 Paola De Cicco
Paola De Cicco Theodore Sanders2
Theodore Sanders2 Giuseppe Cirino
Giuseppe Cirino Kevin J. Maloy
Kevin J. Maloy Angela Ianaro
Angela Ianaro- 1Department of Pharmacy, University of Naples Federico II, Naples, Italy
- 2Sir William Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
Chronic inflammation contributes to tumor initiation in colitis-associated colorectal cancer (CRC). Indeed, inflammatory bowel disease (IBD) patients show an increased risk of developing CRC. Cancer immune evasion is a major issue in CRC and preclinical and clinical evidence has defined a critical role for myeloid-derived suppressor cells (MDSCs) that contribute to tumor growth and progression by suppressing T-cells and modulating innate immune responses. MDSCs comprise a heterogeneous population of immature myeloid cells that can be distinct in two subtypes: CD11b+Ly6G+Ly6Clow with granulocytic phenotype (G-MDSCs) and CD11b+Ly6G−Ly6Chigh with monocytic phenotype (M-MDSCs). Hydrogen sulfide (H2S) is an endogenous gaseous signaling molecule that regulates various physiological and pathophysiological functions. In particular, several studies support its anti-inflammatory activity in experimental colitis and ulcer. However, the role of the H2S pathway in innate immune-mediated IBD has not yet been elucidated. To better define a possible link between MDSCs and H2S pathway in colitis-associated CRC development, we used an innate immune-mediated IBD model induced by infection with the bacterium Helicobacter hepaticus (Hh), closely resembling human IBD. Here, we demonstrated an involvement of MDSCs in colitis development. A significant time-dependent increase of both G-MDSCs and M-MDSCs was observed in the colon and in the spleen of Hh-infected mice. Following, we observed that chronic oral administration of the H2S donor DATS reduced colon inflammation by limiting the recruitment of G-MDSCs in the colon of Hh-infected mice. Thus, we identify the metabolic pathway l-cysteine/H2S as a possible new player in the immunosuppressive mechanism responsible for the MDSCs-promoted colitis-associated cancer development.
Introduction
Colorectal cancer (CRC) is one of the major cause of morbidity and mortality throughout the world. It is the third most common cancer worldwide1. Chronic intestinal inflammation is the primary risk factor for the development of gastrointestinal malignancy (1). A meta-analysis estimates the risk of CRC in ulcerative colitis patients to be 2% after 10 years, 8% after 20 years, and 18% after 30 years of disease (2). Although the exact etiology remains unclear, a multifactorial interaction among immunological, genetic, and environmental factors contribute to the disturbance of homeostasis leading to the generation of an abnormal immune response against the commensal microbiota (3, 4). In particular, loss of the intestinal homeostasis and induction of pathogenic inflammatory response is first related to the aberrant activation of the innate immune system in the gut that consists of intestinal epithelial cells and several leukocyte population (i.e., neutrophils, dendritic cells, monocytes/macrophages, and innate lymphoid cells) (5). Cancer immune evasion is a major issue in CRC and preclinical and clinical evidence has defined a critical role for myeloid-derived suppressor cells (MDSCs) in modulating the innate immune responses by suppressing T cell anti-tumor functions (6). MDSCs consist of a heterogeneous population of immature myeloid cells characterized by co-expression of CD11b and Gr-1 (7). Two distinct subtypes of MDSCs have been identified in tumor-bearing mice: CD11b+Ly6G+Ly6Clow with granulocytic phenotype (G-MDSCs) and CD11b+Ly6G−Ly6Chigh with monocytic phenotype (M-MDSCs). Both M-MDSCs and G-MDSCs exercise their potent immunosuppressive activity by modifying the microenvironment through the depletion of amino acids (arginine, tryptophan, glutamine, and cysteine). So far, the main immunosuppressive mechanisms described are based on l-arginine metabolism via Arginase 1 (ARG1) and inducible nitric oxide synthase 2 (NOS2) and reactive oxygen species production (8–10). In CRC, the blood MDSCs numbers correlate with stage and metastatic burden (11). Similar G-MDSCs and M-MDSCs populations have also been described in inflammatory bowel diseases (IBD). However, their role in both IBD and CRC still needs to be elucidated (12).
Helicobacter species that colonize the lower bowel and biliary tract of mice have been associated with the development of colitis resembling human IBD in susceptible hosts. Furthermore, Helicobacter hepaticus (Hh) infection has been demonstrated to exacerbate the development of cancer at both intestinal and extra-intestinal sites (13). To better define the role of MDSCs in colitis development, we used an innate immune-mediated IBD model induced by infection with Hh. Epithelial hyperplasia and crypt abscesses, associated with a marked granulocyte accumulation within intestinal tissues, characterize this preclinical model closely resembling the human IBD (14). Hh is a gram-negative, spiral-shaped, microaerophilic bacterium that is a common member of the mouse intestinal microbiota found predominantly in the cecum and colon. Although it does not cause invasive infections in most immune competent mouse strains, Hh induces chronic intestinal inflammation in susceptible mice lacking adaptive immune system, such as 129SvEvRag−/− mice, that can eventually progress to colon cancer (13).
Recently, it has been demonstrated that hydrogen sulfide (H2S) promotes the resolution of colitis and enhance ulcer healing (15, 16). H2S is an endogenous gaseous signaling molecule that regulates various physiological and pathophysiological functions. In particular, H2S exhibits several anti-inflammatory effects such as reduction of edema formation and suppression of pro-inflammatory cytokines release (17). In mammals, H2S is endogenously produced from sulfur-containing amino acids, such as l-cysteine (l-cys), mainly by two pyridoxal-5′-phosphate (P5P)-dependent enzymes, cystathionine gamma-lyase (CSE, EC 4.4.1.1), and cystathionine beta-synthase (CBS, EC4.2.1.22). H2S is also generated from dietary sulfate metabolism in the lumen of the large intestine by anaerobic sulfate-reducing bacteria (18). The role of H2S colonic inflammation and cancer has been recently reviewed (18–20). However, the role of hydrogen sulfide pathway in innate immune-mediated IBD has not yet been elucidated. Here, we have evaluated the role of MDSCs and its link with the hydrogen sulfide pathway in the intestinal inflammation development upon stimulation with pathogenic Hh.
Materials and Methods
Animals
129SvEvRag2−/− (Rag2−/−) mice were bred and maintained under specific pathogen-free conditions in an accredited animal facility in the Pathology Services Building at the University of Oxford. Experiments were conducted in accordance with the UK Scientific Procedures Act (1986) under a Project License (PPL) authorized by the UK Home Office Animal Procedures Committee and approved by the Sir William Dunn School Ethical Review Committee.
Induction of Colitis
Helicobacter hepaticus NCI-Frederick isolate 1A (strain 51449) was grown on blood agar plates containing trimethoprim, vancomycin, and polymyxin B under microaerophilic conditions. Cultures were expanded for 3–4 days in TSB (Oxoid) containing 10% FCS (GIBCO BRL) and Helicobacter-free Rag2−/− mice were fed three times on consecutive days with Hh 1A (1.0 × 108 CFU) by oral gavage. Mice were sacrificed at different time points (3 and 6 weeks) after the first Hh inoculation.
In Vivo Drug Treatment
Helicobacter hepaticus-infected Rag2−/− mice received diallyl trisulfide (DATS; 50 mg/kg) or vehicle (PBS) starting at week 4 after the first Hh inoculation. DATS or vehicle were given by oral gavage once a day for 14 days. Treatment groups were mixed in cages to minimize cage effects.
Assessment of Intestinal Inflammation
Mice were sacrificed when symptoms of clinical disease (diarrhea) became apparent in control groups, usually 6 weeks after initiation of experiments. Samples of proximal, mid, and distal colon were immediately fixed in buffered 10% formalin. Four to five microns of paraffin-embedded sections were stained with hematoxylin and eosin, and inflammation was graded according to the following scoring system. Each sample was graded semi-quantitatively from 0 to 3 for four criteria: (1) degree of epithelial hyperplasia and goblet cell depletion; (2) leukocyte infiltration in the lamina propria; (3) area of tissue affected; (4) presence of markers of severe inflammation such as crypt abscesses, submucosal inflammation, and ulcers. Typical features of each grade are as follows: 0 = normal; 1 = mild epithelial hyperplasia; 2 = pronounced hyperplasia with substantial leukocytic infiltrates; 3 = severe hyperplasia severe transmural inflammation, ulceration, crypt abscesses, and severe depletion of goblet cells. Scores for each criterion were added to give an overall inflammation score for each sample of 0–12. The individual scores from the section of proximal, mid, and distal colon were averaged to obtain the total inflammation scores for the colon.
Isolation of Leukocyte Subpopulations from Spleen and Colon
Cell suspensions from spleen and colon from infected and uninfected Rag2−/− mice were prepared as described below. Colons were longitudinally opened, cut into 1-cm pieces, and incubated (three times) in RPMI 1640 with 10% FCS and 5 mM EDTA at 37°C to remove epithelial cells. Tissue was then digested with collagenase VIII/DNase I solution for 45 min at 37°C. The isolated cells were layered on a 30/40/75% Percoll gradient, which was centrifuged for 20 min at 600 g, and the 40/75% interface, containing mostly leukocytes, was recovered. Spleens were crushed in petri dish on filter with syringe. The cells were collected and suspended in ACK for the lysis of red blood cells and then centrifuge at 1,500 rpm for 5 min. Cells were then analyzed using flow cytometry.
Flow Cytometry
Aliquots of 5 × 105 cells were washed in FACS buffer (PBS, 0.1% BSA), incubated with a fixable viability dye and anti-Fc receptor (αCD16/32), and stained using the following panel of monoclonal antibodies to murine cell surface molecules (all from BD Biosciences): PerCP-Cy5.5-conjugated anti-CD11b, PE-conjugated anti-Ly6G, FITC-conjugated anti-Ly6C, and Violet1-conjugated anti-CD45. Cells were washed in FACS buffer and analyzed by Dako Cyan Flow cytometry.
H2S Synthesizing Activity in Colon
Samples from colon of 1 cm from infected and uninfected Rag2−/− mice were taken from the proximal, mid, and distal region, snap frozen in liquid nitrogen and stored at −80°C until the H2S assay was performed. Colon pieces were homogenized in ice-cold 100 mmol/L potassium phosphate buffer (pH = 7.4), sodium orthovanadate 10 mM, PMSF 100 mM, and protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) with a FastPrep™24 homogenizer, and the protein concentration was determined using the Bradford assay. H2S synthesis from colon tissue homogenates was measured in the presence of the exogenous substrate l-cys and the cofactor required by H2S-producing enzymes, P5P and in the presence of the CSE inhibitor (dl-propargylglycine; PAG) or CBS inhibitor (O-carboxymethyl-hydroxylamine hemihydrochloride, CHH). The lysates were added in a reaction mixture (total volume 500 µL) containing P5P (2 mM, 20 µL), l-cys (10 mM, 20 µL), and saline (30 µL) or P5P (2 mM, 20 µL), L-cys (2 mM, 20 µL), and CHH/PAG (30 µL). The reaction was performed in parafilm-sealed Eppendorf tubes and initiated by transferring tubes from ice to a 37°C water bath. After 40 min incubation, zinc acetate 1% (ZnAc; 250 µL) was added to trap any H2S emitted followed by trichloroacetic acid 10% (TCA; 250 µL). Subsequently, N,N-dimethylphenylendiamminesulphate 20 µM (DPD; 133 µL) in 7.2 M HCl and FeCl3 (30 µM, 133 µL) in 1.2 M HCl were added. After 20 min, absorbance values was measured at 670 nm with a microplate reader and H2S concentration was calculated against a calibration curve of NaHS (3.12–250 µM). Results are expressed as nanomoles per milligram protein per minute.
Immunoblot
Cystathionine beta-synthase and CSE intestinal levels was performed by Western blot analysis of colonic tissues of either healthy mice or mice 6 weeks following the induction of colitis. Tissue homogenates were obtained as described above. Equal amounts of protein (40 µg) were loaded onto a 10% gel, subjected to SDS-PAGE, and electro-transferred onto polyvinylidene difluoride (PVDF) membranes (Hybond-P PVDF Membrane, Amersham Biosciences, Buckinghamshire, UK). The membranes were blocked for 2 h in 5% low-fat milk in PBS with 0.1% Tween 20 (PBST) at room temperature. Then, the filters were incubated with the following primary antibodies: CSE (1:500 dilution, Proteintech), CBS (1:500, Novus Biological), and GAPDH (1:1,000 dilution, Santa Cruz Biotechnology) overnight at 4°C. The membranes were washed three times with PBST and then incubated with HRP-conjugated anti-mouse or anti-rabbit IgG (1:2,000, Cell Signaling) for 2 h at room temperature. The immune complexes were detected by the ECL chemiluminescence method (Thermo Fisher Scientific).
RNA Purification and Quantitative Real-Time PCR
Colon samples were snap frozen in liquid nitrogen. Tissue material was homogenized in RLT buffer (QIAGEN) with β-mercaptoethanol using FastPrep™24 homogenizer (MP Biomedicals) with lysing matrix D beads (MP Biomedicals). RNA isolation was performed using the RNeasy kit (QIAGEN). RNA purity and quantification was determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific). cDNA synthesis was performed using the Superscript III reverse transcription kit from Invitrogen. Quantitative real-time PCR for the candidate genes was carried out as described before (21) using the following primer pairs: CBS isoform 1: 5′-CCAGGCACCTGTGGTCAAC-3′ and 3′-GGTCTCGTATTGGATCTGCT-5′; CSE: 5′-TTCCTGCCTAGTTTCCAGCAT-3′, and 3′-GGAAGTCCTGCTTAAATGTGGTG-5′; IL-6: 5′-GAGGATACCACTCCCAACAGACC-3′ and 3′-AAGTGCATCATCGTTGTTCATACA-5′; TNF-α: 5′-TACTGAACTTCGGGGTGATTGGTCC-3′ and 3′-CCTGGTTAGTGGGGCTTCAAGTCAT-5′. Primers for Hprt (Mm01545399_m1), was obtained from Applied Biosystems. cDNA samples were analyzed in triplicate and values were normalized on Hprt expression. Analysis was performed according to the ∆−Ct method.
Quantification of Hh in Cecal Contents
DNA was purified from cecal content taken from Hh-infected mice using the DNA Stool kit (QIAGEN). Hh DNA was quantified using a real-time PCR method based on the cdtB gene, performed with the ABI prism Taqman 7700 sequence detection system (PE Biosystems). Standard curves were constructed using Hh DNA that was purified from bacterial cultures using the DNeasy kit (QIAGEN).
In Vitro Stimulation of Bone Marrow-Derived Macrophages (BMDMs)
Bone marrow cells were isolated from femur and tibia of C57/BL6 mice and were cultured in L929 fibroblast conditioned RPMI 1640 medium (containing 15% L929 supernatant and 10% FCS) for 7 days. The resulting macrophages were stimulated over night with 20 MOI of live Hh. Cells were pre-incubated with DATS (10 µM) 30 min before the stimulation with Hh. Then, the supernatants were collected for the detection of IL-6 and TNFα cytokines by ELISA.
ELISA
IL-6 and TNFα levels in culture supernatants were evaluated using ELISA kits according to the manufacturer’s instruction (DuoSet ELISA, R&D systems, Minneapolis, MN, USA).
Statistical Analysis
Values were expressed as mean ± SEM. Differences between experimental groups were assessed by Student’s t-test or one-way ANOVA, followed by Bonferroni test P < 0.05 was considered statistically significant: *P < 0.05; **P < 0.01; ***P < 0.001.
Results
MDSCs Accumulate in Spleen and in Colon of Hh-Infected Mice
T cell- and B cell-deficient Rag2−/− mice were infected with Hh. As early as 3–6 weeks after Hh infection, mice developed splenomegaly and severe intestinal inflammation (Figures 1A,B). The response was dependent upon the activation of the innate immunity and characterized by a marked epithelial cell hyperplasia, extensive inflammatory infiltrates, and goblet cell depletion. Hh infection of Rag2−/− mice resulted in a profound proliferation of both splenic and intestinal leukocytes as previously shown (14) (data not shown). To verify the presence of MDSCs in the splenic and colon lamina propria lymphoid immune population, we performed a FACS staining for the two MDSCs subpopulation based on differential expression of Ly6C and Ly6G (Figure 1C). In fact, as previously described (9), the mononuclear fraction (M-MDSCs) is characterized by CD11b+Ly6G−Ly6Chigh phenotype and the polimorfononuclear fraction (G-MDSCs) is characterized by CD11b+Ly6G+Ly6Clow phenotype. The progressive intestinal inflammation induced by Hh in Rag2−/− mice induced a significant (P < 0.001), time-dependent increase in the frequency of each fractions both in colon (Figures 1D,E) and spleen lymphoid immune populations (Figures 1F,G). Three weeks after infection, a marked expansion of both G-MDSCs and M-MDSCs was observed. Conversely, after 3 weeks more (at 6 weeks), only the G-MDSC populations resulted increased by about 20%, whereas M-MDSCs remained almost unchanged. These results suggest that the polimorfonuclear fraction might have a pivotal role in colitis development in this inflammatory setting.
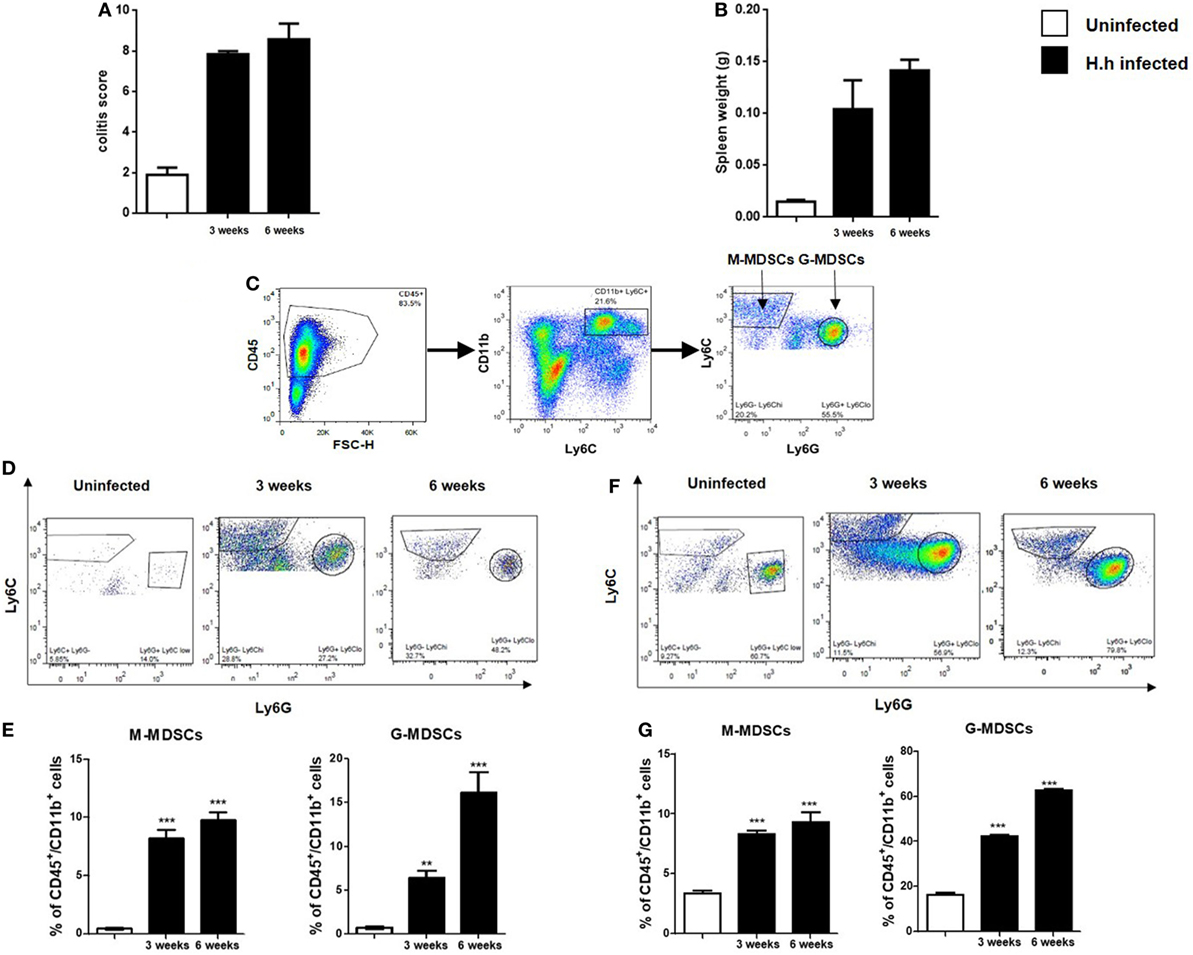
Figure 1. Helicobacter hepaticus (Hh) infection induces myeloid-derived suppressor cells (MDSCs) accumulation. 129SvEv Rag2−/− mice were infected with Hh and sacrificed after 3 or 6 weeks after infection. (A) Scores of colon inflammation assessed by histological analysis. (B) Spleen weights. (C) Representative FACS plots showing the gating strategy used to identify MDSCs subpopulations. Following a leukocytes gate (CD45+), cells were further gated for CD11b+ myeloid cells. Ly6C and Ly6G were used to distinguish M-MDSCs and G-MDSCs. (D–G) Flow cytometric analysis of M-MDSCs and G-MDSCs populations as gated in panel (C) and relative quantification in colon (D,E) and spleen (F,G) of 129SvEv Rag2−/− uninfected or Hh-infected mice. The frequency of G-MDSCs and M-MDSCs significantly increased in time-dependent manner in both colon and spleen. In particular, 6 weeks post-infection, the accumulation of G-MDSCs was about 20- and 30-fold higher in colon and spleen, respectively, as compared to uninfected mice. On the other hand, intestinal or splenic M-MDSCs fraction only increased by 10% in Hh-infected mice 6 weeks post-infection. Data are shown as mean ± SEM (n = 5 per group) (**P < 0.01, ***P < 0.001 vs. uninfected).
Colonic H2S Synthesis Is Markedly Reduced After Hh Infection
Both CBS and CSE are constitutively expressed in the colon of healthy Rag2−/− mice with no significant difference between CBS/CSE protein (Figure 2A) and gene expression (Figure 2B). Thus, we evaluated the metabolic activity of these enzymes by measuring their ability to produce sulfide adding the substrate l-cys and their respective inhibitors. In healthy mice, we found that the preferential CBS inhibitor CHH (3 mmol/L) reduced H2S synthesis by ~65% (P < 0.001, vs. l-cys), whereas the selective CSE inhibitor PAG (10 mmol/L) was ineffective (Figure 2C). These results imply that in the colon of Rag2−/− mice: (i) CBS appears to be the major responsible for colonic H2S synthesis and (ii) change in the CBS activity accounts for H2S production. After 3 and 6 weeks following Hh mouse infection, we observed a significant reduction in the expression of both CBS protein and mRNA (P < 0.01 and P < 0.001 vs. uninfected, respectively; Figures 2A,B). This effect translated into a significant reduction of H2S levels in inflamed colon (Figure 2D). CSE levels were left unchanged (Figures 2A,B).
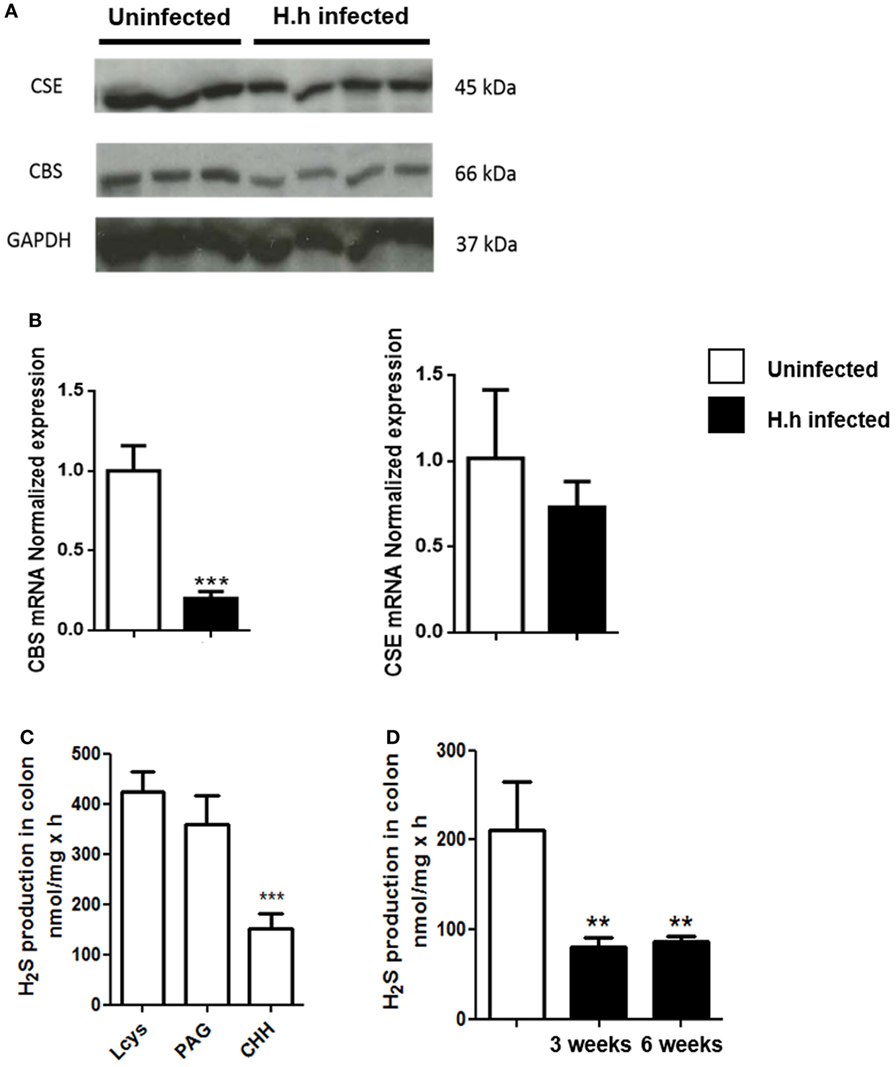
Figure 2. H2S production is significantly reduced during Helicobacter hepaticus (Hh)-induced colitis. 129SvEv Rag2−/− mice were infected with Hh and sacrificed 6 weeks post-infection. (A) Representative western blot and (B) relative CSE and CBS mRNA expression performed on colon samples from uninfected and Hh-infected mice. A significant reduction in the expression of both CBS protein and mRNA was observed in Hh-infected mice. (C) H2S synthesis by the colon of healthy mice was measured in colon homogenates, in the absence or presence of CBS inhibitor [(CHH), 3 mmol/L] or CSE inhibitor (PAG, 10 mmol/L). The substrate for H2S synthesis (l-cys) was present at 4 mmol/L. Only CHH significantly reduced H2S synthesis in tissue from healthy controls (***P < 0.001 vs. l-cys alone). (D) H2S levels measured in the colon of Hh-infected mice 3 or 6 weeks post-infection resulted significantly reduced as compared to uninfected mice. Data are shown as mean ± SEM (n = 5 per group) (**P < 0.01, ***P < 0.001 vs. uninfected).
H2S Donor Reduces Colitis Severity
To support the hypothesis that the reduction of H2S synthesis contributes to the development of Hh-induced colitis, we performed a pharmacological modulation. DATS, a natural long lasting H2S donor (22) was orally administered to Rag2−/− mice previously infected with Hh (1.0 × 108 CFU). Four weeks after infection, a group of mice received DATS at 50 mg/kg while the control group received only the vehicle (PBS) every day for 2 weeks. Mice were analyzed for intestinal inflammation 6 weeks after the first infection with Hh. In DATS-treated group, a significant (P < 0.05) reduction of colon inflammation as compared with the control group was evident. The histological analysis revealed that the reduction in colon inflammation score induced by DATS was more marked in the colon distal part (Figures 3A,B). The anti-inflammatory effect of DATS was not associated with a change in bacterial burden, as we could not detect any differences in cecal Hh colonization levels upon treatment (Figure 4). Thus, to gain insights into the anti-inflammatory mechanism(s) of H2S in this model of colitis-associated cancer, we analyzed the content of pro-inflammatory cytokines. As shown in Figure 5A, the inflammatory response was sustained by high levels of IL-6 and TNF-α mRNA expression in colonic tissues from infected mice, as compared to uninfected mice. In DATS-treated mice, a reduction of the pro-inflammatory cytokines mRNA was observed confirming a key role for these cytokines. This hypothesis is further supported by the in vitro experiments carried out on BM-derived macrophages (BMDMs) stimulated with Hh bacteria (20 MOI). As expected, infection of BMDMs cells with Hh induced a sustained release of IL-6 and TNFα in the supernatants as compared to unstimulated BMDM. Overnight incubation of infected BMDMs cells with DATS (10 µM) significantly (P < 0.001) reduced the release of these cytokines (Figure 5B).
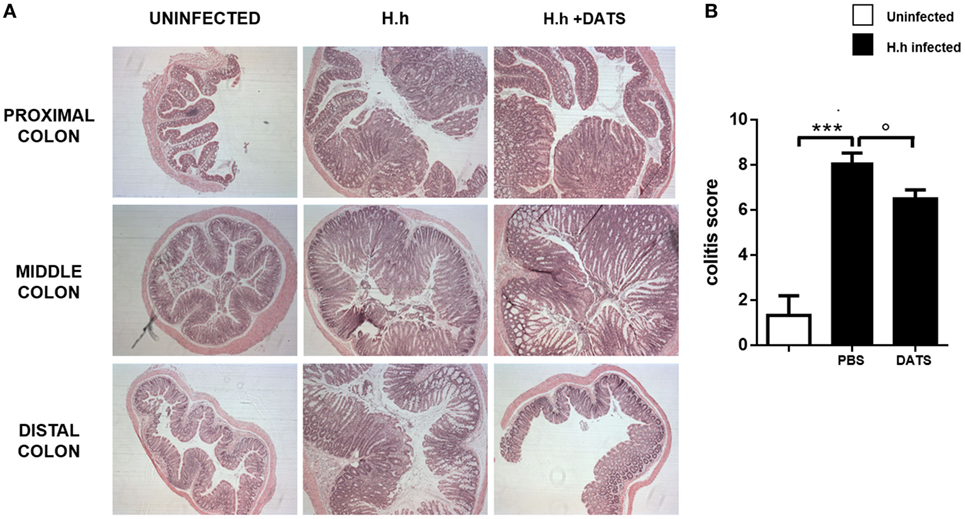
Figure 3. H2S donor reduces the severity of colitis. (A) Representative photomicrographs (magnification ×50) of H&E-stained proximal, middle, and distal colon isolated from healthy mice and from 6 weeks Helicobacter hepaticus-infected mice treated with DATS or only vehicle (PBS) and correspondent (B) inflammation score. DATS significantly reduced inflammation in the colon. Data are shown as mean ± SEM (n = 5 per group) (***P < 0.001 vs. uninfected. °P < 0.05 vs. PBS).
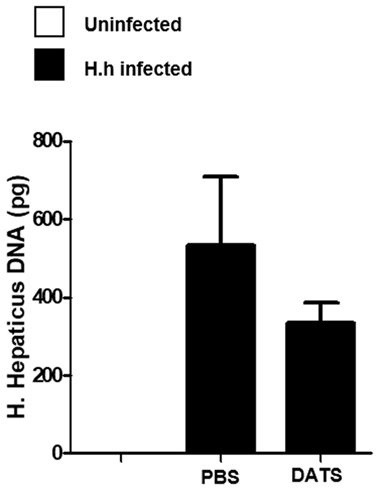
Figure 4. DATS does not affect Helicobacter hepaticus (Hh) colonization levels. Quantification of Hh DNA in cecum content samples from healthy mice and from 6 weeks Hh-infected mice treated with DATS or vehicle (PBS) by using a real-time PCR assay. Data are shown as mean ± SEM (n = 5 per group).
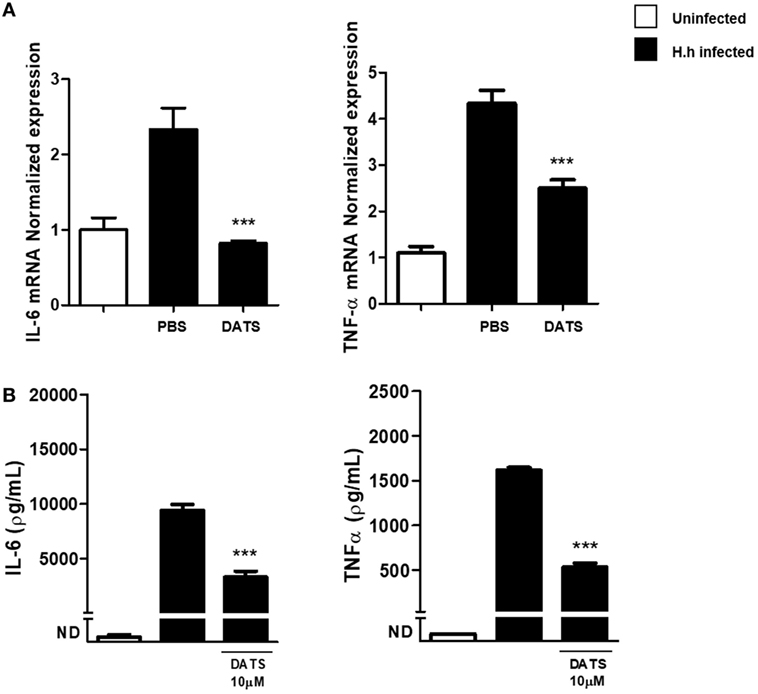
Figure 5. DATS reduces pro-inflammatory cytokines production. (A) Relative expression of TNF-α and IL-6 mRNA within the colons of both uninfected mice and Helicobacter hepaticus (Hh)-infected mice, 6 weeks post-infection, receiving DATS or vehicle (PBS). Expression of cytokines mRNA was determined by real-time quantitative PCR. Data are shown as mean ± SEM (n = 5 per group) (***P < 0.001 vs. PBS). (B) Bone marrow-derived macrophages from uninfected mice were stimulated with live Hh (20 MOI). Incubation of cells with DATS (10 µM) significantly reduced IL-6 and TNFα release. Data are shown as mean ± SEM of three pooled independent experiments (***P < 0.001 vs. Hh-infected).
H2S Reduces the Number of G-MDSCs
To evaluate if the hydrogen sulfide effect on the intestinal inflammation was due to a modification of the innate immune response, we isolated and characterized colon and spleen leukocytes from Hh-infected mice. As expected, Hh-mediated inflammation was associated with an increase in the frequency of both G-MDSCs and M-MDSCs cells in infected mice as compared with uninfected mice. Replenishing hydrogen sulfide, by using as exogenous source the H2S donor DATS, induced a significative reduction both in the frequency (P < 0.05 vs. PBS) and in the absolute number (P < 0.01 vs. PBS) of G-MDSCs in colon (Figure 6A). It has to be noted that hydrogen sulfide addition did not affect G-MDSCs frequencies in the spleen (Figure 6B) suggesting a localized anti-inflammatory effect. Finally, the number of both colon and spleen M-MDSCs were not affected by DATS treatment (Figures 6C,D). Thus, the H2S-induced reduction of Hh-triggered inflammation is related to the modulation of the innate component of the immune system.
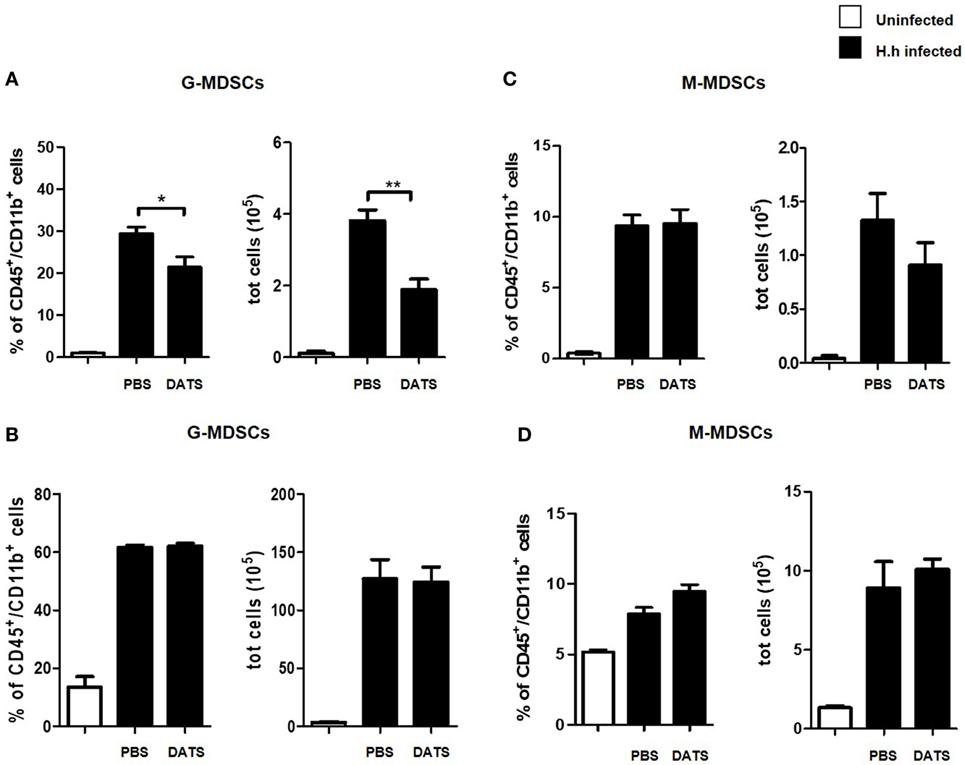
Figure 6. DATS inhibits G-MDSCs accumulation in the colon of Helicobacter hepaticus (Hh)-infected mice. Leukocytes were isolated from the colon and the spleen of 129SvEv Rag2−/− uninfected or Hh-infected 6 weeks post-infection. Infected mice were treated with DATS or vehicle (PBS). G-MDSCs and M-MDSCs frequencies and absolute number were assessed by flow cytometry in colon (A,C) and in spleen (B,D). G-MDSCs frequency and number was reduced in the colon following treatment with DATS. Data are shown as mean ± SEM (n = 5 per group) (*P < 0.05, **P < 0.01, DATS vs. PBS).
Discussion
Infiltration of immune cells, specifically CD11b+ myeloid cells, and their aberrant activation play a central role in carcinogenesis contributing to create a pro-inflammatory microenvironment that enables tumor promotion (23). MDSCs have emerged as key effector cells in tumor microenvironment responsible for tumor progression and metastasis (24). Since pro-inflammatory molecules induce MDSCs, we speculate that Hh infection of Helicobacter-free 129SvEv/Rag2−/− mice could promote MDSC accumulation in colon and spleen facilitating tumor formation. Conflicting data are present in literature regarding MDSCs accumulation and function during colitis in mice. These divergences are likely due to the specific experimental animal model used. Haile et al., using a model of T cell-mediated autoimmune enterocolitis, showed that MDSCs were protective and suppressed development of disease (25). In another study, Zhang and coworkers, using a model of colitis induced by dextran sodium sulfate (DSS), demonstrated that the transfer of splenic DSS-derived CD11b+Gr-1+ MDSCs into a recipient mice suppressed myeloid-lineage cell development in the lamina propria and ameliorated disease parameters (26). There is also evidence supporting a pro-inflammatory role of myeloid cells in experimental IBD. In fact, Guan et al. observed that both CD11b+Ly6C+ and CD11b+Ly6G+ cells increased in spleen and in colonic lamina propria in mice with acute colitis and also correlated with the intestinal inflammation severity (27).
Thus, to better define the role of MDSCs in colitis-associated cancer development, we decided to use an innate immune-mediated IBD model lacking the adaptive immune response. Another benefit of the model chosen is that it closely resembles human IBD since members of the Helicobacteraceae family have been found in the colon of IBD patients (28, 29). The development of colitis in Hh-infected Rag2−/− mice was accompanied by an increase of MDSCs in lymphoid and non-lymphoid tissue in a time-dependent manner. The G-MDSCs subtype was predominant both in the spleen and in the colon of infected mice. This finding is in accordance with the current relevant literature and confirms that during tumor progression the G-MDSC population is predominant (30). Oxidative stress in human IBD correlates with disease activity and represents one of the key features of tumor initiation (31). More recently, a role for hydrogen sulfide in oxidative stress has been defined (32). H2S is an endogenous mediator that exhibits several anti-inflammatory activities and contribute to gastric mucosal defense (15). Its protective role in the resolution of colitis and ulcer healing in rats and mice has been elegantly demonstrated by Wallace and coworkers (15, 16, 33, 34). In the attempt to better define the downstream signaling in this model we have assessed the possible role played by the hydrogen sulfide pathway. It is widely assumed that desulfhydration of l-cys is the major source of H2S in mammals and is catalyzed by the trans-sulfuration pathway enzymes: CBS and CSE. They are both P5P-dependent lyases and generate H2S in many tissues including brain, liver, kidney, ileum, uterus, and placenta. Beside other main reactions, CBS and CSE catalyze the synthesis of cystathionine from l-cysteine and l-homocysteine, generating H2S (35). In addition, a third pathway represented by the enzymes 3-mercaptopyruvate sulphurtransferase (3MST) in conjunction with cysteine aminotransferase, has been found to contribute to H2S production in the brain and in the vascular endothelium of thoracic aorta (36, 37). Finally, very recently, it has been discovered that also cysteinyl-tRNA synthetases 2 (CARS2), a mitochondrial isoform of CARS, are involved in polysulfide production (38). CBS and CSE are currently considered the major H2S enzymatic source in a variety of tissues; we therefore concentrated our attention on these enzymes. However, the possible contribution of the other alternative metabolic pathways in the development of colitis in Hh-infected mice cannot be completely excluded. Our results demonstrated that CBS is the primary H2S-producing enzyme in the colon of both healthy and infected Rag2−/− mice. In this regard, CBS in rat colon do represent the major source of colonic H2S either in healthy state or during inflammation (16). H2S synthesis by the Hh-infected mice colon was reduced by 100-fold after induction of colitis at 3 and 6 weeks, thereby implying a putative protective role for hydrogen sulfide. This reduction of H2S synthesis well fits with the downregulation of CBS protein and mRNA expression in the colon of infected mice during colitis. Hh-mediated inflammation induced an increase in the frequency of G-MDSCs and M-MDSCs cells in colon and in spleen of infected mice as opposite to control mice. To further investigate on the contribution of hydrogen sulfide, we designed an experiment where the exogenous hydrogen sulfide was “furnished” by mean of a donor. Chronic oral administration of the H2S donor DATS did not cause eradication of bacteria or decreased levels of colonization in mice but reduced colon inflammation. It has to be noted that the mechanisms by which H2S is released from DATS have in depth analyzed by several research groups (22, 39–42). In particular, it has been shown that H2S is liberated from DATS in presence of GSH and l-cys (43). However, sulfane sulfur (polysulfides), rather than H2S, are considered the active agent in physiological signaling (44–47). The recent study (38) showing that CBS and CARS2 can also produce sulfane sulfur species directly from cystine and cysteine, respectively, added another tassel in this already very complicated puzzle.
Finally, in order to gain mechanistic insights into this anti-inflammatory effect, we have evaluated changes in the inflammatory phenotype of the immune population in the colon. Treatment with DATS induced a significative reduction both in the frequency and in the absolute number of G-MDSCs in colon without affecting G-MDSCs frequencies in the spleen. This finding implies a localized selective effect of hydrogen sulfide on colon G-MDSCs population.
It has been shown that Hh-induced malignancy in Rag2−/− mice was readily reversible by blocking underlying bacteria-driven inflammation with antibodies directed at TNF-α and IL-6 (48, 49). TNF-α and IL-6 signaling has been proposed as a tumor-promoting mechanism in colitis-associated cancer. In fact, the levels of these pro-inflammatory cytokines increase during inflammatory reactions and are responsible, at least in part, for MDSCs accumulation and for the increase in their suppressive activity (24). In tune with these findings, levels of IL-6 and TNF-α mRNA are increased in colonic tissues from infected mice. Interestingly, hydrogen sulfide replenishment with DATS reduced TNF-α and IL-6 levels as compared to control mice. To further support our hypothesis, we performed experiments on BM-derived macrophages stimulated with Hh bacteria. Indeed, it is known that Hh challenge of ex vivo-cultured BMDMs induces activation of both the NF-κB and ERK pathways (50, 51). Infection of BMDMs cells with Hh induced a sustained release of the pro-inflammatory cytokines IL-6 and TNFα in the supernatants as compared to unstimulated BMDM. Treatment of BMDMs cells with exogenous hydrogen sulfide, by using DATS, reduced cytokines release mimicking the in vivo setting. In conclusion, these results indicate that the metabolic pathway l-cys/H2S can exert a protective role in intestinal Hh-induced inflammation. Dysregulation of H2S homeostasis has also been implicated in numerous pathological conditions and diseases (52). Thus, the reduction of H2S levels reported during colitis might induce metabolic changes in the microenvironment, as the alteration of the redox cellular status, which promotes tumorigenesis. Metabolic reprograming has been suggested as a key hallmark of cancer progression (53). Recent studies have revealed that immune cells possess distinct metabolic characteristics that influence their immunological phenotype and functions and so their contribution to cancer progression (54). Thus, the identification of new metabolic targets could be of great importance in modifying the plasticity of tumor-promoting immune cells and in CRC prevention. Our results demonstrating that MDSCs mediate significant intestinal inflammation upon stimulation with pathogenic Hh assume a therapeutic significance since, targeting MDSCs would be promising treatment option for IBD patient to reduce the risk of CRC and to manage inflammatory symptoms in order to provide an improved quality of life.
Ethics Statement
Experiments were conducted in accordance with the UK Scientific Procedures Act (1986) under a Project License (PPL) authorized by the UK Home Office Animal Procedures Committee and approved by the Sir William Dunn School Ethical Review Committee.
Author Contributions
PC designed, performed the experiments, and analyzed the data; TS performed the experiments; GC revised critically the manuscript; KM and AI supervised all the experiments, revised critically the intellectual contributions to the manuscript, and gave final approval to the publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was funded by Italian Government Grants, PRIN 2012 no: 2012WBSSY4_005.
Footnote
- ^Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx?cancer=colorectal (Accessed: October 25, 2017)
References
1. Ullman TA, Itzkowitz SH. Intestinal inflammation and cancer. Gastroenterology (2011) 140(6):1807–16. doi:10.1053/j.gastro.2011.01.057
2. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut (2001) 48(4):526–35. doi:10.1136/gut.48.4.526
3. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med (2009) 361(21):2066–78. doi:10.1056/NEJMra0804647
4. Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol (2014) 20(1):6–21. doi:10.3748/wjg.v20.i1.6
5. Harrison OJ, Maloy KJ. Innate immune activation in intestinal homeostasis. J Innate Immun (2011) 3(6):585–93. doi:10.1159/000330913
6. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
7. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi:10.1038/nri3175
8. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. l-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol (2003) 24(6):302–6. doi:10.1016/S1471-4906(03)00132-7
9. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111(8):4233–44. doi:10.1182/blood-2007-07-099226
10. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181(8):5791–802. doi:10.4049/jimmunol.181.8.5791
11. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58(1):49–59. doi:10.1007/s00262-008-0523-4
12. Ostanin DV, Bhattacharya D. Myeloid-derived suppressor cells in the inflammatory bowel diseases. Inflamm Bowel Dis (2013) 19(11):2468–77. doi:10.1097/MIB.0b013e3182902b11
13. Fox JG, Ge Z, Whary MT, Erdman SE, Horwitz BH. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol (2011) 4(1):22–30. doi:10.1038/mi.2010.61
14. Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med (2003) 197(1):111–9. doi:10.1084/jem.20021345
15. Wallace JL, Dicay M, McKnight W, Martin GR. Hydrogen sulfide enhances ulcer healing in rats. FASEB J (2007) 21(14):4070–6. doi:10.1096/fj.07-8669com
16. Wallace JL, Vong L, McKnight W, Dicay M, Martin GR. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology (2009) 137(2):569–78, 578.e561. doi:10.1053/j.gastro.2009.04.012
17. Chan MV, Wallace JL. Hydrogen sulfide-based therapeutics and gastrointestinal diseases: translating physiology to treatments. Am J Physiol Gastrointest Liver Physiol (2013) 305(7):G467–73. doi:10.1152/ajpgi.00169.2013
18. Wallace JL, Ianaro A, de Nucci G. Gaseous mediators in gastrointestinal mucosal defense and injury. Dig Dis Sci (2017) 62(9):2223–30. doi:10.1007/s10620-017-4681-0
19. Guo FF, Yu TC, Hong J, Fang JY. Emerging roles of hydrogen sulfide in inflammatory and neoplastic colonic diseases. Front Physiol (2016) 7:156. doi:10.3389/fphys.2016.00156
20. Ianaro A, Cirino G, Wallace JL. Hydrogen sulfide-releasing anti-inflammatory drugs for chemoprevention and treatment of cancer. Pharmacol Res (2016) 111:652–8. doi:10.1016/j.phrs.2016.07.041
21. Panza E, De Cicco P, Ercolano G, Armogida C, Scognamiglio G, Anniciello AM, et al. Differential expression of cyclooxygenase-2 in metastatic melanoma affects progression free survival. Oncotarget (2016) 7(35):57077–85. doi:10.18632/oncotarget.10976
22. Benavides GA, Squadrito GL, Mills RW, Patel HD, Isbell TS, Patel RP, et al. Hydrogen sulfide mediates the vasoactivity of garlic. Proc Natl Acad Sci U S A (2007) 104(46):17977–82. doi:10.1073/pnas.0705710104
23. Waldner MJ, Neurath MF. Mechanisms of immune signaling in colitis-associated cancer. Cell Mol Gastroenterol Hepatol (2015) 1(1):6–16. doi:10.1016/j.jcmgh.2014.11.006
24. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182(8):4499–506. doi:10.4049/jimmunol.0802740
25. Haile LA, von Wasielewski R, Gamrekelashvili J, Kruger C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology (2008) 135(3):871–81, 881.e871–5. doi:10.1053/j.gastro.2008.06.032
26. Zhang J, Wang B, Zhang W, Wei Y, Bian Z, Zhang CY, et al. Protein tyrosine phosphatase 1B deficiency ameliorates murine experimental colitis via the expansion of myeloid-derived suppressor cells. PLoS One (2013) 8(8):e70828. doi:10.1371/journal.pone.0070828
27. Guan Q, Moreno S, Qing G, Weiss CR, Lu L, Bernstein CN, et al. The role and potential therapeutic application of myeloid-derived suppressor cells in TNBS-induced colitis. J Leukoc Biol (2013) 94(4):803–11. doi:10.1189/jlb.0113050
28. Hansen R, Thomson JM, Fox JG, El-Omar EM, Hold GL. Could Helicobacter organisms cause inflammatory bowel disease? FEMS Immunol Med Microbiol (2011) 61(1):1–14. doi:10.1111/j.1574-695X.2010.00744.x
29. Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature (2011) 474(7351):307–17. doi:10.1038/nature10209
30. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun (2016) 7:12150. doi:10.1038/ncomms12150
31. Moura FA, de Andrade KQ, dos Santos JC, Araujo OR, Goulart MO. Antioxidant therapy for treatment of inflammatory bowel disease: does it work? Redox Biol (2015) 6:617–39. doi:10.1016/j.redox.2015.10.006
32. Xie ZZ, Liu Y, Bian JS. Hydrogen sulfide and cellular redox homeostasis. Oxid Med Cell Longev (2016) 2016:6043038. doi:10.1155/2016/6043038
33. Flannigan KL, Agbor TA, Blackler RW, Kim JJ, Khan WI, Verdu EF, et al. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc Natl Acad Sci U S A (2014) 111(37):13559–64. doi:10.1073/pnas.1413390111
34. Paul-Clark M, Elsheikh W, Kirkby N, Chan M, Devchand P, Agbor TA, et al. Profound chemopreventative effects of a hydrogen sulfide-releasing NSAID in the APCMin/+ mouse model of intestinal tumorigenesis. PLoS One (2016) 11(2):e0147289. doi:10.1371/journal.pone.0147289
35. Kabil O, Vitvitsky V, Xie P, Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal (2011) 15(2):363–72. doi:10.1089/ars.2010.3781
36. Shibuya N, Mikami Y, Kimura Y, Nagahara N, Kimura H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J Biochem (2009) 146(5):623–6. doi:10.1093/jb/mvp111
37. Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal (2009) 11(4):703–14. doi:10.1089/ARS.2008.2253
38. Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun (2017) 8(1):1177. doi:10.1038/s41467-017-01311-y
39. Zhao Y, Wang H, Xian M. Cysteine-activated hydrogen sulfide (H2S) donors. J Am Chem Soc (2011) 133(1):15–7. doi:10.1021/ja1085723
40. Zhao Y, Biggs TD, Xian M. Hydrogen sulfide (H2S) releasing agents: chemistry and biological applications. Chem Commun (Camb) (2014) 50(80):11788–805. doi:10.1039/c4cc00968a
41. Liang D, Wu H, Wong MW, Huang D. Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: the reaction pathways and intermediates of glutathione with polysulfides. Org Lett (2015) 17(17):4196–9. doi:10.1021/acs.orglett.5b01962
42. Park CM, Weerasinghe L, Day JJ, Fukuto JM, Xian M. Persulfides: current knowledge and challenges in chemistry and chemical biology. Mol Biosyst (2015) 11(7):1775–85. doi:10.1039/c5mb00216h
43. Cai YR, Hu CH. Computational study of H2S release in reactions of diallyl polysulfides with thiols. J Phys Chem B (2017) 121(26):6359–66. doi:10.1021/acs.jpcb.7b03683
44. Toohey JI. The conversion of H(2)S to sulfane sulfur. Nat Rev Mol Cell Biol (2012) 13(12):803; author reply 803. doi:10.1038/nrm3391-c2
45. Liu C, Zhang F, Munske G, Zhang H, Xian M. Isotope dilution mass spectrometry for the quantification of sulfane sulfurs. Free Radic Biol Med (2014) 76:200–7. doi:10.1016/j.freeradbiomed.2014.08.003
46. Toohey JI, Cooper AJ. Thiosulfoxide (sulfane) sulfur: new chemistry and new regulatory roles in biology. Molecules (2014) 19(8):12789–813. doi:10.3390/molecules190812789
47. DeLeon ER, Gao Y, Huang E, Olson KR. Garlic oil polysulfides: H2S- and O2-independent prooxidants in buffer and antioxidants in cells. Am J Physiol Regul Integr Comp Physiol (2016) 310(11):R1212–25. doi:10.1152/ajpregu.00061.2016
48. Poutahidis T, Haigis KM, Rao VP, Nambiar PR, Taylor CL, Ge Z, et al. Rapid reversal of interleukin-6-dependent epithelial invasion in a mouse model of microbially induced colon carcinoma. Carcinogenesis (2007) 28(12):2614–23. doi:10.1093/carcin/bgm180
49. Erdman SE, Rao VP, Poutahidis T, Rogers AB, Taylor CL, Jackson EA, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A (2009) 106(4):1027–32. doi:10.1073/pnas.0812347106
50. Tomczak MF, Erdman SE, Poutahidis T, Rogers AB, Holcombe H, Plank B, et al. NF-kappa B is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J Immunol (2003) 171(3):1484–92. doi:10.4049/jimmunol.171.3.1484
51. Tomczak MF, Gadjeva M, Wang YY, Brown K, Maroulakou I, Tsichlis PN, et al. Defective activation of ERK in macrophages lacking the p50/p105 subunit of NF-kappaB is responsible for elevated expression of IL-12 p40 observed after challenge with Helicobacter hepaticus. J Immunol (2006) 176(2):1244–51. doi:10.4049/jimmunol.176.2.1244
52. Stein A, Bailey SM. Redox biology of hydrogen sulfide: implications for physiology, pathophysiology, and pharmacology. Redox Biol (2013) 1(1):32–9. doi:10.1016/j.redox.2012.11.006
53. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
Keywords: colitis-associated cancer, colorectal cancer, cystathionine gamma-lyase, cystathionine beta-synthase, Helicobacter hepaticus, hydrogen sulfide, myeloid-derived suppressor cells
Citation: De Cicco P, Sanders T, Cirino G, Maloy KJ and Ianaro A (2018) Hydrogen Sulfide Reduces Myeloid-Derived Suppressor Cell-Mediated Inflammatory Response in a Model of Helicobacter hepaticus-Induced Colitis. Front. Immunol. 9:499. doi: 10.3389/fimmu.2018.00499
Received: 16 January 2018; Accepted: 26 February 2018;
Published: 27 March 2018
Edited by:
Fulvio D’Acquisto, Queen Mary University of London, United KingdomReviewed by:
Peter Nagy, National Institute of Oncology, HungarySheng-Jun Wang, Jiangsu University, China
Copyright: © 2018 De Cicco, Sanders, Cirino, Maloy and Ianaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Ianaro, aWFuYXJvQHVuaW5hLml0