 Gabrielle Bradshaw
Gabrielle Bradshaw Robbie R. Lualhati
Robbie R. Lualhati Cassie L. Albury
Cassie L. Albury Neven Maksemous
Neven Maksemous Deidre Roos-Araujo
Deidre Roos-Araujo Robert A. Smith
Robert A. Smith Miles C. Benton
Miles C. Benton David A. Eccles
David A. Eccles Rod A. Lea
Rod A. Lea Heidi G. Sutherland
Heidi G. Sutherland Larisa M. Haupt
Larisa M. Haupt Lyn R. Griffiths*
Lyn R. Griffiths*
- Genomics Research Centre, School of Biomedical Sciences, Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD, Australia
Background: We investigated the molecular etiology of a young male proband with confirmed immunodeficiency of unknown cause, presenting with recurrent bacterial and Varicella zoster viral infections in childhood and persistent lymphopenia into early adulthood.
Aim: To identify causative functional genetic variants related to an undiagnosed primary immunodeficiency.
Method: Whole genome microarray copy number variant (CNV) analysis was performed on the proband followed by whole exome sequencing (WES) and trio analysis of the proband and family members. A >4 kbp deletion identified by repeated CNV analysis of exome sequencing data along with three damaging missense single nucleotide variants were validated by Sanger sequencing in all family members. Confirmation of the causative role of the candidate gene was performed by qPCR and Western Blot analyses on the proband, family members and a healthy control.
Results: CNV identified our previously reported interleukin 25 amplification in the proband; however, the variant was not validated to be a candidate gene for immunodeficiency. WES trio analysis, data filtering and in silico prediction identified a novel, damaging (SIFT: 0; Polyphen 1; Grantham score: 101) and disease-causing (MutationTaster) single base mutation in the X chromosome (c.511C > T p.Arg171Trp) MSN gene not identified in the UCSC Genome Browser database. The mutation was validated by Sanger sequencing, confirming the proband was hemizygous X-linked recessive (–/T) at this locus and inherited the affected T allele from his non-symptomatic carrier mother (C/T), with other family members (father, sister) confirmed to be wild type (C/C). Western Blot analysis demonstrated an absence of moesin protein in lymphocytes derived from the proband, compared with normal expression in lymphocytes derived from the healthy control, father and mother. qPCR identified significantly lower MSN mRNA transcript expression in the proband compared to an age- and sex-matched healthy control subject in whole blood (p = 0.02), and lymphocytes (p = 0.01). These results confirmed moesin deficiency in the proband, directly causative of his immunodeficient phenotype.
Conclusion: These findings confirm X-linked moesin-associated immunodeficiency in a proband previously undiagnosed up to 24 years of age. This study also highlights the utility of WES for the diagnosis of rare or novel forms of primary immunodeficiency disease.
Introduction
Primary immunodeficiency disorders (PIDs) encompass a diverse group of mostly inherited genetic disorders that compromise immune system function and predispose individuals to recurrent infections and other immune disorders such as autoimmunity, hematological disorders and lymphoid malignancies (1–3). As defined by the International Union of Immunological Societies, there are almost 300 single gene defects described which are causative for the wide range of phenotypes (4), making PIDs a rapidly expanding field of medicine. Loss of function mutations in multiple genes coding for proteins that regulate the actin cytoskeleton are known to cause PID, the most well characterized form being Wiskott-Aldrich (WAS) syndrome (5). This is a rare X chromosome-linked PID where expression and function of the WAS protein results in failure of Arp2/3-mediated actin polymerization causing a combined defect of innate and adaptive immunity associated with microthrombocytopenia, eczema, blood cancers, and increased risk of autoimmunity (5).
Next-generation DNA sequencing technologies such as whole genome sequencing (WGS) and whole exome sequencing (WES) have revolutionized the field of genomics by enabling a high-throughput and increasingly cost-effective method for complex and rare disease diagnosis (6). While WGS is the most comprehensive technique able to identify around 4 million variants within the entire genome, including coding and non-coding regions, WES provides more focus on only protein-coding exons, which harbor around 85% of pathogenic mutations (7, 8). Currently, WES permits lower operating costs (US$800 per sample compared to US$1,500 per sample for WGS) with faster data generation and less complex analysis, however, it is unable to identify structural variants (6, 9). Here, we used WES to elucidate possible disease-causing variants in a case diagnosed previously as immunodeficient of unknown cause.
Background
The proband is a 24-year–old male. At age 7 weeks, he had boils in his groin and axillae. At 18 months of age, he had a prolonged episode of bronchitis for which he received antibiotics but was not hospitalized. He suffered repeated upper respiratory tract infections and ear infections for the next few years. He had two episodes of chicken pox (Varicella) at around 18 months and 3 years. The first episode was severe, with widespread skin lesions, but no secondary infection or hemorrhagic lesions. At about 3 years of age, he had a persistent eye infection, which progressed to periorbital cellulitis, secondary to Pseudomonas aeruginosa infection. Neutropenia was first noted during that episode. Bone marrow aspirate showed normal myeloid maturation, consistent with an immune etiology. No treatment was started at that stage. At 4 years of age, he had an episode of thrombocytopenia, with platelets falling to <5. A further bone marrow aspirate was consistent with an immune mediated thrombocytopenia, and he responded rapidly to intravenous immunoglobulin. He had some further skin infections and paronychiae at this time, and continued to suffer repeated respiratory infections, although not requiring hospitalization. At 6 years 6 months of age, he had his first episode of pneumonia and had several hospitalizations.
At this stage, a diagnosis of combined immunodeficiency was made: he had persistent absolute lymphopenia, normal T-cell receptor Vβ distribution, low numbers of T-cell receptor excision circles (TRECs), no mutation in the common γ chain gene, low IgG, IgA, and IgM. He was negative for HIV by PCR. He started immunoglobulin replacement (IVIg) and G-CSF treatment in early 2000 and has very much improved symptomatically since then, however, lymphocyte counts have remained severely decreased (Tables 1 and 2).
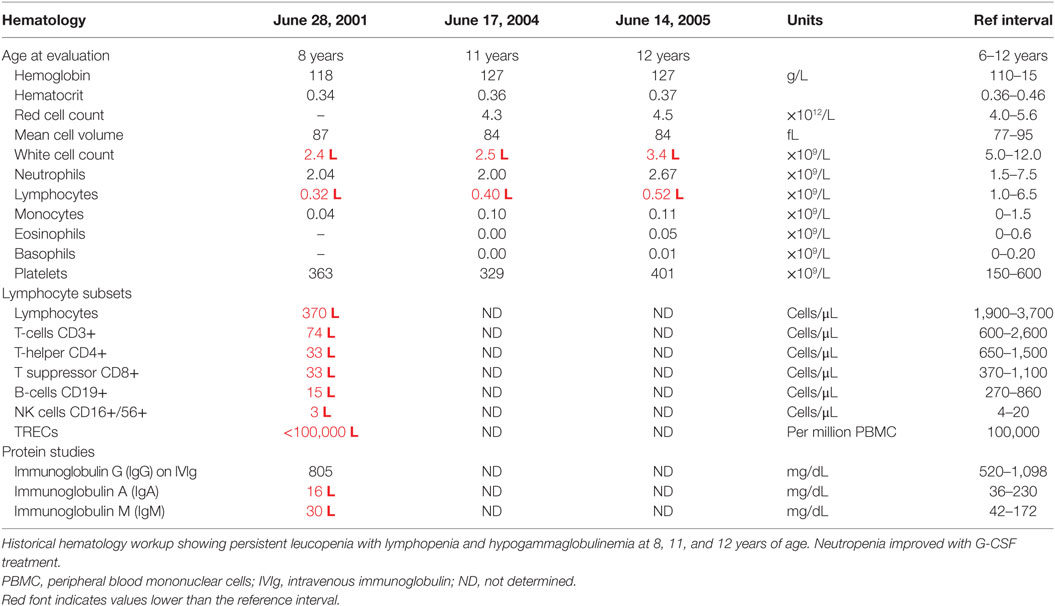
Table 1. Hematology laboratory workup for the proband.
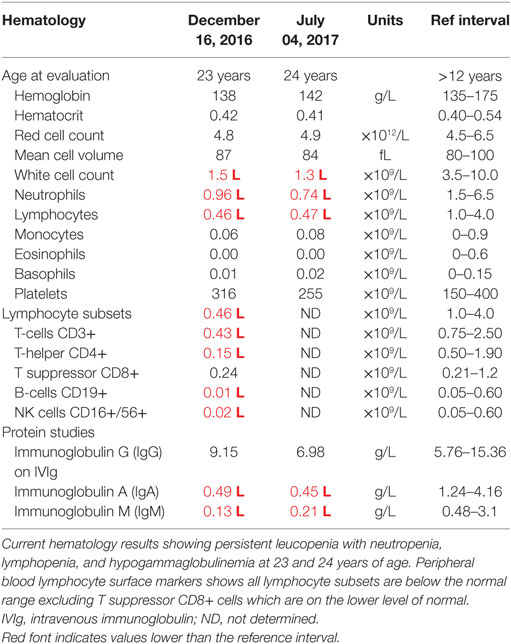
Table 2. Hematology laboratory workup for the proband.
We previously investigated a functional molecular cause for this undiagnosed immunodeficiency through whole genome arrays. Briefly, analysis of Affymetrix 250K SNP microarray data of the case and a matched healthy control subject via a copy number analysis tool identified hyperploidy of a region centromeric to chromosome 14q11.2, mapped over the interleukin 25 (IL25) gene (10). IL-25 (a member of the IL-17 family of cytokines) induction in mice has been previously associated with a T-helper (Th) 2-like pathological immune response (11) and shown to regulate the development of autoimmune inflammation mediated by IL-17-producing cells (12, 13) and was deemed a plausible candidate for further analyses.
Green et al. (10) paired genetic analysis with transcriptional profiling and found a large number of genes associated with Th1/Th2 profiles to be differentially expressed in peripheral blood lymphocytes of the proband, indicative of a Th2 bias confirmed by flow cytometric analysis. T-cells from the proband and control subject were cultured and activated in vitro with qPCR analysis demonstrating higher IL25 expression in the proband when compared to the control. The skewed Th2 immunity hypothesized in the proband was in line with his susceptibility to infections normally cleared by Th1 responses, such as Varicella and Pseudomonas. Further in vitro studies were performed in B-cell line models to measure the effect of IL-25 treatment on cellular proliferation and viability, however when proband and control lymphocytes were treated with exogenous IL-25, no differences were observed (data not shown).
Materials and Methods
Whole Exome Sequencing
Peripheral blood samples were obtained from all participants and genomic DNA (gDNA) extracted using the QIAamp DNA Blood Maxi Kit (Qiagen) according to the manufacturer’s instructions. Exome sequencing of the family trio (case, mother, and father) was performed on the Ion Chef and Ion Proton Next Generation Sequencing platform using the Ion Ampliseq Exome RDY Kit 4 × 2 (Life Technologies). DNA was quantitated using the Agilent 2100 Bioanalyser using the Agilent High Sensitivity DNA kit (Agilent Technologies) with 50 ng gDNA used for library preparation of each family member. Barcode adapters were ligated to each exome library with the Ion Express Barcode Adapters 1-96 Kit (Life Technologies). Using a Qubit 2.0 Fluorometer (Life Technologies), libraries were diluted to a concentration of 100 pM before being clonally amplified on the Ion Chef System using the Ion PI Chip v3 (Life Technologies) prior to loading on an Ion PI Chip v3 for sequencing via semiconduction.
WES Analysis
Raw sequences from each library were aligned to the GRCh37/Hg19 reference genome via the Ion Torrent Server TMAP alignment algorithm. Trio analysis was performed on the Ion Reporter Suite V.5.0 (Life Technologies) where variant annotation identified included single-nucleotide polymorphisms (SNPs) and indels (insertions and deletions) for each exome library. At a total read-depth of 20×, the target base coverage was 94.63%. A combined total of 51,997 variants were identified with 482 of these unique to the proband. Variant filtering then focused on deleterious variants (frameshift insertion and deletion, stoploss, missense, and nonsense) followed by screening using the OMIM public database as a filter for genes related to immunological disorders. 13 identified variants were then further filtered using in silico missense variant effect prediction tools with the following parameters designated as damaging: SIFT < 0.05, PolyPhen > 0.8, and Grantham Score > 100. SIFT and PolyPhen predict both the structural and functional impact of a variant based on properties such as accessible surface area, ligand contacts, solvent accessible area, and change in residue side change volume (14). The Grantham score focuses on the difference between amino acid atomic composition, polarity, and volume where a radical amino acid substitution gives a score above 100 (15). MutationTaster predicts whether a variant is tolerable, polymorphic, or damaging and disease-causing to identify potential disease-causing variants (16). Three final candidate variants were then verified using the Integrative Genome Viewer (IGV) software (Broad Institute). Copy number variant (CNV) analysis was performed on WES data for the case and family members using the same filtering steps as described. WES was used to indicate the general presence of the CNV, which was validated by touchdown PCR and Sanger sequencing. A more detailed description of the methods used can be found in the BMC Bioinformatics paper by Demidov et al. (17). With the ongoing advances in sequencing technology and improvement in alignment techniques for large-scale data analysis, we were able to confirm that IL25 does not lie within the previously identified amplified region on chromosome 14, but rather 4 Mb downstream.
Sanger Sequencing
PCR was performed using forward and reverse primers for the MSN (F: 5′-TTCTCTCCTGCACAGGGACTTT-3′; R: 5′-ATTCACCCTGTAAGGGAAGTGGG-3′), TET2 (F:5′-GCCTGATGGAACAGGATAGAAC-3′;R:5′-TTCCCTTCATACAGGGTATTC-3′), and NLRP8 (F:5′-ATATCCAGCGCCTGATAGCG-3′; R:5′-TCAGGGTGACGGTCAGTT-3′) single nucleotide variants (SNVs). Following amplification, PCR products were treated with Exo-SAP for use in Big Dye Terminator v3.1 sequencing reactions, analyzed on the 3500 Genetic Analyzer (ThermoFisher Scientific, Life Technologies). Sequencing data for each sample chromatogram was assessed using Chromas Lite 2.1.1 software.
Lymphocyte Isolation, Activation, and Expansion In Vitro
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized whole blood from the family trios and a healthy control subject, using the Ficoll-Histopaque centrifugation method (Sigma-Aldrich). Isolated PBMCs were washed in 1X PBS and plated into T-75 culture flasks in RPMI-1640 culture medium (Invitrogen, ThermoFisher) with phytohemagglutinin (PHA) mitogen for up to 24 h to allow monocytes to adhere to the bottom of the flask. After 3 h lymphocytes in suspension were removed and plated into a new T-75 flask with PHA and IL-2 (Sigma-Aldrich). Lymphocytes were allowed to proliferate in RPMI-1640 culture medium at 37°C in 5% CO2 for up to 5 days, and if necessary further expanded after 2 days. Live lymphocyte counts were performed on a hemocytometer and viability assessed by Trypan blue dye. Cell lysates were prepared for qPCR and Western Blot analyses with 4 × 105 to 3 × 106 cells.
Total RNA Extraction, cDNA Synthesis, and qPCR
Whole blood from a healthy control subject and family members was collected in PAXgene™ vacutainer tubes for immediate stabilization of intracellular RNA at collection. Total RNA was extracted using the PreAnalytiX PAXgene™ Blood RNA Kit according to the manufacturer’s instructions. Lymphocytes were harvested in TRIzol and total RNA extracted using the Direct-zol™ RNA MiniPrep kit (Zymo Research). Total RNA from whole blood and lymphocytes was converted to cDNA using the iScript™ cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. Samples for qPCR were assayed in triplicate using the Promega SYBR Green with 18S expression used as an endogenous control for normalization of the data. The ΔΔCt comparison method was used to measure relative gene expression on the QuantStudio™ 7 instrument (ThermoFisher) and analyzed using Student’s T-test to calculate statistical significance (p < 0.05).
Western Blotting
Total protein was extracted from cell lysates using Runx protein-lysis buffer (containing protease and phosphatase inhibitors) as previously described (18). Protein concentration was measured using the Qubit™ Protein Assay Kit (Invitrogen). A Western Blot using 30 µg protein per sample was performed using an anti-MSN primary antibody (ab52490, Abcam) and an HRP-conjugated secondary antibody (anti-Rabbit IgG #7074, Cell Signaling). Sample loading was normalized using HRP-conjugated anti-Beta-actin (#5125S, Cell Signaling). Detection of target protein was carried out with ECL (Clarity™ ECL, Bio-Rad) using the Fusion Spectra chemiluminescent system (Vilber Lourmat, Fisher Biotec) and optical density quantitation assessed using Bio-1D software.
Ethics and Cell Line Validation
This study was performed in accordance with the recommendations of the Queensland University of Technology Human Research Ethics Committee (Approval number 1400000125). All subjects gave written informed consent in accordance with the Declaration of Helsinki. Written informed consent was also obtained from the proband and family members for the publication of this case report. Cell line validations were performed for commercial Toledo non-Hodgkin lymphoma (NHL) B-cell and MCF-7 breast cancer cell lines (data not shown).
Results
Identification and Validation of a SIRPβ1 Deletion
Copy number variant analysis of the WES data identified a large >4 kbp deletion in the SIRPβ1 gene in the proband (chr20:1,576,140–1,580,375 Hg38) not present in other family members. Further inspection suggested the deletion in the proband encompassed exon 2 of the SIRPβ1 gene. Validation of this deletion by touchdown PCR and Sanger sequencing confirmed a deletion of exon 2, which has high sequence homology with another exon in the same gene (data not shown). Further Sanger validation to determine the size of the deletion was discontinued due to high sequence homology in that region. Furthermore, this exon 2 deletion has also been found in a number of healthy individuals (19); and therefore, this variant was considered to be an unlikely pathogenic candidate for the immunodeficiency.
Identification and Validation of a Hemizygous X-Linked MSN Mutation
Filtering of the WES data on the Ion Reporter Suite yielded three variants in three immune function genes TET2, NLRP8 and MSN with damaging SIFT and Polyphen scores predicted to be disease-causing/polymorphic by MutationTaster (Table 3). The TET2 SNP (rs111948941, C > T) has a frequency of 0.15. The homozygous case (T/T) inherited an affected allele from each parent, both of whom are heterozygous (C/T) at this locus. The NLRP8 variant (rs754128390, A > C) is very rare, occurring in <1/10,000 individuals. The proband was identified to be de novo heterozygous (A/C) with both parents homozygous wild type (A/A) at this locus. The MSN variant (c.511C > T, p.Arg171Trp at position chrX:64951012, Hg19) was a novel mutation, not reported in the genomic databases or the literature at the time of query. The proband is hemizygous X-linked for the mutation (-/T), inheriting the affected allele from his heterozygous (C/T) mother. The TET2 SNP and NLRP8 rare variant occur in healthy individuals with no known disease associations, therefore, they were considered unlikely candidates and not analyzed further. WES coverage for the MSN mutation was 145× for the proband, 119× for the father, 150× for the mother, with an average total coverage of 138×. The MSN mutation was validated by Sanger sequencing (Figure 1A) and confirmed using IGV for whole genome and exome data in all four family members (Figure 1B) and as such, considered to be the most likely candidate.
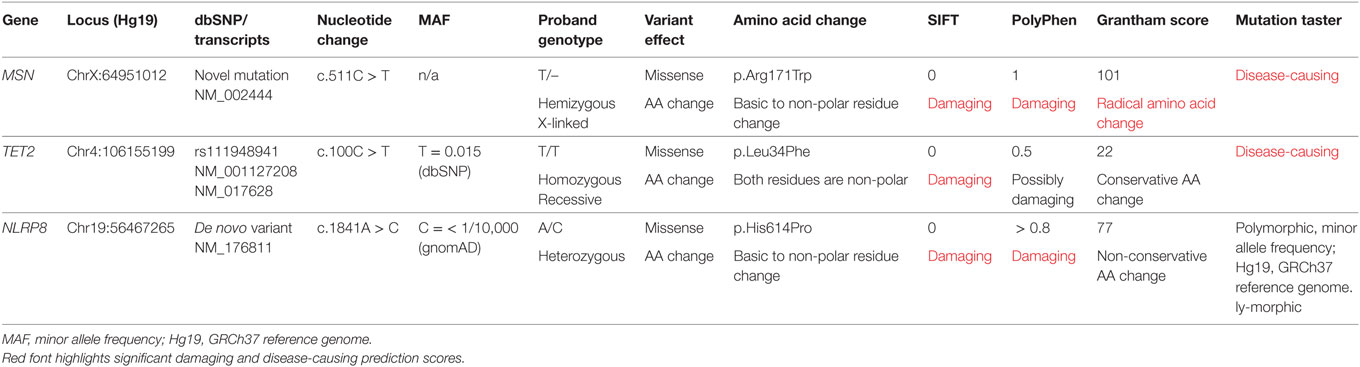
Table 3. Whole exome trio analysis data filtering and in silico prediction tools (SIFT, PolyPhen, Grantham Score, and MutationTaster) identified three damaging and disease-causing SNVs in the proband: MSN C > T, TET2 C > T, and NLRP8 A > C.
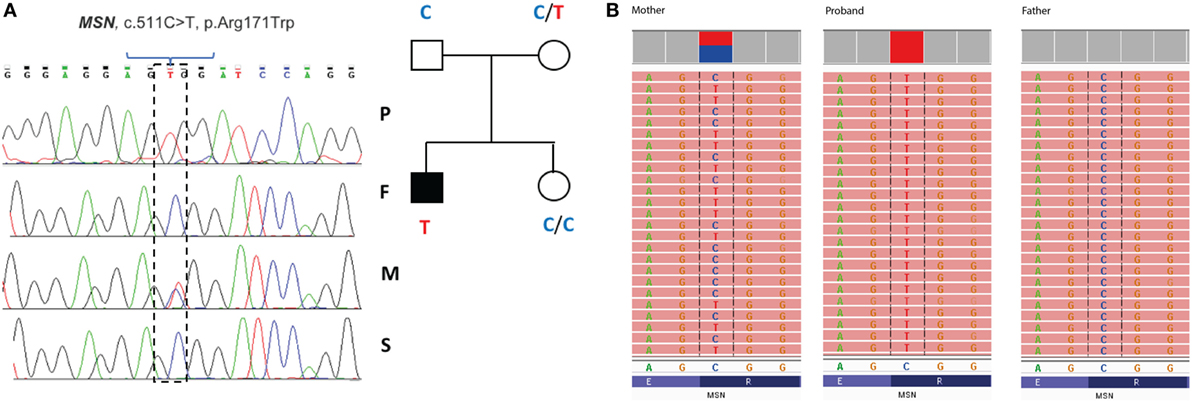
Figure 1. Validation of the R171WMSN mutation in family members. (A) Sanger sequence and pedigree tree diagram for the missense MSN C > T mutation. P, proband; F, father; M, mother; S, sister. Mother is heterozygous C/T marked by a double blue and red peak and proband is hemizygous –/T marked by a single red peak. (B) IGV software showing MSN genotypes for the mother (C/T), proband (–T), and father (C/C), respectively. IGV, integrative genomics viewer.
Protein Detection and Quantification
MSN encodes the moesin protein (MSN) which is a member of the ezrin-radixin-moesin (ERM) family of cell structure-related proteins. Western Blot analysis confirmed the presence of the 68 kDa MSN protein in the Toledo NHL B-cell line (positive control) and absence of MSN in the MCF-7 breast cancer cell line (negative control). MSN was shown to be present in the healthy control, father, mother, and is absent in the proband (Figure 2A). Relative MSN concentrations were normalized against beta-actin concentrations where the MCF-7 cells and the proband show very low MSN concentrations (<0.5 μg; Figure 2B). These data confirm the proband has a MSN protein deficiency, not present in the healthy control or parents, and is the most likely sole genetic cause for the persistent lymphopenia.
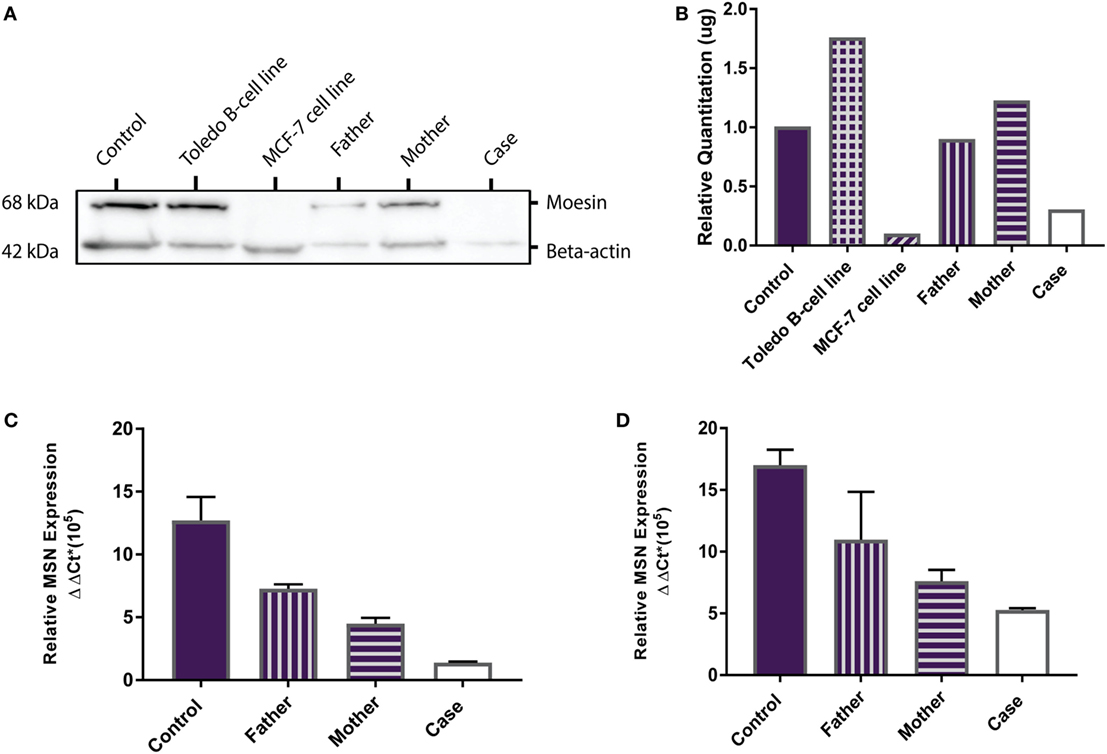
Figure 2. MSN protein detection and MSN gene expression. (A) Western Blot shows absence of the 68 kDa moesin protein in the MCF-7 cell line (negative control, lane 3) and in the proband (lane 6), and presence of moesin in the healthy control subject (lane 1), Toledo B-cell line (positive control, lane 2), father (lane 4), and mother (lane 5). Beta-actin (42 kDa) was used as the loading control. (B) Optical density protein quantitation software showed very low levels of moesin (<0.5 μg) in the MCF-7 cells and the proband, confirming a moesin deficiency. (C) qPCR analysis of MSN mRNA transcript levels in whole blood (PAXgene) from the control subject, father, mother and proband. Error bars depict standard error of the mean (SEM) between triplicates. (D) qPCR analysis of MSN mRNA transcript levels in lymphocytes from the control subject, father, mother, and case. Error bars depict SEM between triplicates.
MSN mRNA Transcript Expression
MSN gene expression was measured in all samples (healthy control, father, mother and proband). In whole blood the proband had significantly lower moesin mRNA expression than the control (p = 0.02), father (p = 0.002) and mother (p = 0.01) and the mother had significantly lower expression than the father (p = 0.01) and control (p = 0.04) (Figure 2C). In isolated lymphocytes, the proband (p = 0.01) and mother (p = 0.004) had significantly lower expression than the control (Figure 2D). Overall expression levels in all participants were higher in lymphocytes than in whole blood, with MSN mainly expressed in lymphocytes, however, it is also expressed in monocytes, neutrophils and platelets. The lower levels observed in the mother compared to the father and control and higher levels than the proband are likely due to the heterozygous C > T mutation in the MSN gene. High levels were observed in the healthy control who was age- and sex-matched to the proband.
Discussion
The variant identified here in the MSN gene is a missense, hemizygous, single-base mutation in the proband. This mutation was not identified in current online databases (UCSC Genome Browser, gnomAD) with no reference number and no disease association reported in OMIM at the time of query (March 2016). A genome-wide association study (GWAS) search in the GWAS Catalog (20) produced no results for disease-trait-associated variants in the MSN gene, or other polymorphisms within the MSN gene.
More recently an OMIM entry for Immunodeficiency-50 was identified describing a novel X-linked moesin-associated immunodeficiency (X-MAID) (21). This was the first study to document a moesin-associated disease in humans and describes six male cases from four unrelated families with the same R171WMSN missense mutation as identified in the proband. A seventh case had a different mutation, p.R553X, which caused a frameshift and truncation of MSN in the F-actin binding domain. The authors show an impact on lymphocyte function in vitro where moesin deficiency impaired proliferation following activation with mitogen, increased adhesion, and reduced migration (21).
Moesin is a member of the ERM family of cell structure-related proteins that regulate the cell’s actin cytoskeleton (5, 22). The mutation identified results in a radical amino acid substitution from basic arginine to non-polar tryptophan within the functional four-point-one, ezrin, radixin, moesin domain. However, interestingly both our study and that of Lagresle-Peyrou et al. suggest that rather than interfering with protein function, the R171WMSN mutation triggers degradation of moesin mRNA in lymphocytes with a loss of expression at the transcript and protein level (21). Moesin has been shown to have a crucial and non-redundant role in lymphocyte homeostasis in mice (23). As such, a lack of moesin protein could prevent efficient lymphocyte migration and egress from lymphoid organs causing persistent absolute lymphopenia in the peripheral blood, as observed in the case. The low TRECs exhibited by the proband at a young age, are an indicator of recent thymic emigrants (24). We compared the Lagresle-Peyrou et al. cases’ (P1-P7) and proband phenotypes in more detail and we surmise the P3 case would be the most similar to the proband in terms of clinical outcomes due to both individuals presenting with no eczema, no Molluscum contagiosum, and presentation of an autoimmune reaction in the form of TTP (P3) and ITP (proband). Considering we cannot confirm whether the P3 case has an IL25 or NLRP8 variant which could play a role in the absence of eczema or M. contagiosum, it would be difficult to determine whether these variants are playing a role in the proband. In addition, as there are no outstanding differences between the proband and all of the other cases, we can be more confident that the R171MSN mutation is the sole molecular cause for this disease in the proband (Table 4).
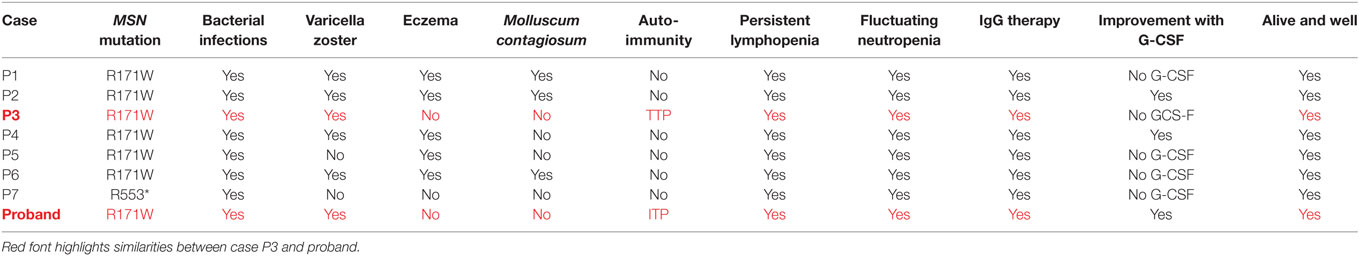
Table 4. Comparison of clinical outcomes for cases P1–P7 (Lagresle-Peyrou et al.) and the proband.
Infants with genetic defects of the immune system that cause severe combined immunodeficiency, or SCID, are effectively identified by population-wide screening practices in the United States (25). Newborns are routinely screened for SCID by using qPCR to quantitate TRECs from DNA extracted from dried blood spots (24); however, this is a laborious assay with a number of limitations. Recently, a newborn case with an R171WMSN mutation was identified by WES (26) when undergoing screening for SCID. Together with our report of an Australian case with X-MAID due to the R171WMSN mutation, this data suggests that although rare, it is a site of recurrent mutation.
Concluding Remarks
Together with our case report, the findings by Lagresle-Peyrou et al. and Delmonte et al. confirm the effectiveness and validity of WES as a diagnostic method for PIDs and SCID, as well as an investigative method for the identification of novel or rare variants causing novel forms of PID not yet been documented. Identification of these rare and novel variants early in the patient’s life will not only aid with swift intervention prior to life-threatening infections, but may also provide possibility for application of gene therapies and personalized treatment options for these patients.
Ethics Statement
This study was performed in accordance with the recommendations of the Queensland University of Technology Human Research Ethics Committee (approval number 1400000125). All subjects gave written informed consent in accordance with the Declaration of Helsinki. Cell line validations were performed for commercial Pfeiffer non-Hodgkin lymphoma (NHL) B-cell and MCF-7 breast cancer cell lines (data not shown).
Author Contributions
GB performed candidate gene validation, designed qPCR primers, performed cell culture, functional experiments and analysis, and wrote the first draft of the manuscript; RRL, NM performed exome sequencing trio analysis; RRL performed candidate gene validations and drafted the exome sequencing method and analysis sections of the manuscript; CA, NM, RAL, MB, DE performed sequencing data analysis and interpretation. DR-A, CA, NM, and RS designed PCR primers for candidate gene validations. HS, LH, and LG conceptualized the study, contributed to data interpretation and edited the final version of the manuscript. LG provided funding support and finalized the manuscript for submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Dr. Rachel Okolicsanyi and Chieh (Jade) Yu for sharing their cell culture expertise and K. M. Taufiql Arif for sharing his Western Blot expertise. We would like to sincerely thank the case and family members, as well as healthy control subjects, who agreed to donate blood and share their medical information for this study.
Funding
Funding for this study has been generously provided by the Herbert family. Funding has also been provided by the GRC Genomics Lymphoma Project fund.
References
1. Rezaei N, Moazzami K, Aghamohammadi A, Klein C. Neutropenia and primary immunodeficiency diseases. Int Rev Immunol (2009) 28(5):335–66. doi:10.1080/08830180902995645
2. Tran H, Nourse J, Hall S, Green M, Griffiths L, Gandhi MK. Immunodeficiency-associated lymphomas. Blood Rev (2008) 22(5):261–81. doi:10.1016/j.blre.2008.03.009
3. Su H. Studying human immunodeficiencies in humans: advances in fundamental concepts and therapeutic interventions. F1000Res (2017) 6:318. doi:10.12688/f1000research.10594.1
4. Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova JL, Chatila T, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol (2015) 35(8):727–38. doi:10.1007/s10875-015-0198-5
5. Burns SO, Zarafov A, Thrasher AJ. Primary immunodeficiencies due to abnormalities of the actin cytoskeleton. Curr Opin Hematol (2017) 24(1):16–22. doi:10.1097/MOH.0000000000000296
6. Seleman M, Hoyos-Bachiloglu R, Geha RS, Chou J. Uses of next-generation sequencing technologies for the diagnosis of primary immunodeficiencies. Front Immunol (2017) 8(847). doi:10.3389/fimmu.2017.00847
7. Rabbani B, Mahdieh N, Hosomichi K, Nakaoka H, Inoue I. Next-generation sequencing: impact of exome sequencing in characterizing Mendelian disorders. J Hum Genet (2012) 57(10):621–32. doi:10.1038/jhg.2012.91
8. Platt C, Geha RS, Chou J. Gene hunting in the genomic era: approaches to diagnostic dilemmas in patients with primary immunodeficiencies. J Allergy Clin Immunol (2014) 134(2):262–8. doi:10.1016/j.jaci.2013.08.021
9. Biesecker LG, Shianna KV, Mullikin JC. Exome sequencing: the expert view. Genome Biol (2011) 12(9):128. doi:10.1186/gb-2011-12-9-128
10. Green MR, Camilleri E, Gandhi MK, Peake J, Griffiths LR. A novel immunodeficiency disorder characterized by genetic amplification of interleukin 25. Genes Immun (2011) 12(8):663–6. doi:10.1038/gene.2011.50
11. Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 Induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity (2001) 15(6):985–95. doi:10.1016/S1074-7613(01)00243-6
12. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med (2007) 204(1):161–70. doi:10.1084/jem.20061738
13. Wang Y-H, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC–activated Th2 memory cells. J Exp Med (2007) 204(8):1837–47. doi:10.1084/jem.20070406
14. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet (2013) 76(7):.20–.27. doi:10.1002/0471142905.hg0720s76
15. Tavtigian SV, Greenblatt MS, Lesueur F, Byrnes GB; IARC Unclassified Genetic Variants Working Group. In silico analysis of missense substitutions using sequence-alignment based methods. Hum Mutat (2008) 29(11):1327–36. doi:10.1002/humu.20892
16. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods (2010) 7(8):575–6. doi:10.1038/nmeth0810-575
17. Demidov G, Simakova T, Vnuchkova J, Bragin A. A statistical approach to detection of copy number variations in PCR-enriched targeted sequencing data. BMC Bioinformatics (2016) 17:429. doi:10.1186/s12859-016-1272-6
18. Oikari LE, Okolicsanyi RK, Qin A, Yu C, Griffiths LR, Haupt LM. Cell surface heparan sulfate proteoglycans as novel markers of human neural stem cell fate determination. Stem Cell Res (2016) 16(1):92–104. doi:10.1016/j.scr.2015.12.011
19. Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, et al. Mapping and sequencing of structural variation from eight human genomes. Nature (2008) 453(7191):56–64. doi:10.1038/nature06862
20. MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res (2017) 45(D1):D896–D901. doi:10.1093/nar/gkw1133
21. Lagresle-Peyrou C, Luce S, Ouchani F, Soheili TS, Sadek H, Chouteau M, et al. X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. J Allergy ClinImmunol (2016) 138(6):1681.e–9.e. doi:10.1016/j.jaci.2016.04.032
22. Fernando H, Martin TA, Douglas-Jones A, Kynaston HG, Mansel RE, Jiang WG. Expression of the ERM family members (ezrin, radixin and moesin) in breast cancer. Exp Ther Med (2010) 1(1):153–60. doi:10.3892/etm_00000025
23. Hirata T, Nomachi A, Tohya K, Miyasaka M, Tsukita S, Watanabe T, et al. Moesin-deficient mice reveal a non-redundant role for moesin in lymphocyte homeostasis. Int Immunol (2012) 24(11):705–17. doi:10.1093/intimm/dxs077
24. Baker MW, Grossman WJ, Laessig RH, Hoffman GL, Brokopp CD, Kurtycz DF, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy ClinImmunol (2009) 124(3):522–7. doi:10.1016/j.jaci.2009.04.007
25. Kwan A, Abraham RS, Currier R, Brower A, Andruszewski K, Abbott JK, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA (2014) 312(7):729–38. doi:10.1001/jama.2014.9132
Keywords: lymphopenia, whole exome sequencing, moesin, MSN, X-linked moesin-associated immunodeficiency
Citation: Bradshaw G, Lualhati RR, Albury CL, Maksemous N, Roos-Araujo D, Smith RA, Benton MC, Eccles DA, Lea RA, Sutherland HG, Haupt LM and Griffiths LR (2018) Exome Sequencing Diagnoses X-Linked Moesin-Associated Immunodeficiency in a Primary Immunodeficiency Case. Front. Immunol. 9:420. doi: 10.3389/fimmu.2018.00420
Received: 01 December 2017; Accepted: 15 February 2018;
Published: 05 March 2018
Edited by:
Antonio Condino-Neto, University of São Paulo, BrazilReviewed by:
Ivan K. Chinn, Baylor College of Medicine, United StatesMichel Massaad, American University of Beirut Medical Center, Lebanon
Copyright: © 2018 Bradshaw, Lualhati, Albury, Maksemous, Roos-Araujo, Smith, Benton, Eccles, Lea, Sutherland, Haupt and Griffiths. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lyn R. Griffiths, bHluLmdyaWZmaXRoc0BxdXQuZWR1LmF1