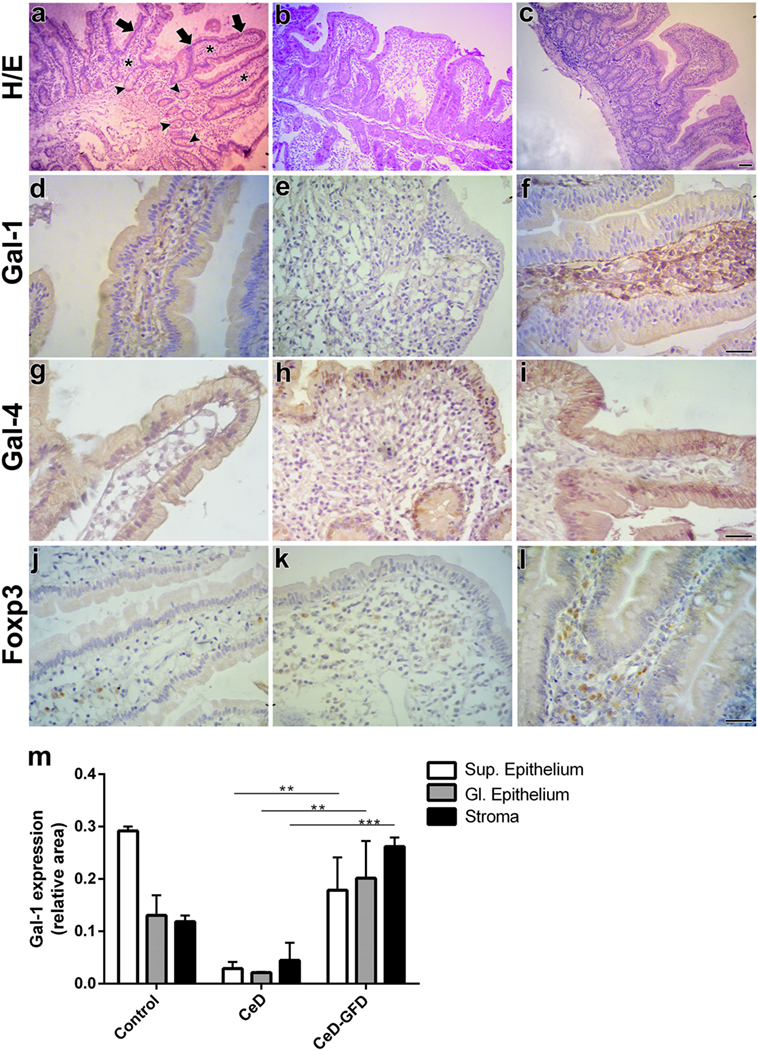
Figure 2. Expression of galectin-1 (Gal-1), Gal-4, and Foxp3 in response to gluten-free diet (GFD) in duodenal biopsies from celiac disease (CeD) patients. Representative micrographs of control subjects, untreated CeD patients and CeD patients subjected to gluten withdrawal (CeD-GFD patients) are shown. (A–C) Hematoxylin/eosin (H/E) staining of paraffin-embedded sections of duodenal biopsies from (A) control subjects, (B) CeD patients, and (C) CeD-GFD patients. Arrows indicate the superficial epithelium, and arrowheads indicate the glandular epithelium while asterisks denote the stroma. Bar = 20 µm. (D–F) Immunohistochemical analysis of Gal-1 expression in duodenal biopsies from control subjects (D), CeD patients (E), and CeD-GFD patients (F). Bar = 20 µm. (G–I) Immunohistochemical analysis of Gal-4 expression in duodenal biopsies from control subjects (G), CeD patients (H), and CeD-GFD patients (I). Bar = 20 µm. (J–L) Immunohistochemical analysis of Foxp3+ cells in biopsies from control subjects (J), CeD patients (K), and CeD-GFD patients (L). Bar = 20 µm. (M) Quantification of Gal-1 expression determined by immunohistochemistry. Bars represent immunostained area corresponding to superficial (Sup) and glandular (Gl) epithelium, and stroma, in paraffin sections from duodenal biopsies from controls, untreated CeD patients and CeD-GFD patients. Evaluation of staining intensity was performed with the Image J software (NIH, Bethesda, MD, USA). One-way ANOVA Tukey test was used for multiple comparisons. **p < 0.01, ***p < 0.001.
Table 2. Analysis of duodenal biopsies from control subjects, untreated CeD patients, and CeD patients subjected to gluten withdrawal.
Overall, while control duodenal biopsies showed moderate Gal-1 staining, and both epithelium and stroma from untreated CeD patients were poorly labeled, CeD-GFD biopsies showed a dramatic increase in Gal-1 immunoreactivity (p < 0.001; Figure 2M), which correlated with normalization of duodenal mucosal structure. Interestingly, the expression of stromal Gal-1 in these patients was not only recovered but also increased in intensity compared with control biopsies (Figures 2D–F,M).
To further characterize the underlying inflammatory response and given the association of Gal-1 with induction of Foxp3+ Tregs, we analyzed the expression of this transcription factor in inflammatory infiltrates (Table 2). Though less accurate in defining human Tregs than mouse Tregs (116), determination of Foxp3 staining is typically considered a reliable indicator of the suppressive tissue microenvironment. An increased number of Foxp3+ cells was observed in CeD-GFD patients (Figures 2J–L), which positively correlated with Gal-1 expression, suggesting activation of a circuit of immunosuppressive events leading to restoration of mucosal homeostasis. Further studies should be aimed at addressing the immunosuppressive potential of this tolerogenic circuit in functional assays.
Our findings suggest that, in response to gluten withdrawal, upregulation of Gal-1 might contribute to restrain the chronic inflammatory response, thus allowing the onset of the recovery process leading to remission of mucosal damage and reestablishment of villi structure. In addition, decreased Gal-1 expression observed in untreated CeD patients compared to control individuals may suggest a role for this lectin in controlling gut homeostasis under physiologic conditions. Interestingly, modulation of Gal-1 expression during CeD development appeared to be specific as no differences were found in the expression of Gal-4, suggesting selective regulation of individual galectins during mucosal inflammation.
Conclusion
The delicate balance between host immunity and tolerance allows the maintenance of gut homeostasis avoiding detrimental intestinal inflammation. Data presented here, resulting both from published information (Figure 1) and new observations (Figure 2), highlight the role of galectins as active players of complex regulatory circuits operating in intestinal mucosal tissue to preserve immune and epithelial homeostasis. While galectins (particularly Gal-1, -2, -3, -4, and -9) may be critical in preserving intestinal homeostasis, an initial set up in which galectins’ expression is altered or the intestinal glycome is reprogrammed may influence development of intestinal inflammation.
To gain insight into the role of Gal-1 in CeD patients, we demonstrated here an increase in Gal-1 expression following GFD that was accompanied by an increased frequency of Foxp3+ cells. The coordinated action of both immunosuppressive mechanisms may occur as synchronized events to generate a tolerogenic milieu in mucosal tissue of treated patients. Since tolerance to gluten peptides would be hard to reestablish under sustained inflammatory conditions, the antigen challenge-free time window (achieved by gluten withdrawal) may allow the development of these immunosuppressive pathways. The subsequent resolution of the inflammatory response may foster the onset of the recovery process, leading to remission of mucosal damage and reestablishment of villi structures.
In line with findings observed in other intestinal inflammatory conditions (76, 77, 84), our observations support the use of Gal-1 agonists to treat severe mucosal inflammation. In addition, Gal-1 may serve as a potential biomarker to follow up CeD progression.
Challenges for the future will embrace the rational manipulation of the Gal-1-glycan axis toward attenuating immune responses in CeD. Studies in Lgals1−/− mice will be necessary to determine a putative role of Gal-1 and its specific ligands in supporting mucosal tolerance to gluten. Moreover, the ability of rGal-1 to suppress intestinal inflammation should also be evaluated in experimental CeD models. In this regard, evidence stemming from the study of experimental models of autoimmunity, chronic inflammation, fetomaternal tolerance, and tumor growth provides fundamental insights into the critical role of this lectin and its specific glycosylated ligands in maintaining and restoring immune tolerance and homeostasis, thus encouraging future implementation of Gal-1-based therapies in CeD patients.
Ethics Statement
Patients and controls were informed in detail about the study, and written consent was obtained. The protocols were approved by Ethics Committees of Hospital “Carlos B. Udaondo.”
Author Contributions
VS acquired data, analyzed and interpreted data, and wrote the manuscript. AQ developed methodology, analyzed and interpreted data, and revised the manuscript. LM analyzed and interpreted data and revised the manuscript. SN analyzed data, managed patients, and revised the manuscript. AC and ES, and EM managed patients and revised the manuscript. KM analyzed and interpreted data and wrote the manuscript. JB and CM conceived and designed the study, analyzed and interpreted data, and revised the manuscript. GR conceived and designed the study, analyzed and interpreted data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the Argentinean Agency for Promotion of Science and Technology (PICT V 2014-3687), University of Buenos Aires and Sales, Bunge & Born and Kenneth Rainin Foundations.
References
1. Blidner AG, Mendez-Huergo SP, Cagnoni AJ, Rabinovich GA. Re-wiring regulatory cell networks in immunity by galectin-glycan interactions. FEBS Lett (2015) 589(22):3407–18. doi:10.1016/j.febslet.2015.08.037
2. Rabinovich GA, Ilarregui JM. Conveying glycan information into T-cell homeostatic programs: a challenging role for galectin-1 in inflammatory and tumor microenvironments. Immunol Rev (2009) 230(1):144–59. doi:10.1111/j.1600-065X.2009.00787.x
3. Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol (2009) 9(5):338–52. doi:10.1038/nri2536
4. Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity (2012) 36(3):322–35. doi:10.1016/j.immuni.2012.03.004
5. Hirabayashi J, Kasai K. The family of metazoan metal-independent beta-galactoside-binding lectins: structure, function and molecular evolution. Glycobiology (1993) 3(4):297–304. doi:10.1093/glycob/3.4.297
6. Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology (2006) 16(11):137r–57r. doi:10.1093/glycob/cwl025
7. Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochim Biophys Acta (2006) 1760(4):616–35. doi:10.1016/j.bbagen.2005.12.020
8. Sundblad V, Croci DO, And Rabinovich GA. Regulated expression of galectin-3, a multifunctional glycan-binding protein, in haematopoietic and non-haematopoietic tissues. Histol Histopathol (2011) 26(2):247–65. doi:10.14670/hh-26.247
9. Sato M, Nishi N, Shoji H, Kumagai M, Imaizumi T, Hata Y, et al. Quantification of galectin-7 and its localization in adult mouse tissues. J Biochem (2002) 131(2):255–60. doi:10.1093/oxfordjournals.jbchem.a003096
10. Gendronneau G, Sidhu SS, Delacour D, Dang T, Calonne C, Houzelstein D, et al. Galectin-7 in the control of epidermal homeostasis after injury. Mol Biol Cell (2008) 19(12):5541–9. doi:10.1091/mbc.e08-02-0166
11. Hotta K, Funahashi T, Matsukawa Y, Takahashi M, Nishizawa H, Kishida K, et al. Galectin-12, an adipose-expressed galectin-like molecule possessing apoptosis-inducing activity. J Biol Chem (2001) 276(36):34089–97. doi:10.1074/jbc.m105097200
12. Yang RY, Hsu DK, Yu L, Chen HY, Liu FT. Galectin-12 is required for adipogenic signaling and adipocyte differentiation. J Biol Chem (2004) 279(28):29761–6. doi:10.1074/jbc.m401303200
13. Gitt MA, Wiser MF, Leffler H, Herrmann J, Xia YR, Massa SM, et al. Sequence and mapping of galectin-5, a beta-galactoside-binding lectin, found in rat erythrocytes. J Biol Chem (1995) 270(10):5032–8. doi:10.1074/jbc.270.10.5032
14. Barres C, Blanc L, Bette-Bobillo P, Andre S, Mamoun R, Gabius HJ, et al. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood (2010) 115(3):696–705. doi:10.1182/blood-2009-07-231449
15. Swaminathan GJ, Leonidas DD, Savage MP, Ackerman SJ, Acharya KR. Selective recognition of mannose by the human eosinophil Charcot-Leyden crystal protein (galectin-10): a crystallographic study at 1.8 A resolution. Biochemistry (1999) 38(46):15406. doi:10.1021/bi995093f
16. Liu FT, Patterson RJ, Wang JL. Intracellular functions of galectins. Biochim Biophys Acta (2002) 1572(2–3):263–73. doi:10.1016/S0304-4165(02)00313-6
17. Thiemann S, Baum LG. Galectins and immune responses-just how do they do those things they do? Annu Rev Immunol (2016) 34:243–64. doi:10.1146/annurev-immunol-041015-055402
18. Patterson RJ, Wang W, Wang JL. Understanding the biochemical activities of galectin-1 and galectin-3 in the nucleus. Glycoconj J (2002) 19(7–9):499–506. doi:10.1023/B:GLYC.0000014079.87862.c7
19. Kubach J, Lutter P, Bopp T, Stoll S, Becker C, Huter E, et al. Human CD4+CD25+ regulatory T cells: proteome analysis identifies galectin-10 as a novel marker essential for their anergy and suppressive function. Blood (2007) 110(5):1550–8. doi:10.1182/blood-2007-01-069229
20. Cooper DN, Barondes SH. Evidence for export of a muscle lectin from cytosol to extracellular matrix and for a novel secretory mechanism. J Cell Biol (1990) 110:1681–91. doi:10.1083/jcb.110.5.1681
21. Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, et al. Oligosaccharide specificity of galectins: a search by frontal affinity chromatography. Biochim Biophys Acta (2002) 1572(2–3):232–54. doi:10.1016/S0304-4165(02)00311-2
22. Stowell SR, Arthur CM, Mehta P, Slanina KA, Blixt O, Leffler H, et al. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J Biol Chem (2008) 283(15):10109–23. doi:10.1074/jbc.m709545200
23. Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci (2015) 128(13):2213–9. doi:10.1242/jcs.151159
24. Elola MT, Blidner AG, Ferragut F, Bracalente C, Rabinovich GA. Assembly, organization and regulation of cell-surface receptors by lectin-glycan complexes. Biochem J (2015) 469(1):1–16. doi:10.1042/BJ20150461
25. Demetriou M, Granovsky M, Quaggin S, Dennis JW. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature (2001) 409(6821):733–9. doi:10.1038/35055582
26. Bonzi J, Bornet O, Betzi S, Kasper BT, Mahal LK, Mancini SJ, et al. Pre-B cell receptor binding to galectin-1 modifies galectin-1/carbohydrate affinity to modulate specific galectin-1/glycan lattice interactions. Nat Commun (2015) 6:6194. doi:10.1038/ncomms7194
27. Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, et al. Regulation of cytokine receptors by golgi N-glycan processing and endocytosis. Science (2004) 306(5693):120–4. doi:10.1126/science.1102109
28. Cerliani JP, Stowell SR, Mascanfroni ID, Arthur CM, Cummings RD, Rabinovich GA. Expanding the universe of cytokines and pattern recognition receptors: galectins and glycans in innate immunity. J Clin Immunol (2011) 31(1):10–21. doi:10.1007/s10875-010-9494-2
29. Cerliani JP, Blidner AG, Toscano MA, Croci DO, Rabinovich GA. Translating the ‘sugar code’ into immune and vascular signaling programs. Trends Biochem Sci (2017) 42(4):255–73. doi:10.1016/j.tibs.2016.11.003
30. Cooper D, Norling LV, Perretti M. Novel insights into the inhibitory effects of galectin-1 on neutrophil recruitment under flow. J Leukoc Biol (2008) 83(6):1459–66. doi:10.1189/jlb.1207831
31. Stowell SR, Karmakar S, Arthur CM, Ju T, Rodrigues LC, Riul TB, et al. Galectin-1 induces reversible phosphatidylserine exposure at the plasma membrane. Mol Biol Cell (2009) 20(5):1408–18. doi:10.1091/mbc.E08-07-0786
32. Rabinovich GA, Sotomayor CE, Riera CM, Bianco I, Correa SG. Evidence of a role for galectin-1 in acute inflammation. Eur J Immunol (2000) 30(5):1331–9. doi:10.1002/(SICI)1521-4141(200005)30:5<1331::AID-IMMU1331>3.0.CO;2-H
33. Correa SG, Sotomayor CE, Aoki MP, Maldonado CA, Rabinovich GA. Opposite effects of galectin-1 on alternative metabolic pathways of L-arginine in resident, inflammatory, and activated macrophages. Glycobiology (2003) 13(2):119–28. doi:10.1093/glycob/cwg010Cwg010
34. Barrionuevo P, Beigier-Bompadre M, Ilarregui JM, Toscano MA, Bianco GA, Isturiz MA, et al. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol (2007) 178(1):436–45. doi:10.4049/jimmunol.178.1.436
35. Malik RK, Ghurye RR, Lawrence-Watt DJ, Stewart HJ. Galectin-1 stimulates monocyte chemotaxis via the p44/42 MAP kinase pathway and a Pertussis toxin-sensitive pathway. Glycobiology (2009) 19(12):1402–7. doi:10.1093/glycob/cwp077
36. Fulcher JA, Hashimi ST, Levroney EL, Pang M, Gurney KB, Baum LG, et al. Galectin-1-matured human monocyte-derived dendritic cells have enhanced migration through extracellular matrix. J Immunol (2006) 177(1):216–26. doi:10.4049/jimmunol.177.1.216
37. Perone MJ, Larregina AT, Shufesky WJ, Papworth GD, Sullivan ML, Zahorchak AF, et al. Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses in naive and activated T cells. J Immunol (2006) 176(12):7207–20. doi:10.4049/jimmunol.176.12.7207
38. Blois SM, Ilarregui JM, Tometten M, Garcia M, Orsal AS, Cordo-Russo R, et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med (2007) 13(12):1450–7. doi:10.1038/nm1680
39. Fulcher JA, Chang MH, Wang S, Almazan T, Hashimi ST, Eriksson AU, et al. Galectin-1 co-clusters CD43/CD45 on dendritic cells and induces cell activation and migration through Syk and protein kinase C signaling. J Biol Chem (2009) 284(39):26860–70. doi:10.1074/jbc.m109.037507
40. Ilarregui JM, Croci DO, Bianco GA, Toscano MA, Salatino M, Vermeulen ME, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol (2009) 10(9):981–91. doi:10.1038/ni.1772
41. Matarrese P, Tinari A, Mormone E, Bianco GA, Toscano MA, Ascione B, et al. Galectin-1 sensitizes resting human T lymphocytes to Fas (CD95)-mediated cell death via mitochondrial hyperpolarization, budding, and fission. J Biol Chem (2005) 280(8):6969–85. doi:10.1074/jbc.M409752200
42. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol (2006) 176(2):778–89. doi:10.4049/jimmunol.176.2.778
43. Toscano MA, Bianco GA, Ilarregui JM, Croci DO, Correale J, Hernandez JD, et al. Differential glycosylation of Th1, Th2 and Th-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol (2007) 8(8):825–34. doi:10.1038/ni1482
44. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol (2006) 18(8):1197–209. doi:10.1093/intimm/dxl060
45. Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood (2007) 109(5):2058–65. doi:10.1182/blood-2006-04-016451
46. Juszczynski P, Ouyang J, Monti S, Rodig SJ, Takeyama K, Abramson J, et al. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc Natl Acad Sci U S A (2007) 104(32):13134–9. doi:10.1073/pnas.0706017104
47. Tabrizi SJ, Niiro H, Masui M, Yoshimoto G, Iino T, Kikushige Y, et al. T cell leukemia/lymphoma 1 and galectin-1 regulate survival/cell death pathways in human naive and IgM+ memory B cells through altering balances in Bcl-2 family proteins. J Immunol (2009) 182(3):1490–9. doi:10.4049/jimmunol.182.3.1490
48. Anginot A, Espeli M, Chasson L, Mancini SJ, Schiff C. Galectin 1 modulates plasma cell homeostasis and regulates the humoral immune response. J Immunol (2013) 190(11):5526–33. doi:10.4049/jimmunol.1201885
49. Croci DO, Morande PE, Dergan-Dylon S, Borge M, Toscano MA, Stupirski JC, et al. Nurse-like cells control the activity of chronic lymphocytic leukemia B cells via galectin-1. Leukemia (2013) 27(6):1413–6. doi:10.1038/leu.2012.315
50. Tsai CM, Wu HY, Su TH, Kuo CW, Huang HW, Chung CH, et al. Phosphoproteomic analyses reveal that galectin-1 augments the dynamics of B-cell receptor signaling. J Proteomics (2014) 103:241–53. doi:10.1016/j.jprot.2014.03.031
51. Ilarregui JM, Bianco GA, Toscano MA, Rabinovich GA. The coming of age of galectins as immunomodulatory agents: impact of these carbohydrate binding proteins in T cell physiology and chronic inflammatory disorders. Ann Rheum Dis (2005) 64(Suppl 4):iv96–103. doi:10.1136/ard.2005.044347
52. Liu FT, Rabinovich GA. Galectins: regulators of acute and chronic inflammation. Ann N Y Acad Sci (2010) 1183:158–82. doi:10.1111/j.1749-6632.2009.05131.x
53. Ge XN, Ha SG, Greenberg YG, Rao A, Bastan I, Blidner AG, et al. Regulation of eosinophilia and allergic airway inflammation by the glycan-binding protein galectin-1. Proc Natl Acad Sci U S A (2016) 113(33):E4837–46. doi:10.1073/pnas.1601958113
54. Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; a potential mechanism of tumor-immune privilege. Cancer Cell (2004) 5(3):241–51. doi:10.1016/S1535-6108(04)00024-8
55. Soldati R, Berger E, Zenclussen AC, Jorch G, Lode HN, Salatino M, et al. Neuroblastoma triggers an immunoevasive program involving galectin-1-dependent modulation of T cell and dendritic cell compartments. Int J Cancer (2011) 131(5):1131–41. doi:10.1002/ijc.26498
56. Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, et al. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res (2011) 71:4423–31. doi:10.1158/0008-5472.CAN-10-4157
57. Croci DO, Salatino M, Rubinstein N, Cerliani JP, Cavallin LE, Leung HJ, et al. Disrupting galectin-1 interactions with N-glycans suppresses hypoxia-driven angiogenesis and tumorigenesis in Kaposi’s sarcoma. J Exp Med (2012) 209:1985–2000. doi:10.1084/jem.20111665
58. Cedeno-Laurent F, Watanabe R, Teague JE, Kupper TS, Clark RA, Dimitroff CJ. Galectin-1 inhibits the viability, proliferation, and Th1 cytokine production of nonmalignant T cells in patients with leukemic cutaneous T-cell lymphoma. Blood (2012) 119(15):3534–8. doi:10.1182/blood-2011-12-396457
59. Kuo PL, Huang MS, Cheng DE, Hung JY, Yang CJ, Chou SH. Lung cancer-derived galectin-1 enhances tumorigenic potentiation of tumor-associated dendritic cells by expressing heparin-binding EGF-like growth factor. J Biol Chem (2012) 287:9753–64. doi:10.1074/jbc.M111.321190
60. Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Méndez-Huergo SP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res (2013) 73:1107–17. doi:10.1158/0008-5472.CAN-12-2418
61. Croci DO, Cerliani JP, Dalotto-Moreno T, Mendez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, et al. Glycosylation-dependent lectin-receptor interactions preserve angiogenesis in anti-VEGF refractory tumors. Cell (2014) 156(4):744–58. doi:10.1016/j.cell.2014.01.043
62. Martinez-Bosch N, Fernandez-Barrena MG, Moreno M, Ortiz-Zapater E, Munne-Collado J, Iglesias M, et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res (2014) 74:3512–24. doi:10.1158/0008-5472.CAN-13-3013
63. Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS, et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget (2016) 7(19):27584–98. doi:10.18632/oncotarget.8488
64. Rabinovich GA, Daly G, Dreja H, Tailor H, Riera CM, Hirabayashi J, et al. Recombinant galectin-1 and its genetic delivery suppress collagen-induced arthritis via T cell apoptosis. J Exp Med (1999) 190(3):385–98. doi:10.1084/jem.190.3.385
65. Mari ER, Rasouli J, Ciric B, Moore JN, Conejo-Garcia JR, Rajasagi N, et al. Galectin-1 is essential for the induction of MOG35-55-based intravenous tolerance in experimental autoimmune encephalomyelitis. Eur J Immunol (2016) 46(7):1783–96. doi:10.1002/eji.201546212
66. Perone MJ, Bertera S, Shufesky WJ, Divito SJ, Montecalvo A, Mathers AR, et al. Suppression of autoimmune diabetes by soluble galectin-1. J Immunol (2009) 182(5):2641–53. doi:10.4049/jimmunol.0800839
67. Toscano MA, Commodaro AG, Ilarregui JM, Bianco GA, Liberman A, Serra HM, et al. Galectin-1 suppresses autoimmune retinal disease by promoting concomitant Th2- and T regulatory-mediated anti-inflammatory responses. J Immunol (2006) 176(10):6323–32. doi:10.4049/jimmunol.176.10.6323
68. Pérez CV, Gómez LG, Gualdoni GS, Lustig L, Rabinovich GA, Guazzone VA. Dual roles of endogenous and exogenous galectin-1 in the control of testicular immunopathology. Sci Rep (2015) 5:12259. doi:10.1038/srep12259
69. Santucci L, Fiorucci S, Rubinstein N, Mencarelli A, Palazzetti B, Federici B, et al. Galectin-1 suppresses experimental colitis in mice. Gastroenterology (2003) 124(5):1381–94. doi:10.1016/S0016-5085(03)00267-1
70. Pang M, He J, Johnson P, Baum LG. CD45-mediated fodrin cleavage during galectin-1 T cell death promotes phagocytic clearance of dying cells. J Immunol (2009) 182(11):7001–8. doi:10.4049/jimmunol.0804329
71. Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity (2012) 37(2):249–63. doi:10.1016/j.immuni.2012.05.023
72. Nio J, Kon Y, Iwanaga T. Differential cellular expression of galectin family mRNAs in the epithelial cells of the mouse digestive tract. J Histochem Cytochem (2005) 53(11):1323–34. doi:10.1369/jhc.5a6685.2005
73. Nio-Kobayashi J, Takahashi-Iwanaga H, Iwanaga T. Immunohistochemical localization of six galectin subtypes in the mouse digestive tract. J Histochem Cytochem (2009) 57(1):41–50. doi:10.1369/jhc.2008.952317
74. Kavanaugh D, Kane M, Joshi L, Hickey RM. Detection of galectin-3 interaction with commensal bacteria. Appl Environ Microbiol (2013) 79(11):3507–10. doi:10.1128/aem.03694-12
75. Stowell SR, Arthur CM, Dias-Baruffi M, Rodrigues LC, Gourdine JP, Heimburg-Molinaro J, et al. Innate immune lectins kill bacteria expressing blood group antigen. Nat Med (2010) 16(3):295–301. doi:10.1038/nm.2103
76. Muglia C, Mercer N, Toscano MA, Schattner M, Pozner R, Cerliani JP, et al. The glycan-binding protein galectin-1 controls survival of epithelial cells along the crypt-villus axis of small intestine. Cell Death Dis (2011) 2:e163. doi:10.1038/cddis.2011.44
77. Papa Gobbi R, De Francesco N, Bondar C, Muglia C, Chirdo F, Rumbo M, et al. A galectin-specific signature in the gut delineates Crohn’s disease and ulcerative colitis from other human inflammatory intestinal disorders. Biofactors (2016) 42(1):93–105. doi:10.1002/biof.1252
78. Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med (2011) 365(18):1713–25. doi:10.1056/nejmra1102942
79. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet (2012) 380(9853):1590–605. doi:10.1016/s0140-6736(12)60026-9
80. Ordas I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet (2012) 380(9853):1606–19. doi:10.1016/s0140-6736(12)60150-0
81. Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol (2013) 13(5):321–35. doi:10.1038/nri3430
82. Nalle SC, Turner JR. Intestinal barrier loss as a critical pathogenic link between inflammatory bowel disease and graft-versus-host disease. Mucosal Immunol (2015) 8(4):720–30. doi:10.1038/mi.2015.40
83. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi:10.1038/nri3661
84. Muglia CI, Gobbi RP, Smaldini P, Delgado ML, Candia M, Zanuzzi C, et al. Inflammation controls sensitivity of human and mouse intestinal epithelial cells to galectin-1. J Cell Physiol (2016) 231(7):1575–85. doi:10.1002/jcp.25249
85. Simovic Markovic B, Nikolic A, Gazdic M, Bojic S, Vucicevic L, Kosic M, et al. Galectin-3 plays an important pro-inflammatory role in the induction phase of acute colitis by promoting activation of NLRP3 inflammasome and production of IL-1beta in macrophages. J Crohns Colitis (2016) 10(5):593–606. doi:10.1093/ecco-jcc/jjw013
86. Hsu DK, Yang RY, Pan Z, Yu L, Salomon DR, Fung-Leung WP, et al. Targeted disruption of the galectin-3 gene results in attenuated peritoneal inflammatory responses. Am J Pathol (2000) 156:1073–83. doi:10.1016/S0002-9440(10)64975-9
87. Jensen-Jarolim E, Gscheidlinger R, Oberhuber G, Neuchrist C, Lucas T, Bises G, et al. The constitutive expression of galectin-3 is downregulated in the intestinal epithelia of Crohn’s disease patients, and tumour necrosis factor alpha decreases the level of galectin-3-specific mRNA in HCT-8 cells. Eur J Gastroenterol Hepatol (2002) 14:145–52. doi:10.1097/00042737-200202000-00008
88. Muller S, Schaffer T, Flogerzi B, Fleetwood A, Weimann R, Schoepfer AM, et al. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm Bowel Dis (2006) 12(7):588–97. doi:10.1097/01.mib.0000225341.37226.7c
89. Lippert E, Gunckel M, Brenmoehl J, Bataille F, Falk W, Scholmerich J, et al. Regulation of galectin-3 function in mucosal fibroblasts: potential role in mucosal inflammation. Clin Exp Immunol (2008) 152:285–97. doi:10.1111/j.1365-2249.2008.03618.x
90. Lippert E, Stieber-Gunckel M, Dunger N, Falk W, Obermeier F, Kunst C. Galectin-3 Modulates Experimental Colitis. Digestion (2015) 92:45–53. doi:10.1159/000431312
91. Tsai HF, Wu CS, Chen YL, Liao HJ, Chyuan IT, Hsu PN. Galectin-3 suppresses mucosal inflammation and reduces disease severity in experimental colitis. J Mol Med (Berl) (2016) 94(5):545–56. doi:10.1007/s00109-015-1368-x
92. Kolios G. Animal models of inflammatory bowel disease: how useful are they really? Curr Opin Gastroenterol (2016) 32(4):251–7. doi:10.1097/mog.0000000000000287
93. Paclik D, Danese S, Berndt U, Wiedenmann B, Dignass A, Sturm A. Galectin-4 controls intestinal inflammation by selective regulation of peripheral and mucosal T cell apoptosis and cell cycle. PLoS One (2008) 3(7):e2629. doi:10.1371/journal.pone.0002629
94. Paclik D, Berndt U, Guzy C, Dankof A, Danese S, Holzloehner P, et al. Galectin-2 induces apoptosis of lamina propria T lymphocytes and ameliorates acute and chronic experimental colitis in mice. J Mol Med (Berl) (2008) 286:1395–406. doi:10.1007/s00109-007-0290-2
95. Paclik D, Lohse K, Wiedenmann B, Dignass AU, Sturm A. Galectin-2 and -4, but not galectin-1, promote intestinal epithelial wound healing in vitro through a TGF-beta-independent mechanism. Inflamm Bowel Dis (2008) 14(10):1366–72. doi:10.1002/ibd.20499
96. Delacour D, Gouyer V, Zanetta JP, Drobecq H, Leteurtre E, Grard G, et al. Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. J Cell Biol (2005) 169(3):491–501. doi:10.1083/jcb.200407073
97. Stechly L, Morelle W, Dessein AF, Andre S, Grard G, Trinel D, et al. Galectin-4-regulated delivery of glycoproteins to the brush border membrane of enterocyte-like cells. Traffic (2009) 10(4):438–50. doi:10.1111/j.1600-0854.2009.00882.x
98. Nishida A, Nagahama K, Imaeda H, Ogawa A, Lau CW, Kobayashi T, et al. Inducible colitis-associated glycome capable of stimulating the proliferation of memory CD4+ T cells. J Exp Med (2012) 209(13):2383–94. doi:10.1084/jem.20112631
99. Hokama AL, Mizoguchi E, Sugimoto K, Shimomura Y, Tanaka Y, Yoshida M, et al. Induced reactivity of intestinal CD4(+) T cells with an epithelial cell lectin, galectin-4, contributes to exacerbation of intestinal inflammation. Immunity (2004) 20:681–93. doi:10.1016/j.immuni.2004.05.009
100. Commins SP. Mechanisms of oral tolerance. Pediatr Clin North Am (2015) 62(6):1523–9. doi:10.1016/j.pcl.2015.07.013
101. Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol (2013) 13(4):294–302. doi:10.1038/nri3407
102. Sollid LM. Molecular basis of celiac disease. Annu Rev Immunol (2000) 18:53–81. doi:10.1146/annurev.immunol.18.1.53
103. Abraham G, Tye-Din JA, Bhalala OG, Kowalczyk A, Zobel J, Inouye M. Accurate and robust genomic prediction of celiac disease using statistical learning. PLoS Genet (2014) 10(2):e1004137. doi:10.1371/journal.pgen.1004137
104. Romanos J, Rosen A, Kumar V, Trynka G, Franke L, Szperl A, et al. Improving coeliac disease risk prediction by testing non-HLA variants additional to HLA variants. Gut (2014) 63(3):415–22. doi:10.1136/gutjnl-2012-304110
105. Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest (2007) 117(1):41–9. doi:10.1172/jci30253
106. Koning F. Celiac disease: quantity matters. Semin Immunopathol (2012) 34(4):541–9. doi:10.1007/s00281-012-0321-0
107. Nilsen EM, Jahnsen FL, Lundin KE, Johansen FE, Fausa O, Sollid LM, et al. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology (1998) 115(3):551–63. doi:10.1016/S0016-5085(98)70134-9
108. Kelly CP, Bai JC, Liu E, Leffler DA. Advances in diagnosis and management of celiac disease. Gastroenterology (2015) 148(6):1175–86. doi:10.1053/j.gastro.2015.01.044
109. Rubio-Tapia A, Hill ID, Kelly CP, Calderwood AH, Murray JA. ACG clinical guidelines: diagnosis and management of celiac disease. Am J Gastroenterol (2013) 108(5):656–76;quiz677. doi:10.1038/ajg.2013.79
110. Bardella MT, Fraquelli M, Quatrini M, Molteni N, Bianchi P, Conte D. Prevalence of hypertransaminasemia in adult celiac patients and effect of gluten-free diet. Hepatology (1995) 22(3):833–6. doi:10.1002/hep.1840220322
111. Meresse B, Ripoche J, Heyman M, Cerf-Bensussan N. Celiac disease: from oral tolerance to intestinal inflammation, autoimmunity and lymphomagenesis. Mucosal Immunol (2009) 2(1):8–23. doi:10.1038/mi.2008.75
112. De Re V, Simula MP, Cannizzaro R, Pavan A, De Zorzi MA, Toffoli G, et al. Galectin-10, eosinophils, and celiac disease. Ann N Y Acad Sci (2009) 1173:357–64. doi:10.1111/j.1749-6632.2009.04627.x
113. Chen X, Song CH, Liu ZQ, Feng BS, Zheng PY, Li P, et al. Intestinal epithelial cells express galectin-9 in patients with food allergy that plays a critical role in sustaining allergic status in mouse intestine. Allergy (2011) 66(8):1038–46. doi:10.1111/j.1398-9995.2011.02585.x
114. De Kivit S, Saeland E, Kraneveld AD, Van De Kant HJ, Schouten B, Van Esch BC, et al. Galectin-9 induced by dietary synbiotics is involved in suppression of allergic symptoms in mice and humans. Allergy (2012) 67(3):343–52. doi:10.1111/j.1398-9995.2011.02771.x
115. De Kivit S, Kraneveld AD, Knippels LM, Van Kooyk Y, Garssen J, Willemsen LE. Intestinal epithelium-derived galectin-9 is involved in the immunomodulating effects of nondigestible oligosaccharides. J Innate Immun (2013) 5(6):625–38. doi:10.1159/000350515
116. Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4+FoxP3+ regulatory T cells. Int Immunopharmacol (2011) 11(10):1489–96. doi:10.1016/j.intimp.2011.05.018
Keywords: celiac disease, galectin-1, galectins, glycans, gut inflammation, inflammatory bowel disease
Citation: Sundblad V, Quintar AA, Morosi LG, Niveloni SI, Cabanne A, Smecuol E, Mauriño E, Mariño KV, Bai JC, Maldonado CA and Rabinovich GA (2018) Galectins in Intestinal Inflammation: Galectin-1 Expression Delineates Response to Treatment in Celiac Disease Patients. Front. Immunol. 9:379. doi: 10.3389/fimmu.2018.00379
Received: 02 December 2017; Accepted: 12 February 2018;
Published: 01 March 2018
Edited by:
Bernd Lepenies, University of Veterinary Medicine, GermanyReviewed by:
Celso A. Reis, Universidade do Porto, PortugalPilar Navarro, Institut Hospital del Mar d’Investigacions Mèdiques, Spain
Copyright: © 2018 Sundblad, Quintar, Morosi, Niveloni, Cabanne, Smecuol, Mauriño, Mariño, Bai, Maldonado and Rabinovich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel A. Rabinovich, Z2FicmllbC5yQGlieW1lLmNvbmljZXQuZ292LmFy, Z2FieXJhYmlAZ21haWwuY29t
†These authors have contributed equally to this work.
‡These authors have jointly supervised this work.