 Linda Ziani1,2,3
Linda Ziani1,2,3 Salem Chouaib
Salem Chouaib Jerome Thiery
Jerome Thiery- 1INSERM, UMR 1186, Villejuif, France
- 2Gustave Roussy Cancer Campus, Villejuif, France
- 3Faculty of Medicine, University Paris Sud, Le Kremlin Bicêtre, France
Among cells present in the tumor microenvironment, activated fibroblasts termed cancer-associated fibroblasts (CAFs), play a critical role in the complex process of tumor-stroma interaction. CAFs, one of the prominent stromal cell populations in most types of human carcinomas, have been involved in tumor growth, angiogenesis, cancer stemness, extracellular matrix remodeling, tissue invasion, metastasis, and even chemoresistance. During the past decade, these activated tumor-associated fibroblasts have also been involved in the modulation of the anti-tumor immune response on various levels. In this review, we describe our current understanding of how CAFs accomplish this task as well as their potential therapeutic implications.
Introduction
It is now well admitted that tumor progression and metastasis formation do not only depend on cancer cell genetic and epigenetic defects but are also controlled by the tumor microenvironment (TME) (1, 2). The TME or stroma is composed of cells from endothelial, mesenchymal, and hematopoietic origins embedded in a complex extracellular matrix (ECM), which enter into a dynamic crosstalk with tumor cells, suitable for tumor growth. Consequently, different elements such as angiogenesis, hypoxia, ECM remodeling, interstitial pressure, metabolism changes have received recent attention as key determinants of the TME modifying cancer cell behavior and disease progression, with potential clinical applications (2, 3). Moreover, the TME is also clearly involved in shaping the cellular fate of tumor-infiltrating lymphocytes and the efficacy of the anti-tumor immune response. Indeed, during tumor progression, tumor cells proliferate under adverse host conditions and use several survival strategies to block the action of key regulators/effectors of the immune response and to circumvent anti-tumor defenses (4–6). Besides the several known classical strategies used by tumor cells to escape immune surveillance (such as down regulation of antigen expression, resistance to cell-mediated lysis or expression/secretion of immunosuppressive molecules), it should be noted that tumor cell evasion from immunosurveillance is also under the control of the TME complexity (7–9). The ability of tumors to orchestrate an immunosuppressive microenvironment is dependent on several mechanisms ultimately leading to the inhibition of various immune effector cells [such as cytotoxic T cell (CTL) or natural killer (NK) cells] or to the recruitment and stimulation in the TME of immunosuppressive cells [such as regulatory T cells (Tregs), type II macrophages or myeloid-derived suppressor cells (MDSCs)]. In particular, among the stromal cells, activated fibroblasts that share similarities with fibroblasts stimulated by acute or chronic inflammatory signals, activated during a wound healing process and observed during tissue fibrosis, also known as myofibroblasts, play a critical role in the complex process of tumor cell-stroma interaction (10–13) and have emerged as important regulators of the anti-tumor immune response (14–16). Here, we will discuss the different mechanisms involved in the immuno-suppressive capabilities of activated fibroblasts in the TME, as well as their potential application for therapeutic intervention, especially in the field of cancer immunotherapy.
Origin of Activated Fibroblasts in the TME and Role in Cancer Progression
Fibroblasts are spindle-shaped, non epithelial (cytokeratin−, E-cadherin−), non endothelial (CD31−) and non-immune (CD45−) cells of a mesenchymal lineage origin (vimentin+). In normal tissue, fibroblasts are usually considered as resting/quiescent cells with negligible metabolic and transcriptional activities (11), but with the ability to respond to growth factors to become activated. During this activation process, fibroblasts exhibit contractile activity, exert physical forces to modify tissue architecture, acquire proliferation and migration properties and become transcriptionally active leading to the secretion of several factors (cytokines, chemokines, etc.) and ECM components (17–19). The ability of resting fibroblasts to become activated was first observed in the context of wound healing (20) and subsequently in pathologic conditions such as acute or chronic inflammation or tissue fibrosis (a chronic wound healing response) (17, 21). This chronic tissue repair response also occurs in the context of cancer, considered as a “wound that never heals” (22). Indeed, emergence and/or accumulation of cancer cells in a given tissue represent a tissue injury, imitating a chronic wound healing response toward the tumor cells, also known as tumor fibrosis or desmoplastic reaction (23). Consequently, major players in tumor fibrotic microenvironment include activated fibroblasts, termed cancer-associated fibroblasts (CAFs), which represent one of the most abundant stromal cell types of several carcinomas including breast, prostate, pancreatic, esophageal, and colon cancers while CAFs are less abundant, but still present, in other neoplasias including ovarian, melanoma, or renal tumors (24). For example, in pancreatic cancer, 60–70% of the tumor tissue is composed of a desmoplastic stroma characterized by extensive collagen deposition and activated CAFs (25).
Several studies have clearly demonstrated that cancer cells can recruit and activate tissue resident fibroblasts in the stroma (26, 27). This phenomenon is mainly dependent on growth factors released by the cancer cells and also by infiltrating immune cells. In particular, transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF) secreted by tumor cells are key determinants of fibroblast activation and proliferation within the TME (28–31). Moreover, the secretion of interleukin (IL)-1β (IL-1β) by immune cells in early neoplasia has emerged as an initiator of nuclear factor-κB signaling in fibroblasts involved in their education and production of pro-tumorogenic and pro-inflammatory factors (32). Furthermore, emerging data suggest that the irreversible activation of CAFs might be driven by epigenetic alterations (33–36). Nevertheless, CAFs can also originate from other cell populations than resident fibroblasts through different mechanisms and depending on the tissue analyzed. Several other local sources of CAFs have been thus suggested. In breast, kidney, lung, and liver carcinomas, a portion of CAFs have been shown to potentially differentiate from epithelial cells via an epithelial-to-mesenchymal transition (EMT) (37, 38). A related process, termed endothelial-to-mesenchymal transition has been involved in the trans-differentiation of endothelial cells to a cell population with a CAF-like phenotype (39). Other cells linked to blood vessels, named pericytes, can trans-differentiate into CAFs in a PDGF-dependent manner (40). Moreover, in breast cancer, adipocytes were shown to differentiate in CAFs (41, 42). Finally, in liver and pancreas tumors, stellate cells, normally involved in organ regeneration, are involved in fibrosis preceding the occurrence of tumors, making them a possible source of CAFs (43, 44). Beyond these local sources, more distant one can be involved in CAFs recruitment/differentiation in the TME. In particular, mesenchymal stem cells, normally residing in the bone marrow, can be attracted in the TME to become an important source of CAFs (42, 45–48). Similarly, fibrocytes, a circulating mesenchymal cell population arising from monocytes precursors which are recruited to sites of chronic inflammation, can differentiate into CAFs after their recruitment into the TME (46, 49).
These various sources represent an important determinant that contributes to the heterogeneity of CAFs (Figure 1) and makes them difficult to distinguish from other cell types present in TME. In this context, morphology and spatial distribution are key determinants in order to identify fibroblasts in a resting or activated state (11). Different markers, which are lower or not expressed by their normal counterparts, can also be used to identify activated fibroblasts such as α-smooth muscle actin (α-SMA), fibroblast-specific protein-1 (FSP-1; also called S100A4), fibroblast-activation protein (FAP), PDGF receptors (PDGFR) α or β, neuron-glial antigen-2 (NG2), periostin (POSTN), podoplanin (PDPN), tenascin-C (TNC), desmin, CD90/THY1, or discoidin domain-containing receptor 2 (DDR2) (24, 50–57). However, it is crucial to note that none of these markers is specific for normal or activated fibroblasts, and that many activated fibroblasts may not express all of these markers at the same time, most likely reflecting the high degree of heterogeneity of CAFs in the TME, as well as possible different and opposite functions in the context of specific TMEs (24). It is indeed conceivable that, depending of the context, quiescent fibroblasts or the other cell types mentioned above might be capable of differentiating into distinct subsets of functional CAFs, with possible diverse functions, either pro- or anti-tumorigenic, as observed for type I and type II macrophages (11, 58). In other words, even if a large body of literature currently supports the tumor-promoting effect of CAFs, some evidence also suggests that CAFs might also restrain tumor growth. For example, the depletion of α-SMA+ CAFs in pancreatic cancer accelerates tumor growth, induces immunosuppression by increasing the number of CD4+Foxp3+ Tregs in tumors and reduces survival (59). Similarly, the deletion of sonic hedgehog, a soluble ligand overexpressed by neoplastic cells in pancreatic ductal adenocarcinoma which drives the formation of a fibroblast-rich desmoplastic stroma, increases the aggressiveness of tumors (60). Nevertheless, for simplicity, we will focus the following part of this review on the tumor-promoting and immunosuppressive capabilities of CAFs, unless otherwise stated.
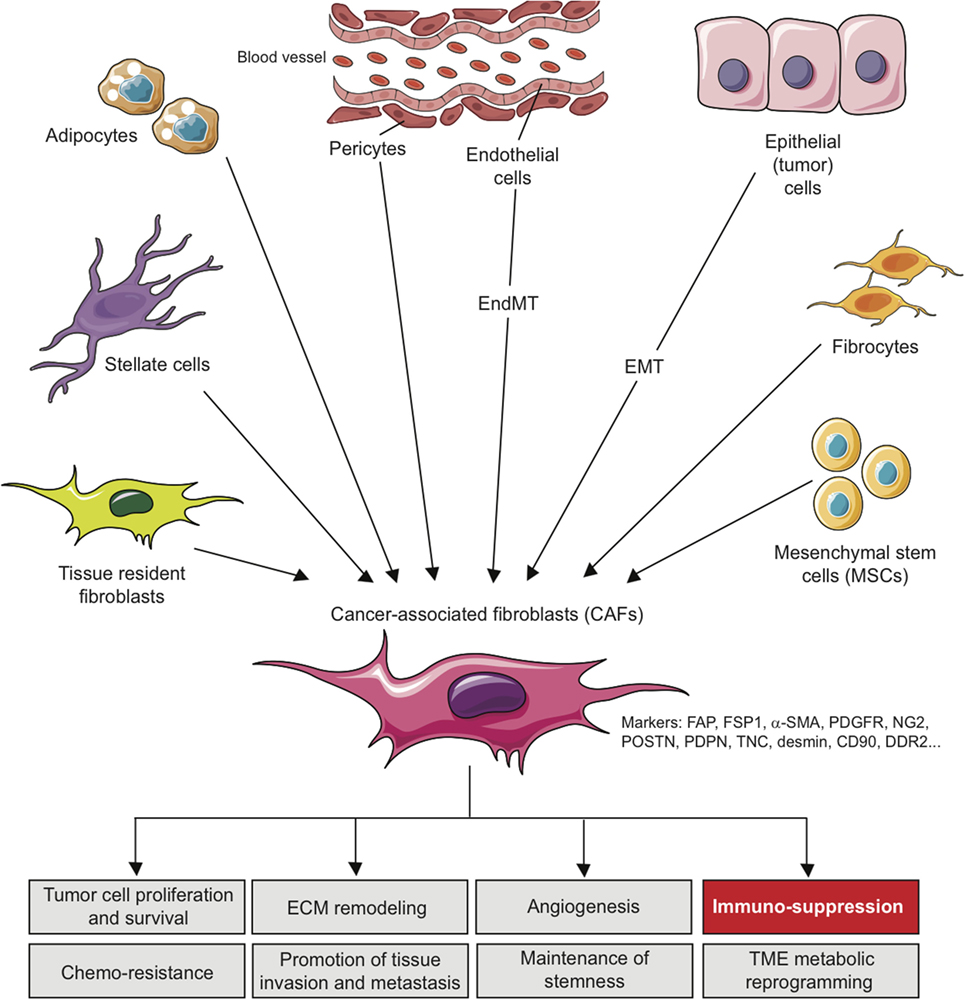
Figure 1. Origins of cancer-associated fibroblasts in the tumor microenvironment (TME) and role in cancer progression. CAFs can originate from diverse cell populations through different mechanisms and depending on the tissue analyzed. Local sources of CAFs include activated tissue resident fibroblasts, trans-differentiated epithelial or endothelial cells resulting from an epithelial-to-mesenchymal transition (EMT) or an endothelial-to-mesenchymal transition (EndMT), trans-differentiated pericytes, adipocytes or stellate cells. Beyond those local sources, more distant one can be involved in CAFs recruitment/differentiation in the TME, including mesenchymal stem cells, normally residing in the bone marrow, and fibrocytes. The acquisition of a CAF phenotype is associated with the potential expression of a variety of CAF-related markers as indicated. In the TME, CAFs can affect several processes leading to tumor growth, as indicated, including immuno-suppression.
In the tumor stroma, CAFs interact with tumor cells and other cell types and as a sign of their activation secrete several factors such as ECM proteins (e.g., collagens), ECM-remodeling enzymes such matrix metallo-proteinases (MMPs), proteoglycans (e.g., laminin, fibronectin), chemokines [e.g., C-X-C motif chemokine ligand 2 (CXCL2), CXCL12/SDF1, chemokine ligand 2 (CCL2/MCP-1), and CCL5/Rantes], vascularization promoting factors [e.g., vascular endothelial growth factor (VEGF)] and other factors/proteins which affect tumor cells proliferation, invasiveness, survival, cancer cell metabolism, and stemness [e.g., TGF-β, EGF, FGF, hepatocyte growth factor (HGF)]. Consequently, CAFs have been involved in tumor growth, cancer cell survival, angiogenesis, maintenance of cancer stemness, ECM remodeling, tissue invasion, metastasis, metabolic reprograming of the TME and even chemoresistance [see Ref. (10–13, 24, 61) for review] (Figure 1). During the past few years, these activated tumor-associated fibroblasts have also been involved in the modulation of the anti-tumor immune response by the secretion of immunosuppressive and pro-inflammatory factors, chemokines, and chemical mediators in the TME. As such, CAFs can potentially affect both innate and adaptive antitumor immune response and consequently tumor progression.
CAF-Mediated Regulation of the Innate Anti-Tumor Immune Response
As mentioned above, several studies including gene signature or mass spectrometry analysis (62–66) have shown that CAFs exhibit a particular immunomodulatory secretome including, but not limited to, CXCL1, CXCL2, CXCL5, CXCL6/GCP-2, CXCL8, CXCL9, CXCL10, CXCL12/SDF1, CCL2/MCP-1, CCL3, CCL5/Rantes, CCL7, CCL20, CCL26, IL-1β, IL-6, IL-10, VEGF, TGF-β, indoleamine-2,3-dioxygenase (IDO), prostaglandin (PG) E2 (PGE2), tumor necrosis factor (TNF) or nitric oxide (NO). This secretion profile is thought to be a major player in shaping the TME, with multiple roles in tumor progression, but beyond its role on tumor cells, this CAFs-related secretome can potentially regulate the innate immune response in several ways (Figure 2).
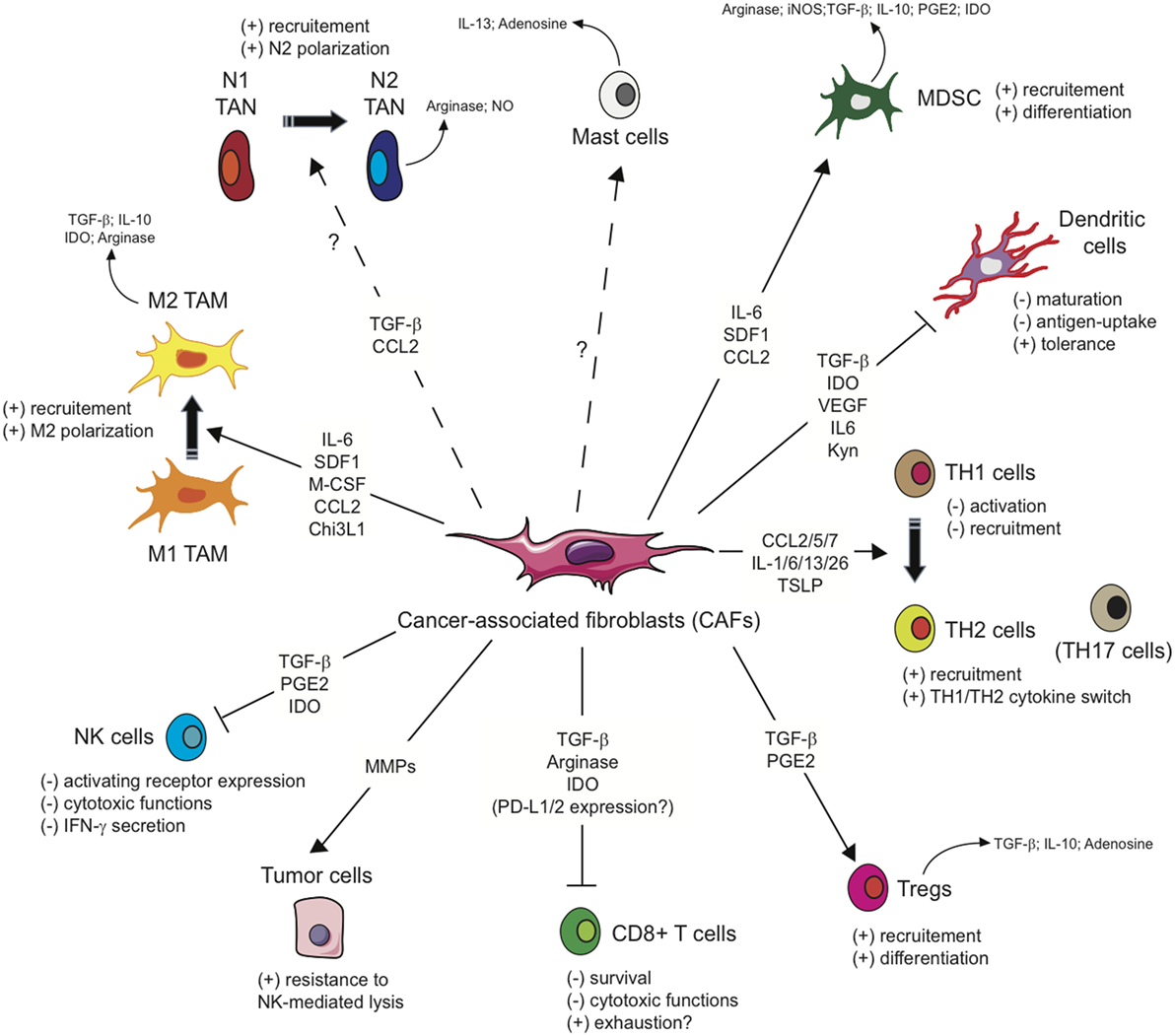
Figure 2. Influence of cancer-associated fibroblasts on the regulation and function of immune cells involved in the antitumor immune response. Due to their secretion of the indicated cytokines, chemokines, or other soluble factors, cancer-associated fibroblasts (CAFs) shape the tumor microenvironment and influence both the innate and adaptive anti-tumor immune response. CAFs favor the recruitment of innate immune cells, such as tumor-associated macrophages (TAM) or potentially tumor-associated neutrophils (TAN), and their acquisition of an immunosuppressive phenotype (M2 and N2, respectively), affect cytotoxic function and cytokine production of natural killer (NK) cells, as well as the susceptibility of tumor cells to NK-mediated lysis, and activate mast cells with a potential immunosuppressive phenotype. CAFs favor the recruitment and differentiation of myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) and interfere with the maturation and function or dendritic cells. CAFs have also the potential ability to influence CD4+ Helper T (TH) lymphocytes, favoring tumor-promoting TH2 and TH17 responses, and reduce the activation, functions, and survival of CD8+ cytotoxic T cells.
In particular, CAFs are important players affecting another major stromal component within tumors, known as tumor-associated macrophages (TAMs) (67). Macrophages are mainly classified into two distinct types: “classically” activated (M1 or type I) and “alternatively” activated (M2 or type II) macrophages. M1 macrophages produce high amounts of pro-inflammatory cytokines and reactive oxygen species and have the capacity to orchestrate a TH1 anti-tumor immune response. On the opposite, M2 macrophages play a significant role in tumor progression, promote tissue repair and angiogenesis, and are characterized by the production of immuno-suppressive factors such as IL10, Arginase, IDO and TGF-β, which inhibit cytotoxic CD8+ T cell-mediated immune response in the TME (67). At least in some settings, CAFs actively promote the recruitment of monocytes to the TME and their differentiation toward M2 macrophages (68). In particular, the secretion of CXCL12/SDF1, macrophage colony-stimulating factor (M-CSF also known as CSF-1), IL-6, and CCL2/MCP-1 by CAFs actively promotes the recruitment of monocytes to the TME and their differentiation into a M2 immunosuppressive phenotype (69–74). It was also recently shown that Chitinase-3-like-1 (Chi3L1; YKL-40 in humans), a secreted glycoprotein involved in several diseases including chronic inflammatory conditions, fibrotic disorders and various types of cancer, is highly expressed in CAFs isolated from mammary tumors and pulmonary metastases in mice, and in the stromal compartment of human breast carcinomas, and enhances macrophage migration in the TME and their expression of an M2-like gene signature (75). Finally, the expression of both CAF (α-SMA+, FSP1+, and FAP+) and M2 macrophages (CD163+ and DC-SIGN+) markers is associated with the poor clinical outcome of colorectal cancer and oral squamous cell carcinoma patients (76, 77), suggesting an association between these two cell types.
Cancer-associated fibroblasts are also potentially involved in the recruitment of neutrophils into the TME, notably through the secretion of CXCL1, CXCL2, CXCL5, CXCL6, CXCL8, and CCL2. Tumor-associated neutrophils (TANs) have been linked to a poorer prognosis for patients with renal and pancreatic cancer; gastric, hepatocellular, colorectal, head and neck carcinomas, and melanoma (78). TAN-derived factors promote tumor cell proliferation, migration, and invasion, and also induce tumor vascularization by the production of pro-angiogenic factors. Moreover, the production of Arginase 1 (Arg 1) and NO by TANs in response to CXCL8 signaling has been linked to the inhibition of T cell functions (79, 80). Nevertheless, recent studies have suggested that TANs can be polarized to an N1 anti-tumoral or N2 pro-tumoral phenotype in the TME, as observed for TAMs. N1 neutrophils are induced upon TGF-β blockade and express immuno-activating cytokines and chemokines, low levels of Arg 1, and are able to kill cancer cells. On the opposite, N2 neutrophils are characterized by expression of CXCR4, VEGF, and MMP9 and are induced following exposure to high TGF-β levels (81) and inhibit CD8+ T cell function by several mechanisms (82). At this point, it is thus uncertain whether CAFs can recruit TANs and drive them to an N2 phenotype in the TME, and whether this recruitment/polarization of TANs participates to the immuno-suppressive activity of CAFs.
Another cell population has also been implicated in the complex CAFs-TME interaction. Mast cells, derived from CD34+/CD117+ pluripotent hematopoietic stem cells, are tissue resident sentinel cells that, upon activation, release a wide spectrum of chemokines and cytokines (83). Interestingly, it was demonstrated in pancreatic tumors that a complex interaction between mast cells and stellate cells (often described as CAF precursors) is able to activate mast cells, which in turn enhance CAF proliferation by their secretion of IL-13 and tryptase, favoring tumor growth (84). Of note, activated mast cells could not only increase tumor progression but might also alter the anti-tumor immune response. For example the release of free adenosine (85) or IL-13 by mast cells might, respectively, inhibit T cell function and promote M2 polarization (83, 86, 87). Mast cells can also promote the generation of highly suppressive MDSCs and Tregs in the TME (88, 89). However, whether CAF-mast cell interactions are linked to the immuno-suppressive capabilities of CAFs is also not clearly established and requires further investigations.
Finally, CAFs can also affect the activity of major innate effector cells, NK cells, which participate to the early immune response through their cytotoxic activity and contribute to the adaptive immune response by the secretion of cytokines and by the promotion of antigen-presenting cell maturation. As previously mentioned, CAFs are thought to be an important source of TGF-β in the TME (90, 91). TGF-β has been involved in the decrease of NK cell activation and cytotoxic activity (92). In this regard, TGF-β-induced miR-183 inhibits DAP12 transcription (a key accessory protein for relaying signals by NK cell receptors) and reduces the expression of the NK-activating receptor NKp30 and NK Group 2D (NKG2D) (93–95), resulting in a weak NK cell cytotoxic activity in the TME. TGF-β also reduces IFN-γ secretion by NK cells, which is important for stimulating effector CD4+ TH1 cells that are required for clearing tumors, notably by repressing T-bet expression through Smad 3 (96–98). Moreover, studies involving melanoma, hepatocellular, and colorectal carcinoma-derived fibroblasts have shown that CAFs can decrease the expression of several NK activating receptors (including NKp30, NKp44, and NKG2D) on the NK cell surface, as well as perforin and granzyme B expression, through the secretion of PGE2 and/or IDO (99–101) leading to an attenuated cytotoxic activity of NK cells against their tumor target cells. We also recently demonstrated that CAFs isolated from melanoma decrease the susceptibility of melanoma tumor cells to NK cell-mediated lysis through the secretion of active MMPs which cleave two ligands of the NK-activating receptor NKG2D, MHC class I-related chain (MIC)-A and MIC-B, at the surface of the tumor cells and consequently decrease the NKG2D-dependent cytotoxic activity of NK cells against melanoma tumor cells, as well as their secretion of IFN-γ (102).
In conclusion, due to their secretion of cytokines, chemokines, or other soluble factors, CAFs shape the TME and favor the recruitment of innate immune cells, such as monocytes or neutrophils, and their acquisition of an immunosuppressive phenotype, but also affect cytotoxic function and cytokine production of NK cells.
CAF-Mediated Regulation of the Adaptive Anti-Tumor Immune Response
Based on the immunomodulatory secretome mentioned above, CAFs might also interfere with the adaptive anti-tumor immune response at different levels, leading to a disruption of T cell function in the TME (Figure 2).
In the TME, dendritic cells (DCs), the most important antigen-presenting cell population, have a pivotal role for the activation of T cell-mediated anti-tumor immunity (103). DC biology can potentially be affected by the CAF secretome in several ways. In particular, CAF-derived TGF-β can affect DC function (96). In response to TGF-β, DCs downregulate the expression of MHC class II molecules and of the co-stimulatory molecules CD40, CD80, and CD86, which are necessary for efficient antigen presentation, and of TNF-α, IFN-γ, and IL-12, that promote T cell recruitment and survival. The resulting immature or tolerogenic DCs alter CD8+ cytotoxic T cell activation and the TH1 polarization of CD4+ helper T (TH) cell populations and also promote the formation of CD4+FoxP3+ Treg cells that potently inhibit the function of other T cells (104, 105). CAFs can also secrete IL-6 and could affect DC functions through this way. Indeed, IL-6-mediated activation of the STAT3 pathway has been involved in the alteration of the DC maturation, disabling T cell activation and inducing T cell anergy and immune tolerance (106–108). Fibroblast-produced IL-6 was also reported to favor the emergence of TAMs from monocytes at the expense of DCs (69). Expression of tryptophan 2,3-dioxygenase (TDO2) by CAFs isolated from lung cancer also promotes tryptophan degradation in kynurenines (Kyn) that inhibits DCs differentiation and functions (109). Finally, CAF-derived VEGF, in addition to its pro-angiogenic effect, has multiple immunoregulatory roles (110). In particular, VEGF inhibits DC generation and maturation (111–114), notably by reducing their MHC class II expression and their ability to take up antigens.
The role of CAFs in regulating T cell activity and function in the TME has also been suggested by several studies. As mentioned earlier, CAFs can be an important source of TGF-β in the TME, which may act on both CD8+ and CD4+ T cells (96, 105). For example, TGF-β promotes cell death of effector CD8+ T cells by inhibiting expression of the pro-survival protein Bcl-2 (115). TGF-β also directly alters cytotoxic CD8+ T cell function by inhibiting the expression of key genes involved in their cytototoxic activity, including perforin, granzymes A and B, Fas ligand, and IFN-γ (116, 117). Furthermore, CAFs could also impair T cell proliferation and effector function through other mechanisms (118), notably depending on their production of metabolic reprogramming factors. The secretion by CAFs of IDO1 (119, 120), an immuno-regulatory enzyme, might contribute to immuno-suppression, tolerance, and tumor escape by catabolizing tryptophan degradation into kynurenines (Kyn), creating an immunosuppressive TME resulting in T-cell anergy and apoptosis through depletion of tryptophan and accumulation of immunosuppressive tryptophan catabolites (121, 122). Similarly, the secretion by CAFs of Arginase 2 (Arg 2), an enzyme metabolizing l-Arginine to l-Ornithine and urea, might participate to the deprivation of Arginine in the TME, which is in normal conditions important for T cell proliferation and functions (123). In this regard, pancreatic cancer suffering patients with CAFs expressing high levels of Arg 2, especially in hypoxia-inducible factor (HIF)-1α positive hypoxic zones, demonstrate a poor clinical outcome (124). CAFs can also secrete galectins, a class of carbohydrate binding proteins that have a high affinity for β galactosides (125, 126), which possess immunoregulatory properties (127) such as, for Galectin-1, induction of apoptosis of activated T cells by binding the glycoprotein receptors CD7, CD43, and CD45 on the cell surface (128, 129). Finally, the secretion of CXCL12/SDF-1 by CAFs from lung and pancreatic tumors can contribute to the exclusion of T cells from the cancer cell proximity (130).
Cancer-associated fibroblasts have also the potential ability to influence CD4+ Helper T (TH) lymphocytes, switching them from anti-tumor to pro-tumor cells. CD4+ TH cells can differentiate into multiple sublineages with different functions and cytokine secretion profiles, which in turn can induce, maintain or regulate antitumor immune responses (131). Schematically, naïve CD4+ T cells can differentiate into TH1 cells mainly secreting IFN-γ and promoting CD8+ T cell-dependent immune response, or into TH2 cells mainly secreting IL-4 and orchestrating humoral immunity. In terms of antitumor immune responses, the superior effects of TH1 cells are thought to be the result of the production of large amounts of IFN-γ, as well as chemokines, which enhance the priming and expansion of antitumor CD8+ cells and help to recruit NK cells and type I macrophages to tumor sites. A third major effector population of CD4+ T cells that could be derived from naïve CD4+ T cells was also shown to exist. These cells, designated TH17 cells (132, 133), are characterized by the production of IL-17 and IL-22 and might have, at least under some circumstances, pro-tumor and immunosuppressive functions in the TME (134), even if this particular point remains highly controversial. Finally, under tolerogenic conditions, naïve CD4+ T cell precursors can differentiate into inducible Tregs that upregulate the expression of the FoxP3 transcription factor (135). Depending on the tumor type, Tregs can be highly enriched in the TME, limiting antitumor immune responses and promoting immunological ignorance of cancer cells, especially through the secretion of immunosuppressive cytokines (TGF-β, IL-10…) (136). In the TME, the presence of CAFs and their secretion of CCL2, CCL5, and CCL17 as well as the polarizing cytokines IL-1, IL-6, IL-13, and IL-26 can favor a tumor promoting TH2 and TH17 immune response, as the expense of tumor protective TH1 response (32, 137–139). For example, in a murine model of breast tumor, the elimination of CAFs in vivo by a DNA vaccine targeting FAP resulted in a shift of the immune TME from a TH2 to a TH1 polarization. This shift was characterized by an increased expression of IL-2 and IL-7, an increased of CD8+ T cell population, and a diminished recruitment of TAM, MDSC, and Tregs (139). Moreover, in pancreatic cancer, the secretion of thymic stromal lymphopoietin (TSLP) by CAFs has been associated with a TH2 cell polarization through myeloid DC conditioning (140). As a main source of TGF-β in the TME, CAFs can also promote Tregs recruitment and differentiation (141). Of note, it has been suggested that CAFs and Tregs enter to a cross-talk via their reciprocal expression of TGF-β, increasing both CAFs activation and Tregs activity. In this regard, FoxP3+ Tregs coexisting with CAFs are correlated with a poor outcome in lung adenocarcinoma (142). Moreover, it was shown that the expression of cyclo-oxygenase-2 (COX-2) by CAFs in lung or pancreatic cancers leads to their secretion of PGE-2, which plays an essential role in Tregs functionality by inducing FoxP3 expression (143, 144).
Cancer-associated fibroblasts in the TME can also interfere with the T cell-dependent immune response by modulating MDSCs. MDSCs are a heterogeneous population of immature myeloid cells that accumulate during pathologic conditions, such as cancer (145, 146). The main factors involved in MDSC-mediated immune suppression include the secretion of Arginase, iNOS, TGF-β, IL-10, PGE2 and IDO, regulating DC and T cell functions, as well as NK cells and macrophages. It has been demonstrated that CAFs isolated from pancreatic tumors drive monocyte precursors toward an MDSC phenotype, in a STAT3-dependent manner, through their secretion of IL-6 (72, 147). Similarly, CAFs from hepatic carcinomas attract monocytes to the TME by their secretion of CXCL12/SDF1 and induce their differentiation into MDSCs through IL-6-mediated STAT3 activation (148), thus altering T cell proliferation and functions, as well as the patients overall survival. Pancreatic stellate cells (described as CAFs precursors) also produce MDSC-promoting cytokines (IL-6, VEGF, M-CSF) and chemokines (CXCL12/SDF1, CCL2/MCP-1) and similarly promote differentiation of MDSCs in a STAT3-dependent manner (72). In a murine liver tumor model, it was also shown that FAP+ CAFs are a major source of CCL2 and that fibroblastic STAT3-CCL2 signaling promotes tumor growth by enhancing the recruitment of MDSCs, which also predicts poor prognosis of patients with intrahepatic cholangiocarcinoma (149).
Finally, an interesting but still controversial point was recently raised based on the observation that CAFs from colon and lung cancers or from melanoma might express programmed death-ligand-1 (PD-L1) and/or PD-L2 (150–152). PD-L1 and PD-L2 are members of the B7 family of co-stimulatory/co-inhibitory molecules expressed by a wide range of cancer cells and engage their receptor programmed death receptor 1 (PD1) expressed on T-cells, strongly counteracting TCR signaling and CD28-co-stimulation (153), resulting in the inhibition of T cell activation, proliferation, and functions. As such, therapeutic antibodies that block PD-L1/PD1 interactions between cancer cells and T cells have recently received great attention because of their capacity to reverse T cell exhaustion in response to persistent antigen stimulation and to improve the immune control of cancer in a variety of tumor types, including melanoma, lung, and renal cell carcinomas (154). As mentioned above, it was shown that myofibroblasts/CAFs from colon cancer expressed PD-L1 and PD-L2 and negatively regulate CD4+ TH cell proliferative response (152). Similarly, CAFs isolated from lung carcinoma were shown to constitutively express PD-L1 and PD-L2, which can be upregulated by IFN-γ, and negatively regulate tumor-associated CD8+ T cell activation (151). In melanoma, PD-L1 expression on CAFs seems to be dependent of IL-1α/β secreted by melanoma tumor cells and melanocytes and could participate to the suppression of melanoma-specific CD8+ T cells (150). However, most of these discoveries rely on CAFs isolation and in vitro experiments, with potential artifacts (155), and clearly require further investigations to determine the physiological relevance of potential PD-L1/L2 expression by CAFs on their immunosuppressive capabilities in vivo.
In conclusion, the CAF secretome can shape the T cell-dependent antitumor immune response by affecting several populations such as DCs, MDSCs, by switching CD4+ TH lymphocytes from a TH1 to a TH2 phenotype, by affecting Tregs and TH17 cells, by affecting CD8+ T cell functions or eventually by expressing some ligands of immune checkpoint receptors.
Indirect Effect of CAFs on Anti-Tumor Immune Response
As mentioned earlier, CAF activation in the TME results in a remodeling of the ECM through deposition of several components and by proteolytic degradation, which in turn affect tumor behavior (18, 156, 157). For example, increased ECM rigidity resulting from thickening and linearization of collagen fibers has been shown to regulate tumor growth and metastasis (158, 159). This modified ECM protein network is also presumed to restrict access of immune cells to cancer cells, serving as a physical barrier at least in some models (160, 161). As such, CAF-modified ECM might be involved in T cell exclusion from the proximity of cancer cells, which has been shown as a dominant immunosuppressive mechanism in multiple cancers and a predictor of patient clinical outcome (160). In this regard, in pancreatic tumor models, it has been proposed that when fibrosis is extensive, the “scar-like” ECM may act as a barrier for CTL infiltration into tumors (162). It was also found that focal adhesion kinase [FAK; a crucial signaling protein that is activated by numerous stimuli and functions as a biosensor to control cell motility (163)] activity is elevated in human pancreatic ductal adenocarcinoma tissues and correlates with high levels of fibrosis and poor CD8+ CTL infiltration (164). Similarly, in lung cancers, CAFs could restrict CD4+ and CD8+ T cells motility. Indeed, it was observed an active T cell motility in loose fibronectin and collagen regions, whereas T cells poorly migrate in dense matrix areas. Furthermore, aligned fibers in perivascular regions and around tumor epithelial cell regions dictate the migratory trajectory of T cells and restricted them from entering tumor islets (165, 166). Finally, interactions between tumor cells and the surrounding modified ECM have been involved as primary forces driving the EMT process. Consequently, the imbalanced biomechanical force at the tumor-stroma interface is an important player initiating EMT (167), which can subsequently lead to tumor cells escaping from T cell-mediated lysis after their acquisition of a mesenchymal-like phenotype (168–170). Thus, in the region where the ECM has been extensively modified by CAFs, an EMT process could protect tumor cells from T cell-mediated destruction.
The CAF-mediated remodeling of the ECM might also affect other immune population than T cells. For example, CAFs have been identified as an important source of hyaluronan, also called hyaluronic acid, a component of the ECM which promotes TAM recruitment, as the genetic ablation of the hyaluronan synthase strongly diminishes their presence within the TME (171). In pancreatic and breast cancers, it was also found that extensive deposition of type I collagen, which can be highly secreted by CAFs, improves TAM infiltration (172), with a potential effect of the ECM composition on their M2 polarization (173, 174). The high levels of CAF-secreted collagen I in tumors could also activate leukocyte-associated Ig-like receptor (LAIR)-1, a collagen-receptor that inhibits immune cell function upon collagen binding (175). Nevertheless, the regulation of macrophages polarization by the ECM composition, as well as its effect on, but not limited to, MDSC, neutrophils, or DCs is still poorly understood.
In addition to the extensive remodeling of the ECM, CAFs might also indirectly regulate the anti-tumor immune response by participating in the emergence of hypoxic stress within the TME. Indeed, in tumors with a high level of fibrosis, tumor tissues are often poorly oxygenated, with a limited number of functional blood vessels, resulting in the presence of zones with a low oxygen pressure called “hypoxic zones” (16, 176, 177). Even if, as mentioned above, CAFs are described as regulators of angiogenesis through the secretion of pro-angiogenic factors, such as VEGF or through the recruitment of endothelial progenitors in the tumor through the release of SDF-1 in the TME (178), the blood vessels present in the TME are poorly functional and leaky. The resulting leaky vessels not only trigger a high interstitial fluid pressure in the TME which affect immune cell transmigration from the vessels to the TME (179), but also affect oxygen availability and acidification of the TME (180). In other words, by their global action on the TME, the presence of CAFs might participate to abnormal angiogenesis and to the creation of hypoxic zones that contribute to the immunosuppressive network within the TME. Indeed, hypoxia has been found to impair the antitumor immune response by several mechanisms (181–184), such as alteration of NK and T cell activation and effector functions, induction of PD-L1 expression on MDSCs via HIF-1α transcription factor, and attraction of TAMs or Tregs to the tumor bed. Furthermore, hypoxic tumor cells secrete factors including TGF-β and PDGF that promote conversion of precursor cell types into CAFs (185), and it was also shown that stromal fibroblasts synergize with hypoxic stress to enhance melanoma aggressiveness (186). This indicates a potential role of hypoxia in the CAFs activation, either by directly acting on CAFs or indirectly by acting on tumor cells, or in their function in the TME. Thus, one may consider that hypoxia not only promotes CAFs activation but might also increase their immunosuppressive properties, even if this last particular point needs to be clarified.
Overall, CAFs might indirectly affect the anti-tumor immune response, with many described and not yet elucidated distinct possibilities, such as the modification of the ECM, vasculature or architecture of the tumors, which make this field very challenging.
Targeting CAFs to Improve Anti-Tumor Immune Response and Immunotherapy
Given the fact that CAFs impair the anti-tumor immunity (and more generally exert pro-tumorigenic effects) by several mechanisms, the design of pre-clinical or clinical studies in order to target these cells in the TME is very seductive to amplify the antitumor immune response and to develop “anti-CAF”-based immunotherapeutic approaches. Such studies can be envisioned based on agents directly targeting CAF specific proteins (e.g., FAP…) and signaling pathways involved in CAF activation (e.g., TGF-β, PDGF, FGF…) or less specifically targeting CAF-secreted factors. Potential therapies aiming at targeting CAFs or reversing the CAF “state,” as well as the ongoing clinical trials have been extensively reviewed in Ref. (18).
Recently, anti-CAF therapies have been mainly focused on FAP (187). A pioneer study has shown, in a transgenic mouse model in which FAP-expressing cells can be ablated, that the depletion of FAP-expressing cells cause rapid hypoxic necrosis of both Lewis lung carcinoma and stromal cells in immunogenic tumors by a process involving IFN-γ and TNF-α, which have previously been shown to be involved in CD8+ T cell-dependent killing of tumor cells (188). The development of chimeric antigen receptor (CAR) T cells targeting FAP has also shown promising results in murine models (189–191) and in malignant pleural mesothelioma patient derived xenograft models (192). A recent study has also demonstrated in two murine melanoma models that depleting FAP+ stromal cells from the TME upon vaccination with an adenoviral-vector reduced the frequencies and functions of immunosuppressive cells, resulting in prolonged survival of melanoma-bearing mice associated with a robust CD8+ T cell response (193). Similarly, in LL2 (murine lung cancer), CT26 (murine colon cancer), and B16F10 (murine melanoma) models, a whole-tumor cell vaccine modified to express FAP seems to induce antitumor immunity against both tumor cells and CAFs and enhances the infiltration of CD8+ T lymphocytes and decreases the accumulation of immunosuppressive cells in the TME (194). Nevertheless, it should be noted that, in addition to CAFs, FAP can be expressed by cells present in several tissues, including multipotent bone marrow stem cells or skeletal muscles. As such, another study has shown that adoptive transfer of FAP-reactive CAR-T cells into mice bearing a variety of subcutaneous tumors mediated limited antitumor effects and induced significant cachexia (a syndrome of progressive weight loss, anorexia, and persistent erosion of body muscle mass) and lethal bone toxicities in two murine strains (195). Thus, these lethal bone toxicity and cachexia observed after CAR T cell-based immunotherapy targeting FAP highlight cautions against its use as a universal target.
As such, targeting the CAF “secretome” or activation pathways, in order to revert the CAF “state,” might be a safer alternative to abrogate, at least partly and probably less specifically, their immunosuppressive role in the TME. In this regard, a recent publication demonstrated that targeting CXCL12 from FAP-expressing CAFs with AMD3100 (Plerixafor) synergizes with anti-PD-L1 immunotherapy in pancreatic cancer (130). Similarly, other proteins secreted by CAFs could be also targeted in order to restrain the immunosuppressive capabilities of these cells, such as IL-6 or TGF-β, using multiple inhibitors (18). For example, trihydroxyphenolic compounds were identified as potent blockers of TGF-β1 in the presence of active lysyl oxidase-like 2 (LOXL2; a member of mammalian copper-dependent LOX enzymes only expressed by fibroblasts or cancer cells and involved in intra- and intermolecular covalent collagen cross-links), and induce potent blockade of pathological collagen accumulation in vivo (196). Thus, these compounds might interfere with the T cell exclusion mediated by the CAF-dependent ECM remodeling previously mentioned, even if this particular point is still hypothetical. The use of Tranilast (Rizaben) (a known suppressor of fibroblast proliferation and TGF-β secretion) has also demonstrated a synergistic effect with a DC-based vaccine in C57BL/6 mice bearing syngeneic E-G7 lymphoma, LLC1 Lewis lung cancer or B16F1 melanoma (197). Another example is retinoic acid, a small molecular derivative of vitamin A, which inhibits IL-6 and ECM production by CAFs (198), potentially affecting their immunosuppressive properties. Nevertheless, more studies are clearly needed to identify other potential therapeutic agents targeting CAFs and/or their immunosuppressive network, which might be use in combination with the current or future anti-tumor immunotherapeutic approaches.
Concluding Remarks
Despite their relative abundance in tumors, fibroblasts have been ignored over decades, but their crucial role has now emerged in the fields of tumor biology and oncology. CAFs have pleiotropic functions in tumor growth and participate to the inflammatory phenotype of the TME by releasing a variety of chemokines, cytokines, and other factors leading to the alteration of the antitumor immune response. Nevertheless, this complex immunosuppressive network related to the “secretome” of CAFs is still poorly understood, even if extensive efforts allowed apprehending their role in both the innate and the adaptive immune response. Of note, the notion that the CAF-specific secretome modulates the anti-tumor immune response often relies on studies limited to cells expanded in vitro. Future challenging studies using preclinical models will be thus needed in order to define more precisely the functional list of CAF-derived factors that exert an immunomodulatory role in the context of the TME complexity in vivo. This is crucial in order to fully understand the global regulation of the antitumor immune response and might also lead to the identification of novel potential therapeutic targets with the ability to increase the efficiency of anti-tumor immunotherapeutic approaches. In particular, targeting the CAFs or their secretome may probably not induce a complete tumor cell death by itself, but it will help to reduce immune effector cell dysfunctions as well as the recruitment of immunosuppressive cells, thus releasing the “brake” for a more effective immune response in combination with therapy targeting immune checkpoints (e.g., anti-CTLA4, anti-PD1/PD-L1 antibodies) or other mechanisms impairing the anti-tumor immune response in patients (199).
Author Contributions
JT wrote the manuscript. LZ and SC participate to helpful discussion and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are supported by INSERM, INCA, and the French “Ligue Nationale Contre Le Cancer” (LNCC-Equipe Labéllisée). LZ was supported by a PhD training fellowship from the french “Ligue Nationale Contre Le Cancer.” We acknowledge further work that was done by our colleagues in the fields of CAF biology and immunosuppression and apologize if some citations are missing due to space limitations.
References
1. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci (2012) 125(Pt 23):5591–6. doi:10.1242/jcs.116392
2. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19(11):1423–37. doi:10.1038/nm.3394
3. Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, et al. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med (2015) 13:45. doi:10.1186/s12916-015-0278-7
4. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res (2015) 21(4):687–92. doi:10.1158/1078-0432.CCR-14-1860
5. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi:10.1038/nature21349
6. Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene (2008) 27(45):5894–903. doi:10.1038/onc.2008.268
7. Chouaib S, Janji B, Tittarelli A, Eggermont A, Thiery JP. Tumor plasticity interferes with anti-tumor immunity. Crit Rev Immunol (2014) 34(2):91–102. doi:10.1615/CritRevImmunol.2014010183
8. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14(10):1014–22. doi:10.1038/ni.2703
9. Hamai A, Benlalam H, Meslin F, Hasmim M, Carre T, Akalay I, et al. Immune surveillance of human cancer: if the cytotoxic T-lymphocytes play the music, does the tumoral system call the tune? Tissue Antigens (2010) 75(1):1–8. doi:10.1111/j.1399-0039.2009.01401.x
10. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res (2011) 1(4):482–97.
11. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer (2016) 16(9):582–98. doi:10.1038/nrc.2016.73
12. Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’ – more than meets the eye. Trends Mol Med (2013) 19(8):447–53. doi:10.1016/j.molmed.2013.05.004
13. Valcz G, Sipos F, Tulassay Z, Molnar B, Yagi Y. Importance of carcinoma-associated fibroblast-derived proteins in clinical oncology. J Clin Pathol (2014) 67(12):1026–31. doi:10.1136/jclinpath-2014-202561
14. Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res (2014) 2(3):187–93. doi:10.1158/2326-6066.CIR-14-0002
15. Harper J, Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol (2014) 25:69–77. doi:10.1016/j.semcancer.2013.12.005
16. Jiang H, Hegde S, DeNardo DG. Tumor-associated fibrosis as a regulator of tumor immunity and response to immunotherapy. Cancer Immunol Immunother (2017) 66(8):1037–48. doi:10.1007/s00262-017-2003-1
17. Darby IA, Laverdet B, Bonte F, Desmouliere A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Invest Dermatol (2014) 7:301–11. doi:10.2147/CCID.S50046
18. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev (2016) 30(9):1002–19. doi:10.1101/gad.279737.116
19. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol (2002) 3(5):349–63. doi:10.1038/nrm809
20. Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia (1971) 27(5):549–50. doi:10.1007/BF02147594
21. Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmouliere A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair (2012) 5(Suppl 1):S5. doi:10.1186/1755-1536-5-S1-S5
22. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med (1986) 315(26):1650–9. doi:10.1056/NEJM198612253152606
23. Ohtani H. Stromal reaction in cancer tissue: pathophysiologic significance of the expression of matrix-degrading enzymes in relation to matrix turnover and immune/inflammatory reactions. Pathol Int (1998) 48(1):1–9. doi:10.1111/j.1440-1827.1998.tb03820.x
24. Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med (2014) 211(8):1503–23. doi:10.1084/jem.20140692
25. Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol (2012) 9(8):454–67. doi:10.1038/nrgastro.2012.115
26. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A (2010) 107(46):20009–14. doi:10.1073/pnas.1013805107
27. Vicent S, Sayles LC, Vaka D, Khatri P, Gevaert O, Chen R, et al. Cross-species functional analysis of cancer-associated fibroblasts identifies a critical role for CLCF1 and IL-6 in non-small cell lung cancer in vivo. Cancer Res (2012) 72(22):5744–56. doi:10.1158/0008-5472.CAN-12-1097
28. Bronzert DA, Pantazis P, Antoniades HN, Kasid A, Davidson N, Dickson RB, et al. Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc Natl Acad Sci U S A (1987) 84(16):5763–7. doi:10.1073/pnas.84.16.5763
29. Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res (2001) 264(1):169–84. doi:10.1006/excr.2000.5133
30. Kuzet SE, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res (2016) 365(3):607–19. doi:10.1007/s00441-016-2467-x
31. Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res (2001) 61(2):550–5.
32. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell (2010) 17(2):135–47. doi:10.1016/j.ccr.2009.12.041
33. Du H, Che G. Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol Lett (2017) 13(1):3–12. doi:10.3892/ol.2016.5451
34. Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res (2015) 25(5):588–603. doi:10.1038/cr.2015.51
35. Vizoso M, Puig M, Carmona FJ, Maqueda M, Velasquez A, Gomez A, et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis (2015) 36(12):1453–63. doi:10.1093/carcin/bgv146
36. Zeisberg EM, Zeisberg M. The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J Pathol (2013) 229(2):264–73. doi:10.1002/path.4120
37. Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest (2002) 110(3):341–50. doi:10.1172/JCI0215518
38. Radisky DC, Kenny PA, Bissell MJ. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem (2007) 101(4):830–9. doi:10.1002/jcb.21186
39. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res (2007) 67(21):10123–8. doi:10.1158/0008-5472.CAN-07-3127
40. Hosaka K, Yang Y, Seki T, Fischer C, Dubey O, Fredlund E, et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc Natl Acad Sci U S A (2016) 113(38):E5618–27. doi:10.1073/pnas.1608384113
41. Jotzu C, Alt E, Welte G, Li J, Hennessy BT, Devarajan E, et al. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst) (2010) 33(2):61–79. doi:10.1155/2010/695162
42. Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One (2012) 7(2):e30563. doi:10.1371/journal.pone.0030563
43. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi:10.1084/jem.20162024
44. Okabe H, Hayashi H, Nakagawa S, Imai K, Nitta H, Arima K, et al. Inducible factors for cancer-associated fibroblasts in liver cancer versus myofibroblasts in inflammatory liver disease. Histol Histopathol (2016) 31(2):141–8. doi:10.14670/HH-11-668
45. Direkze NC, Alison MR. Bone marrow and tumour stroma: an intimate relationship. Hematol Oncol (2006) 24(4):189–95. doi:10.1002/hon.788
46. Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res (2004) 64(23):8492–5. doi:10.1158/0008-5472.CAN-04-1708
47. Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun (2003) 309(1):232–40. doi:10.1016/S0006-291X(03)01544-4
48. Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, Ganesan S, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res (2008) 68(11):4331–9. doi:10.1158/0008-5472.CAN-08-0943
49. Koliaraki V, Pallangyo CK, Greten FR, Kollias G. Mesenchymal cells in colon cancer. Gastroenterology (2017) 152(5):964–79. doi:10.1053/j.gastro.2016.11.049
50. Corsa CA, Brenot A, Grither WR, Van Hove S, Loza AJ, Zhang K, et al. The action of discoidin domain receptor 2 in basal tumor cells and stromal cancer-associated fibroblasts is critical for breast cancer metastasis. Cell Rep (2016) 15(11):2510–23. doi:10.1016/j.celrep.2016.05.033
51. De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J (2004) 18(9):1016–8. doi:10.1096/fj.03-1110fje
52. Kelly T, Huang Y, Simms AE, Mazur A. Fibroblast activation protein-alpha: a key modulator of the microenvironment in multiple pathologies. Int Rev Cell Mol Biol (2012) 297:83–116. doi:10.1016/B978-0-12-394308-8.00003-0
53. Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J Biol Chem (1999) 274(51):36505–12. doi:10.1074/jbc.274.51.36505
54. Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol (1995) 130(2):393–405. doi:10.1083/jcb.130.2.393
55. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther (2006) 5(12):1640–6. doi:10.4161/cbt.5.12.3354
56. True LD, Zhang H, Ye M, Huang CY, Nelson PS, von Haller PD, et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod Pathol (2010) 23(10):1346–56. doi:10.1038/modpathol.2010.122
57. Yurugi Y, Wakahara M, Matsuoka Y, Sakabe T, Kubouchi Y, Haruki T, et al. Podoplanin expression in cancer-associated fibroblasts predicts poor prognosis in patients with squamous cell carcinoma of the lung. Anticancer Res (2017) 37(1):207–13. doi:10.21873/anticanres.11308
58. Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol (2014) 4:62. doi:10.3389/fonc.2014.00062
59. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi:10.1016/j.ccr.2014.04.005
60. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25(6):735–47. doi:10.1016/j.ccr.2014.04.021
61. Paraiso KH, Smalley KS. Fibroblast-mediated drug resistance in cancer. Biochem Pharmacol (2013) 85(8):1033–41. doi:10.1016/j.bcp.2013.01.018
62. Bagordakis E, Sawazaki-Calone I, Macedo CC, Carnielli CM, de Oliveira CE, Rodrigues PC, et al. Secretome profiling of oral squamous cell carcinoma-associated fibroblasts reveals organization and disassembly of extracellular matrix and collagen metabolic process signatures. Tumour Biol (2016) 37(7):9045–57. doi:10.1007/s13277-015-4629-y
63. De Boeck A, Hendrix A, Maynard D, Van Bockstal M, Daniels A, Pauwels P, et al. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics (2013) 13(2):379–88. doi:10.1002/pmic.201200179
64. Ge S, Mao Y, Yi Y, Xie D, Chen Z, Xiao Z. Comparative proteomic analysis of secreted proteins from nasopharyngeal carcinoma-associated stromal fibroblasts and normal fibroblasts. Exp Ther Med (2012) 3(5):857–60. doi:10.3892/etm.2012.483
65. Lin ZY, Chuang YH, Chuang WL. Cancer-associated fibroblasts up-regulate CCL2, CCL26, IL6 and LOXL2 genes related to promotion of cancer progression in hepatocellular carcinoma cells. Biomed Pharmacother (2012) 66(7):525–9. doi:10.1016/j.biopha.2012.02.001
66. Torres S, Bartolome RA, Mendes M, Barderas R, Fernandez-Acenero MJ, Pelaez-Garcia A, et al. Proteome profiling of cancer-associated fibroblasts identifies novel proinflammatory signatures and prognostic markers for colorectal cancer. Clin Cancer Res (2013) 19(21):6006–19. doi:10.1158/1078-0432.CCR-13-1130
67. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi:10.1038/nrclinonc.2016.217
68. Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One (2017) 12(7):e0182039. doi:10.1371/journal.pone.0182039
69. Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol (2000) 1(6):510–4. doi:10.1038/82763
70. Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene (2014) 33(19):2423–31. doi:10.1038/onc.2013.191
71. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol (2014) 5:489. doi:10.3389/fimmu.2014.00489
72. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73(10):3007–18. doi:10.1158/0008-5472.CAN-12-4601
73. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget (2017) 8(5):8633–47. doi:10.18632/oncotarget.14374
74. Zhang J, Chen L, Xiao M, Wang C, Qin Z. FSP1+ fibroblasts promote skin carcinogenesis by maintaining MCP-1-mediated macrophage infiltration and chronic inflammation. Am J Pathol (2011) 178(1):382–90. doi:10.1016/j.ajpath.2010.11.017
75. Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene (2017) 36(31):4457–68. doi:10.1038/onc.2017.65
76. Fujii N, Shomori K, Shiomi T, Nakabayashi M, Takeda C, Ryoke K, et al. Cancer-associated fibroblasts and CD163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance. J Oral Pathol Med (2012) 41(6):444–51. doi:10.1111/j.1600-0714.2012.01127.x
77. Herrera M, Herrera A, Dominguez G, Silva J, Garcia V, Garcia JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci (2013) 104(4):437–44. doi:10.1111/cas.12096
78. Ocana A, Nieto-Jimenez C, Pandiella A, Templeton AJ. Neutrophils in cancer: prognostic role and therapeutic strategies. Mol Cancer (2017) 16(1):137. doi:10.1186/s12943-017-0707-7
79. Powell DR, Huttenlocher A. Neutrophils in the tumor microenvironment. Trends Immunol (2016) 37(1):41–52. doi:10.1016/j.it.2015.11.008
80. Shaul ME, Fridlender ZG. Neutrophils as active regulators of the immune system in the tumor microenvironment. J Leukoc Biol (2017) 102(2):343–9. doi:10.1189/jlb.5MR1216-508R
81. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi:10.1016/j.ccr.2009.06.017
82. Leliefeld PH, Koenderman L, Pillay J. How neutrophils shape adaptive immune responses. Front Immunol (2015) 6:471. doi:10.3389/fimmu.2015.00471
83. Varricchi G, Galdiero MR, Loffredo S, Marone G, Iannone R, Granata F. Are mast cells MASTers in cancer? Front Immunol (2017) 8:424. doi:10.3389/fimmu.2017.00424
84. Ma Y, Hwang RF, Logsdon CD, Ullrich SE. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res (2013) 73(13):3927–37. doi:10.1158/0008-5472.CAN-12-4479
85. Marquardt DL, Gruber HE, Wasserman SI. Adenosine release from stimulated mast cells. Proc Natl Acad Sci U S A (1984) 81(19):6192–6. doi:10.1073/pnas.81.19.6192
86. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol (2016) 29:7–16. doi:10.1016/j.coph.2016.04.001
87. Martinez-Nunez RT, Louafi F, Sanchez-Elsner T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J Biol Chem (2011) 286(3):1786–94. doi:10.1074/jbc.M110.169367
88. Danelli L, Frossi B, Pucillo CE. Mast cell/MDSC a liaison immunosuppressive for tumor microenvironment. Oncoimmunology (2015) 4(4):e1001232. doi:10.1080/2162402X.2014.1001232
89. Yang Z, Zhang B, Li D, Lv M, Huang C, Shen GX, et al. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS One (2010) 5(1):e8922. doi:10.1371/journal.pone.0008922
90. San Francisco IF, DeWolf WC, Peehl DM, Olumi AF. Expression of transforming growth factor-beta 1 and growth in soft agar differentiate prostate carcinoma-associated fibroblasts from normal prostate fibroblasts. Int J Cancer (2004) 112(2):213–8. doi:10.1002/ijc.20388
91. Zhuang J, Lu Q, Shen B, Huang X, Shen L, Zheng X, et al. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep (2015) 5:11924. doi:10.1038/srep11924
92. Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol (2017) 8:930. doi:10.3389/fimmu.2017.00930
93. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U S A (2003) 100(7):4120–5. doi:10.1073/pnas.0730640100
94. Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U S A (2014) 111(11):4203–8. doi:10.1073/pnas.1319269111
95. Park YP, Choi SC, Kiesler P, Gil-Krzewska A, Borrego F, Weck J, et al. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the gammac cytokines and TGF-beta1. Blood (2011) 118(11):3019–27. doi:10.1182/blood-2011-04-346825
96. Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol (2010) 10(8):554–67. doi:10.1038/nri2808
97. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol (2005) 6(6):600–7. doi:10.1038/ni1197
98. Trotta R, Dal Col J, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-beta utilizes SMAD3 to inhibit CD16-mediated IFN-gamma production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol (2008) 181(6):3784–92. doi:10.4049/jimmunol.181.6.3784
99. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A (2009) 106(49):20847–52. doi:10.1073/pnas.0906481106
100. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett (2012) 318(2):154–61. doi:10.1016/j.canlet.2011.12.020
101. Li T, Yi S, Liu W, Jia C, Wang G, Hua X, et al. Colorectal carcinoma-derived fibroblasts modulate natural killer cell phenotype and antitumor cytotoxicity. Med Oncol (2013) 30(3):663. doi:10.1007/s12032-013-0663-z
102. Ziani L, Safta-Saadoun TB, Gourbeix J, Cavalcanti A, Robert C, Favre G, et al. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget (2017) 8(12):19780–94. doi:10.18632/oncotarget.15540
103. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity (2014) 40(5):642–56. doi:10.1016/j.immuni.2014.04.016
104. Raker VK, Domogalla MP, Steinbrink K. Tolerogenic dendritic cells for regulatory T cell induction in man. Front Immunol (2015) 6:569. doi:10.3389/fimmu.2015.00569
105. Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol (2014) 32:51–82. doi:10.1146/annurev-immunol-032713-120257
106. Kitamura H, Kamon H, Sawa S, Park SJ, Katunuma N, Ishihara K, et al. IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity (2005) 23(5):491–502. doi:10.1016/j.immuni.2005.09.010
107. Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, et al. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci (2017) 108(10):1947–52. doi:10.1111/cas.13332
108. Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol (2004) 173(6):3844–54. doi:10.4049/jimmunol.173.6.3844
109. Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS, et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget (2016) 7(19):27584–98. doi:10.18632/oncotarget.8488
110. Li YL, Zhao H, Ren XB. Relationship of VEGF/VEGFR with immune and cancer cells: staggering or forward? Cancer Biol Med (2016) 13(2):206–14. doi:10.20892/j.issn.2095-3941.2015.0070
111. Dikov MM, Ohm JE, Ray N, Tchekneva EE, Burlison J, Moghanaki D, et al. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J Immunol (2005) 174(1):215–22. doi:10.4049/jimmunol.174.1.215
112. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood (1998) 92(11):4150–66.
113. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med (1996) 2(10):1096–103. doi:10.1038/nm1096-1096
114. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol (1998) 160(3):1224–32.
115. Sanjabi S, Mosaheb MM, Flavell RA. Opposing effects of TGF-beta and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity (2009) 31(1):131–44. doi:10.1016/j.immuni.2009.04.020
116. Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol (2005) 174(9):5215–23. doi:10.4049/jimmunol.174.9.5215
117. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8(5):369–80. doi:10.1016/j.ccr.2005.10.012
118. Takahashi H, Sakakura K, Kawabata-Iwakawa R, Rokudai S, Toyoda M, Nishiyama M, et al. Immunosuppressive activity of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Cancer Immunol Immunother (2015) 64(11):1407–17. doi:10.1007/s00262-015-1742-0
119. Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res (2014) 16(4):410. doi:10.1186/s13058-014-0410-1
120. Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood (2004) 103(12):4619–21. doi:10.1182/blood-2003-11-3909
121. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ (2002) 9(10):1069–77. doi:10.1038/sj.cdd.4401073
122. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72(21):5435–40. doi:10.1158/0008-5472.CAN-12-0569
123. Timosenko E, Hadjinicolaou AV, Cerundolo V. Modulation of cancer-specific immune responses by amino acid degrading enzymes. Immunotherapy (2017) 9(1):83–97. doi:10.2217/imt-2016-0118
124. Ino Y, Yamazaki-Itoh R, Oguro S, Shimada K, Kosuge T, Zavada J, et al. Arginase II expressed in cancer-associated fibroblasts indicates tissue hypoxia and predicts poor outcome in patients with pancreatic cancer. PLoS One (2013) 8(2):e55146. doi:10.1371/journal.pone.0055146
125. He XJ, Tao HQ, Hu ZM, Ma YY, Xu J, Wang HJ, et al. Expression of galectin-1 in carcinoma-associated fibroblasts promotes gastric cancer cell invasion through upregulation of integrin beta1. Cancer Sci (2014) 105(11):1402–10. doi:10.1111/cas.12539
126. Tang D, Gao J, Wang S, Ye N, Chong Y, Huang Y, et al. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumour Biol (2016) 37(2):1889–99. doi:10.1007/s13277-015-3942-9
127. Rabinovich GA, Toscano MA. Turning ‘sweet’ on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol (2009) 9(5):338–52. doi:10.1038/nri2536
128. Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature (1995) 378(6558):736–9. doi:10.1038/378736a0
129. Stillman BN, Hsu DK, Pang M, Brewer CF, Johnson P, Liu FT, et al. Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J Immunol (2006) 176(2):778–89. doi:10.4049/jimmunol.176.2.778
130. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110(50):20212–7. doi:10.1073/pnas.1320318110
131. Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res (2014) 2(2):91–8. doi:10.1158/2326-6066.CIR-13-0216
132. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol (2005) 6(11):1133–41. doi:10.1038/ni1261
133. Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol (2010) 10(4):248–56. doi:10.1038/nri2742
134. Guery L, Hugues S. Th17 cell plasticity and functions in cancer immunity. Biomed Res Int (2015) 2015:314620. doi:10.1155/2015/314620
135. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
136. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol (2012) 3:51. doi:10.3389/fimmu.2012.00051
137. Barnas JL, Simpson-Abelson MR, Brooks SP, Kelleher RJ Jr, Bankert RB. Reciprocal functional modulation of the activation of T lymphocytes and fibroblasts derived from human solid tumors. J Immunol (2010) 185(5):2681–92. doi:10.4049/jimmunol.1000896
138. Haniffa MA, Wang XN, Holtick U, Rae M, Isaacs JD, Dickinson AM, et al. Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. J Immunol (2007) 179(3):1595–604. doi:10.4049/jimmunol.179.3.1595
139. Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One (2009) 4(11):e7965. doi:10.1371/journal.pone.0007965
140. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med (2011) 208(3):469–78. doi:10.1084/jem.20101876
141. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
142. Kinoshita T, Ishii G, Hiraoka N, Hirayama S, Yamauchi C, Aokage K, et al. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci (2013) 104(4):409–15. doi:10.1111/cas.12099
143. Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc’h N, Zeng G, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol (2005) 175(3):1483–90. doi:10.4049/jimmunol.175.3.1483
144. Sharma S, Yang SC, Zhu L, Reckamp K, Gardner B, Baratelli F, et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+ CD25+ T regulatory cell activities in lung cancer. Cancer Res (2005) 65(12):5211–20. doi:10.1158/0008-5472.CAN-05-0141
145. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
146. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37(3):208–20. doi:10.1016/j.it.2016.01.004
147. Kim JH, Oh SH, Kim EJ, Park SJ, Hong SP, Cheon JH, et al. The role of myofibroblasts in upregulation of S100A8 and S100A9 and the differentiation of myeloid cells in the colorectal cancer microenvironment. Biochem Biophys Res Commun (2012) 423(1):60–6. doi:10.1016/j.bbrc.2012.05.081
148. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36(8):1090–101. doi:10.1038/onc.2016.273
149. Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res (2016) 76(14):4124–35. doi:10.1158/0008-5472.CAN-15-2973
150. Khalili JS, Liu S, Rodriguez-Cruz TG, Whittington M, Wardell S, Liu C, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res (2012) 18(19):5329–40. doi:10.1158/1078-0432.CCR-12-1632
151. Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ Jr, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol (2007) 178(9):5552–62. doi:10.4049/jimmunol.178.9.5552
152. Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology (2008) 135(4):1228–1237, 1237.e1–2. doi:10.1053/j.gastro.2008.07.016
153. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2017). doi:10.1038/nri.2017.108
154. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol (2016) 7:550. doi:10.3389/fimmu.2016.00550
155. Ghebeh H, Dermime S. Comment on "Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells". J Immunol (2007) 179(2):732; author reply 3. doi:10.4049/jimmunol.179.2.732
156. Fullar A, Dudas J, Olah L, Hollosi P, Papp Z, Sobel G, et al. Remodeling of extracellular matrix by normal and tumor-associated fibroblasts promotes cervical cancer progression. BMC Cancer (2015) 15:256. doi:10.1186/s12885-015-1272-3
157. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep (2014) 15(12):1243–53. doi:10.15252/embr.201439246
158. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol (2012) 196(4):395–406. doi:10.1083/jcb.201102147
159. Sangaletti S, Chiodoni C, Tripodo C, Colombo MP. The good and bad of targeting cancer-associated extracellular matrix. Curr Opin Pharmacol (2017) 35:75–82. doi:10.1016/j.coph.2017.06.003
160. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science (2015) 348(6230):74–80. doi:10.1126/science.aaa6204
161. Sorokin L. The impact of the extracellular matrix on inflammation. Nat Rev Immunol (2010) 10(10):712–23. doi:10.1038/nri2852
162. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res (2014) 20(13):3422–33. doi:10.1158/1078-0432.CCR-13-2972
163. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer (2014) 14(9):598–610. doi:10.1038/nrc3792
164. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med (2016) 22(8):851–60. doi:10.1038/nm.4123
165. Salmon H, Donnadieu E. Within tumors, interactions between T cells and tumor cells are impeded by the extracellular matrix. Oncoimmunology (2012) 1(6):992–4. doi:10.4161/onci.20239
166. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122(3):899–910. doi:10.1172/JCI45817
167. Torzilli PA, Bourne JW, Cigler T, Vincent CT. A new paradigm for mechanobiological mechanisms in tumor metastasis. Semin Cancer Biol (2012) 22(5–6):385–95. doi:10.1016/j.semcancer.2012.05.002
168. Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res (2013) 73(8):2418–27. doi:10.1158/0008-5472.CAN-12-2432
169. Akalay I, Janji B, Hasmim M, Noman MZ, Thiery JP, Mami-Chouaib F, et al. EMT impairs breast carcinoma cell susceptibility to CTL-mediated lysis through autophagy induction. Autophagy (2013) 9(7):1104–6. doi:10.4161/auto.24728
170. Terry S, Buart S, Tan TZ, Gros G, Noman MZ, Lorens JB, et al. Acquisition of tumor cell phenotypic diversity along the EMT spectrum under hypoxic pressure: consequences on susceptibility to cell-mediated cytotoxicity. Oncoimmunology (2017) 6(2):e1271858. doi:10.1080/2162402X.2016.1271858
171. Kobayashi N, Miyoshi S, Mikami T, Koyama H, Kitazawa M, Takeoka M, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Res (2010) 70(18):7073–83. doi:10.1158/0008-5472.CAN-09-4687
172. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Int Biol Quant Biosci Nano Macro (2015) 7(10):1120–34. doi:10.1039/c5ib00040h
173. McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A (2013) 110(43):17253–8. doi:10.1073/pnas.1308887110
174. Stahl M, Schupp J, Jager B, Schmid M, Zissel G, Muller-Quernheim J, et al. Lung collagens perpetuate pulmonary fibrosis via CD204 and M2 macrophage activation. PLoS One (2013) 8(11):e81382. doi:10.1371/journal.pone.0081382
175. Lebbink RJ, van den Berg MC, de Ruiter T, Raynal N, van Roon JA, Lenting PJ, et al. The soluble leukocyte-associated Ig-like receptor (LAIR)-2 antagonizes the collagen/LAIR-1 inhibitory immune interaction. J Immunol (2008) 180(3):1662–9. doi:10.4049/jimmunol.180.3.1662
176. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest (2007) 117(12):3810–20. doi:10.1172/JCI30487
177. Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol (2009) 296(3):G582–92. doi:10.1152/ajpgi.90368.2008
178. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell (2005) 121(3):335–48. doi:10.1016/j.cell.2005.02.034
179. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol (2015) 15(11):692–704. doi:10.1038/nri3908
180. Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol (2014) 9:47–71. doi:10.1146/annurev-pathol-012513-104720
181. Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene (2017) 36(4):439–45. doi:10.1038/onc.2016.225
182. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol (2015) 6:482. doi:10.3389/fimmu.2015.00482
183. Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol (2017) 8:270. doi:10.3389/fimmu.2017.00270
184. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. a review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol (2015) 309(9):C569–79. doi:10.1152/ajpcell.00207.2015
185. Giaccia AJ, Schipani E. Role of carcinoma-associated fibroblasts and hypoxia in tumor progression. Curr Top Microbiol Immunol (2010) 345:31–45. doi:10.1007/82_2010_73
186. Comito G, Giannoni E, Di Gennaro P, Segura CP, Gerlini G, Chiarugi P. Stromal fibroblasts synergize with hypoxic oxidative stress to enhance melanoma aggressiveness. Cancer Lett (2012) 324(1):31–41. doi:10.1016/j.canlet.2012.04.025
187. Jiang GM, Xu W, Du J, Zhang KS, Zhang QG, Wang XW, et al. The application of the fibroblast activation protein alpha-targeted immunotherapy strategy. Oncotarget (2016) 7(22):33472–82. doi:10.18632/oncotarget.8098
188. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (2010) 330(6005):827–30. doi:10.1126/science.1195300
189. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther (2013) 21(8):1611–20. doi:10.1038/mt.2013.110
190. Lo A, Wang LS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res (2015) 75(14):2800–10. doi:10.1158/0008-5472.CAN-14-3041
191. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi:10.1158/2326-6066.CIR-13-0027
192. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med (2013) 11:187. doi:10.1186/1479-5876-11-187
193. Zhang Y, Ertl HC. Depletion of FAP+ cells reduces immunosuppressive cells and improves metabolism and functions CD8+T cells within tumors. Oncotarget (2016) 7(17):23282–99. doi:10.18632/oncotarget.7818
194. Chen M, Xiang R, Wen Y, Xu G, Wang C, Luo S, et al. A whole-cell tumor vaccine modified to express fibroblast activation protein induces antitumor immunity against both tumor cells and cancer-associated fibroblasts. Sci Rep (2015) 5:14421. doi:10.1038/srep14421
195. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210(6):1125–35. doi:10.1084/jem.20130110
196. Wei Y, Kim TJ, Peng DH, Duan D, Gibbons DL, Yamauchi M, et al. Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. J Clin Invest (2017) 127(10):3675–88. doi:10.1172/JCI94624
197. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci (2015) 106(2):134–42. doi:10.1111/cas.12584
198. Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, et al. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer Lett (2014) 345(1):132–9. doi:10.1016/j.canlet.2013.12.006
Keywords: cancer, tumor microenvironment, cancer-associated fibroblasts, immune suppression, immunotherapy
Citation: Ziani L, Chouaib S and Thiery J (2018) Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 9:414. doi: 10.3389/fimmu.2018.00414
Received: 13 November 2017; Accepted: 14 February 2018;
Published: 01 March 2018
Edited by:
Khashayarsha Khazaie, Mayo Clinic College of Medicine & Science, United StatesReviewed by:
Fabian Benencia, Ohio University, United StatesAlessandro Poggi, Ospedale Policlinico San Martino, Italy
Copyright: © 2018 Ziani, Chouaib and Thiery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerome Thiery, amVyb21lLnRoaWVyeUBndXN0YXZlcm91c3N5LmZy