 Reiner K. W. Mailer
Reiner K. W. Mailer- 1Institute of Clinical Chemistry and Laboratory Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Cardiovascular Medicine Unit, Department of Medicine, Karolinska Insititutet, Stockholm, Sweden
FOXP3 is the lineage-defining transcription factor of CD4+ CD25+ regulatory T cells. While many aspects of its regulation, interaction, and function are conserved among species, alternatively spliced FOXP3 isoforms are expressed only in human cells. This review summarizes current knowledge about alternative splicing of FOXP3 and the specific functions of FOXP3 isoforms in health and disease. Future perspectives in research and the therapeutic potential of manipulating alternative splicing of FOXP3 are discussed.
Introduction
The immune system’s ability to tolerate structures recognized as self or non-pathogenic non-self is mediated by immunosuppressive mechanisms. Arguably the most effective suppressors of immune responses are CD4+ CD25+ regulatory T (Treg) cells (1–5). Phenotype and function of human Treg cells depend on the expression of their lineage-defining master transcription factor forkhead box P3 (FOXP3) (6). Ectopic expression of FOXP3 via retroviral transduction induces a Treg cell-like expression profile in human and, across species barriers, in mouse CD4+ CD25− T cells (7, 8). Vice versa the absence of functional FOXP3 is causative for severe autoimmune diseases and allergy in patients suffering from immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome (9, 10). In mice, the homologous transcription factor Foxp3 (indicated as murine protein by the lower case letters; a consensus followed by most, but not all researchers) exerts the same function and truncated Foxp3 protein resulting from of a 2 bp insertion, that generates a frameshift and premature stop codon, leads to the scurfy phenotype, similar to IPEX syndrome (11). Detailed analysis over the last decades has been given a comprehensive view about mechanisms that regulate expression and protein functions of FOXP3/Foxp3 for Treg cell-mediated immunosuppression in health and disease (12). Here, a comparative overview is given at first, to apprehend the specific effects facilitated by human FOXP3 isoforms.
FOXP3 Versus Foxp3
FOXP3 transcripts consist of a 5′-untranslated region (exon-1 in humans and exons-2b, -2a, and -1 in mice, in front of the start codon in exon 1) and 11 protein-encoding exons (Figure 1A). Proteins translated from murine and human transcripts share 86.5% amino acid sequence identity. In both species, distinct functional domains (N-terminal proline-rich region, zinc finger, leucine zipper, and forkhead domain) were identified (Figure 1B). FOXP3 binds to GTAAACA motifs via the winged-helix structure of the c-terminal forkhead domain and requires dimerization of the transcription factor by the leucine zipper, whereas the N-terminal 181 amino acids prevent DNA binding in an autoinhibitory fashion (13). Foxp3 regulates gene expression at several hundred DNA-binding sites identified throughout the genome (14–16). Despite the similarities to murine Foxp3, >50% of DNA regions bound by human FOXP3 are species-specific (17) and were not detected in studies analyzing mouse CD4+ CD25+ Treg cells (15) or FoxP3 overexpressing cell lines (16). FOXP3 allows transcriptional repression and transcriptional activation as part of a multi-protein complex whose composition likely diversifies the affinity to and the mode of interaction with DNA-binding sites (Figure 1C). Moreover, in cooperation with FOXP3-associated chromatin-modifying enzymes FOXP3 stabilizes epigenetically the Treg cell phenotype and function. Several excellent reviews summarize FoxP3-bound cofactors and the corresponding FoxP3 binding regions (18–20). However, it should be noted that identified cofactors may be species- and context-specific as posttranscriptional and posttranslational modifications can affect the complex formation (see below).
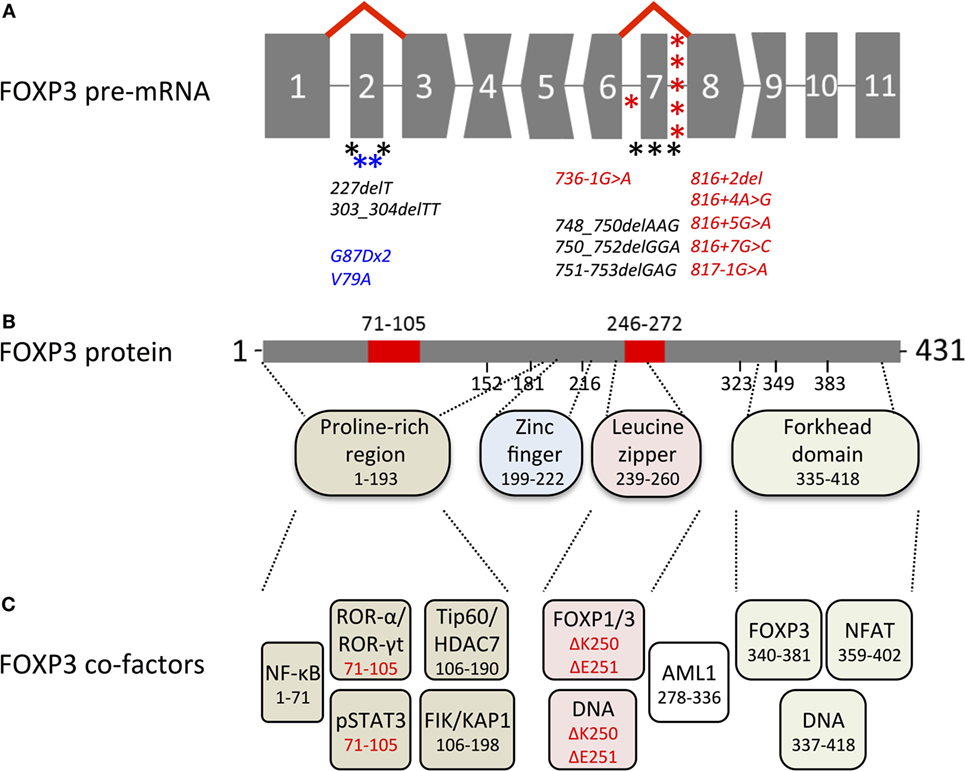
Figure 1. Overview of genetic, transcriptional, and functional features of human FOXP3. (A) Structure of the FOXP3 pre-mRNA. Codons interrupted by introns are represented by convex and concave shapes for 1 and 2 nucleotides overhang, respectively. Alternative splicing of exon 2 and exon 7 is displayed by red lines; asterisks mark immunodysregulation, polyendocrinopathy, enteropathy, X-linked mutations that target alternatively spliced exons (black) or introns predicted to affect alternative splicing (red) and carcinoma mutations in alternatively spliced exons (blue). (B) FOXP3 protein with alternatively spliced exons (red), numbers indicate the region or the first amino acid of a new exon; dashed lines indicate regions of functional domains. (C) Cofactors binding FOXP3 functional domains, color-coded as in (B) numbers indicate the binding region, isoform-specific interactions highlighted in red.
Transcriptional Regulation of FOXP3
Due to the importance of FOXP3 for immunoregulation, intensive research efforts have been undertaken to elucidate potentially therapeutic mechanisms that induce FOXP3 transcription in CD4+ T cells (21). Species comparison identified several conserved non-coding sequences (CNS) that regulate FOXP3 mRNA transcription through chromatin modifications (22). Initiation of FOXP3 expression is controlled by CNS3, which is located at intron 1 and facilitates indispensable c-Rel-mediated transcription in Treg precursor cells (23–25). Followed by this pioneer element, FOXP3 expression is further enhanced and stabilized by CNS2. This region is located distal to the promoter in intron-1 and controls the heritable maintenance of FOXP3 expression through various transcription factors, including FOXP3 itself. The accessibility to CNS2 is ensured through demethylation of its CpG islands, termed Treg cell-specific demethylated region (TSDR), that has been used to assess the Treg cell stability qualitatively and quantitatively (26, 27). In addition, CNS1, at the proximal site of intron-1, is associated with transforming growth factor-β1 (TGF-β1)-mediated FOXP3 induction. Thus, the transcriptional and epigenetic landscape of Treg cells appears to be a mosaic independently generated by T cell antigen receptor (TCR) stimulation (28, 29), FOXP3 expression (30), and TGFβ1-mediated effects (31). In line with this, comprehensive studies utilizing an array of conditional knockout mice revealed that constant Foxp3 expression (32) as well as continuous stimulation of the TCR and CD28 co-stimulation (33–35) is required for Treg cell function.
Activation-Induced FOXP3 Expression
Importantly, anti-CD3/anti-CD28 stimulation of naïve human CD4+ T cells is sufficient to induce FOXP3 expression transiently but lacks strong immunosuppressive capacity (36–40), whereas in mice the differentiation of suppressive pTreg cells in vivo or iTreg cells in vitro depends on the presence of TGF-β1 during TCR ligation (41). In human naïve T cells, however, TGF-β1 treatment fails to stabilize FOXP3 expression and to induce phenotype and function comparable to Treg cells (42). Nevertheless, activation-induced FOXP3 expression in non-Treg cells has been shown to restrain proliferation and cytokine expression intrinsically (43), suggesting that activation-induced FOXP3 slows down T cell responses to enable further immunomodulatory mechanisms. This implies that FOXP3 expression among human CD4+ T cells is less sufficient to delineate the suppressive Treg cell population. These divergent FOXP3 functions in comparison to murine Foxp3 may be related with another important difference between mice and men, i.e., the generation of human FOXP3 isoforms through alternative splicing, as FOXP3 isoforms are variably expressed and differ in their ability to imprint the Treg cell phenotype.
Alternative Splicing
Alternative splicing is an important mechanism to diversify protein functions and has been observed as a common concept in multiple T cell proteins to adapt to TCR stimulation (44). During the synthesis of pre-mRNA by RNA polymerase II in the nucleus, a multi-protein complex, termed the spliceosome, removes introns, and combines exons to generate mature mRNAs (45, 46). This posttranscriptional modification process relies on the identification of consensus sequences at the 5′- and 3′-splice site by ribonucleoproteins. This step is regulated by exonic or intronic enhancer/silencer auxiliary elements, which represent promising targets for antisense gene therapy with splice-shifting oligonucleotides (47). Exons not recognized by the spliceosome are not inserted into the mRNA and isoforms derived from this alternatively spliced transcript lack the encoded protein region. Following the splicing process, exon–exon junction sites on the mRNA remain bound to the exon junction complex until the first translating ribosome removes these protein assemblies. This facilitates the degradation of incorrectly spliced mRNAs, in which intron retention or exon skipping occurred erroneously, via nonsense-mediated mRNA decay. This quality control mechanism detects premature translation–termination codons generated by a frameshift (stop codons that are preceding exon junction complexes on the mRNA) and degrades the faulty mRNA.
Alternative Splicing of FOXP3
Importantly, the structure of the FOXP3 gene limits the amount of possible exon deletions (Figure 1A), as only exon 2 and exon 7 have blunt 5′- and 3′- ends, whereas codon-interrupting sequences would cause frameshifts in FOXP3 isoforms with exon deletion (Δ indicates deleted exon in the transcript/protein) Δ3, Δ4, Δ6, Δ8, and Δ9. Deletion of exon 1 would erase the transcription start site, whereas Δ5 and Δ10, albeit possible without frameshift, would omit DNA-interacting sites (zinc finger and forkhead domain, respectively). Interestingly, the gene architecture of murine FoxP3 (429 aa) is identical to that in humans, suggesting that alternative splicing of coding exons could also take place in mice. However, neither FoxP3 isoforms nor differential usage of non-coding alternative exons was found in lymphoid tissues or in phenotypically distinct CD4+ CD25low hepatic Treg cells (48, 49), whereas in rat (429 aa), isoforms with alternative N-terminal region (429 + additional 59 aa, GenBank accession: BAJ05811.1) or C-terminal region (322 + alternative 13 aa, GenBank accession: BAJ05812) have been reported. Taken together, splicing control of the transcription factor appears to be species-specific and no model organism is currently known that expresses analogs to human FOXP3 splice variants in CD4+ T cells: full-length FOXP3 (FOXP3fl) and exon 2 (FOXP3Δ2) as well as exon 2 and exon 7 (FOXP3Δ2Δ7) lacking isoforms (8, 50). The exact molecular mechanism of alternative splicing of FOXP3 pre-mRNA has not been revealed yet, but the analysis of intronic sequences (intron 2 and intron 7) with splice factor binding prediction software points toward heterogeneous nuclear ribonucleoproteins that might act as intronic splicing silencers (51). However, since the skipping of these exons is not quantitatively correlated, it is likely that several different factors affect alternative splicing exon-specifically. Furthermore, the current concept for the change of isoform ratios envisages that alternatively spliced exons are more likely included into the nascent transcripts when RNA polymerase II elongation is slowed down through methylation of CpG islands and/or histone deacetylation (52, 53). Along this line, initial FOXP3 transcription of “closed” chromatin induced by TCR stimulation and mediated by c-Rel/CNS3 would favor FOXP3fl expression. In contrast, FOXP3 transcription of “open” chromatin in bona fide Treg cells mediated by TSDR/CNS2 would favor exon skipping. Further research will be necessary to answer these questions and to pinpoint factors that regulate alternative splicing of FOXP3.
All Exons Combined
FOXP3 Isoform Ratios in Disease
The longest FOXP3 transcript translates into FOXP3fl and allocates 20–30% of the total FOXP3 expression (mRNA and protein) in human CD4+ CD25+ Treg cells (8, 54, 55). We have recently demonstrated that the inclusion of exon 2 among cellular FOXP3 isoforms expressed by CD4+ T cells correlates with TCR stimulation in vitro (56). This activation-induced FOXP3 isoform profile has been detected by co-stainings with two different anti-FOXP3 antibody clones that recognize exon 2 and a non-spliced region of FOXP3 to calculate a ratio of either fluorescence intensity per individual cell. This flow cytometric approach revealed a dose-dependent increase of the average (FOXP3 exon 2/FOXP3 total) ratio in CD4+ T cells treated with anti-CD3 antibodies, low-density lipoprotein or phorbolester. Besides these relative changes in both FOXP3low non-Treg cells as well as FOXP3high Treg cells, the total FOXP3+ T cell population is stained proportionally with both antibodies (56, 57), unless alternative splicing of exon 2 is artificially enforced by splice-shifting antisense oligonucleotides (ASO) (54). This strongly indicates that CD4+ T cells naturally express FOXP3 isoforms lacking exon 2 only in combination with FOXP3fl, but not alone. Along this line, gating analyzes to describe cell populations stained positively for FOXP3 total but negatively for FOXP3 exon 2 may be misleading and define FOXP3 isoform ratios improperly, as described for peripheral blood from patients with giant cell arteritis (58), vasculitis (59), or anti-phospholipid syndrome (60). Thus, instead of population analysis, it appears that isoform expression ratios calculated per cell represent a more liable and accurate parameter to investigate the relative expression of FOXP3 exon 2 by flow cytometry.
Moreover, the expression of FOXP3 isoforms has been assessed in immunoblots using an isoform-sensitive anti-FOXP3 antibody clone. Chen et al. have shown that FOXP3 expression in CD4+ CD25+ T cells negatively correlated with C–C chemokine ligand 3 serum concentrations derived from peripheral blood of psoriasis patients (61). Although not analyzed separately, it appears that the (upper FOXP3fl/lower FOXP3Δ2 band) ratio is decreasing in remission, whereas relatively more FOXP3fl is detected in patients with refractory lesions or chronic disease development. At this stage, it is unclear whether the altered FOXP3 isoform ratio in psoriasis depends on activated non-Treg cells or impaired Treg cells. However, in the same report FOXP3 expression has been shown to depend on the AKT pathway, which is activated downstream of the TCR and abrogates FOXP3-mediated suppression if constitutively activated (62). Strong activation reduced, whereas weak activation of the AKT pathway increased the [upper FOXP3fl/lower FOXP3Δ2 band] ratio in CD4+ CD25+ T cells in vitro (61). This corroborates our recent findings that TCR stimulation promotes an activation-induced FOXP3 isoform profile (56).
In addition, the association of impaired FOXP3 isoform ratios with disease has been investigated in several studies through quantification of FOXP3 splice variants by real-time PCR. In rheumatoid arthritis patients, the increase of FOXP3 transcripts including exon 2 exceeds that of FOXP3 transcripts excluding exon 2 in isolated CD4+ T cells from blood (2.3- to 3.2-fold) and synovial fluid (3.6-fold) (63–65). Moreover, this shift is associated with increased expression of activation marker CD25, decreased expression of Treg cell marker CTLA-4 and can be reversed through decoy TNF-α receptor treatment. Likewise, inclusion of exon 2 in FOXP3 mRNAs significantly increases in blood from coronary artery disease patients compared to healthy controls (56) and the same trend is seen in blood from patients with autoantibodies (66), rheumatoid arthritis (67), and biopsies from inflammatory bowel disease patients (57).
Taken together, these results suggest that the relative increase of FOXP3fl correlates with de novo induction of FOXP3 in activated CD4+ T cell and is associated with autoimmune and inflammatory diseases. Additional studies are required to investigate FOXP3 isoform patterns in resting, effector and memory subpopulations of Treg and non-Treg cells.
FOXP3fl-Specific Functions
The presence of exon 2 in FOXP3fl enables isoform-specific functions. Interestingly, exon 2 harbors a nuclear export signal (NES), which promotes the relocation of FOXP3fl into the cytoplasm following its TCR stimulation-dependent expression in CD4+ CD25− T cells (68). In the same report, Magg et al., have shown that targeted mutation of FOXP3’s NES promotes Treg cell-associated gene transcription and suppressive capacity. Thus, nuclear localization of FOXP3fl supports the Treg cell phenotype, whereas cytoplasmic FOXP3fl facilitates additional isoform-specific functions, such as exon 2-specific associations with cofactors. Importantly, an interaction that has been demonstrated for human tumor-induced Treg cells is the association of signal transducers and activators of transcription 3 (STAT3) with the β-sheet region of exon 2 in FOXP3fl (69). In these cells, FOXP3fl binds to phosphorylated STAT3 to promote IL-10 transcription. Moreover, FOXP3fl recruits cytoplasmic histone acetyltransferase-1 into the nucleus to control epigenetic chromatin modifications at promoter sites. It is, therefore, tempting to speculate that this mechanism also contributes to the accessibility of CNS regions to imprint FOXP3 transcription upon induction.
Another FOXP3fl-specific interaction is the association of exon 2 with Th17 cell lineage-defining transcription factors, i.e., retinoid acid receptor-related orphan receptors (ROR)-α and ROR-γt (70, 71). The LXXLL motif of FOXP3 exon 2 has been identified to interact with ROR-α and the expression of FOXP3fl is necessary to block ROR-γt-mediated Th17 cell differentiation in retroviral transduction experiments. Therefore, it has been proposed that human Th17 cells might predominantly express FOXP3Δ2. However, ex vivo analysis of Th17 cells from patients with Th17-related diseases reveals that IL-17 expression is rather associated with FOXP3fl (57, 72). This suggests that FOXP3 isoform-specific functions can be obscured upon overexpression in vitro (73), because retro- or lentiviral gene transfer into primary T cells results in multiple infections of target cells, random integration sites, a marked overexpression of transduced genes under the control of viral promoters and activation-induced expression of endogenous FOXP3. To avoid interference by these factors, we analyzed FOXP3 isoform-specific functions by splice-shifting ASOs. Notably, we have found that these synthetic polymers effectively inhibit the inclusion of alternatively spliced exons in endogenous FOXP3 mRNAs and that lack of exon 2 does not promote Th17 cell differentiation (54). Moreover, IL-17 mRNA does not correlate with FOXP3 transcripts expressing or lacking exon 2 in biopsies obtained from patients with Crohn’s disease or peripheral blood from patients with coronary artery disease (54, 56). Thus, the FOXP3fl-ROR-interaction is dispensable for Th17 cell differentiation blockade and may rather abrogate the FOXP3fl-mediated control of cytokine expression (74).
So far, only few FOXP3fl-specific functions have been reported. As mentioned above, FOXP3fl increases the p-STAT3-dependent transcription of IL-10 (69). In addition, FOXP3fl induces the expression of PIM2, a serine/threonine kinase that is involved in the phosphorylation of murine Foxp3 and the expansion of human FOXP3fl-transduced T cells (75, 76). Both mechanisms await further investigation to clarify their impact on human Treg cell functions. In general, the Treg cell phenotype that is induced by FOXP3fl or FOXP3Δ2 expression in naïve CD4+ T cells appears to be overlapping, as NFAT/NF-κB-mediated transcriptional control, Treg cell marker expression as well as suppressive capacity is comparable in transduced human and murine T cells (8, 50, 55). However, the isoform-specific spatial and temporal expression of FOXP3fl indicates that the amount of FOXP3fl may influence the T cell fate in vivo.
Exon 2 Skipping
Around 70% of FOXP3 transcripts in human CD4+ CD25+ T cells are expressed as the alternative splice variant FOXP3Δ2 (8, 54, 55). FOXP3Δ2 lacks the NES in exon 2 and is less prone for relocation into the cytoplasm compared to FOXP3fl (68). Despite the predominant expression of this isoform in Treg cells, relatively few studies have taken alternative splicing of human FOXP3 into account and investigated individual isoforms independently. Thus, specific features of FOXP3Δ2, such as DNA occupancy and cofactor binding to the exon1–exon3 interface, remain largely unknown. Moreover, the coexpression of FOXP3 isoforms and the possibility that a multiprotein complex may contain different FOXP3 isoforms could hamper the assignment of isoform-specific functions in Treg cells from healthy donors. Furthermore, the detection of FOXP3 isoforms relies on suitable isoform-sensitive anti-FOXP3 antibody clones that bind specific epitopes. Although alternative splicing can only occur at exon 2 and exon 7 (see above), De Rosa et al. have reported four different bands by immunoblot analysis of iTreg cells (77). In this paper, the interference with metabolic pathways regulates the amount of induced FOXP3; FOXP3 isoform ratios, however, remain constant in real-time PCR and FOXP3fl-specific immunoblots with respect to the total FOXP3 expression. In contrast, the quantification of unidentified bands obtained from immunoblots utilizing anti-FOXP3 antibody clone PCH101 have been interpreted to depict different FOXP3 isoform ratios that may have compromised the conclusion. Thus, the FOXP3 isoform ratio of iTregs cells in comparison to ex vivo isolated Treg cells or in vitro generated effector T cells awaits further investigation.
IPEX Mutations Targeting FOXP3 Exon 2
However, evidence for the function of FOXP3 exons can be deduced from reported IPEX mutations that affect alternative splicing or the protein sequence of exons (78). A cluster of IPEX mutations targeting the 5′ splice donor site of intron 1 (position 210 + 1/+2 nt) is predicted to cause aberrant splicing of exon 1 (47). However, two mutations are identified within exon 2 that lead to frameshifts and premature stop codons (227delT and 303_304delTT) (79–84). While the mutated FOXP3fl transcript is likely to be degraded by nonsense-mediated mRNA decay, alternative splicing of exon 2 should produce mRNA that encodes for functional FOXP3Δ2. Surprisingly, no Treg cells expressing FOXP3Δ2 alone have been detected in these IPEX patients by flow cytometry using isoform-sensitive anti-FOXP3 antibodies (80, 82). Compared to IPEX patients of similar age with diminished FOXP3 expression due to a mutation in the forkhead domain, the mutation of exon 2 caused a strong increase of CD45RO expression in peripheral CD4+ T cells (80). Moreover, FOXP3 profiles of the Treg cell phenotype population (CD4+ CD25+ CD127−) derived from IPEX patients and IPEX patients’ mothers have been used to reveal functional differences between specific mutations (82). CD4+ CD25+ CD127− T cells, that are FOXP3+ in healthy controls, lack FOXP3 expression in patients with a point mutation in the forkhead domain. The IPEX patient’s mother, who carries the wild type allel and the IPEX mutation on her respective X chromosomes, has about 50% FOXP3− T cells within the Treg cell phenotype population, suggesting that the IPEX mutation in T cells with inactivated wild type X-chromosome does not impair their development. In contrast, no FOXP3− T cells are found in the CD4+ CD25+ CD127− population from the female carrier of the IPEX mutation in exon 2. This suggests that FOXP3Δ2 lacks the ability to promote its own transcription during development and that imprinting of the CD4+ CD25+ CD127− FOXP3+ Treg cell phenotype requires the presence of a functional non-spliced FOXP3 N-terminus.
Exon 7 Skipping
Another alternative splice variant, FOXP3Δ2Δ7, allocates 1–3% of the total FOXP3 mRNA in human CD4+ CD25+ Treg cells (8, 54). The alternatively spliced isoform FOXP3Δ2Δ7 was first described in peripheral blood and was thought to reduce T cell activation (85). Later characterization showed that in contrast to FOXP3fl- and FOXP3Δ2-transduced murine CD4+ T cells, FOXP3Δ2Δ7 expression does not induce the Treg cell phenotype and fail to suppress proliferation of responder cells (8). These results have also been confirmed for human T cells in lentiviral FOXP3Δ2Δ7 transduction experiments and found that specifically in T cells this isoform is strictly expressed in the nucleus through the deletion of both NES located in exon 2 and exon 7 (68). Moreover, transgenic expression of the artificial murine equivalent, FoxP3δ2δ7, does not provide the ability for immune regulation and FoxP3δ2δ7 knock-in mice develop a scurfy-like lymphoproliferative disease (86).
IPEX Mutations Targeting FOXP3 Exon 7
The importance of exon 7 for the proper function of the transcription factor is highlighted by many IPEX mutations that target the coding sequence (37, 82, 87–92) as well as the flanking sequences intron 6 (92) and intron 7 (93–98), that control alternative splicing of exon 7 (Figure 1A). The mutations located in intronic sequences were predicted to facilitate aberrant splicing of exon 7 (78). In line with that, FOXP3 mRNA lacking exon 7 has been detected in two patients with IPEX mutation 816 + 4A > G or 816 + 7G > C (95) and FOXP3 expression, albeit reduced, has been reported for IPEX mutations 816 + 7G > C and 817 − 1 G > A (95–97). Interestingly, malignant Treg cells from a Sézary syndrome patient expressed high amounts of FOXP3Δ2Δ7 and to a lesser extent FOXP3Δ2, but lacked FOXP3fl expression, indicating that FOXP3Δ2Δ7 fails to suppress the impaired activation and proliferation status (99).
FOXP3 exon 7 is part of the leucine zipper domain (Figure 1B) and the IPEX mutations ΔLys250 and ΔGlu251 have been shown to prevent FOXP3 homodimerization or FOXP1 heterodimerization (88, 100). However, the forkhead domain alone has been reported to form a stable domain-swapped dimer with DNA (101) and dimerization of FOXP3Δ2Δ7 with other isoforms has been intact in HEK293T cells (8). Of note, no FOXP3 expression has been detected in CD4+ T cells from FOXP3ΔGlu251 IPEX patients by flow cytometry using isoform-sensitive anti-FOXP3 antibodies (91), whereas patient-derived cell lines and transfected Jurkat or HEK293T cells readily express both mutants, human FOXP3ΔGlu251 or murine FoxP3ΔGlu250 (88, 102). These data highlight the cell type-specific expression, interaction and translocation of FOXP3, and indicate that in T cells impaired formation of homo-/heterodimers not only diminishes FOXP3 functions but also destabilizes its expression in total. In line with this notion, competing posttranslational modifications of the lysine residues in exon 7 have been found to support complex formation through acetylation or protein degradation through ubiquitination (103–105). The absence of exon 7 potentially abrogates this control mechanism and may lead to a swift change of the FOXP3 isoform ratio in conditions that are unfavorable for Treg cells. By this, the relatively low expression of FOXP3Δ2Δ7 may exert its dominant-negative effect to modulate Treg cell functions (8). The precise mechanism deployed to inhibit the immunosuppressive isoforms FOXP3fl and FOXP3Δ2 is unknown so far. FOXP3Δ2Δ7 has been shown to reduce NFAT- and NF-κB-mediated gene transcription as well as to interact with AML1 similar to the other FOXP3 isoforms (8). Based on this, it is plausible that FOXP3Δ2Δ7 neutralizes and sequestrates many cofactors while missing out a crucial component to facilitate immunosuppression, which could encompass the interaction with FOXP1 (88).
Exon 7 Skipping in Th17 Cells
Interestingly, pro-inflammatory stimuli can decrease the function of human Treg cells and induce mixed phenotype profiles. In particular, Th17 cells share some developmental trails with Treg cells and sustain FOXP3 expression to inhibit Th1 cell differentiation (43). Inflammatory bowel disease is associated with the accumulation of FOXP3+ T cells and these cells have been found to preferentially express IL-17 or IL-17 in combination with TNF-α (57). Notably, Treg/Th17 cell plasticity is triggered by IL-1β/IL-2 signaling (106), conditions that also increase the expression of FOXP3 transcripts lacking exon 7 (54). Moreover, deleting exon 2 and exon 7, but not exon 2 alone, from FOXP3 pre-mRNA via splice-shifting ASOs promotes the differentiation of naïve T cells into Th17 cells (54). Thus, FOXP3Δ2Δ7 affects Th17 cell differentiation and may contribute to altered FOXP3Δ2/FOXP3total ratios observed in blood from Hashimoto’s thyroiditis patients or in intestinal biopsies from celiac disease patients (107, 108). Compared to healthy controls, FOXP3 transcripts lacking exon 7 increase in synovial fluid of rheumatoid arthritis as well as in peripheral blood and biopsies from Crohn’s disease patients (54, 63). Furthermore, the expression of IL17A mRNA and FOXP3 transcripts lacking exon 7 correlates in Crohn’s disease biopsies and peripheral blood from coronary artery disease patients (54, 56), indicating that skipping of exon 7 is a common event for IL-17 expression in diverse settings. Further studies will have to elucidate whether FOXP3 isoforms also play a role for IL-17-releasing non-CD4+ cells, e.g., FOXP3+ CD8+ T cells and recently described FOXP3+ eosinophils (109–111).
Taken together, alternative splicing of FOXP3 exon 7 abrogates the suppressive function of the transcription factor and promotes Th17 cell differentiation. The isoform FOXP3Δ2Δ7 may compete for cofactors and prevent complex formation; therefore, exon 7 represents a promising target to counteract other splice variants.
FOXP3 Isoforms in Cancer
FOXP3 expression is not exclusively restricted to immune cells as mRNA and protein is also present in epithelial cells (112). Moreover, absence, mutation, or splice defects of FOXP3 have been reported in various types of carcinoma (113–116). FOXP3 represses the expression of different oncogenes (113–116), while overexpression of FOXP3, but not FOXP3ΔGlu251, decreases tumor cell proliferation (116, 117). Interestingly, some malignancies affected alternative splicing of FOXP3 (99, 113, 118–120). Of note, the numbering of FOXP3 exons differs between immunology and oncology literature. (The first exon is labeled exon-1 and exon 1 in immunological and oncological context, respectively.) Compared to FOXP3fl and FOXP3Δ2 expressing primary mammary epithelial cells, FOXP3fl is absent in breast cancer cell lines and increased exon skipping deleted exon 2, exon 7, or combinations of exon 2 and exon 3, or exon 2 and exon 7 (113). Moreover, the elevated expression of FOXP3Δ2 in bladder cancer causes chemotherapy resistance, facilitates development of more aggressive tumors and correlates inversely with overall survival (119). Furthermore, the FOXP3 gene is directly hit by somatic mutations, some of which target exon 2 (G87Dx2 and V79A) (113, 120). Analysis of the V79A mutation, found in prostate cancer, revealed that mutated FOXP3fl fails to localize within the chromatin fraction, while wild-type FOXP3Δ2 lacks repression of MYC transcription and colony formation in spite of chromatin localization (120). Thus, biased expression of FOXP3Δ2 in epithelial cancers suggests that this isoform lacks the ability for cell cycle control and epigenetic regulation.
Conclusion
In summary, cumulative evidence suggests that the expression of FOXP3fl is promoted by TCR signaling in CD4+ T cells and that higher FOXP3fl ratios in peripheral blood indicate inflammatory conditions. FOXP3fl is an intrinsic suppressor of proliferation in T cells and epithelial cells and exerts specific functions that are linked with epigenetic modifications imprinting the Treg cell lineage development. FOXP3Δ2 is more spread to the nuclear compartment than FOXP3fl, maintains primarily the suppressive phenotype in Treg cells and requires a pre-established Treg cell phenotype for stable expression. In contrast, nuclear FOXP3Δ2Δ7 correlates with IL-17 expression in peripheral blood and restricts the function of other FOXP3 isoforms via competition for cofactors (Figure 2).
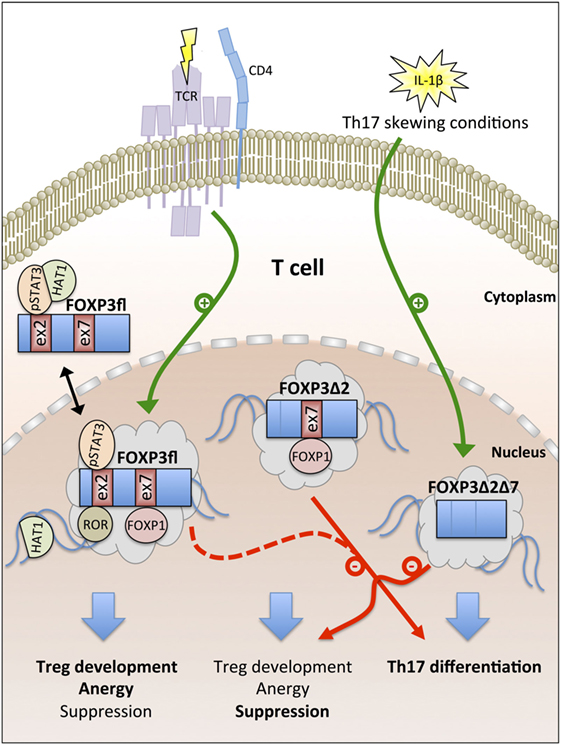
Figure 2. Overview of FOXP3 isoform functions. T cell antigen receptor (TCR) signaling promotes the generation of the full-length FOXP3 isoform FOXP3fl. Nuclear export signals (NES) in FOXP3fl enable recruitment of cytoplasmic cofactors [e.g., phosphorylated signal transducer and activator of transcription 3 (pSTAT3) and histone acetyltransferase-1 (HAT1)]. In the nucleus, exon 2 and exon 7 of FOXP3fl bind to retinoid acid receptor-related orphan receptors (ROR) and forkhead box P1 (FOXP1), respectively. FOXP3fl induces Treg cell development, T cell anergy, and suppression. FOXP3Δ2 lacks a NES in exon 2 and supports Treg cell-mediated suppression but fails to interact with pSTAT3 and ROR; both FOXP3fl and FOXP3Δ2 inhibit Th17 cell differentiation. IL-1β signaling induces the generation of FOXP3Δ2Δ7 that lacks NES in exon 2 and exon 7. FOXP3Δ2Δ7 promotes Th17 cell differentiation and acts as dominant-negative inhibitor of FOXP3fl and FOXP3Δ2.
Future Perspectives
The generation of isoforms has a strong impact on functional properties of human FOXP3 and the analysis of the FOXP3 isoform profile helps to assess the cellular status. Thus, utilized anti-human FOXP3 antibody clones need to be specified to clarify whether FOXP3 isoform expression has been considered. The development of additional anti-FOXP3 antibodies specific for alternatively spliced regions and epitope mapping for existent antibody clones will improve future investigations of FOXP3-expressing cells, as there are currently no monoclonal anti-FOXP3 antibodies available that recognize exon 7 or Δ7 and the array of applications for Δ2-specific antibodies is limited (Table 1). However, real-time PCR techniques to quantify alternative splicing of exon 2 and exon 7 have provided insight into activation-induced and Th17-related FOXP3 isoform expression ratios in healthy and inflammatory conditions at cell population levels. Moreover, a novel approach that calculates the splicing ratio of FOXP3 exon 2 through co-stainings with antibodies recognizing exon 2 and a non-spliced region of FOXP3 by flow cytometry offers the possibility to observe differential alternative splicing on a cellular level. This method may help to elucidate FOXP3 isoform expression during thymic development and in peripheral T cell subpopulations in future studies.
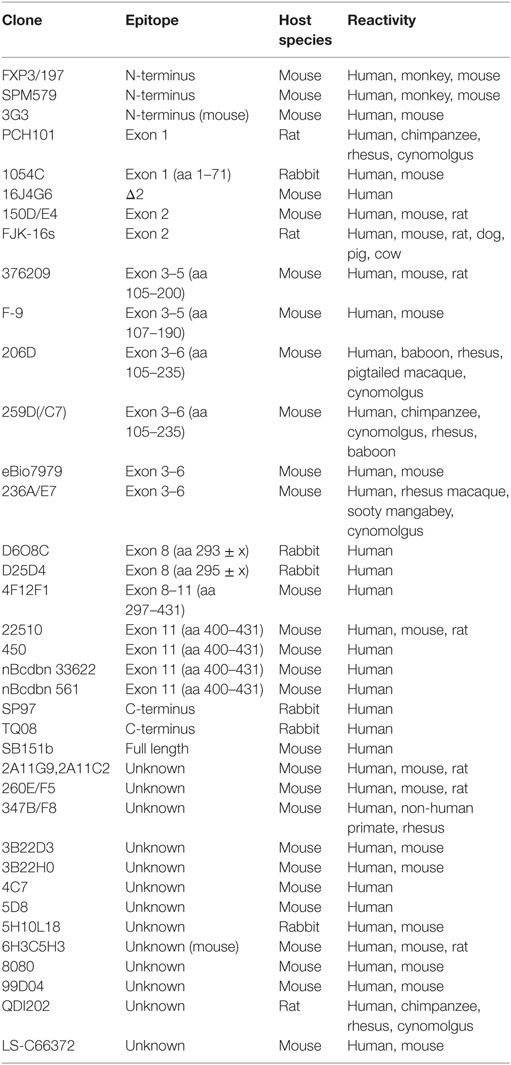
Table 1. Epitopes of monoclonal anti-human FOXP3 antibody clones.
While alternative splicing of FOXP3 appears to add another layer of complexity to Treg cell biology, it can be of potential therapeutic value. First, the availability of whole transcriptome sequencing data will reveal associations of FOXP3 splice variants with clinical diseases allowing faster diagnosis. Second, genetic disorders with impaired alternative splicing have previously been treated with ASOs in clinical trials (121). Along this line, ASOs that interfere with FOXP3 pre-mRNA splicing may become a potent tool to affect cellular functions, as alternative splicing of FOXP3 regulates development, proliferation, lineage stability and suppressive capacity of FOXP3+ cells. Thus, manipulation of alternative splicing to generate therapeutic FOXP3 isoform profiles could allow novel treatments to modulate Treg cell suppression, to control Th17 cell differentiation as well as to inhibit carcinoma proliferation in the future.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author has filed a patent application on FOXP3 isoform modification and declares no conflict of interest.
Acknowledgments
I like to thank Dr. Thomas Renné for critical reading of this manuscript.
Funding
This work was supported by the German Research Foundation (SFB 841, TP B08 and SFB 877, TP A11).
References
1. Levings MK, Sangregorio R, Roncarolo MG. Human CD25+CD4+ T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med (2001) 193:1295–302. doi:10.1084/jem.193.11.1295
2. Dieckmann D, Plottner H, Berchtold S, Berger T, Schuler G. Ex vivo isolation and characterization of CD4+CD25+ T cells with regulatory properties from human blood. J Exp Med (2001) 193:1303–10. doi:10.1084/jem.193.11.1303
3. Jonuleit H, Schmitt E, Stassen M, Tuettenberg A, Knop J, Enk AH. Identification and functional characterization of human CD4+CD25+ T cells with regulatory properties isolated from peripheral blood. J Exp Med (2001) 193:1285–94. doi:10.1084/jem.193.11.1285
4. Ng WF, Duggan PJ, Ponchel F, Matarese G, Lombardi G, Edwards AD, et al. Human CD4+CD25+ cells: a naturally occurring population of regulatory T cells. Blood (2001) 98:2736–44. doi:10.1182/blood.V98.9.2736
5. Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol (2001) 167:1245–53. doi:10.4049/jimmunol.167.3.1245
6. Roncador G, Brown PJ, Maestre L, Hue S, Martínez-Torrecuadrada JL, Ling KL, et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. Eur J Immunol (2005) 35:1681–91. doi:10.1002/eji.200526189
7. Yagi H, Nomura T, Nakamura K, Yamazaki S, Kitawaki T, Hori S, et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol (2004) 16:1643–56. doi:10.1093/intimm/dxh165
8. Mailer RK, Falk K, Rötzschke O. Absence of leucine zipper in the natural FOXP3Delta2Delta7 isoform does not affect dimerization but abrogates suppressive capacity. PLoS One (2009) 4:e6104. doi:10.1371/journal.pone.0006104
9. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi:10.1038/83707
10. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27:20–1. doi:10.1038/83713
11. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi:10.1038/83784
12. Kitagawa Y, Sakaguchi S. Molecular control of regulatory T cell development and function. Curr Opin Immunol (2017) 49:64–70. doi:10.1016/j.coi.2017.10.002
13. Koh KP, Sundrud MS, Rao A. Domain requirements and sequence specificity of DNA binding for the forkhead transcription factor FOXP3. PLoS One (2009) 4:e8109. doi:10.1371/journal.pone.0008109
14. Rudra D, deRoos P, Chaudhry A, Niec RE, Arvey A, Samstein RM, et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol (2012) 13:1010–9. doi:10.1038/ni.2402
15. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature (2007) 445:936–40. doi:10.1038/nature05563
16. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature (2007) 445:931–5. doi:10.1038/nature05478
17. Sadlon TJ, Wilkinson BG, Pederson S, Brown CY, Bresatz S, Gargett T, et al. Genome-wide identification of human FOXP3 target genes in natural regulatory T cells. J Immunol (2010) 185:1071–81. doi:10.4049/jimmunol.1000082
18. Ramsdell F, Ziegler SF. FOXP3 and scurfy: how it all began. Nat Rev Immunol (2014) 14:343–9. doi:10.1038/nri3650
19. Lozano T, Casares N, Lasarte JJ. Searching for the Achilles heel of FOXP3. Front Oncol (2013) 3:294. doi:10.3389/fonc.2013.00294
20. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol (2012) 30:531–64. doi:10.1146/annurev.immunol.25.022106.141623
21. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17:703–17. doi:10.1038/nri.2017.75
22. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463:808–12. doi:10.1038/nature08750
23. Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, et al. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity (2009) 31:932–40. doi:10.1016/j.immuni.2009.10.006
24. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med (2009) 206:3001–14. doi:10.1084/jem.20091411
25. Long M, Park SG, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity (2009) 31:921–31. doi:10.1016/j.immuni.2009.09.022
26. Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol (2007) 5:e38. doi:10.1371/journal.pbio.0050038
27. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38:1654–63. doi:10.1002/eji.200838105
28. Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell (2012) 151:153–66. doi:10.1016/j.cell.2012.06.053
29. Ohkura N, Hamaguchi M, Morikawa H, Sugimura K, Tanaka A, Ito Y, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity (2012) 37:785–99. doi:10.1016/j.immuni.2012.09.010
30. Bettini ML, Pan F, Bettini M, Finkelstein D, Rehg JE, Floess S, et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity (2012) 36:717–30. doi:10.1016/j.immuni.2012.03.020
31. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol (2008) 9:632–40. doi:10.1038/ni.1607
32. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature (2007) 445:771–5. doi:10.1038/nature05543
33. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15:1070–8. doi:10.1038/ni.3004
34. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity (2014) 41:722–36. doi:10.1016/j.immuni.2014.10.012
35. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest (2013) 123:580–93. doi:10.1172/JCI65013
36. Walker MR, Kasprowicz DJ, Gersuk VH, Bènard A, Van Landeghen M, Buckner JH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25– T cells. J Clin Invest (2003) 112:1437–43. doi:10.1172/JCI19441
37. Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A (2006) 103:6659–64. doi:10.1073/pnas.0509484103
38. Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol (2007) 19:345–54. doi:10.1093/intimm/dxm014
39. Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naïve human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood (2007) 110:2983–90. doi:10.1182/blood-2007-06-094656
40. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol (2007) 37:129–38. doi:10.1002/eji.200636435
41. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells: their development, stability, and applications. Trends Immunol (2016) 37:803–11. doi:10.1016/j.it.2016.08.012
42. Schmidt A, Eriksson M, Shang MM, Weyd H, Tegnér J. Comparative analysis of protocols to induce human CD4+Foxp3+ regulatory T cells by combinations of IL-2, TGF-beta, retinoic acid, rapamycin and butyrate. PLoS One (2016) 11:e0148474. doi:10.1371/journal.pone.0148474
43. McMurchy AN, Gillies J, Gizzi MC, Riba M, Garcia-Manteiga JM, Cittaro D, et al. A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood (2013) 121:1265–75. doi:10.1182/blood-2012-05-431023
44. Martinez NM, Lynch KW. Control of alternative splicing in immune responses: many regulators, many predictions, much still to learn. Immunol Rev (2013) 253:216–36. doi:10.1111/imr.12047
45. Berget SM, Moore C, Sharp PA. Spliced segments at the 5’ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci U S A (1977) 74:3171–5.
46. Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell (1977) 12:1–8. doi:10.1016/0092-8674(77)90180-5
47. Havens MA, Duelli DM, Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA (2013) 4:247–66. doi:10.1002/wrna.1158
48. Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol (2006) 24:209–26. doi:10.1146/annurev.immunol.24.021605.090547
49. Mailer RKW, Gisterå A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia induces differentiation of regulatory T cells in the liver. Circ Res (2017) 120:1740–53. doi:10.1161/CIRCRESAHA.116.310054
50. Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, et al. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest (2005) 115:3276–84. doi:10.1172/JCI24685
51. Piva F, Giulietti M, Nocchi L, Principato G. SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics (2009) 25:1211–3. doi:10.1093/bioinformatics/btp124
52. Nieto Moreno N, Giono LE, Cambindo Botto AE, Muñoz MJ, Kornblihtt AR. Chromatin, DNA structure and alternative splicing. FEBS Lett (2015) 589:3370–8. doi:10.1016/j.febslet.2015.08.002
53. Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exonrecognition. Cell Res (2013) 23:1256–69. doi:10.1038/cr.2013.110
54. Mailer RK, Joly AL, Liu S, Elias S, Tegner J, Andersson J. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep (2015) 5:14674. doi:10.1038/srep14674
55. Aarts-Riemens T, Emmelot ME, Verdonck LF, Mutis T. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(-) cells. Eur J Immunol (2008) 38:1381–90. doi:10.1002/eji.200737590
56. Lundberg AK, Jonasson L, Hansson GK, Mailer RKW. Activation-induced FOXP3 isoform profile in peripheral CD4+ T cells is associated with coronary artery disease. Atherosclerosis (2017) 267:27–33. doi:10.1016/j.atherosclerosis.2017.10.026
57. Lord JD, Valliant-Saunders K, Hahn H, Thirlby RC, Ziegler SF. Paradoxically increased FOXP3+ T cells in IBD do not preferentially express the isoform of FOXP3lacking exon 2. Dig Dis Sci (2012) 57:2846–55. doi:10.1007/s10620-012-2292-3
58. Miyabe C, Miyabe Y, Strle K, Kim ND, Stone JH, Luster AD, et al. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann Rheum Dis (2017) 76:898–905. doi:10.1136/annrheumdis-2016-210070
59. Free ME, Bunch DO, McGregor JA, Jones BE, Berg EA, Hogan SL, et al. Patients with antineutrophil cytoplasmic antibody-associated vasculitis have defective Treg cell function exacerbated by the presence of a suppression-resistant effector cell population. Arthritis Rheum (2013) 65:1922–33. doi:10.1002/art.37959
60. Jakiela B, Iwaniec T, Plutecka H, Celinska-Lowenhoff M, Dziedzina S, Musial J. Signs of impaired immunoregulation and enhanced effector T-cell responses in the primary antiphospholipid syndrome. Lupus (2016) 25:389–98. doi:10.1177/0961203315618267
61. Chen L, Wu J, Pier E, Zhao Y, Shen Z. mTORC2-PKBα/Akt1 Serine 473 phosphorylation axis is essential for regulation of FOXP3 Stability by chemokine CCL3 in psoriasis. J Invest Dermatol (2013) 133:418–28. doi:10.1038/jid.2012.333
62. Crellin NK, Garcia RV, Levings MK. Altered activation of AKT is required for the suppressive function of human CD4+CD25+ T regulatory cells. Blood (2007) 109:2014–22. doi:10.1182/blood-2006-07-035279
63. Ryder LR, Bartels EM, Woetmann A, Madsen HO, Odum N, Bliddal H, et al. FoxP3 mRNA splice forms in synovial CD4+ T cells in rheumatoid arthritis and psoriatic arthritis. APMIS (2012) 120:387–96. doi:10.1111/j.1600-0463.2011.02848.x
64. Ryder LR, Ryder LP, Bartels EM, Woetmann A, Madsen HO, Ødum N, et al. Differential effects of decoy receptor- and antibody-mediated tumour necrosis factor blockage on FoxP3 expression in responsive arthritis patients. APMIS (2013) 121:337–47. doi:10.1111/apm.12004
65. Ryder LR, Woetmann A, Madsen HO, Ødum N, Ryder LP, Bliddal H, et al. Expression of full-length and splice forms of FoxP3 in rheumatoid arthritis. Scand J Rheumatol (2010) 39:279–86. doi:10.3109/03009740903555374
66. Link M, Salur L, Kisand K, Rajasalu T, Tillmann V, Uibo R. Higher FoxP3 mRNA expression in peripheral blood mononuclear cells of GAD65 or IA-2 autoantibody-positive compared with autoantibody-negative persons. APMIS (2008) 116:896–902. doi:10.1111/j.1600-0463.2008.00889.x
67. Suzuki K, Setoyama Y, Yoshimoto K, Tsuzaka K, Abe T, Takeuchi T. Decreased mRNA expression of two FOXP3 isoforms in peripheral blood mononuclear cells from patients with rheumatoid arthritis and systemic lupus erythematosus. Int J Immunopathol Pharmacol (2011) 24:7–14. doi:10.1177/039463201102400102
68. Magg T, Mannert J, Ellwart JW, Schmid I, Albert MH. Subcellular localization of FOXP3 in human regulatory and nonregulatory T cells. Eur J Immunol (2012) 42:1627–38. doi:10.1002/eji.201141838
69. Hossain DM, Panda AK, Manna A, Mohanty S, Bhattacharjee P, Bhattacharyya S, et al. FoxP3 acts as a cotranscription factor with STAT3 in tumor-induced regulatory T cells. Immunity (2013) 39:1057–69. doi:10.1016/j.immuni.2013.11.005
70. Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol (2008) 180:4785–92. doi:10.4049/jimmunol.180.7.4785
71. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453:236–40. doi:10.1038/nature06878
72. Free ME, Su MA. Reply: to PMID 23553415. Arthritis Rheum (2013) 65:3316–8. doi:10.1002/art.38211 PMID 23553415,
73. Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, et al. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol (2006) 18:1197–209. doi:10.1093/intimm/dxl060
74. Chellappa S, Hugenschmidt H, Hagness M, Line PD, Labori KJ, Wiedswang G, et al. Regulatory T cells that co-express RORγt and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. Oncoimmunology (2015) 5:e1102828. doi:10.1080/2162402X.2015.1102828
75. Deng G, Nagai Y, Xiao Y, Li Z, Dai S, Ohtani T, et al. Pim-2 kinase influences regulatory T cell function and stability by mediating Foxp3 protein N-terminal phosphorylation. J Biol Chem (2015) 290:20211–20. doi:10.1074/jbc.M115.638221
76. Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol (2008) 180:5794–8. doi:10.4049/jimmunol.180.9.5794
77. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol (2015) 16:1174–84. doi:10.1038/ni.3269
78. d’Hennezel E, Bin Dhuban K, Torgerson T, Piccirillo CA. The immunogenetics of immune dysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet (2012) 49:291–302. doi:10.1136/jmedgenet-2012-100759
79. Kobayashi I, Shiari R, Yamada M, Kawamura N, Okano M, Yara A, et al. Novel mutations of FOXP3 in two Japanese patients with immune dysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX). J Med Genet (2001) 38:874–6. doi:10.1136/jmg.38.12.874
80. Fuchizawa T, Adachi Y, Ito Y, Higashiyama H, Kanegane H, Futatani T, et al. Developmental changes of FOXP3-expressing CD4+CD25+ regulatory T cells and their impairment in patients with FOXP3 gene mutations. Clin Immunol (2007) 125:237–46. doi:10.1016/j.clim.2007.08.004
81. Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, et al. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care (2009) 32:111–6. doi:10.2337/dc08-1188
82. Otsubo K, Kanegane H, Kamachi Y, Kobayashi I, Tsuge I, Imaizumi M, et al. Identification of FOXP3-negative regulatory T-like (CD4(+)CD25(+)CD127(low)) cells in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Clin Immunol (2011) 141:111–20. doi:10.1016/j.clim.2011.06.006
83. Moudgil A, Perriello P, Loechelt B, Przygodzki R, Fitzerald W, Kamani N. Immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: an unusual cause of proteinuria in infancy. Pediatr Nephrol (2007) 22:1799–802. doi:10.1007/s00467-007-0532-0
84. Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood (2007) 109:383–5. doi:10.1182/blood-2006-05-025072
85. Smith EL, Finney HM, Nesbitt AM, Ramsdell F, Robinson MK. Splice variants of human FOXP3 are functional inhibitors of human CD4+ T-cell activation. Immunology (2006) 119:203–11. doi:10.1111/j.1365-2567.2006.02425.x
86. Joly AL, Liu S, Dahlberg CI, Mailer RK, Westerberg LS, Andersson J. Foxp3 lacking exons 2 and 7 is unable to confer suppressive ability to regulatory T cells in vivo. J Autoimmun (2015) 63:23–30. doi:10.1016/j.jaut.2015.06.009
87. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet (2002) 39:537–45. doi:10.1136/jmg.39.8.537
88. Li B, Samanta A, Song X, Iacono KT, Brennan P, Chatila TA, et al. FOXP3 is a homo-oligomer and a component of a supramolecular regulatory complex disabled in the human XLAAD/IPEX autoimmune disease. Int Immunol (2007) 19:825–35. doi:10.1093/intimm/dxm043
89. Hashimura Y, Nozu K, Kanegane H, Miyawaki T, Hayakawa A, Yoshikawa N, et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Nephrol (2009) 24:1181–6. doi:10.1007/s00467-009-1119-8
90. López SI, Ciocca M, Oleastro M, Cuarterolo ML, Rocca A, de Dávila MT, et al. Autoimmune hepatitis type 2 in a child with IPEX syndrome. J Pediatr Gastroenterol Nutr (2011) 53:690–3. doi:10.1097/MPG.0b013e3182250651
91. Moes N, Rieux-Laucat F, Begue B, Verdier J, Neven B, Patey N, et al. Reduced expression of FOXP3 and regulatory T-cell function in severe forms of early-onset autoimmune enteropathy. Gastroenterology (2010) 139:770–8. doi:10.1053/j.gastro.2010.06.006
92. Halabi-Tawil M, Ruemmele FM, Fraitag S, Rieux-Laucat F, Neven B, Brousse N, et al. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br J Dermatol (2009) 160:645–51. doi:10.1111/j.1365-2133.2008.08835.x
93. Passerini L, Olek S, Di Nunzio S, Barzaghi F, Hambleton S, Abinun M, et al. Forkhead box protein 3 (FOXP3) mutations lead to increased TH17 cell numbers and regulatory T-cell instability. J Allergy Clin Immunol (2011) 128:1376.e–9.e. doi:10.1016/j.jaci.2011.09.010
94. Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, et al. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol (2008) 122:1105.e–12.e. doi:10.1016/j.jaci.2008.09.027
95. Harbuz R, Lespinasse J, Boulet S, Francannet C, Creveaux I, Benkhelifa M, et al. Identification of new FOXP3 mutations and prenatal diagnosis of IPEX syndrome. Prenat Diagn (2010) 30:1072–8. doi:10.1002/pd.2613
96. Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders J, et al. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol (2010) 126:1000–5. doi:10.1016/j.jaci.2010.05.021
97. Heltzer ML, Choi JK, Ochs HD, Sullivan KE, Torgerson TR, Ernst LM. A potential screening tool for IPEX syndrome. Pediatr Dev Pathol (2007) 10:98–105. doi:10.2350/06-07-0130.1
98. Tsuda M, Torgerson TR, Selmi C, Gambineri E, Carneiro-Sampaio M, Mannurita SC, et al. The spectrum of autoantibodies in IPEX syndrome is broad and includes anti-mitochondrial autoantibodies. J Autoimmun (2010) 35:265–8. doi:10.1016/j.jaut.2010.06.017
99. Krejsgaard T, Gjerdrum LM, Ralfkiaer E, Lauenborg B, Eriksen KW, Mathiesen AM, et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sézary syndrome. Leukemia (2008) 22:2230–9. doi:10.1038/leu.2008.224
100. Song X, Li B, Xiao Y, Chen C, Wang Q, Liu Y, et al. Structural and biological features of FOXP3 dimerization relevant to regulatory T cell function. Cell Rep (2012) 1:665–75. doi:10.1016/j.celrep.2012.04.012
101. Chen Y, Chen C, Zhang Z, Liu CC, Johnson ME, Espinoza CA, et al. DNA binding by FOXP3 domain-swapped dimer suggests mechanisms of long-range chromosomal interactions. Nucleic Acids Res (2015) 43:1268–82. doi:10.1093/nar/gku1373
102. Chae WJ, Henegariu O, Lee SK, Bothwell AL. The mutant leucine-zipper domain impairs both dimerization and suppressive function of Foxp3 in T cells. Proc Natl Acad Sci U S A (2006) 103:9631–6. doi:10.1073/pnas.0600225103
103. Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc Natl Acad Sci U S A (2007) 104:4571–6. doi:10.1073/pnas.0700298104
104. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115:965–74. doi:10.1182/blood-2009-02-207118
105. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CP, Pals CE, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity (2013) 39:259–71. doi:10.1016/j.immuni.2013.05.018
106. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL-1beta and IL-2 convert human Treg into T(H)17 cells. Clin Immunol (2009) 131:298–307. doi:10.1016/j.clim.2008.12.008
107. Kristensen B, Hegedüs L, Madsen HO, Smith TJ, Nielsen CH. Altered balance between self-reactive T helper (Th)17 cells and Th10 cells and between full-length forkhead box protein 3 (FoxP3) and FoxP3 splice variants in Hashimoto’s thyroiditis. Clin Exp Immunol (2015) 180:58–69. doi:10.1111/cei.12557
108. Serena G, Yan S, Camhi S, Patel S, Lima RS, Sapone A, et al. Proinflammatory cytokine interferon-γ and microbiome-derived metabolites dictate epigenetic switch between forkhead box protein 3 isoforms in coeliac disease. Clin Exp Immunol (2017) 187:490–506. doi:10.1111/cei.12911
109. Kaur G, Goodall JC, Jarvis LB, Hill Gaston JS. Characterisation of Foxp3 splice variants in human CD4+ and CD8+ T cells – identification of Foxp3Δ7 in human regulatory T cells. Mol Immunol (2010) 48:321–32. doi:10.1016/j.molimm.2010.07.008
110. Lingblom C, Wallander J, Ingelsten M, Bergquist H, Bove M, Saalman R, et al. Eosinophils from eosinophilic oesophagitis patients have T cell suppressive capacity and express FOXP3. Clin Exp Immunol (2017) 187:455–65. doi:10.1111/cei.12898
111. Guerra ES, Lee CK, Specht CA, Yadav B, Huang H, Akalin A, et al. Central Role of IL-23 and IL-17 producing eosinophils as immunomodulatory effector cells in acute pulmonary aspergillosis and allergic asthma. PLoS Pathog (2017) 13:e1006175. doi:10.1371/journal.ppat.1006175
112. Chen GY, Chen C, Wang L, Chang X, Zheng P, Liu Y. Cutting edge: broad expression of the FoxP3 locus in epithelial cells: a caution against early interpretation of fatal inflammatory diseases following in vivo depletion of FoxP3-expressing cells. J Immunol (2008) 180:5163–6. doi:10.4049/jimmunol.180.8.5163
113. Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell (2007) 129:1275–86. doi:10.1016/j.cell.2007.04.034
114. Zuo T, Liu R, Zhang H, Chang X, Liu Y, Wang L, et al. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J Clin Invest (2007) 117:3765–73. doi:10.1172/JCI32538
115. Liu R, Wang L, Chen G, Katoh H, Chen C, Liu Y, et al. FOXP3 up-regulates p21 expression by site-specific inhibition of histone deacetylase 2/histonedeacetylase 4 association to the locus. Cancer Res (2009) 69:2252–9. doi:10.1158/0008-5472.CAN-08-3717
116. Li W, Wang L, Katoh H, Liu R, Zheng P, Liu Y. Identification of a tumor suppressor relay between the FOXP3 and the hippo pathways in breast and prostate cancers. Cancer Res (2011) 71:2162–71. doi:10.1158/0008-5472.CAN-10-3268
117. Zhang HY, Sun H. Up-regulation of Foxp3 inhibits cell proliferation, migration and invasion in epithelial ovarian cancer. Cancer Lett (2010) 287:91–7. doi:10.1016/j.canlet.2009.06.001
118. Ebert LM, Tan BS, Browning J, Svobodova S, Russell SE, Kirkpatrick N, et al. The regulatory T cell-associated transcription factor FoxP3 is expressed by tumor cells. Cancer Res (2008) 68:3001–9. doi:10.1158/0008-5472.CAN-07-5664
119. Zhang H, Prado K, Zhang KX, Peek EM, Lee J, Wang X, et al. Biased expression of the FOXP3Δ3 isoform in aggressive bladder cancer mediates differentiation and cisplatin chemotherapy resistance. Clin Cancer Res (2016) 22:5349–61. doi:10.1158/1078-0432.CCR-15-2581
120. Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, et al. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer Cell (2009) 16:336–46. doi:10.1016/j.ccr.2009.08.016
Keywords: FOXP3, isoform, alternative splicing, FOXP3Δ2, FOXP3Δ2Δ7, Treg cells, Th17 cells, antisense oligonucleotides
Citation: Mailer RKW (2018) Alternative Splicing of FOXP3—Virtue and Vice. Front. Immunol. 9:530. doi: 10.3389/fimmu.2018.00530
Received: 07 December 2017; Accepted: 28 February 2018;
Published: 13 March 2018
Edited by:
Paulo Vieira, Institut Pasteur, FranceReviewed by:
Antonio Bandeira, Center national de la recherche scientifique (CNRS), FranceXing Chang, Shanghai Institutes for Biological Sciences (CAS), China
Copyright: © 2018 Mailer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reiner K. W. Mailer, ci5tYWlsZXJAdWtlLmRl