 Viktor Fleming1,2
Viktor Fleming1,2 Xiaoying Hu1,2
Xiaoying Hu1,2 Rebekka Weber1,2
Rebekka Weber1,2 Vasyl Nagibin1,2Christopher Groth1,2
Vasyl Nagibin1,2Christopher Groth1,2 Peter Altevogt1,2Jochen Utikal1,2
Peter Altevogt1,2Jochen Utikal1,2 Viktor Umansky1,2*
Viktor Umansky1,2*
- 1Skin Cancer Unit, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 2Department of Dermatology, Venereology and Allergology, University Medical Center Mannheim, Heidelberg University, Mannheim, Germany
The immune system has many sophisticated mechanisms to balance an extensive immune response. Distinct immunosuppressive cells could protect from excessive tissue damage and autoimmune disorders. Tumor cells take an advantage of those immunosuppressive mechanisms and establish a strongly immunosuppressive tumor microenvironment (TME), which inhibits antitumor immune responses, supporting the disease progression. Myeloid-derived suppressor cells (MDSC) play a crucial role in this immunosuppressive TME. Those cells represent a heterogeneous population of immature myeloid cells with a strong immunosuppressive potential. They inhibit an antitumor reactivity of T cells and NK cells. Furthermore, they promote angiogenesis, establish pre-metastatic niches, and recruit other immunosuppressive cells such as regulatory T cells. Accumulating evidences demonstrated that the enrichment and activation of MDSC correlated with tumor progression, recurrence, and negative clinical outcome. In the last few years, various preclinical studies and clinical trials targeting MDSC showed promising results. In this review, we discuss different therapeutic approaches on MDSC targeting to overcome immunosuppressive TME and enhance the efficiency of current tumor immunotherapies.
Introduction
Immunosuppression is a hallmark of most cancer entities and is pivotal for cancer growth and progression (1, 2). In recent years, accumulating data highlighted myeloid-derived suppressor cells (MDSC) as one of the main driver of an immunosuppressive tumor microenvironment (TME) (3). Their accumulation and activation correlated with tumor progression, metastasis, and recurrence of many types of tumors. In addition, the efficacy of immunotherapy was negatively correlated with an increased MDSC frequency and activity (4, 5). Therefore, targeting MDSC becomes a promising treatment approach to overcome tumor progression and tumor-mediated immunosuppression.
Myeloid-derived suppressor cells represent a heterogeneous population of immature myeloid cells (IMC) that fail to terminally differentiate and exhibit a strong capacity to suppress the functions of T and NK cells (6–9). Under healthy conditions, IMC differentiate into macrophages, dendritic cells (DCs), or granulocytes. During an acute inflammation, IMC expand and differentiate mainly into monocytes and activated neutrophils (7). This process, known as myelopoiesis, is essential to protect the host from pathological conditions. In contrast to acute inflammation, chronic inflammation and cancer are characterized by a persistent release of signals of low stimulatory intensity (10–12). Although these stimuli still activate myelopoiesis, the accumulating IMC fail to completely differentiate into activated neutrophils and monocytes. Instead, the long-term inflammatory signals create conditions for the expansion and activation of MDSC (13, 14). They migrate to the site of inflammation, lymphoid organs, and pre-metastatic niches and promote tumor progression by immunological and non-immunological mechanisms (15). Figure 1 illustrates the biology and functions of MDSC during tumor progression.
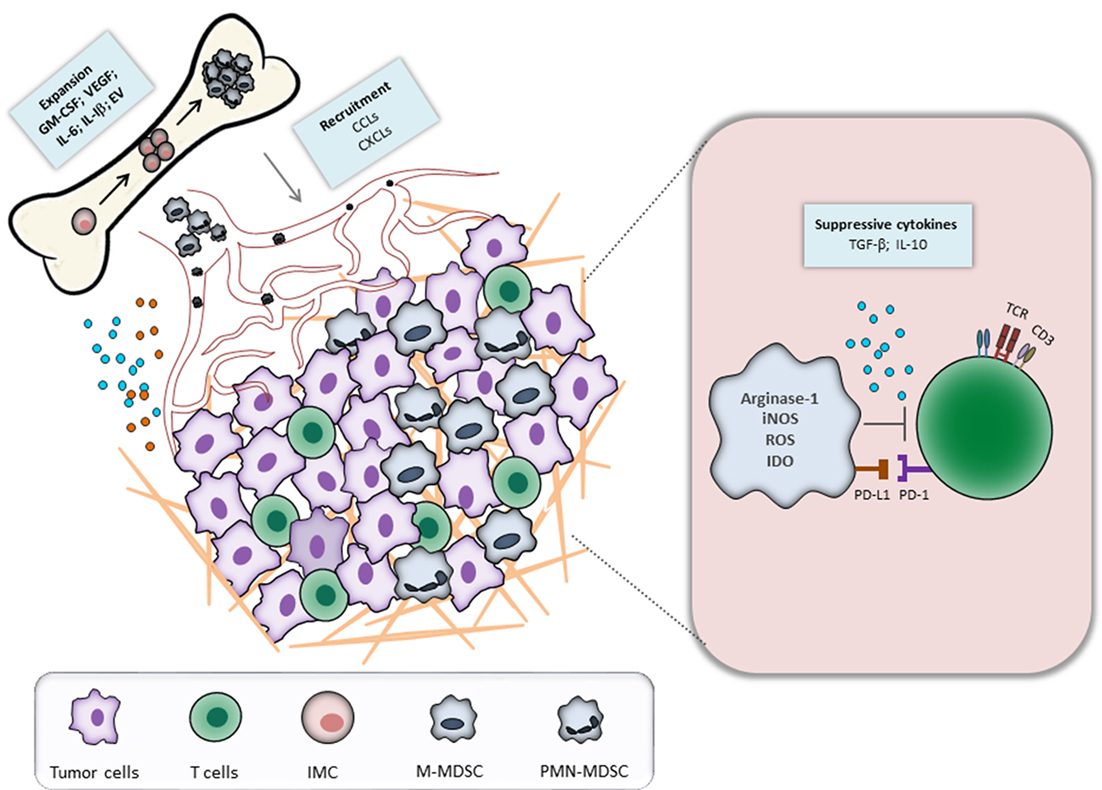
Figure 1. Myeloid-derived suppressor cells (MDSC) recruitment and activation during tumor progression. Tumor and immune cells constantly release inflammatory mediators, leading to the dysregulation of normal myelopoiesis and to the conversion of immature myeloid cells (IMC) into MDSC in the bone marrow. The latter cells expand and migrate to the tumor site through the interaction between CCR and respective chemokines (CCL). In the tumor microenvironment, MDSC are activated and strongly inhibit an antitumor reactivity of T cells via various mechanisms.
Phenotype of MDSC
Myeloid-derived suppressor cells consist of two major subpopulations, which are traditionally described by their phenotypical and morphological characteristics. The first population is called monocytic MDSC (M-MDSC), whereas the second is polymorphonuclear MDSC (PMN-MDSC) (8), which was previously known as granulocytic MDSC (6). Both MDSC subsets can be found under pathological conditions in the bone marrow, spleen, lung, peripheral blood, and tumor tissue; in most cancer entities, PMN-MDSC represent more than 80% of all MDSC (16). In mice, M-MDSC are defined as CD11b+Ly6G−Ly6Chigh and share phenotypical and morphological characteristics with monocytes. PMN-MDSC are described as CD11b+Ly6GhighLy6Clow cells and resemble neutrophils (16, 17). In human, M-MDSC are defined as CD11b+CD14+CD15−HLA-DRlow/− cells. Due to the low or absence of the HLA-DR expression, M-MDSC they can be distinguished from monocytes. Human PMN-MDSC are characterized as CD11b+CD14−CD15+HLA-DR− or CD11b+CD14−CD66b+ (17, 18). In addition, a subset of more immature human MDSC characterized as Lin− (including CD3, CD14, CD15, CD19, CD56) HLA-DR−CD33+ cells was defined as early-stage MDSC (eMDSC) (17). At the moment, the mouse equivalent of eMDSC is not clearly determined. Recently, a new marker for human PMN-MDSC has been proposed; they were found to express lectin-type oxidized LDL receptor-1 (LOX-1) that can discriminate them from neutrophils (19).
Conversion of IMC into MDSC by Tumor-Derived Extracellular Vesicles (EV)
Expansion and activation of MDSC could be stimulated by many soluble factors, which are predominately released within the TME by tumor and immune cells (20). Specifically, granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte CSF, macrophage CSF, stem cell factor, transforming growth factor (TGF)-β, tumor necrosis factor (TNF)-α, vascular endothelial growth factor (VEGF), prostaglandin E2, cyclooxygenase 2, S100A9, S100A8, interleukin (IL)-1β, IL-6, and IL-10 are considered to be crucial for MDSC expansion (6, 8, 21–23). Furthermore, tumor cells can stimulate the secretion of these inflammatory mediators by cancer-associated fibroblasts and vice versa leading to an autocrine loop, which promotes tumor growth by converting myeloid cells into MDSC (20).
In addition to soluble inflammatory factors, tumor-derived EV could contribute to the generation of MDSC. EV consist of microvesicles that are created by the outward budding of the plasma membrane and exosomes, which are generated through the endosomal system (24). Due to their phospholipid bilayer, EV are stable vehicles to carry biological active molecules (25). It was shown that tumor-derived EV are predominately taken up by MDSC (26). After the uptake of EV derived from a Lewis lung carcinoma (LLC) and glioma, MDSC displayed an increased expression of immunosuppressive molecules like arginase-1 (ARG1) and programmed death ligand 1 (PD-L1) (26). Filipazzi et al. (27) demonstrated that CD14+ monocytes lost the expression of HLA-DR and acquired an immunosuppressive activity upon EV uptake. In contract, EV from healthy donors were not able to convert monocytes into MDSC-like cells (27). Several studies showed that EV trigger toll-like receptor (TLR) signaling in myeloid cells. THP-1 monocytic cell line showed increased production of inflammatory molecules like IL-1β, IL-6, and TNF-α upon the EV treatment, which was due to TLR2 and TLR4 signaling (28, 29). Chalmin et al. (30) demonstrated that tumor-derived EV triggered the expansion and activation of murine and human MDSC via HSP72 that stimulated TLR2 signaling. Furthermore, by using the B16 transplantable melanoma model, it was shown that tumor EV could facilitate formation of metastasis through the transfer of the Met receptor tyrosine kinase to bone marrow cells (31). As the bone marrow cells were not further characterized, it is conceivable that such melanoma-derived EV converted bone marrow-derived IMC into potent MDSC.
Immunosuppression Induced by MDSC
Myeloid-derived suppressor cells use a broad range of suppressive molecules to inhibit antitumor reactivity of immune cells, supporting thereby tumor growth and metastasis. By inhibiting the activity of tumor-infiltrating lymphocytes, MDSC show their extraordinary potential of silencing the immune response (6–11, 16–18, 32, 33). One of the main immunosuppressive mediators is ARG1, which is an essential enzyme for the urea cycle (34, 35). It converts l-arginine into l-ornithine and urea, leading to the depletion of l-arginine. The lack of l-arginine causes a translational blockade in infiltrating T cells leading to cell cycle arrest in G0-G1 (36). Furthermore, T cells become anergic due to the downregulation of the T cell receptor (TCR) ζ-chain, which is essential for TCR signaling (37). Besides ARG1, MDSC express also of inducible nitric oxide synthase (iNOS), which also catabolize l-arginine. The main product of the reaction is nitric oxide (NO) that could induce T cell anergy (16) and nitrosylate important mediators of the IL-2 pathway (38). MDSC express also elevated levels of indoleamine 2,3-dioxygenase (IDO) that degrade l-tryptophan into N-formylkynurenine. The lack of tryptophan results in the cell cycle arrest in T cells and induces anergy (39). Moreover, tryptophan starvation is known to drive the differentiation of CD4+ T cells into immunosuppressive regulatory T cells (Treg) (40). Kynurenine and 3-hydroxykynurenine, the products of IDO activity, exert also immunosuppressive functions, inhibiting effector T cell survival and proliferation (41). In addition, kynurenine drives the differentiation of CD4+ T cells into Treg and induces apoptosis in thymocytes (42, 43). Kynurenine was also reported to dampen NK cell function and proliferation (44). Furthermore, reactive oxygen species (ROS) produced by MDSC in high concentrations were shown to induce T cell apoptosis (9, 11, 16) In addition, ROS was demonstrated to downregulate the expression of TCR ζ-chain, leading to impaired TCR signaling (10, 16, 17). Reacting with NO, ROS form peroxynitrite, which nitrosylates the TCR, resulting in T cell anergy (45). MDSC also secrete immunosuppressive cytokines and growth factors such as TGF-β and IL-10 that reduce antitumor activity of effector T cells and recruit Treg (46, 47).
It has been recently described that MDSC could exert their immunosuppressive effects via upregulation of PD-L1 (48, 49). Upon the binding of PD-L1 to the PD-1 receptor expressed on T cells, they become anergic, losing their ability to produce interferon (IFN)-γ and IL-2 (48). Moreover, MDSC were shown to express the death receptor CD95 and induce T cell apoptosis via CD95 ligand expressed on activated T cells (50).
Non-Immunological Ways of Promoting Tumor Progression
In addition to the establishment of an immunosuppressive TME, MDSC could promote tumor progression by non-immunological mechanisms (51). In particular, MDSC produce large amounts of matrix metalloproteinases (MMP), especially MMP9, which process the extracellular matrix and basal membrane and enable the tumor to leave the tissue, to enter the blood stream, and migrate to the site of later metastasis (52). It was shown that the pre-metastatic niche is performed before the tumor cells enter the blood stream (53). This process is still not fully understood but studies have confirmed that MDSC play an essential role (9, 54). It was found that MDSC accumulated in pre-metastatic niches with the help of monocyte chemoattractant protein-1 that dampens the activity of NK cells, which are also preferably found in the pre-metastatic niche (55). In addition, it was reported that MDSC produce MMP9 within the pre-metastatic niche, facilitating the penetration of metastatic cells (56). A further hallmark of tumor progression is angiogenesis that is crucial for the nutrition, vasculature, and dissemination of the tumor (57). MDSC promote angiogenesis by secreting elevated levels of VEGF and basic fibroblast growth factor (bFGF) (58). It was reported that blocking of angiogenesis resulted in the inhibition of tumor migration and formation of metastasis (59).
Correlation between Tumor Burden, Resistance to Immunotherapy, and MDSC
The expansion of MDSC has been demonstrated in many types of human tumors (6, 7). Moreover, elevated levels of MDSC were found not only in solid tumors but also in blood of non-Hodgkin lymphoma and multiple myeloma patients (18). Importantly, the frequency of circulating MDSC was found to correlate with the disease stage. It was reported that patients with stage III and IV hepatocellular carcinoma, melanoma, non-small cell lung cancer, pancreatic, esophageal, gastric, and bladder cancer had higher frequencies of MDSC in the peripheral blood as compared to stage I and II patients (60–63). In addition, an association between MDSC numbers and clinical response to radio-, chemo-, and immunotherapy was reported (64). Several recent studies described that in melanoma patients treated with the immune checkpoint inhibitor, ipilimumab, decreased amounts and immunosuppressive functionality of both M- and PMN-MDSC correlated with beneficial therapeutic effects (65–68). Altogether, these studies show that MDSC could be not only promising biomarkers for the survival of patients and the treatment efficacy but also could serve as a valuable target in combined immunotherapy of cancer patients.
MDSC Targeting in Cancer
In recent years, increasing numbers of preclinical and clinical studies were performed to target MDSC with beneficial effects, resulting in the tumor growth inhibition and the survival prolongation. The MDSC modulation was achieved by (i) the inhibition of their immunosuppressive activity; (ii) the blockade of MDSC recruitment to the tumor site; and (iii) the regulation of myelopoiesis and/or depletion of MDSC in the tumor-bearing hosts (Figure 2). Ongoing clinical trials are summarized in Table 1.
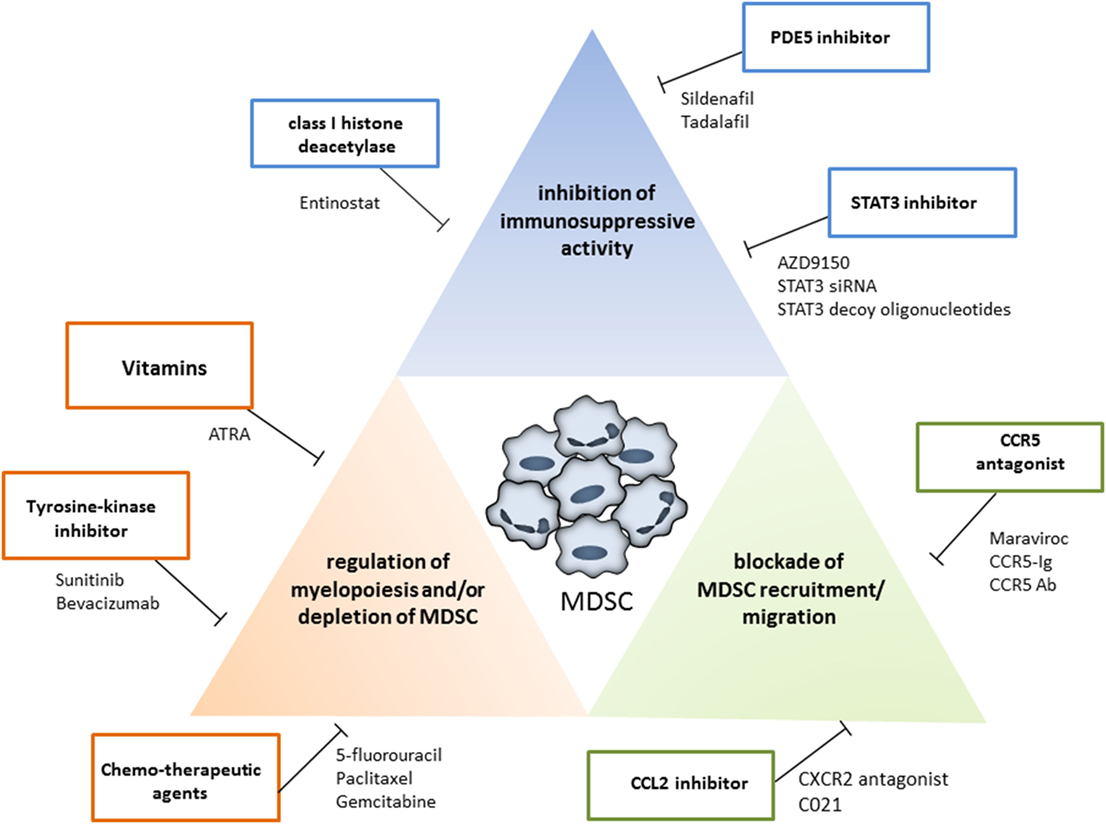
Figure 2. Strategies for myeloid-derived suppressor cells (MDSC) targeting. The MDSC modulation could be achieved by the inhibition of their immunosuppressive activity (blue box), by the blockade of MDSC recruitment to the tumor site (green box), and by the regulation of myelopoiesis and/or depletion of MDSC (red box). Examples for each therapeutic approach are shown.
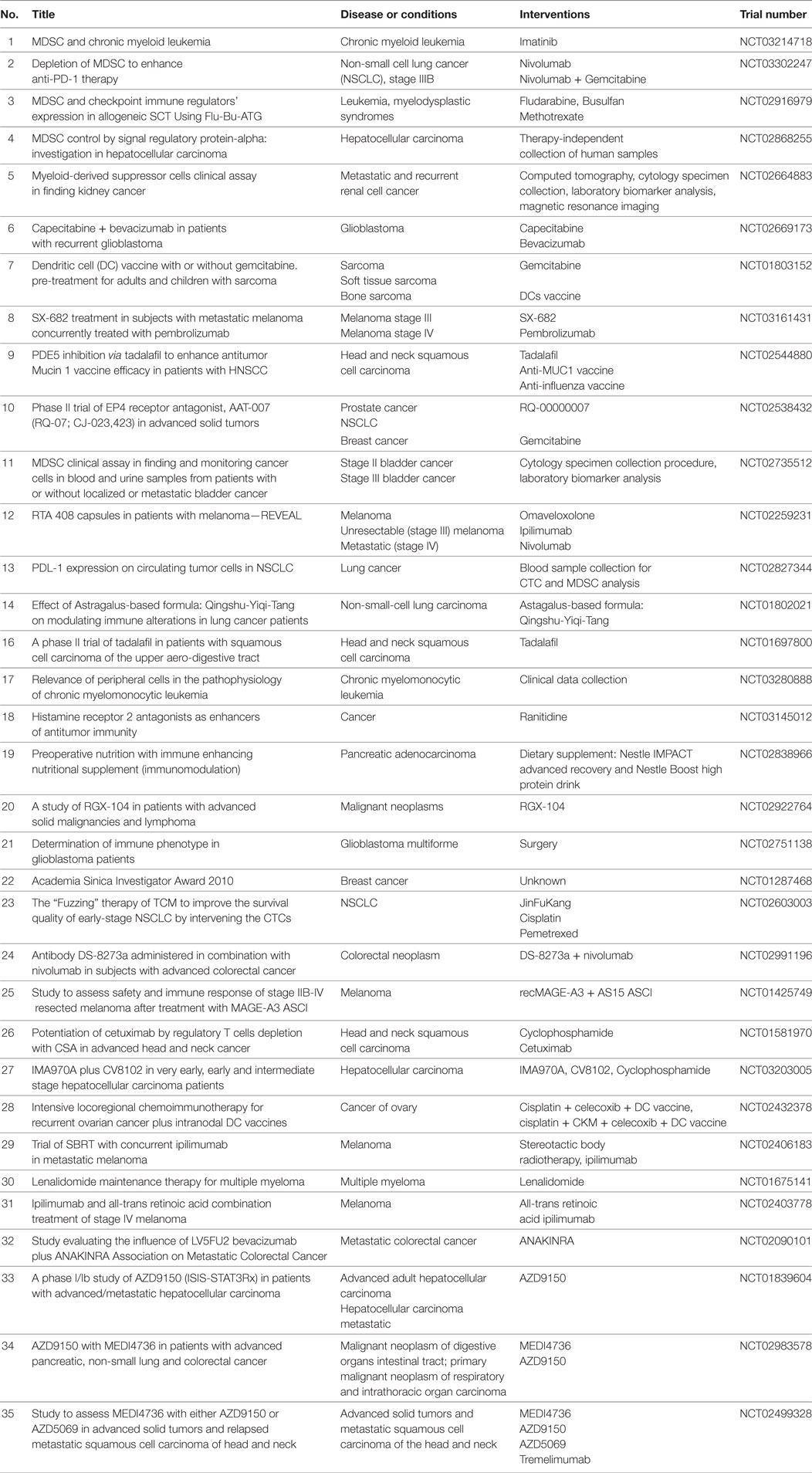
Table 1. Ongoing clinical trials to target myeloid-derived suppressor cells (MDSC) in cancer patients.
Inhibition of MDSC-Mediated Immunosuppression
In preclinical mouse models, it has been demonstrated that inhibitors of phosphodiesterase-5, sildenafil, and tadalafil significantly inhibited the MDSC functions by the downregulation of iNOS and ARG1 activities, leading to the activation of antitumor immunity and the prolongation of survival of tumor-bearing mice (69–71). Recent clinical trials with tadalafil in patients with head and neck squamous cell carcinoma and melanoma confirmed this positive effect (72–74). It was shown that decreased amounts of MDSC and their immunosuppressive pattern correlated with an increased T cell reactivity and improved clinical outcome of advanced cancer patients.
A class I histone deacetylase inhibitor, entinostat, has been recently evaluated in several preclinical tumor models for its ability to affect MDSC functions (75, 76). The authors demonstrated that entinostat reduced the expression of ARG1, iNOS, and COX2 in both M- and PMN-MDSC subsets. In addition, they observed a strong reduction of tumor-infiltrating macrophages, suggesting a strong effect of this drug on the innate immunity. Interestingly, the combination of entinostat with anti-PD-1 antibodies significantly increased survival and delayed tumor growth in mice with LLC and renal cell carcinoma as compared to the treatment with anti-PD-1 antibodies alone. A combined therapy with nivolumab and entinostat in renal cell carcinoma patients is now planned.
A further promising way to target MDSC is the blockade of the activation of STAT3, which is a main transcription factor for immunosuppressive activity in myeloid cells (77). In the past, a number of clinical trials have been performed to target STAT3 with small molecular inhibitors with a limited efficacy and broad side effects (78). Recently, a new possibility to target STAT3 has been tested, in which STAT3 siRNA or decoy oligonucleotides were used to interfere with STAT3 mRNA (78). At the moment, several STAT3 oligonucleotide inhibitors, in particular AZD9150, were applied in the combination with immune checkpoint inhibitors in the frame of the phase I/II clinical trial (Table 1). In another approach, STAT3 siRNA or decoy oligonucleotides were coupled to CpG oligonucleotides, which are well-known agonists of TLR9 (79, 80). By this technique, a selective delivery of the drugs to TLR9-positive cells was ensured. Upon the treatment, TLR9-expressing myeloid cells (in particular, PMN-MDSC) displayed a decreased immunosuppressive activity, whereas TLR9-positive tumor cells lost the resistance to apoptosis via the STAT3 signaling (79, 80).
A further possibility to target MDSC is the modulation their metabolic pathways (81, 82). It was shown that tumor-infiltrating MDSC displayed an upregulation of the fatty acid translocase, CD36, which resulted in an increased uptake and oxidation of fatty acids. Accumulated lipids were reported to further increase an immunosuppressive capacity of MDSC in a STAT3- and STAT5-dependent manner (83). Pharmacological inhibition of the fatty acid oxidation decreased the immunosuppressive capacity of MDSC and in combination with low-dose chemotherapy and adoptive cellular therapy resulted in antitumor effect in LLC and colon adenocarcinoma mouse models (81).
Blocking MDSC Trafficking
Myeloid-derived suppressor cells exhibit their main immunosuppressive activity within the TME. Therefore, intensive investigations were performed to block the migration of MDSC to the tumor site. Chemokine receptors are a key driving force for the migration of immune cells (84). Myeloid cells (in particular, MDSC) express C-X-C motif chemokine receptor (CXCR) 2 (85). The main ligands for CXCR2 are C-C motif chemokine ligand (CCL)2 and CCL5, which are elevated in the TME (86, 87). To block the CXCR2-CCL2 interaction, tumor-bearing mice were treated with the chemotherapeutic drug docetaxel combination with a CXCR2 antagonist, showing a significant therapeutic effect (88).
Another chemokine receptor CCR5, which is expressed on a broad spectrum of immune cells (84), interacts with its ligands CCL3, CCL4, and CCL5 (89). Interestingly, the patients with a mutated CCR5 variant were reported to be resistant to the prostate cancer development (90). Furthermore, CCR5 has a critical role in tumor progression since it has been shown that the CCR5–CCL5 axis supported tumor growth, invasion, and migration of MDSC to the tumor site (87, 91). By targeting the CCR5-CCR5 ligand interaction, tumor growth and invasiveness could be suppressed in pancreatic, colorectal, prostate, and breast cancer (92–94).
In a spontaneous Ret transgenic mouse melanoma model, we have demonstrated that the tumor progression correlated with the accumulation of CCR5+ MDSC in the TME that displayed significantly stronger immunosuppressive capacity than their CCR5− counterpart (87). By blocking the CCR5–CCR5 ligand interaction with a mCCR5-Ig fusion protein, the survival of melanoma bearing mice was significantly improved as compared to the control group. Importantly, it was also shown that the frequency of CCR5+ M-MDSC and CCR5+ PMN-MDSC was increased in the peripheral blood of melanoma patients and that CCR5+ M-MDSC accumulated in melanoma lesions (87). Similar to the situation in melanoma bearing mice, CCR5+ MDSC from melanoma patients displayed an increased immunosuppressive pattern compared to the CCR5− MDSC subset. Taken together, targeting CCR5 on MDSC could be applied not only to prevent the MDSC migration and accumulation in the TME but also to reduce MDSC immunosuppressive functions in cancer patients (87, 91).
Depletion of MDSC
The number of MDSC in tumor-bearing hosts could be reduced by (i) the normalization of myelopoiesis, (ii) the inhibition of the conversion of IMC into MDSC, and (iii) the differentiation of MDSC into mature myeloid cells like DC or macrophages. All-trans retinoic acid (ATRA) seems to be a very promising agent for these approaches. ATRA is a vitamin A derivative binding to the retinoic acid receptor. By blocking the retinoic acid signal transduction, MDSC could differentiate into DC and macrophages (95). In addition, it was described that administration of ATRA led to the downregulation of ROS production in MDSC by activating the extracellular-signal regulated kinase (ERK)1/2 pathway (96). In a completed clinical trial, ATRA was applied in metastatic renal carcinoma patients in combination with the IL-2 administration (97). The frequency of MDSC was significantly decreased, and the ratio between DC and MDSC was much higher than in the untreated group. In a second clinical trial with late stage small cell lung cancer patients, ATRA was used together with a DC vaccine against p53 (98). The outcome confirmed the inhibitory effect of ATRA on the frequency of circulating MDSC. The combination of the DC vaccine and ATRA resulted in the development of p53-specific CD8+ T cells. It should be mentioned that ATRA was used in many other clinical trials with inhibitory effects on tumor progression; however, MDSC were not evaluated in these trials, and the positive effect was linked to other mechanisms.
Since tumor-derived EV were reported to induce the conversion of non-immunosuppressive IMC into MDSC and further activated their immunosuppressive functions (26, 27), the inhibitors of the EV release from tumor cells were tested in mice-bearing CT26 colon carcinoma (30). It was demonstrated that the treatment of these mice with dimethyl amiloride or omeprazole reduced EV content in serum that was associated with the reduction of MDSC expansion and immunosuppressive activity (30).
Clinical trials with tyrosine kinase inhibitors (such as sunitinib) revealed that these agents could target MDSC. Since sunitinib could block VEGF and c-kit signaling, which are involved in the generation of MDSC (99), its effect on MDSC from cancer patients was evaluated. Sunitinib treatment of metastatic renal cell carcinoma patients was reported to decrease the number of circulating MDSC (100, 101). Interestingly, M-MDSC from treated patients displayed a reduced STAT3 activation and ARG1 expression that was accompanied with an elevated activity and proliferation of CD8 T cells. However, no significant prolongation of the overall survival was observed.
Other chemotherapeutics such as gemcitabine and 5-fluorouracil were shown to induce selectively apoptosis of MDSC in the spleen and TME in several mouse tumor models (102–104). Interestingly, both chemotherapeutic agents displayed no significant effect on the frequencies of T cells, NK cells, DC, and B cells. It was also shown that gemcitabine reduced the frequency MDSC and Treg as well TGF-β1 level in the peripheral blood of pancreatic cancer patients (103). Similar to the preclinical observation, gemcitabine has no effect in effector T cells. In a clinical trial, gemcitabine treatment of pancreatic cancer patients resulted in a dramatic decrease in PMN-MDSC (103). An application of 5-fluorouracil in the preclinical mouse model and colorectal cancer patients affected MDSC, leading to the immune recovery and tumor regression (104). Administration of another chemotherapeutic, docetaxel, induced a decrease of tumor burden in a preclinical mouse model of mammary carcinoma (105). This beneficial effect was accompanied by the conversion of MDSC into a M1-like cells characterized by the upregulation of CCR7 (105). The effect of doxorubicin on MDSC in mammary cancer models was also investigated (106). The treatment of these mice with doxorubicin led to the reduction of MDSC frequencies in the spleen, peripheral blood, and tumors. Furthermore, the immunosuppressive activity of residual MDSC was impaired. The depletion of MDSC resulted in the enhancement of granzyme B and IFN-γ production by effector T and NK cells (106). Moreover, this study demonstrated that MDSC isolated from patients were also sensitive to doxorubicin treatment in vitro (106).
Using Ret transgenic melanoma mouse model, we demonstrated that the administration of ultra-low, non-cytotoxic doses of paclitaxel induced the reduction of MDSC numbers and immunosuppressive functions (107). This effect was associated with an inhibition of the p38 MAPK pathway as well as the production of TNF-α and S100A9 in MDSC. Treated mice showed elevated activity of CD8 T cells, which correlated with the prolongation of mouse survival (107). In addition, it was reported that the treatment of MDSC in vitro with ultra-low concentrations of paclitaxel stimulated their differentiation into DC (108).
Future Perspectives
Tumor cells developed multiple mechanisms to evade the immune system and to progress. One of the key mechanisms is the establishment of an immunosuppressive TME, where MDSC play a crucial role. By altering MDSC function and biology, various preclinical and clinical studies showed a beneficial effect. This suggests that MDSC targeting could be a promising strategy to apply together with existing immunotherapeutic strategies such as boosting the immune system by vaccination or negative immune checkpoint inhibitors. Thus, combining gemcitabine with a DNA vaccination induced a strong antitumor immune response accompanied by a reduced self-tolerance in a preclinical HER2-expressing mouse tumor model (109). Furthermore, another preclinical study showed that the administration of sunitinib with an HPV vaccination resulted in a tumor-free survival in 75% mice in the HPV-expressing tumor model (110). In addition, a clinical trial was initiated in stage IV melanoma patients, by whom ATRA was applied together with ipilimumab (111). This trial and many other starting combinatorial approaches will help to develop an efficient strategy for the treatment of cancer patients.
Author Contributions
VF: writing, review, and revision of the manuscript and revision of the figures. XH: preparing the figures. RW, PA, and JU: review and revision of the manuscript. VN: preparing the table. CG: revision of the manuscript. VU: writing, review, and revision of the manuscript and revision of the table and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the German Research Council (RTG2099 to JU, VU) and the DKFZ-MOST Cooperation in Cancer Research (CA181 to VU). This work was kindly backed by the COST Action BM1404 Mye-EUNITER (www.mye-euniter.eu). COST is supported by the EU Framework Program Horizon 2020.
Abbreviations
ARG1, arginase-1; ATRA, all-trans retinoic acid; bFGF, basic fibroblast growth factor; CD, cluster of differentiation; CCL, C-C motif chemokine ligand; CCR, C-C motif receptor; COX, cyclooxygenase; CXCL, C-X-C motif ligand; CXCR, C-X-C motif receptor; DC, dendritic cell; ERK, extracellular-signal regulated kinase; EV, extracellular vesicles; HSP, heat shock protein; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, interleukin; iNOS, inducible nitric oxide synthase; LLC, Lewis lung carcinoma; LOX, lectin-type oxidized low-density lipoprotein receptor; IMC, immature myeloid cells; M, monocytic; MCP, monocyte chemoattractant protein; MDSC, myeloid-derived suppressor cells; MMP, matrix metalloproteinases; NO, nitric oxide; NSCLC, non-small cell lung cancer; PD-1, programmed death receptor; PD-L1, programmed death ligand 1; PMN, polymorphonuclear; ROS, reactive oxygen species; STAT, signal transducer and activator of transcription; TCR, T cell receptor; TGF, transforming growth factor; TLR, toll-like receptor; TME, tumor microenvironment; TNF, tumor necrosis factor; Treg, regulatory T cells; VEGF, vascular endothelial growth factor.
References
1. Ostrand-Rosenberg S. Immune surveillance: a balance between protumor and antitumor immunity. Curr Opin Genet Dev (2008) 18:11–8. doi:10.1016/j.gde.2007.12.007
2. Cavallo F, De Giovanni C, Nanni P, Forni G, Lollini PL. 2011: the immune hallmarks of cancer. Cancer Immunol Immunother (2011) 60:319–26. doi:10.1007/s00262-010-0968-0
3. Umansky V, Sevko A. Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron (2013) 6:169–77. doi:10.1007/s12307-012-0126-7
4. Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother (2015) 64:1609–21. doi:10.1007/s00262-015-1766-5
5. Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangere V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predict the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res (2016) 76:5241–52. doi:10.1158/0008-5472.CAN-15-3164
6. Gabrilovich D, Nagaraj S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
7. Gabrilovich D. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5:3–8. doi:10.1158/2326-6066
8. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res (2015) 128:95–139. doi:10.1016/bs.acr.2015.04.002
9. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi:10.1016/j.it.2016.01.004
10. Meirow Y, Kanterman J, Baniyash M. Paving the road to tumor development and spreading: myeloid-derived suppressor cells are ruling the fate. Front Immunol (2015) 6:523. doi:10.3389/fimmu.2015.00523
11. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
12. Ray A, Chakraborty K, Ray P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front Cell Infect Microbiol (2013) 3:52. doi:10.3389/fcimb.2013.00052
13. Loftus TJ, Mohr AM, Moldawer LL. Dysregulated myelopoiesis and hematopoietic function following acute physiologic insult. Curr Opin Hematol (2018) 25(1):37–43. doi:10.1097/MOH.0000000000000395
14. Ueha S, Shand FH, Matsushima K. Myeloid cell population dynamics in healthy and tumor-bearing mice. Int Immunopharmacol (2011) 11:783–8. doi:10.1016/j.intimp.2011.03.003
15. Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol (2010) 185:5828–34. doi:10.4049/jimmunol.0903636
16. Gabrilovich D, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
17. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Mandruzzato S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
18. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci (2014) 1319:47–65. doi:10.1111/nyas.12469
19. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol (2016) 1(2):aaf8943. doi:10.1126/sciimmunol.aaf8943
20. Umansky V, Blattner C, Gebhardt C, Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel) (2016) 4:36. doi:10.3390/vaccines4040036
21. Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi:10.1186/2051-1426-1-10
22. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest (2015) 125:3356–64. doi:10.1172/JCI80005
23. Ugel S, De Sanctis F, Mandruzzato S, Bronte V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest (2015) 125:3365–76. doi:10.1172/JCI80006
24. Isola AL, Chen S. Exosomes: the messengers of health and disease. Curr Neuropharmacol (2017) 15:157–65. doi:10.2174/1570159X14666160825160421
25. Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: insights and new perspectives. Cancer Res (2017) 77(23):6480–8. doi:10.1158/0008-5472.CAN-17-0994
26. Ridder K, Sevko A, Heide J, Rupp AK, Macas J, Starmann J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology (2015) 4:e1008371. doi:10.1080/2162402X.2015.1008371
27. Filipazzi P, Bürdek M, Villa A, Rivoltini L, Huber V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin Cancer Biol (2012) 22:342–9. doi:10.1016/j.semcancer.2012.02.005
28. Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C, et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via toll-like receptor signaling. J Biol Chem (2013) 288:36691–702. doi:10.1074/jbc.M113.512806
29. Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D, et al. Macrophage immunomodulation by breast cancer-derived exosomes requires Toll-like receptor 2-mediated activation of NF-κ B. Sci Rep (2014) 4:5750. doi:10.1038/srep05750
30. Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, et al. Membrane associated Hsp72 from tumor derived exosomes mediates STAT3 dependent immunosuppressive function of mouse and human myeloid derived suppressor cells. J Clin Invest (2010) 120:457–71. doi:10.1172/JCI40483
31. Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, et al. Melanoma exosomes educate bone marrow progenitor cells. Nat Med (2012) 18:883–91. doi:10.1038/nm.2753
32. Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res (2008) 68:2561–3. doi:10.1158/0008-5472.CAN-07-6229
33. Umansky V, Blattner C, Fleming V, Hu X, Gebhardt C, Altevogt P, et al. Myeloid-derived suppressor cells and tumor escape from immune surveillance. Semin Immunopathol (2017) 39:295–305. doi:10.1007/s00281-016-0597-6
34. Bronte V, Serafini P, De Stanto C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol (2003) 170:270–8. doi:10.4049/jimmunol.170.1.270
35. Bronte V, Zanovello P. Regulation of immune responses by l-arginine metabolism. Nat Rev Immunol (2005) 5:641–54. doi:10.1038/nri1668
36. Rodriguez PC, Quiceno DG, Ochoa AC. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–74. doi:10.1182/blood-2006-06-031856
37. Baniyash M. TCR ζ-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol (2004) 4:675–87. doi:10.1038/nri1434
38. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol (2002) 168:689–95. doi:10.4049/jimmunol.168.2.689
39. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72:5435–40. doi:10.1158/0008-5472.CAN-12-0569
40. Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity (2005) 22:633–42. doi:10.1016/j.immuni.2005.03.013
41. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med (2002) 196:459–68. doi:10.1084/jem.20020121
42. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol (2010) 185:3190–8. doi:10.4049/jimmunol.0903670
43. Fallarino F, Grohmann U, Vacca C, Orabona C, Spreca A, Fioretti MC, et al. T cell apoptosis by kynurenines. Adv Exp Med Biol (2003) 527:183–90. doi:10.1007/978-1-4615-0135-0_21
44. Della CM, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite l-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood (2006) 108:4118–25. doi:10.1182/blood-2006-03-006700
45. Hardy LL, Wick DA, Webb JR. Conversion of tyrosine to the inflammation-associated analog 3′-nitrotyrosine at either TCR- or MHC-contact positions can profoundly affect recognition of the MHC class i-restricted epitope of lymphocytic choriomeningitis virus glycoprotein 33 by CD8 T cells. J Immunol (2008) 180:5956–62. doi:10.4049/jimmunol.180.9.5956
46. Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol (2008) 83:1136–44. doi:10.1189/jlb.090761
47. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179:977–83. doi:10.4049/jimmunol.179.2.977
48. Berger KN, Pu JJ. PD-1 pathway and its clinical application: a 20 year journey after discovery of the complete human PD-1 gene. Gene (2017) 638:20–5. doi:10.1016/j.gene.2017.09.050
49. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211:781–90. doi:10.1084/jem.20131916
50. Sinha P, Chornoguz O, Clements VK, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S. Myeloid-derived suppressor cells express the death receptor Fas and apoptose in response to T cell-expressed FasL. Blood (2011) 117:5381–90. doi:10.1182/blood-2010-11-321752
51. Safarzadeh E, Orangi M, Mohammadi H, Babai F, Baradaran B. Myeloid-derived suppressor cells: important contributors to tumor progression and metastasis. J Cell Physiol (2018) 233(4):3024–36. doi:10.1002/jcp.26075
52. Baniyash M. Myeloid-derived suppressor cells as intruders and targets: clinical implications in cancer therapy. Cancer Immunol Immunother (2016) 65:857–67. doi:10.1007/s00262-016-1849-y
53. Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol (2011) 21:139–46. doi:10.1016/j.semcancer.2011.01.002
54. Kitamura T, Qian B, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol (2015) 15:73–86. doi:10.1038/nri3789
55. Sceneay J, Parker BS, Smyth MJ, Möller A. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology (2013) 2:e22355. doi:10.4161/onci.22355
56. Jacob A, Prekeris R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front Cell Dev Biol (2015) 3:4. doi:10.3389/fcell.2015.00004
57. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med (1971) 285:1182–6. doi:10.1056/NEJM197111182852108
58. Shen P, Wang A, He M, Wang Q, Zheng S. Increased circulating Lin−/low CD33+ HLA-DR− myeloid-derived suppressor cells in hepatocellular carcinoma patients. Hepatol Res (2014) 44:639–50. doi:10.1111/hepr.12167
59. Pan W, Sun Q, Wang Y, Wang J, Cao S, Ren X. Highlights on mechanisms of drugs targeting MDSCs: providing a novel perspective on cancer treatment. Tumor Biol (2015) 36:3159–69. doi:10.1007/s13277-015-3363-9
60. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol (2001) 166:678–89. doi:10.4049/jimmunol.166.1.678
61. Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother (2011) 60:1419–30. doi:10.1007/s00262-011-1028-0
62. Eruslanov E, Neuberger M, Daurkin I, Perrin GQ, Algood C, Dahm P, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer (2012) 130:1109–19. doi:10.1002/ijc.26123
63. Jiang H, Gebhardt C, Umansky L, Beckhove P, Schulze TJ, Utikal J, et al. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int J Cancer (2015) 136:2352–60. doi:10.1002/ijc.29297
64. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58:49–59. doi:10.1007/s00262-008-0523-4
65. Pico de Coaña Y, Poschke I, Gentilcore G, Mao Y, Nyström M, Hansson J, et al. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their arginase1 production. Cancer Immunol Res (2013) 1:158–62. doi:10.1158/2326-6066.CIR-13-0016
66. Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin Cancer Res (2015) 21:5453–9. doi:10.1158/1078-0432.CCR-15-0676
67. Santegoets SJ, Stam AG, Lougheed SM, Gall H, Jooss K, Sacks N, et al. Myeloid derived suppressor and dendritic cell subsets are related to clinical outcome in prostate cancer patients treated with prostate GVAX and ipilimumab. J Immunother Cancer (2014) 2:31. doi:10.1186/s40425-014-0031-3
68. Sade-Feldman M, Kanterman J, Klieger Y, Ish-Shalom E, Olga M, Saragovi A, et al. Clinical significance of circulating CD33+CD11b+HLA-DR- myeloid cells in patients with stage iv melanoma treated with ipilimumab. Clin Cancer Res (2016) 22:5661–72. doi:10.1158/1078-0432.CCR-15-3104
69. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203:2691–702. doi:10.1084/jem.20061104
70. Meyer C, Sevko A, Ramacher M, Bazhin AV, Falk CS, Osen W, et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc Natl Acad Sci U S A (2011) 108:17111–6. doi:10.1073/pnas.1108121108
71. Lin S, Wang J, Wang L, Wen J, Guo Y, Qiao W, et al. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am J Cancer Res (2017) 7:41–52.
72. Califano J, Khan Z, Noonan K, Rudraraju L, Zhang Z, Wang H, et al. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res (2015) 21:30–8. doi:10.1158/1078-0432.CCR-14-1716
73. Weed DT, Vella JL, Reis IM, De la Fuente AC, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin Cancer Res (2015) 21:39–48. doi:10.1158/1078-0432.CCR-14-1711
74. Hassel JC, Jiang H, Bender C, Winkler J, Sevko A, Shevchenko I, et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic melanoma (TaMe). Oncoimmunology (2017) 6:e1326440. doi:10.1080/2162402X.2017.1326440
75. Orillion A, Hashimoto A, Damayanti N, Shen L, Adelaiye-Ogala R, Arisa S, et al. Entinostat neutralizes myeloid derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin Cancer Res (2017) 23:5187–201. doi:10.1158/1078-0432.CCR-17-0741
76. Kim K, Skora AD, Li Z, Liu Q, Tam AJ, Blosser RL, et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc Natl Acad Sci U S A (2014) 111:11774–9. doi:10.1073/pnas.1410626111
77. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9:798–809. doi:10.1038/nrc2734
78. Kortylewski M, Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol Immunother (2017) 66:979–88. doi:10.1007/s00262-017-1966-2
79. Spinetti T, Spagnuolo L, Mottas I, Secondini C, Treinies M, Rüegg C, et al. TLR7-based cancer immunotherapy decreases intratumoral myeloid-derived suppressor cells and blocks their immunosuppressive function. Oncoimmunology (2016) 5:e1230578. doi:10.1080/2162402X.2016.1230578
80. Zhang Q, Hossain DM, Duttagupta P, Moreira D, Zhao X, Won H, et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood (2016) 127:1687–700. doi:10.1182/blood-2015-08-665604
81. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res (2015) 3:1236–47. doi:10.1158/2326-6066.CIR-15-0036
82. Al-Khami AA, Rodriguez PC, Ochoa AC. Energy metabolic pathways control the fate and function of myeloid immune cells. J Leukoc Biol (2017) 102:369–80. doi:10.1189/jlb.1VMR1216-535R
83. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology (2017) 30:e1344804. doi:10.1080/2162402X.2017.1344804
84. Homey B, Müller A, Zlotnik A. Chemokines: agents for the immunotherapy of cancer? Nat Rev Immunol (2002) 2:175–84. doi:10.1038/nri748
85. Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, et al. Monocytic CCR2+ myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res (2012) 72:876–86. doi:10.1158/0008-5472.CAN-11-1792
86. Izhak L, Wildbaum G, Weinberg U, Shaked Y, Alami J, Dumont D, et al. Predominant expression of CCL2 at the tumor site of prostate cancer patients directs a selective loss of immunological tolerance to CCL2 that could be amplified in a beneficial manner. J Immunol (2010) 184:1092–101. doi:10.4049/jimmunol.0902725
87. Blattner C, Fleming V, Weber R, Himmelhan BS, Altevogt P, Gebhardt C, et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res (2018) 78:157–67. doi:10.1158/0008-5472.CAN-17-0348
88. Izhak L, Wildbaum G, Zohar Y, Anunu R, Klapper L, Elkeles A, et al. A novel recombinant fusion protein encoding a 20-amino acid residue of the third extracellular (E3) domain of CCR2 neutralizes the biological activity of CCL2. J Immunol (2009) 183:732–9. doi:10.4049/jimmunol.0802746
89. Combadiere C, Ahuja SK, Tiffany HL, Murphy PM. Cloning and functional expression of CC CKR5, a human monocyte CC chemokine receptor selective for MIP-1(alpha), MIP-1(beta), and RANTES. J Leukoc Biol (1996) 60:147–52. doi:10.1002/jlb.60.1.147
90. Balistreri CR, Carruba G, Calabrò M, Campisi I, Di Carlo D, Lio D, et al. CCR5 proinflammatory allele in prostate cancer risk: a pilot study in patients and centenarians from Sicily. Ann N Y Acad Sci (2009) 1155:289–92. doi:10.1111/j.1749-6632.2008.03691.x
91. Umansky V, Blattner C, Gebhardt C, Utikal J. CCR5 in recruitment and activation of myeloid-derived suppressor cells in melanoma. Cancer Immunol Immunother (2017) 66:1015–23. doi:10.1007/s00262-017-1988-9
92. Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res (2003) 63:8360–5.
93. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol (2009) 182:1746–55. doi:10.4049/jimmunol.182.3.1746
94. Velasco-Velázquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, et al. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res (2012) 72:3839–50. doi:10.1158/0008-5472.CAN-11-3917
95. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res (2007) 67:11021–8. doi:10.1158/0008-5472.CAN-07-2593
96. Li Y, Wongsiriroj N, Blaner WS. The multifaceted nature of retinoid transport and metabolism. Hepatobiliary Surg Nutr (2014) 3:126–39. doi:10.3978/j.issn.2304-3881.2014.05.04
97. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res (2006) 66:9299–307. doi:10.1158/0008-5472.CAN-06-1690
98. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother (2013) 6:909–18. doi:10.1007/s00262-013-1396-8
99. Kodera Y, Katanasaka Y, Kitamura Y, Tsuda H, Nishio K, Tamura T, et al. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res (2011) 13(3):R66. doi:10.1186/bcr2903
100. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15:2148–57. doi:10.1158/1078-0432.CCR-08-1332
101. Nayak D, Roth TL, Mcgavern DB. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res (2010) 21:4073–85. doi:10.1158/1078-0432.CCR-14-2742
102. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70:3052–61. doi:10.1158/0008-5472.CAN-09-3690
103. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFβ-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med (2016) 14:282. doi:10.1186/s12967-016-1037-z
104. Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, et al. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res (2014) 74:6022–35. doi:10.1158/0008-5472.CAN-14-0657
105. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res (2010) 16:4583–94. doi:10.1158/1078-0432.CCR-10-0733
106. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res (2014) 74:104–18. doi:10.1158/0008-5472.CAN-13-1545
107. Sevko A, Michels T, Vrohlings M, Umansky L, Beckhove P, Kato M, et al. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J Immunol (2013) 190:2464–71. doi:10.4049/jimmunol.1202781
108. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol (2012) 9:292–300. doi:10.3109/1547691X.2011.642418
109. Danishmalik SN, Sin JI. Therapeutic tumor control of HER2 DNA vaccines is achieved by an alteration of tumor cells and tumor microenvironment by gemcitabine and anti-Gr-1 Ab treatment in a HER2-expressing tumor model. DNA Cell Biol (2017) 6:801–11. doi:10.1089/dna.2017.3810
110. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells – an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4:e954829. doi:10.4161/21624011.2014.954829
Keywords: myeloid-derived suppressor cells, immunosuppression, cancer immunotherapy, tumor microenvironment, therapeutic targeting
Citation: Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, Utikal J and Umansky V (2018) Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 9:398. doi: 10.3389/fimmu.2018.00398
Received: 30 November 2017; Accepted: 13 February 2018;
Published: 02 March 2018
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Susanna Mandruzzato, Università degli Studi di Padova, ItalyNicolas Larmonier, Université de Bordeaux, France
Copyright: © 2018 Fleming, Hu, Weber, Nagibin, Groth, Altevogt, Utikal and Umansky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viktor Umansky, di51bWFuc2t5QGRrZnouZGU=