 Ana Cardoso1,2,3,4,5,6,7
Ana Cardoso1,2,3,4,5,6,7 Antonio Gil Castro6,7‡Ana Catarina Martins1,2‡Guilhermina M. Carriche1,2†Valentine Murigneux8,9†Isabel Castro6,7Ana Cumano3,4,5
Antonio Gil Castro6,7‡Ana Catarina Martins1,2‡Guilhermina M. Carriche1,2†Valentine Murigneux8,9†Isabel Castro6,7Ana Cumano3,4,5 Paulo Vieira3,4,5§
Paulo Vieira3,4,5§ Margarida Saraiva1,2*§
Margarida Saraiva1,2*§
- 1i3S – Instituto de Investigação e Inovação em Saúde, Porto, Portugal
- 2IBMC – Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal
- 3Department of Immunology, Unité Lymphopoièse, Institut Pasteur, Paris, France
- 4University Paris Diderot, Sorbonne Paris Cité, Cellule Pasteur, Paris, France
- 5INSERM U1223, Paris, France
- 6ICVS, University of Minho, Braga, Portugal
- 7ICVS/3B’s – PT Government Associate Laboratory, Braga, Portugal
- 8Department of Immunology, Unité Intégrité du génome, immunité et cancer, Institut Pasteur, Paris, France
- 9Department of Genomes and Genetics, Unité Intégrité du génome, immunité et cancer, Institut Pasteur, Paris, France
Inflammatory bowel disease encompasses a group of chronic-inflammatory conditions of the colon and small intestine. These conditions are characterized by exacerbated inflammation of the organ that greatly affects the quality of life of patients. Molecular mechanisms counteracting this hyperinflammatory status of the gut offer strategies for therapeutic intervention. Among these regulatory molecules is the anti-inflammatory cytokine interleukin (IL)-10, as shown in mice and humans. Indeed, IL-10 signaling, particularly in macrophages, is essential for intestinal homeostasis. We sought to investigate the temporal profile of IL-10-mediated protection during chemical colitis and which were the underlying mechanisms. Using a novel mouse model of inducible IL-10 overexpression (pMT-10), described here, we show that mice preconditioned with IL-10 for 8 days before dextran sulfate sodium (DSS) administration developed a milder colitic phenotype. In IL-10-induced colitic mice, Ly6C cells isolated from the lamina propria showed a decreased inflammatory profile. Because our mouse model leads to transcription of the IL-10 transgene in the bone marrow and elevated seric IL-10 concentration, we investigated whether IL-10 could imprint immune cells in a long-lasting way, thus conferring sustained protection to colitis. We show that this was not the case, as IL-10-afforded protection was only observed if IL-10 induction immediately preceded DSS-mediated colitis. Thus, despite the protection afforded by IL-10 in colitis, novel strategies are required, specifically to achieve long-lasting protection.
Introduction
Inflammatory bowel disease (IBD) comprises a complex group of inflammatory conditions of the gastrointestinal tract (1) affecting an increasing number of patients worldwide (2–4). Both forms of IBD, Crohn’s disease (CD) and ulcerative colitis (UC), result from alterations in the immune homeostasis of the intestinal tissue leading to local uncontrolled inflammation (5, 6). The gut is a very particular site in terms of immune repertoire and regulation, as even in homeostatic conditions constant exposure to antigens occurs (7). Thus, the maintenance of intestinal homeostasis, primarily carried out by intestinal macrophages, requires a constant and fine-tuned balance between the state of tolerance and inflammation (8). In the gut environment, macrophages encounter a plethora of stimuli, from dietary antigens to commensal bacteria, yet, due to their unique tissue-specific characteristics, remain tolerant (9). In the predisease stage, the epithelial or mucosal barriers become compromised allowing bacteria from the luminal side to invade the lamina propria of the gut (10). This event triggers an acute inflammatory response due to the activation of immune cells by direct contact with bacterial products (10). The induced inflammation results either in elimination of the foreign bacterial incursion or in an exacerbated immune response that can result in tissue damage. The damage caused by deregulated inflammation will perpetuate the activation of effector cells and ultimately lead to the clinical onset of IBD (10, 11).
Epidemiological studies have shown that the etiology of IBD is multifactorial, with genetic predisposition, dysfunctional intestinal barrier and imbalances of the microbiome all contributing to this condition (12–15). Genome-wide association studies revealed that the main genetic alterations associated with IBD are found in genes encoding proteins linked to innate or adaptive immunity, such as the nucleotide-binding oligomerization domain-containing protein 2, Janus kinase (JAK) 2, and tumor necrosis factor superfamily 15 (16–18). Other alterations are associated with molecules involved in leukocyte trafficking, regulation of barrier function and secretion of defensins (17). Two reports associate loss-of-function mutations in interleukin (IL)-10 or IL-10R subunits with severe IBD (19, 20). These mutations result in severe enterocolitis, with onset before one year of age, and unresponsiveness to immunosuppressive therapies. The only available therapy for these patients is immune reconstitution with hematopoietic stem cells (21–23). Although complete loss-of-function mutations in IL-10 and IL-10R strongly correlate with IBD, they have an extremely low occurrence rate (19, 24). The most frequent mutations affecting the IL-10 genes associated with IBD are in fact single-nucleotide polymorphisms associated with low expression of this molecule (25). However, harboring such mutations does not always translate in low serum levels of IL-10 (23) during the disease stage. This is likely due to the significant increase on the number of IL-10-producing myeloid cells in CD patients (26–29), to the extent that elevated serum levels of IL-10 correlate with disease activity in CD (30–32).
The role of IL-10 in intestinal inflammation is also seen in the mouse model, as IL-10-deficient mice develop microbiome-dependent spontaneous enterocolitis (33). Furthermore, mice with macrophage restricted IL-10R deficiency also develop a spontaneous colitic profile (34), stressing the critical role of the monocyte/macrophage axis in the immunologic events leading to IBD. Interestingly, it has been shown, in a model of infection that IL-10 can exert a direct effect on monocytes/macrophages subsets, leading to changes in their inflammatory profile and survival (35). Moreover, IL-10 has been shown to confer protection from hyperinflammatory states by the induction of the JAK1/STAT3 signaling pathway that suppresses expression of proinflammatory mediators and activates expression of anti-inflammatory genes (36).
Taking into account the results obtained in murine models of IL-10 perturbation, the genetic correlation established in humans, and the anti-inflammatory properties of IL-10, this cytokine emerged as a very promising candidate for IBD therapy. However, in IBD patients IL-10-based therapy has not resulted in substantial clinical improvements (37). The main caveats in these clinical trials were the subcutaneous route of administration and the concentration of the recombinant molecule that did not ensure that IL-10 levels reached the mucosal sites, pointing out the importance of novel-locally targeted therapeutic strategies. Furthermore, IL-10 administration to IL-10-deficient murine models only protected from colitis if administered before disease establishment (38).
In this study, we report a novel mouse model of IL-10 overexpression (the pMT-10 mouse) and use it to better explore the mechanisms of immune regulation elicited by IL-10 in the context of intestinal inflammation. We show that a short period of IL-10 overexpression prior to the induction of colitis ameliorates the disease outcome, despite the presence of CD11b+ Ly6C+ cells in the gut, previously associated with the development of detrimental inflammation. As compared to control animals that do not overexpress IL-10, Ly6C cells isolated from the gut lamina propria of colitic pMT-10 mice showed a decreased inflammatory profile. Thus, we propose that IL-10 overexpression impaired the response of these cells to the stimulus. In addition to the local effect of IL-10 in controlling exacerbated immune responses, our model allows for the study of IL-10 in imprinting de novo generated and circulating monocytes. This is because, constant IL-10 expression is found in specific tissues, in pMT-10 mice, culminating in a systemic effect. Therefore, IL-10 is likely to affect other important compartments, such as the bone marrow (BM) and spleen. IL-10-afforded protection was only seen if IL-10 triggering immediately preceded dextran sulfate sodium (DSS)-induced colitis, thus calling for novel strategies that sustain the effect of IL-10 to offer long-lasting protection.
Materials and Methods
Ethics Statement
In Portugal, all animal experiments were performed in strict accordance with recommendations of the European Union Directive 2010/63/EU and previously approved by Portuguese National Authority for Animal Health–Direção Geral de Alimentação e Veterinária (DGAV). Mice were euthanized by CO2 inhalation with efforts to minimize suffering.
In France, all animal procedures were approved by the Pasteur Institute Safety Committee and conducted according to French and European Community Institutional guidelines.
Animals
The study involved the use of the following 7–14-week-old female mice: wild-type C57BL/6j, pMT-10-IL-10 inducible mice, and pMT-10 crossed with IL-10Rα-deficient mice (39) (pMT-10.IL-10Rα−/−). Food was ad libitum for all animals.
Generation of pMT-10 Mice
pMT10 mice were generated by A. Gil Castro and Paulo Vieira. Mouse IL-10 cDNA was cloned into the p169ZT vector, which carries a sheep metalloprotein (MT) 1a promoter, a β-globin splice site and a SV40 polyadenylation (polyA) signal. The resulting vector—pMT-10 (see Figure 1A)—was then injected into C57BL/6j eggs and transgenic founders were identified by PCR using MT and IL-10-specific primers. IL-10 overexpression was induced by feeding the mice a 2% sucrose solution with 50 mM of zinc (Zn) sulfate.
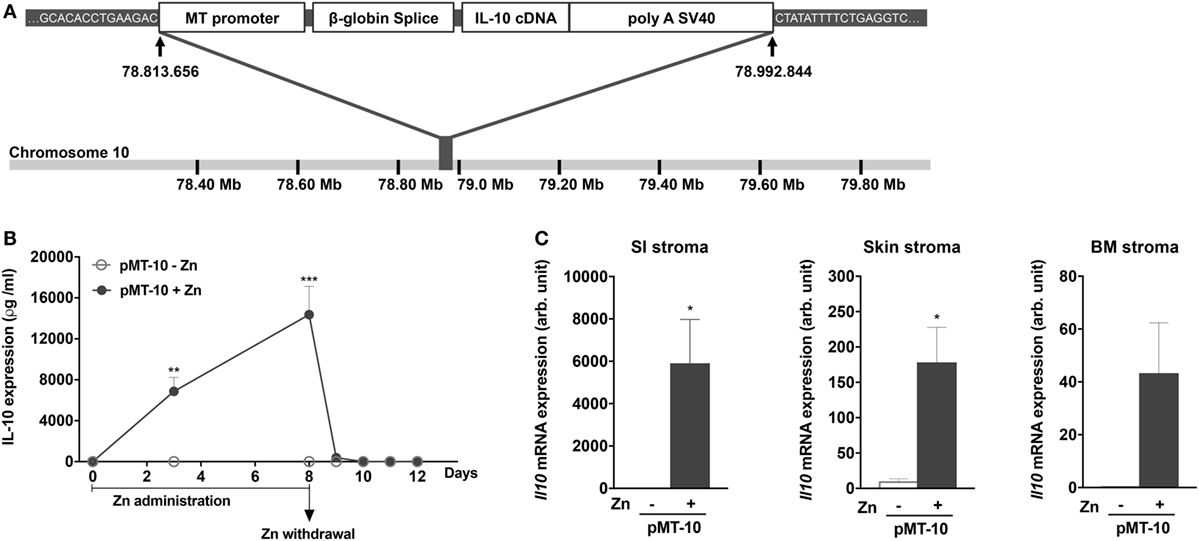
Figure 1. A novel mouse model for inducible interleukin (IL)-10 expression: pMT-10 mice. (A) Schematic representation showing the targeting vector and insertion site. (B) Kinetics of IL-10 overexpression in the serum at different time points post Zn administration and Zn withdrawal. pMT-10 mice were fed with normal (pMT-10-Zn) or Zn-enriched (pMT-10 + Zn) water and at the indicated time points blood was harvested and the amount of IL-10 in serum measured by immunoassay. (C) qRT-PCR identified CD45− TER119− cell subsets from skin, bone marrow, and small intestine (SI) as the main producers of IL-10 in pMT-10 mice fed for 8 days with Zn-enriched water. In both (B,C), each point or bar represents the mean ± SEM for three independent mice. Data were analyzed with (B) two-way analysis of variance (Sidak’s multiple comparisons test) or (C) Student’s t-test, *p < 0.05; **p < 0.01; ***p < 0.001.
DSS-Induced Colitis
Mice were fed for 8 days with 3% DSS (TdB consultancy) in the drinking water, and were monitored, daily, for weight loss and disease progression. Colitis progression was measured by the Disease Activity Index (DAI), as previously described [Table 1 (40)].
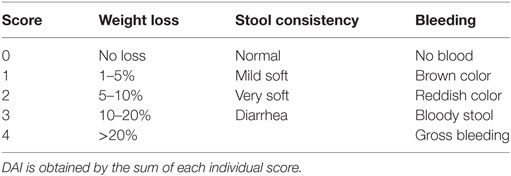
Table 1. Disease Activity Index (DAI) parameters.
Assessment of Intestinal Inflammation
Mice were euthanized on day 8 post-DSS administration or earlier if the symptoms of clinical disease (significant weight loss or diarrhea) became apparent. Samples from colons were immediately fixed in 4% paraformaldehyde. Then, 5 µm paraffin-embedded sections were stained with hematoxylin and eosin, and inflammation was assessed in a blinded fashion using a previously described system [Table 2 (41)]. Samples were graded semiquantitatively from 0 to 3 for the four following criteria: (i) degree of epithelial hyperplasia and goblet depletion; (ii) leukocyte infiltration in the lamina propria; (iii) area of tissue affected; and (iv) the presence of markers of severe inflammation such as crypt abscesses submucosal inflammation and ulcers. For each sample, criteria scores were added to give an overall inflammation score of 0–12.
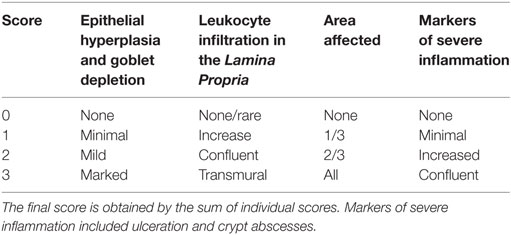
Table 2. Parameters for histological analysis of colitis severity.
Cytokine Quantification
Interleukin-10 concentration in the serum was quantified using a commercially available ELISA kit (R&D systems).
Preparation of Cell Suspensions
Isolation of non-hematopoietic cells (CD45− TER119−) or hematopoietic (CD45+) BM cells followed standard protocols. Briefly, hematopoietic BM cells were extracted by flushing the femurs and tibias with 2 mL of HBBS repeatedly. To obtain the BM non-hematopoietic cells, the bone fragments were incubated in RPMI medium with Liberase TL (0.5 mg/ml; Roche) for 30 min at 37°C. To help dissociation of non-hematopoietic cells from the bone, after each incubation period, the femurs and tibias were flushed with RPMI. We repeated this step three times after which we flushed the bones one last time, harvested the cell suspensions and added 1 volume of RPMI containing 10% FCS. Small intestine (SI), non-hematopoietic cells were isolated as previously described (42). Skin non-hematopoietic cells were isolated from ear samples. Samples were harvested and the epidermis exposed by separating the external layers. Epidermis was incubated for 45 min at 37°C with Liberase HL (0.5 mg/ml; Roche) and DNase I (1 U/mL; Invitrogen). After 45 min, non-hematopoietic cells were dissociated from the tissue by mechanical disruption, collected, washed in HBSS containing 10% FCS and recovered. At the end, non-hematopoietic cells were sort-purified by excluding all CD45+ TER119+ cells (Figure S1A in Supplementary Material). Cell suspensions for all other organs were obtained by mechanical disruption.
Lamina propria leukocytes (LPLs) were prepared as previously described (43). Briefly, LPLs were harvested, dissociated and resuspended in Hank’s Balanced Solution (HBSS) supplemented with 1% fetal calf serum (FCS; Gibco). To isolate LPLs, the colon was flushed with phosphate-buffered saline (PBS; Gibco), opened and cut into 1 cm pieces. To eliminate epithelial cells these fragments were incubated at 37°C in Ca- and Mg-free PBS containing 10% FCS and 5.0 mM EDTA under strong agitation for 30 min. For LPL isolation, the remaining fragments were incubated in RPMI medium with Liberase TL (0.5 mg/ml; Roche) for 30 min at 37°C. To complete the digestion, the suspension was repeatedly passed through a 10 ml syringe for 5 min, filtered through a 40 μm cell strainer (BD Bioscience) and collected by centrifugation. The cell pellet was resuspended in 44% Percoll (GE Healthcare), laid over 67% Percoll, and centrifuged at 600 g for 20 min at 20°C. Cells at the interface were collected, washed in HBSS containing 1% FCS and recovered.
Antibodies
Antibodies were conjugated to fluorochromes (FITC, PE, PECy7, APC, APCCy7, Pacific Blue, and BV711) and were specific for the following mouse antigens: CD3 (145-2C11; Biolegend), CD11b (M1/70; Sony), CD11c (HL3; Biolegend), CD19 (6D5; Sony), CD45.2 (104; Biolegend), Ly6C (Hk1.4; eBioscience), Ly6G (RB6-8C5; BD Pharmingen), CD45 (30F11; Sony), and TER119 (Ter119; BD Pharmingen).
Cell Sorting and Multiplex Real Time-PCR Analysis
CD45− TER119− or CD45+ cells were sort-purified based on the expression of CD45 and TER119 using an Aria sorter (BD). Dead cells were eliminated by exclusion with propidium iodide (PI). mRNA from sorted cells was extracted using RNeasy Micro kit (Qiagen) and converted into cDNA by reverse transcription with PrimeScript RT Reagent kit (Takara, Clonetech). qRT-PCR was performed using Taqman primers (see Table S1 in Supplementary Material for references) and Taqman Universal Master Mix (Applied Biosystems). qRT-PCR reactions were performed on a ABI 7300 thermocycler (Applied Biosystems).
Lamina propria leukocytes were FACS-sort purified based on the expression of CD45.2, CD11b and Ly6C, using an Aria III sorter (BD). Cells expressing CD3, CD19, CD11c, and Ly6G were excluded. Dead cells were eliminated by exclusion with PI. CD45.2+ CD11b+ Ly6C+ cells were sorted directly into a mix of 9 µl of CellsDirect One-Step qRT-PCR kit (Life Technologies), containing a mixture of diluted primers (0.05× final concentration, see Table S1 in Supplementary Material for references). Preamplified cDNA (18 cycles), was obtained according to the manufacturer’s instructions and was diluted 1:5 in TE buffer (pH = 8; Ambion). The sample mixture was as follows: diluted cDNA (2.9 µl), Sample Loading Reagent (0.32 µl; Fluidigm), and Taqman Universal PCR Master Mix (3.5 µl; Applied Biosystems). The assay mixture was as follow: Assay Loafing Reagent (Fluidigm) and Taqman Mix. A 48 × 48 Dynamic Array integrated fluidic circuit (IFC; Fluidigm) was primed with control line fluid, and the chip was loaded with assays and samples with and X IFC Controller (Fluidigm). The experiments were run on a BioMark HD (Fluidigm) for 40 cycles. Gene expression was normalized for Hprt and assessed by the 2ΔCt method.
Statistical Analysis
Statistical analysis was performed with the Student’s t-test or two-way analysis of variance as indicated in the figure legends. The analysis was performed with Prism Software (GraphPad). Graphs containing errors bars show means ± SEM. Statistical significance is represented as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Generation of a Novel Mouse Model of IL-10 Overexpression
To study the biological impact of IL-10 overexpression in different settings, we engineered a novel mouse model to allow for inducible IL-10 expression, the pMT-10 mouse (44). For this, a construct containing the IL-10 cDNA under the control of the inducible sheep MT promoter was introduced in the genome of wild-type BL/6 mice (Figure 1A). Whole genome sequencing revealed a single insertion of the transgene in chromosome 10, between positions 78.813.656 and 78.992844 bp (Figure 1A). We estimated, by qRT-PCR, the number of copies of the transgene to be 50–100 (data not shown). The MT promoter is activated in the presence of 50 mM of Zn in the organism, administered in the drinking water. Kinetic analysis of IL-10 in the serum of pMT-10 mice fed with Zn-enriched water showed a rapid increase of circulating IL-10 (Figure 1B). Indeed, as soon as day 3 after IL-10 induction, the levels of this cytokine in the serum were very high (7–12 ng/ml) (Figure 1B). Moreover, suspending Zn administration led to a sharp drop in IL-10 in sera in only 24 h, to below detection levels in only 48 h (Figure 1B). As expected, circulating IL-10 was undetectable in pMT-10 mice fed with normal water (Figure 1B). Transcriptional analysis of different organs and cellular compartments of induced pMT-10 mice revealed that the expression of the exogenous IL-10 cDNA was restricted to CD45−TER119− cells from the SI, skin and, to a less extent, BM (Figure 1C). IL-10 induction was not detected in the other organs analyzed (liver, spleen, kidney, choroid plexus, lung, and colon) nor in hematopoietic cells isolated from the BM (Figure S1B in Supplementary Material). Thus, the pMT-10 mouse model allows for timely controlled IL-10 overexpression in specific anatomic locations, accompanied by a strong increase of the levels of this cytokine in the serum.
DSS-Induced Colitis Is Ameliorated in IL-10 Preexposed Mice
Despite the clear link between low levels of IL-10 and susceptibility to colitis in human (45) as well as in mouse models (33, 34), administration of IL-10 to treat this condition showed only limited effects (37). A possible reason may be the poor accessibility of IL-10 to the site of inflammation. In this context, and in view of the high expression seen in the SI of induced mice, the pMT-10 mouse model offers an opportunity to further address the effects of IL-10 expression in the gut in the context of colitis. For this, we used the DSS experimental model, a highly reliable and reproducible way of causing UC-like symptoms in the mouse model by inducing acute inflammation with the recruitment of inflammatory cells (46).
We investigated the impact of IL-10 overexpression prior to DSS-induced colitis. For this, pMT-10 mice were induced to overexpress IL-10 for 8 days, before initiation of DSS administration (Figure 2A). As controls, non-induced pMT-10 or BL6 mice fed with control or Zn-enriched water were used. In our experimental setting, wild-type BL/6 mice started to show signs of disease from days 4 to 5 after administration of 3% DSS in the drinking water (Figure 2B). Control pMT-10 mice showed a progression of the DAI very similar to BL/6 mice (Figure 2B). As compared to pMT-10 or BL6 fed with control water, mice preconditioned with IL-10 showed significantly lower DAI after day 5 (Figure 2B) indicating that IL-10 conferred partial protection. Zn administration to BL6 mice prior to DSS, resulted in partial protection. Indeed, by day 7 of DSS administration, a significantly lower DAI was observed in BL/6 mice fed with Zn-enriched water as compared to BL6 control (Figure 2B). Nevertheless, the maximal protection was observed for pMT-10 mice overexpressing IL-10, which suggests a synergistic effect of IL-10 and Zn in the amelioration of the disease (Figure 2B). The DAI encompasses three scores, one of which is the weight loss. Relatively to control animals, both pMT-10 and BL/6 mice fed with Zn prior to DSS administration showed less weight loss (Figure S2 in Supplementary Material). pMT-10 mice preexposed to IL-10 showed the least reduction in colon length as compared to all control groups (pMT-10 or BL/6 fed with control water, and BL/6 fed with Zn-enriched water – Figure 2C), in line with maximal protection being conferred by IL-10. Histologic analysis of the organ, comprising the analysis of inflammatory infiltrates, architectural distortion (crypt shortening and branching) and ulceration, showed an improvement in pMT-10 mice preexposed to IL-10 as compared to BL/6 preexposed to Zn (Figures 2D,E). However, despite a reduction in the histological score of some pMT-10 mice preexposed to IL-10, the overall group did not reach statistical significance when compared to the other control groups (Figures 2D,E).
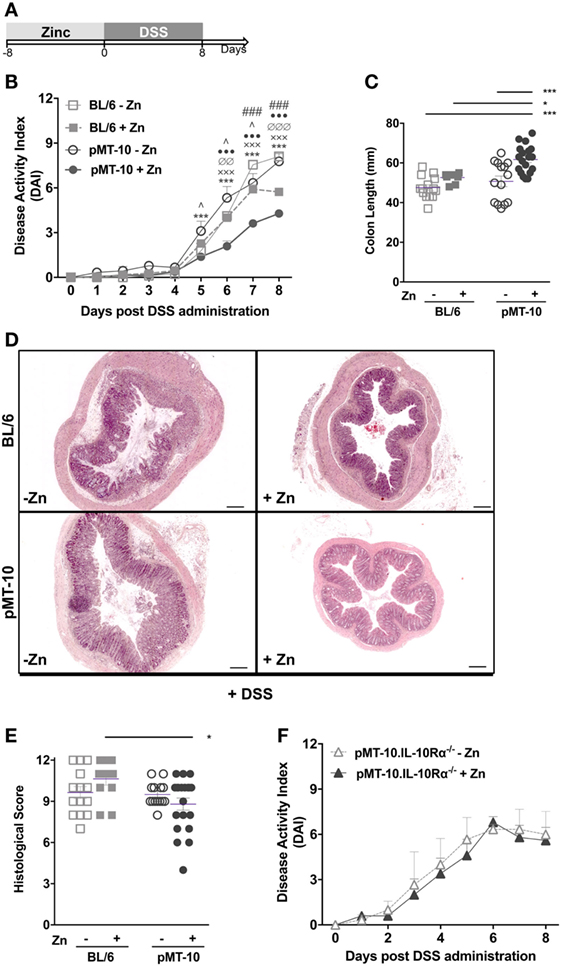
Figure 2. Dextran sulfate sodium (DSS)-induced pathology is ameliorated by preexposure to interleukin (IL)-10. (A) BL/6, pMT-10, and pMT-10.IL-10Rα−/− mice were fed for 8 days with normal (BL/6-Zn, pMT-10-Zn, and pMT-10.IL-10Rα−/− Zn, respectively) or Zn-enriched (BL/6 + Zn, pMT-10 + Zn, and pMT-10.IL-10Rα−/− + Zn) water, followed by 8 days of 3% DSS administration also in the drinking water. (B,F) Disease progression based on DAI parameters was registered every day for 8 days. Each point represents the mean ± SEM for three to five independent mice, in two independent experiments. (C) Colon length measurement at day 8 of DSS administration. (D) Representative H&E-stained sections of large bowel at 40× magnification (scale bar = 200 µm). (E) Colitis scores derived from evaluation of colon and cecum from either group. Each dot represents one independent animal; represented is also mean ± SEM. Data were analyzed with (B,F) two-way analysis of variance (Sidak’s multiple comparisons test) or (C,E) Student’s t-test. (B) # compare BL/6-Zn against BL/6 + Zn; ^ compare BL/6-Zn against pMT-10-Zn; • compare BL/6-Zn against pMT-10 + Zn; ∅ compare BL/6 + Zn against pMT-10-Zn; x compare BL/6 + Zn against pMT-10 + Zn, * compare pMT-10-Zn against pMT-10 + Zn. One symbol, p < 0.05; two symbols, p < 0.01; three symbols, p < 0.001.
Thus, our data showed that IL-10 overexpression prior to intestinal insult afforded a significant degree of protection from DSS-induced colitis. Additionally, our data also suggest a synergistic effect of IL-10 and Zn in the amelioration of the disease. To investigate this issue, we repeated the experiment using pMT-10 mice crossed with IL-10Rα−/− mice. Since pMT-10.IL-10Rα−/− double mutant mice are unresponsive to IL-10, the effects observed would only be due to Zn administration. In these mice, we observed an accelerated disease progression upon DSS administration, with an elevated DAI score as early as day 3, in line with the known role of IL-10 in controlling the disease (Figure 2F). In these mice, Zn administration did not confer protection against DSS-induced colitis (Figure 2F). Taken together, our findings support the notion that the protection conferred by Zn requires IL-10 signaling.
Preexposure to IL-10 Promotes a More Controlled Inflammatory Response
Previous studies have shown that monocytes and macrophages are the major effector subsets of colonic inflammation (34, 47). Mice with macrophage-specific IL-10R deficiency develop a spontaneous colitic profile, emphasizing the importance of IL-10 in regulating the macrophage response to prevent uncontrolled inflammation (34). Thus, we next investigated whether IL-10 ameliorated DSS-induced colitis by restricting the monocyte/macrophage response. Considering that Zn administration also improved the outcome of DSS-induced colitis in BL/6 mice, we compared the transcriptional profile of monocytes/macrophages from BL/6 or pMT-10 mice preexposed to Zn and subjected to DSS administration for 4 days. We chose this time point, since signs of colitis induced by DSS in both BL/6 and pMT-10 mice only become obvious after day 4 of DSS administration. Thus, BL/6 and pMT-10 mice were fed with Zn-enriched water for 8 days and then received DSS for 4 days (Figure 3A). At this time point, Ly6C+ cells from each mouse from the different groups were FACS purified (Figure 3B). Expression of 22 genes (Table S1 in Supplementary Material) associated with the uncontrolled immune response developed in IBD were analyzed by multiplex RT-PCR. All samples, from both groups, expressed three house-keeping genes (Hprt, Actb, and Gapdh). Of the 22 genes analyzed, we failed to detect expression of 9 (Il4, Il9, Il12α, Il12β, Il13, Il17, Il23, Ifnγ, and Cx3cl1) in Ly6C+ cells isolated from the lamina propria in both groups. We detected expression of the 10 remaining genes in Ly6C+ cells, in Zn-fed BL/6 and pMT-10 mice after DSS administration (Figure 3C), but no expression in the absence of insult (data not shown). Thus, Ly6C+ cells alter their expression profile in response to DSS insult. Most interestingly, on day 4 post-DSS administration, Ly6C+ cells isolated from induced pMT-10 mice presented an overall less inflammatory profile than those isolated from BL/6 mice (Figure 3C). In the case of Tnfα and Cd86, the differences observed between the two mouse groups were statistically significant (Figure 3C). In all, these findings suggest that exposure to IL-10 before DSS induction acts by preventing an inflammatory profile in Ly6C+ cells. Of note, the frequency of inflammatory macrophages recruited to the inflamed gut was similar between the two groups, and the same was true for CD3 T cells and CD19 B cells, showing that IL-10 overexpression does not impact the recruitment of immune cells to the gut (Figure 3D).
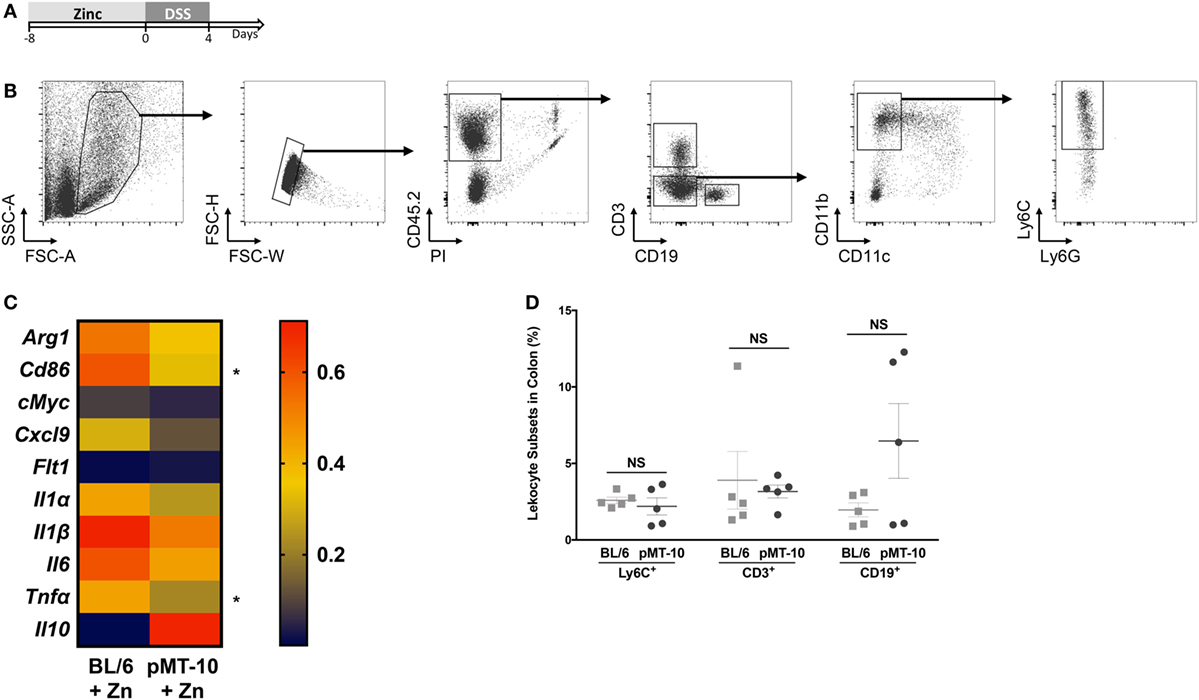
Figure 3. Ly6C+ cells preexposed to interleukin (IL)-10 reveal a less inflammatory profile upon DSS-induced colitis than those preexposed to Zn. (A) pMT-10 or BL/6 mice were fed with Zn-enriched water for 8 days, followed by 4 days of 3% DSS administration. (B) At the end of the DSS treatment, Lamina propria leukocytes (LPLs) were isolated and Ly6C+ cells sort-purified. Shown is the gating strategy for Ly6C+ cells purification. (C) Sort-purified Ly6C+ cells (n = 25 cells) were analyzed by qRT-PCR for a total of 22 genes using the BioMark HD system. Samples were normalized for Hprt expression. Represented is the expression heatmap compiling the genes which expression was detected in either mouse group. Each heatmap rectangle represents the mean of gene expression obtained for cells isolated from five independent mice. (D) The frequency of the different leukocyte subsets was determined upon staining of LPLs for Ly6C+ cell sorting. Each dot represents one independent animal; represented is also mean ± SEM. Data were analyzed with Student’s t-test, *p < 0.05.
IL-10 Protection against DSS-Induced Colitis Is Not Long Lasting
In our mouse model, IL-10 is also overexpressed in the BM and is found at high levels in the serum, possibly creating an anti-inflammatory environment that could precondition de novo generated or circulating monocytes. In this setting, the circulating monocytes could thus be educated to be less responsive once recruited to the colon during DSS-induced colitis. To study this possibility, we combined a period of IL-10 overexpression with a resting period of 7 or 21 days prior to DSS administration (Figure 4A). As shown in Figure 1B, the levels of IL-10 return to basal ones as soon as 24 h after the zinc administration is suspended. Thus, after a 7-day rest, the circulating levels of IL-10 are normalized. Furthermore, we chose the 21 days time-point, because previous reports showing that resident intestinal macrophages have a life span of approximately 3 weeks (48). After 7-day rest period, the protection in DAI afforded by previous IL-10 exposure was partly lost (Figure 4B), being less pronounced than when no resting was performed and only observed at later time-points. After the 21-day resting period, the protection in DAI of pMT-10 previously overexpressing IL-10 was completely ablated (Figure 4B). Furthermore, independently of the resting period, no differences were observed between the experimental groups in what concerned colon length (Figure 4C) or histology (Figures 4D,E). Thus, we conclude that IL-10 overexpression, over a period of 8 days, does not confer long-lasting protection against intestinal inflammation.
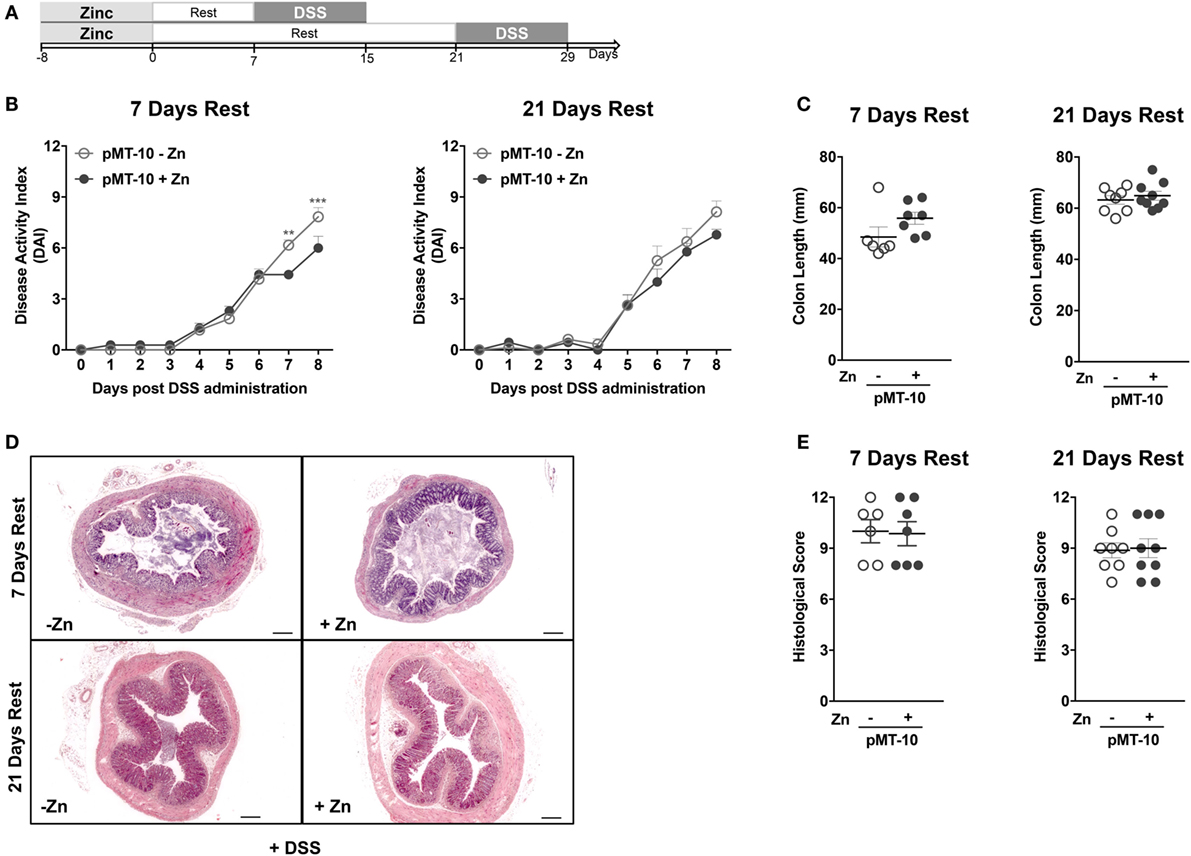
Figure 4. The interleukin-10 protection conferred against DSS-induced colitis is not long lasting. (A) pMT-10 mice were fed with control (pMT-10-Zn) or Zn-enriched (pMT-10 + Zn) water for 8 days, followed by a 7- or 21-day resting period where only normal water was available, and by 8 days of 3% DSS. (B) Disease progression based on Disease Activity Index (DAI) parameters was registered every day for 8 days. (C) Colon length measurement at day 8 of DSS administration. (D) Representative H&E-stained sections of large bowel at 40× magnification (scale bar = 200 µm). (E) Colitis scores derived from evaluation of colon and cecum from both groups. Each dot represents one independent animal, in two independent experiments; represented is also mean ± SEM. Data were analyzed with (B) two-way analysis of variance (Sidak’s multiple comparisons test) or (C,E) Student’s t-test.
Discussion
Despite the fact that IBD is a treatable condition, there are many limitations to the therapeutic approaches currently available (5). Major obstacles in this context are the heterogeneity of the disease, which implies that dosage and schedule may differ across disease conditions, and the requirement of a localized action of the therapeutic agent. In view of the strong immune component associated with disease development, it is not surprising that therapeutic manipulations of the immune response have been widely sought approaches to tackle IBD. Indeed, a commonly used therapy for IBD is the administration of anti-tumor necrosis factor alpha (TNF-α) antibodies (49). However, in line with the above mentioned limitations, up to one-third of IBD patients do not respond to this therapy, and those who respond eventually develop some degree of intolerance to the medication (50). In this context, several animal models of IBD, both spontaneous and experimentally induced (such as DSS), were developed to investigate the role of various factors on the pathogenesis of the disease and to evaluate the different therapeutic options. A molecule that has been widely studied in the context of IBD is IL-10. This cytokine keeps intestinal inflammation in check by exerting a direct effect on monocyte/macrophage populations (34). Thus, it is not surprising that IL-10-based therapies have been tested in IBD. However, both in human (51–54) and mouse models (38), administration of IL-10 did not significantly improve intestinal inflammation, perhaps in part due to the fact that administered IL-10 did not reach the inflamed tissue.
In this study, we report a novel transgenic mouse model of inducible IL-10 overexpression, the pMT-10 mice, in which high IL-10 transcription is observed in the intestine, skin and BM. Upon induction of the transgene, high levels of IL-10 are detected also in the serum. Taking advantage of this novel mouse model, we investigated the dynamics of IL-10 afforded protection during DSS-induced colitis. We found that induction of IL-10 prior to DSS administration impacted the progression of colitis. We show that a short period of IL-10 overexpression before the induction of colitis ameliorated the disease outcome, despite the presence of CD11b+ Ly6C+ cells in the gut, previously associated with the development of detrimental inflammation. However, in comparison to control animals that do not overexpress IL-10, Ly6C cells isolated from the lamina propria of colitic pMT-10 mice showed a decreased inflammatory profile. Thus, we hypothesize that IL-10 overexpression impairs the response of these cells to the insult, reaffirming both the critical role of these cells on intestinal inflammation (47) and that of IL-10 in regulating their inflammatory responses (34). In line with a previously described protective role for Zn in the context of intestinal inflammation (55, 56), we show some effect of Zn in reducing the severity of colitis, which occurred both later and to a lower extent than that observed for the combined condition Zn + IL-10. In addition, the protective effect of Zn failed to overcome the exacerbated colitis observed in mice that did not respond to endogenous IL-10. The mechanistic bases underlying the protection conferred by Zn alone remain unknown. One possibility is that Zn may contribute to diminish the amount of free radical species generated during acute colitis which contribute to protein, DNA chain and lipid damage (57). In any case, as IBD patients often present a Zn deficiency and respond well to Zn supplementation therapy (58), the exploitation of combined IL-10 and Zn therapies may be worth considering. In line with this, the benefits of combined Zn and anti-TNF therapy were previously described (55).
The pMT-10 mouse model allows for local, as well as systemic, IL-10 overexpression. The fact that we detected increased transcription of the IL-10 transgene in the BM and elevated levels of seric IL-10 led us to hypothesize that preexposure to IL-10 might induce long-lasting transcriptional changes in circulating monocytes, for example through epigenetic imprinting. If this were the case, we might be able to educate these cells to gain long-lasting tolerance to DSS-induced colitis. Our data obtained after a 1- or 3-week rest post-IL-10 exposure show that recent IL-10 exposure is required for maximal protection. Thus, the protective effects of IL-10 were not sustained over time, implying that IL-10 presence at the time of insult is necessary to prevent colitis. Therefore, inducing IL-10 expression in our mouse model at the beginning of disease would be of interest. Unfortunately, we were unable to explore this possibility because the Zn necessary to activate the transgene precipitates in the presence of DSS, when both are provided in the drinking water. In addition, we were unable to induce high levels of IL-10 expression in the serum of mice fed with a Zn-enriched diet and ethical issues prevented us from attempting to induce sustained high levels of IL-10 by frequent gavage or i.p. injections of Zn-containing preparations.
In conclusion, we herein present a novel mouse model of inducible IL-10 overexpression. We also show the potential of this model for the study of the IL-10 biology in the specific setting of DSS-induced colitis. Our data further support the protective role for IL-10 in intestinal inflammation, showing that this cytokine delays disease progression even when delivered before DSS administration. However, the effect is not long-lasting, which calls for alternative approaches to prevent IBD.
Ethics Statement
In Portugal, all animal experiments were performed in strict accordance with recommendations of the European Union Directive 2010/63/EU and previously approved by Portuguese National Authority for Animal Health–Direção Geral de Alimentação e Veterinária (DGAV). Mice were euthanized by CO2 inhalation with efforts to minimize suffering. In France, all animal procedures were approved by the Pasteur Institute Safety Committee and conducted according to French and European Community Institutional guidelines. Mice were euthanized by CO2 inhalation with efforts to minimize suffering.
Author Contributions
AC, AM, and GC performed the experiments. AGC and PV made the pMT-10 mice. VM sequenced the genome of the pMT-10 mouse. AC, AM, AGC, IC, AC, PV, and MS planned the experiments and analyzed data. AC, AGC, PV, and MS wrote the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Werner Müller (Faculty of Biology, Medicine and Health, University of Manchester, United Kingdom) for providing the IL-10R-deficient mice. We also thank the excellent support given by the animal house workers at IBMC-i3S.
Funding
We acknowledge the Portuguese Foundation for Science and Technology (FCT) for providing a PhD grant to AC (SFRH/BD/84704/2012). This article is a result of the project Norte-01-0145-FEDER-000012—Structured program on bioengineered therapies for infectious diseases and tissue regeneration, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (FEDER). The MS lab is also financed by a FCT-ANR grant (FCTANR/BIM-MEC/0007/2013). This work was also backed by the COST Action BM1404 European Network of Investigators Triggering Exploratory Research on Myeloid Regulatory Cells (http://www.mye-euniter.eu), which is supported by the Horizon 2020—EU Framework Program Research and Innovation Programme. MS is a FCT Associate Investigator. AGC lab: This work was developed under the scope of the project NORTE-01-0145-FEDER-000013, supported by the Northern Portugal Regional Operational Programme (NORTE 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (FEDER); by the project NORTE-01-0145-FEDER-000023, supported by the Northern Portugal Regional Operational Programme (NORTE 2020), under the Portugal 2020 Partnership Agreement, through FEDER; and by FEDER, through the Competitiveness Factors Operational Programme (COMPETE), and by National funds, through the Foundation for Science and Technology (FCT), under the scope of the project POCI-01-0145-FEDER-007038. PV is funded by ANR, through the project MYELOTEN (ANR-13-ISV1-0003-01).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/articles/10.3389/fimmu.2018.00400/full#supplementary-material.
References
1. Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest (2007) 117(3):514–21. doi:10.1172/JCI30587
2. Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology (2011) 140(6):1785–94. doi:10.1053/j.gastro.2011.01.055
3. Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology (2004) 126(6):1504–17. doi:10.1053/j.gastro.2004.01.063
4. Shivananda S, Lennard-Jones J, Logan R, Fear N, Price A, Carpenter L, et al. Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease (EC-IBD). Gut (1996) 39(5):690–7. doi:10.1136/gut.39.5.690
5. Schirbel A, Fiocchi C. Inflammatory bowel disease: established and evolving considerations on its etiopathogenesis and therapy. J Dig Dis (2010) 11(5):266–76. doi:10.1111/j.1751-2980.2010.00449.x
6. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature (2007) 448(7152):427–34. doi:10.1038/nature06005
7. Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol (2014) 14(10):667–85. doi:10.1038/nri3738
8. Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol (2013) 34(4):162–8. doi:10.1016/j.it.2013.02.001
9. Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol (2013) 6(3):498–510. doi:10.1038/mi.2012.89
10. Xu XR, Liu CQ, Feng BS, Liu ZJ. Dysregulation of mucosal immune response in pathogenesis of inflammatory bowel disease. World J Gastroenterol (2014) 20(12):3255–64. doi:10.3748/wjg.v20.i12.3255
11. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol (2014) 14(5):329–42. doi:10.1038/nri3661
12. Montgomery SM, Morris DL, Pounder RE, Wakefield AJ. Asian ethnic origin and the risk of inflammatory bowel disease. Eur J Gastroenterol Hepatol (1999) 11(5):543–6. doi:10.1097/00042737-199905000-00013
13. Carr I, Mayberry JF. The effects of migration on ulcerative colitis: a three-year prospective study among Europeans and first- and second-generation South Asians in Leicester (1991–1994). Am J Gastroenterol (1999) 94(10):2918–22. doi:10.1016/S0002-9270(99)00494-3
14. Helman J. Congenital familial acheiria associated with an interventricular septal defect and divarication of the upper abdominal rectus muscles. S Afr Med J (1979) 56(23):982.
15. Orholm M, Munkholm P, Langholz E, Nielsen OH, Sorensen TI, Binder V. Familial occurrence of inflammatory bowel disease. N Engl J Med (1991) 324(2):84–8. doi:10.1056/NEJM199101103240203
16. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474(7351):307–17. doi:10.1038/nature10209
17. Van Limbergen J, Radford-Smith G, Satsangi J. Advances in IBD genetics. Nat Rev Gastroenterol Hepatol (2014) 11(6):372–85. doi:10.1038/nrgastro.2014.27
18. Shih DQ, Targan SR, McGovern D. Recent advances in IBD pathogenesis: genetics and immunobiology. Curr Gastroenterol Rep (2008) 10(6):568–75. doi:10.1007/s11894-008-0104-x
19. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med (2009) 361(21):2033–45. doi:10.1056/NEJMoa0907206
20. Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity (2014) 40(5):706–19. doi:10.1016/j.immuni.2014.03.011
21. Aithal GP, Craggs A, Day CP, Welfare M, Daly AK, Mansfield JC, et al. Role of polymorphisms in the interleukin-10 gene in determining disease susceptibility and phenotype in inflamatory bowel disease. Dig Dis Sci (2001) 46(7):1520–5. doi:10.1023/A:1010604307776
22. Amre DK, Mack DR, Morgan K, Israel D, Lambrette P, Costea I, et al. Interleukin 10 (IL-10) gene variants and susceptibility for paediatric onset Crohn’s disease. Aliment Pharmacol Ther (2009) 29(9):1025–31. doi:10.1111/j.1365-2036.2009.03953.x
23. Wang AH, Lam WJ, Han DY, Ding Y, Hu R, Fraser AG, et al. The effect of IL-10 genetic variation and interleukin 10 serum levels on Crohn’s disease susceptibility in a New Zealand population. Hum Immunol (2011) 72(5):431–5. doi:10.1016/j.humimm.2011.02.014
24. Glocker EO, Frede N, Perro M, Sebire N, Elawad M, Shah N, et al. Infant colitis – it’s in the genes. Lancet (2010) 376(9748):1272. doi:10.1016/S0140-6736(10)61008-2
25. Reuss E, Fimmers R, Kruger A, Becker C, Rittner C, Hohler T. Differential regulation of interleukin-10 production by genetic and environmental factors – a twin study. Genes Immun (2002) 3(7):407–13. doi:10.1038/sj.gene.6363920
26. Braat H, Peppelenbosch MP, Hommes DW. Interleukin-10-based therapy for inflammatory bowel disease. Expert Opin Biol Ther (2003) 3(5):725–31. doi:10.1517/14712598.3.5.725
27. Melgar S, Yeung MM, Bas A, Forsberg G, Suhr O, Oberg A, et al. Over-expression of interleukin 10 in mucosal T cells of patients with active ulcerative colitis. Clin Exp Immunol (2003) 134(1):127–37. doi:10.1046/j.1365-2249.2003.02268.x
28. Meresse B, Rutgeerts P, Malchow H, Dubucquoi S, Dessaint JP, Cohard M, et al. Low ileal interleukin 10 concentrations are predictive of endoscopic recurrence in patients with Crohn’s disease. Gut (2002) 50(1):25–8. doi:10.1136/gut.50.1.25
29. Schreiber S, Heinig T, Thiele HG, Raedler A. Immunoregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology (1995) 108(5):1434–44. doi:10.1016/0016-5085(95)90692-4
30. Kucharzik T, Stoll R, Lugering N, Domschke W. Circulating antiinflammatory cytokine IL-10 in patients with inflammatory bowel disease (IBD). Clin Exp Immunol (1995) 100(3):452–6. doi:10.1111/j.1365-2249.1995.tb03721.x
31. Ljuca F, Gegic A, Salkic NN, Pavlovic-Calic N. Circulating cytokines reflect mucosal inflammatory status in patients with Crohn’s disease. Dig Dis Sci (2010) 55(8):2316–26. doi:10.1007/s10620-009-1016-9
32. Mitsuyama K, Tomiyasu N, Takaki K, Masuda J, Yamasaki H, Kuwaki K, et al. Interleukin-10 in the pathophysiology of inflammatory bowel disease: increased serum concentrations during the recovery phase. Mediators Inflamm (2006) 2006(6):26875. doi:10.1155/MI/2006/26875
33. Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell (1993) 75(2):263–74. doi:10.1016/0092-8674(93)80068-P
34. Zigmond E, Bernshtein B, Friedlander G, Walker CR, Yona S, Kim KW, et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity (2014) 40(5):720–33. doi:10.1016/j.immuni.2014.03.012
35. Nguyen HH, Tran BT, Muller W, Jack RS. IL-10 acts as a developmental switch guiding monocyte differentiation to macrophages during a murine peritoneal infection. J Immunol (2012) 189(6):3112–20. doi:10.4049/jimmunol.1200360
36. Shouval DS, Ouahed J, Biswas A, Goettel JA, Horwitz BH, Klein C, et al. Interleukin 10 receptor signaling: master regulator of intestinal mucosal homeostasis in mice and humans. Adv Immunol (2014) 122:177–210. doi:10.1016/B978-0-12-800267-4.00005-5
37. Marlow GJ, van Gent D, Ferguson LR. Why interleukin-10 supplementation does not work in Crohn’s disease patients. World J Gastroenterol (2013) 19(25):3931–41. doi:10.3748/wjg.v19.i25.3931
38. Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest (1996) 98(4):1010–20. doi:10.1172/JCI118861
39. Pils MC, Pisano F, Fasnacht N, Heinrich JM, Groebe L, Schippers A, et al. Monocytes/macrophages and/or neutrophils are the target of IL-10 in the LPS endotoxemia model. Eur J Immunol (2010) 40(2):443–8. doi:10.1002/eji.200939592
40. De Fazio L, Cavazza E, Spisni E, Strillacci A, Centanni M, Candela M, et al. Longitudinal analysis of inflammation and microbiota dynamics in a model of mild chronic dextran sulfate sodium-induced colitis in mice. World J Gastroenterol (2014) 20(8):2051–61. doi:10.3748/wjg.v20.i8.2051
41. Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity (2008) 28(4):559–70. doi:10.1016/j.immuni.2008.02.019
42. Stzepourginski I, Eberl G, Peduto L. An optimized protocol for isolating lymphoid stromal cells from the intestinal lamina propria. J Immunol Methods (2015) 421:14–9. doi:10.1016/j.jim.2014.11.013
43. Uhlig HH, Coombes J, Mottet C, Izcue A, Thompson C, Fanger A, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J Immunol (2006) 177(9):5852–60. doi:10.4049/jimmunol.177.9.5852
44. Mesquita AR, Correia-Neves M, Roque S, Castro AG, Vieira P, Pedrosa J, et al. IL-10 modulates depressive-like behavior. J Psychiatr Res (2008) 43(2):89–97. doi:10.1016/j.jpsychires.2008.02.004
45. Engelhardt KR, Grimbacher B. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol (2014) 380:1–18. doi:10.1007/978-3-662-43492-5_1
46. Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol (2012) 2012:718617. doi:10.1155/2012/718617
47. Farache J, Zigmond E, Shakhar G, Jung S. Contributions of dendritic cells and macrophages to intestinal homeostasis and immune defense. Immunol Cell Biol (2013) 91(3):232–9. doi:10.1038/icb.2012.79
48. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi:10.1016/j.immuni.2012.12.001
49. Melmed GY, Targan SR. Future biologic targets for IBD: potentials and pitfalls. Nat Rev Gastroenterol Hepatol (2010) 7(2):110–7. doi:10.1038/nrgastro.2009.218
50. Peyrin-Biroulet L, Desreumaux P, Sandborn WJ, Colombel JF. Crohn’s disease: beyond antagonists of tumour necrosis factor. Lancet (2008) 372(9632):67–81. doi:10.1016/S0140-6736(08)60995-2
51. Colombel JF, Rutgeerts P, Malchow H, Jacyna M, Nielsen OH, Rask-Madsen J, et al. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut (2001) 49(1):42–6. doi:10.1136/gut.49.1.42
52. Fedorak RN, Gangl A, Elson CO, Rutgeerts P, Schreiber S, Wild G, et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology (2000) 119(6):1473–82. doi:10.1053/gast.2000.20229
53. Schreiber S, Fedorak RN, Nielsen OH, Wild G, Williams CN, Nikolaus S, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology (2000) 119(6):1461–72. doi:10.1053/gast.2000.20196
54. van Deventer SJ, Elson CO, Fedorak RN. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Crohn’s Disease Study Group. Gastroenterology (1997) 113(2):383–9. doi:10.1053/gast.1997.v113.pm9247454
55. Barollo M, Medici V, D’Inca R, Banerjee A, Ingravallo G, Scarpa M, et al. Antioxidative potential of a combined therapy of anti TNFalpha and Zn acetate in experimental colitis. World J Gastroenterol (2011) 17(36):4099–103. doi:10.3748/wjg.v17.i36.4099
56. Chen BW, Wang HH, Liu JX, Liu XG. Zinc sulphate solution enema decreases inflammation in experimental colitis in rats. J Gastroenterol Hepatol (1999) 14(11):1088–92. doi:10.1046/j.1440-1746.1999.02013.x
57. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol (2007) 39(1):44–84. doi:10.1016/j.biocel.2006.07.001
Keywords: interleukin-10, macrophages, inflammation, colitis, therapy
Citation: Cardoso A, Gil Castro A, Martins AC, Carriche GM, Murigneux V, Castro I, Cumano A, Vieira P and Saraiva M (2018) The Dynamics of Interleukin-10-Afforded Protection during Dextran Sulfate Sodium-Induced Colitis. Front. Immunol. 9:400. doi: 10.3389/fimmu.2018.00400
Received: 11 August 2017; Accepted: 13 February 2018;
Published: 01 March 2018
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases, United StatesReviewed by:
Robson Coutinho-Silva, Universidade Federal do Rio de Janeiro, BrazilCinzia Fionda, Sapienza Università di Roma, Italy
Copyright: © 2018 Cardoso, Gil Castro, Martins, Carriche, Murigneux, Castro, Cumano, Vieira and Saraiva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margarida Saraiva, bWFyZ2FyaWRhLnNhcmFpdmFAaWJtYy51cC5wdA==
†Present address: Guilhermina M. Carriche, Institute of Infection Immunology, TWINCORE, Centre for Experimental and Clinical Infection Research, a Joint Venture between the Medical School Hannover (MHH) and the Helmholtz Centre for Infection Research (HZI), Hannover, Germany;
Valentine Murigneux, UQ Diamantina Institute, Translational Research Institute, University of Queensland, Brisbane, QLD, Australia
‡These authors have contributed equally for this work.
§Co-senior authors.