 Maha Ahmed Al-Mozaini1,2
Maha Ahmed Al-Mozaini1,2 Anthony G. Tsolaki1
Anthony G. Tsolaki1 Munirah Abdul-Aziz1,3
Munirah Abdul-Aziz1,3 Suhair M. Abozaid1,2
Suhair M. Abozaid1,2 Mohammed N. Al-Ahdal2
Mohammed N. Al-Ahdal2 Ansar A. Pathan1Valarmathy Murugaiah1Evgeny M. Makarov1
Ansar A. Pathan1Valarmathy Murugaiah1Evgeny M. Makarov1 Anuvinder Kaur1
Anuvinder Kaur1 Robert B. Sim3
Robert B. Sim3 Uday Kishore1*
Uday Kishore1* Lubna Kouser1*
Lubna Kouser1*
- 1College of Health and Life Sciences, Brunel University London, London, United Kingdom
- 2Department of Infection and Immunity, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia
- 3Department of Biochemistry, Oxford University, Oxford, United Kingdom
Mycobacterium tuberculosis can proficiently enter macrophages and diminish complement activation on its cell surface. Within macrophages, the mycobacterium can suppress macrophage apoptosis and survive within the intracellular environment. Previously, we have shown that complement regulatory proteins such as factor H may interfere with pathogen–macrophage interactions during tuberculosis infection. In this study, we show that Mycobacterium bovis BCG binds properdin, an upregulator of the complement alternative pathway. TSR4+5, a recombinant form of thrombospondin repeats 4 and 5 of human properdin expressed in tandem, which is an inhibitor of the alternative pathway, was also able to bind to M. bovis BCG. Properdin and TSR4+5 were found to inhibit uptake of M. bovis BCG by THP-1 macrophage cells in a dose-dependent manner. Quantitative real-time PCR revealed elevated pro-inflammatory responses (TNF-α, IL-1β, and IL-6) in the presence of properdin or TSR4+5, which gradually decreased over 6 h. Correspondingly, anti-inflammatory responses (IL-10 and TGF-β) showed suppressed levels of expression in the presence of properdin, which gradually increased over 6 h. Multiplex cytokine array analysis also revealed that properdin and TSR4+5 significantly enhanced the pro-inflammatory response (TNF-α, IL-1β, and IL-1α) at 24 h, which declined at 48 h, whereas the anti-inflammatory response (IL-10) was suppressed. Our results suggest that properdin may interfere with mycobacterial entry into macrophages via TSR4 and TSR5, particularly during the initial stages of infection, thus affecting the extracellular survival of the pathogen. This study offers novel insights into the non-complement related functions of properdin during host–pathogen interactions in tuberculosis.
Introduction
Properdin is an upregulator of the alternative pathway of complement activation. In one of the three pathways of the complement system, the alternative pathway, the activation of the major complement opsonin, C3, is driven by a complex serine protease, C3bBb, also called the C3 convertase, which is bound to the surface of the complement-activating target. To form C3bBb, factor B associates with C3b in the presence of Mg2+ and factor D, a serine protease, which cleaves factor B into Bb and Ba fragments producing a C3 convertase C3bBb (1). This complex, which has a half-life of 90 s, is stabilized by the binding of properdin, which increases the half-life by 5- to 10-fold (2). Furthermore, C3b molecules are generated by C3bBb and deposited near to the surface-bound convertase leading to the opsonization of the target and formation of C5 convertase, producing C5a and C5b, leading on to the lytic pathway and cell lysis (3).
The monomer of human properdin (53 kDa) has a flexible rod-like structure with a length of 26 nm and a diameter of 2.5 nm, composed of seven thrombospondin type I repeats (TSR). Each TSR is of about 60 amino acids, typically containing six conserved cysteine residues: these occur in TSR1–TSR6 (4–6), but the N-terminal domain, TSR0, is truncated. TSR4 is crucial for binding to C3bBb and TSR5 for binding to C3bBb, suggesting that both TSRs may be important for stabilizing the C3 convertase complex (5). Recently, TSR4+5, expressed as a double domain, has been shown to bind to properdin ligands such as C3b and inhibit the alternative complement pathway (7). These studies demonstrate the important role that these TSRs may play in the alternative pathway and in their interaction with pathogens.
Properdin circulates in plasma at a concentration of about 4–25 µg/ml existing as cyclic oligomers, dimer, trimer, and tetramer in a ratio of 26:54:20 (4). The dissociation and reassociation of properdin upon denaturation–renaturation cycles stimulated by guanidine or low pH indicates properdin ratio stability in solution (8). The interaction between properdin monomers involves the N-terminal end of one monomer and the C-terminal end of another (9). Properdin can also bind to microbial surfaces of several pathogens, including Neisseria gonorrhoeae (10), Salmonella typhimurium lipopolysaccharide (LPS), Neisseria meningitidis lipooligosaccharide (11), and Chlamydia pneumoniae (12). Binding of properdin to microbial surfaces results in the recruitment of fluid phase C3b, inducing assembly of C3 convertase C3bBb and causing further deposition of C3b on the pathogen surface (13–15), subsequently generating a C5 convertase, MAC formation, and cell lysis. At 10 µg/ml, recombinant properdin enhanced complement deposition on N. meningitidis and S. pneumoniae and dramatically enhanced serum lysis of these bacteria, and in the mouse model, significantly reduced bacteremia and increased survival rates (16).
Although Mycobacterium tuberculosis and its close-relative Mycobacterium bovis have significant interaction with components of the innate immune system, e.g., toll-like receptors, complement, surfactant proteins SP-A and SP-D (17), the initial stages of tuberculosis pathogenesis remain poorly understood. Many bacteria have evolved mechanisms to evade immune responses: by inhibiting complement activation by proteolytic cleavage of complement proteins, having their own complement inhibitors (18), or binding complement regulatory proteins like factor H (15, 19).
Mycobacterium tuberculosis is a highly specialized intracellular pathogen and may exploit complement proteins to enhance its uptake by macrophages. Although it has been shown that M. tuberculosis can activate all three pathways of the complement system (20, 21), it is unclear how the pathogen uses complement proteins in tuberculosis pathogenesis. M. tuberculosis has been shown to bind to complement receptors (CR) CR1, CR3, and CR4 and gain entry into macrophages (22–24). There is also evidence that enhanced phagocytosis of M. tuberculosis by human alveolar and monocyte-derived macrophages results from C3 opsonization (24). The ability of M. tuberculosis to bind to CR3 non-opsonically has also been shown which may be important for bacterial invasion when complement is sparse, for example, in the lung (25). Properdin has recently been considered as a pattern recognition receptor (PRR) on its own, i.e., binding to recognition patterns without need for prior deposition of C3b or C3bBb (26–28). Therefore, we investigated the role of properdin in tuberculosis pathogenesis, by using the model organism M. bovis BCG.
Here, we show, for the first time, that properdin and recombinant form of TSR4+5 expressed as a two-module protein binds to M. bovis BCG, demonstrating its role as a soluble PRR. Properdin and TSR4+5 were found to inhibit the uptake of M. bovis BCG by macrophages during phagocytosis, altering the pro- and anti-inflammatory cytokine response, and thus, possibly shaping the adaptive immune response in tuberculosis pathogenesis.
Materials and Methods
Purification of Native Properdin
The affinity columns, IgG Sepharose and anti-properdin monoclonal antibody Sepharose, were prepared as described previously (7). The IgG-Sepharose column was prepared from human non-immune IgG (~26 mg IgG/ml of Sepharose) coupled to CNBr-activated Sepharose (GE Healthcare, UK). For preparation of the anti-human properdin column, CNBr-activated Sepharose (GE Healthcare Life Sciences, UK) was used to couple to anti-properdin mouse monoclonal antibody (2 mg/ml). One liter of human plasma (TCS Biosciences) containing 5 mM EDTA was filtered through Whatman filter paper before applying to IgG Sepharose to deplete C1q (which would otherwise have bound to the IgG on the anti-properdin Sepharose). The column was washed with three bed volume of HEPES buffer (10 mM HEPES, 140 mM NaCl, 0.5 mM EDTA, and pH 7.4). Plasma was then applied to the monoclonal anti-properdin column and washed with the same HEPES buffer. Bound properdin was eluted with 3 M MgCl2 and the peak fractions were dialyzed against HEPES buffer overnight at 4°C. Contaminants were further removed by applying the pooled protein fractions to a HiTrap Q FF-Sepharose (GE Healthcare) ion-exchange column, followed by washing the column with three bed volumes of 50 mM Tris–HCl, pH 7.5, 50 mM NaCl, and 5 mM EDTA. Properdin did not bind to the Q Sepharose column and appeared in the flow-through free from contaminants as demonstrated by SDS-PAGE.
For the size exclusion chromatography analysis, 50 µl of the proteins at the concentrations varying from 0.3 mg/mL to 1.0 mg/mL were applied to a TSKgel G2000SWXL, 5 µm, 7.8 × 300 mm column (Tosoh Bioscience). The column was equilibrated with buffer containing 50 mM sodium phosphate, pH7.0 and 300 mM NaCl at the flow rate 0.3 ml/min using SCL-10Avp HPLC system (Shimadzu). The absorbance was detected at 230 and 280 nm. The Bio-Rad Gel Filtration Standard (Cat # 151-1901) were used for the protein molecular weight calibration of the column.
Expression and Purification of TSR4+5
The recombinant maltose-binding protein (MBP) fusion proteins MBP-TSR4+5, MBP-TSR4, or MBP-TSR5 were expressed in Escherichia coli as described previously (7, 29). The E. coli BL21 bacterial cells (Life Technologies) were grown in 1 L of Luria–Bertani medium with 100 µg/ml ampicillin, shaking at 37°C until an optical density at 600 nm (OD600) of between 0.6 and 0.8 was reached. Protein expression was then induced in the bacterial cell culture with 0.4 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich) for 3 h shaking at 37°C. The cells were then pelleted at 4,500 rpm, 4°C for 10 min, lysed using 50 ml lysis buffer [20 mM Tris–HCl, pH 8.0, 0.5 M NaCl, 1 mM EDTA, 0.25% v/v Tween 20, 5% v/v glycerol, 100 µg/ml lysozyme (Sigma-Aldrich), and 0.1 mM phenylmethanesulfonyl fluoride (Sigma-Aldrich)], and incubated for 1 h at 4°C on a rotary shaker. The cell lysate was then sonicated using a Soniprep 150 (MSE, London, UK) at 60 Hz for 30 s with an interval of 2 min (12 cycles) and then centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was diluted five-fold with buffer A (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.25% v/v Tween 20) and passed through an amylose resin column (25 ml bed) (New England Biolabs) that was equilibrated in buffer A. The affinity column was washed with buffer A without Tween 20 and with 1 M NaCl, 20 mM Tris–HCl, pH 8.0, 1 mM EDTA, followed by buffer B (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA). The MBP-TSR4+5 fusion protein was eluted with 100 ml of buffer B containing 10 mM maltose (Sigma-Aldrich) (affinity elution buffer). Trace contaminants were further removed by applying the fusion protein to a DEAE Sepharose column. Thus, the affinity purified fusion protein in affinity elution buffer was applied to the ion-exchange (5 ml bed) column and washed with three column volumes of low salt buffer containing 50 mM Tris–HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, at pH 7.5. After extensive washing with low salt buffer, the fusion protein eluted at 0.2 M NaCl using a NaCl gradient (50 mM to 1 M). The peak elutions were then passed through Pierce™ High Capacity Endotoxin Removal Resin (Qiagen) to remove LPS. Endotoxin levels were determined using the QCL-1000 Limulus amebocyte lysate system (Lonza), and the assay was linear over a range of 0.1–1.0 EU/ml (10 EU = 1 ng of endotoxin). The endotoxin levels were less than 4 pg/µg of the MBP-TSR4+5.
Mycobacterial Cell Culture
Mycobacterium bovis BCG (Pasteur strain) were grown in liquid culture using Middlebrook 7H9 media (Sigma-Aldrich), supplemented with 0.2% (v/v) glycerol, 0.05% (v/v) Tween-80, and 10% (v/v) albumin dextrose catalase (ADC) (BD BBL, Becton Dickinson). Green fluorescent protein (GFP)-expressing M. bovis BCG (Danish Strain 1331) containing the pGFPHYG2 plasmid was a kind gift from B. Robertson, Imperial College, London, UK. GFP-M. bovis BCG was grown in the above conditions/media but with the addition of 50 µg/ml of hygromycin to maintain the plasmid. Cultures were incubated at 37°C with agitation (~120 rpm) for 7–10 days until the bacteria had reached the exponential growth phase at OD600nm = 0.60–1.00.
Assay of Human Properdin and TSR4+5 Binding to Mycobacteria
Mycobacterium bovis BCG, harvested and washed in PBS, was adjusted to a concentration of 1.25 × 109 cells/ml in PBS (OD600 = 1 equates to approximately 1 × 109 cell/ml). Then 200 µl of bacterial suspension was dispensed into individual microtiter wells of a 96-well plate (Maxisorp™, NUNC). Plates were incubated at 4°C overnight and washed with buffer 1 [10 mM HEPES pH 7.5, 140 mM NaCl, 0.5 mM EDTA, and 100 µg/ml hen ovalbumin (Sigma-Aldrich)]. Wells were blocked for 2 h at 37°C with buffer 1 + 10% (w/v) Marvel Dried Milk powder.
Human properdin (up to 50 µg/ml) or TSR4+5 (up to 30 µg/ml) were added, in two-fold serial dilutions (100 µl/well) in buffer 1 and incubated for 2 h at 37°C. Individual TSR4 and TSR5 proteins, MBP and BSA were used as negative controls. Microtiter wells were washed three times with buffer 1. Mouse anti-properdin monoclonal antibody (1.19 mg/ml) diluted 1/2,500 in buffer 1 (29) was added to the wells containing properdin. Mouse anti-MBP monoclonal antibody (Sigma-Aldrich) was added to wells containing TSR4+5, TSR4 and TSR5, diluted 1/5,000 in buffer 1, and incubated for 1 h at 37°C. For the BSA negative control, mouse anti-BSA monoclonal antibody (Sigma-Aldrich) was used (1/5,000 dilution). Plates were washed an additional three times in buffer 1 and then incubated with goat anti-mouse IgG-horseradish peroxidase conjugate (Sigma-Aldrich), diluted 1/5,000 in buffer 1. The substrate p-nitrophenol phosphate (Sigma-Aldrich) was then added to each well, and the plates read at 405 nm.
Fluorescence Microscopy for TSR4+5 Binding to Mycobacteria
Mycobacterium bovis BCG bacteria (approximately 106 cells) were spotted on poly-l-lysine coated microscope slides (Sigma-Aldrich) and incubated at 37°C for cells to adhere. After washing three times with PBS, bacterial cells were then fixed with 4% paraformaldehyde for 5 min. Slides were washed three times with PBS and then incubated at 37°C for 1 h with 0, 1, or 10 μg/ml of TSR4+5, or 10 µg/ml of BSA (negative control) in buffer 1. Slides were washed three times with PBS, and then the primary monoclonal antibody (mouse anti-MBP) added at 1/500 dilution and incubated for 1 h at room temperature. After washing three times with PBS, goat anti-mouse conjugated with AlexaFluor488 (1/500 dilution) was added as the secondary antibody and incubated for 1 h at room temperature. Slides were then washed three times with PBS and mounted with antifade (Citifluor AF3) PBS solution and viewed using a LeicaDM4000 Fluorescence microscope. Images were processed using Image J.1
Phagocytosis Assay
THP-1 macrophage cells were cultured in RPMI-1640 (Gibco) (RPMI) containing 10% (v/v) fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM l-glutamine (Sigma-Aldrich), 100 U/ml penicillin (Sigma-Aldrich), 100 µg/ml streptomycin (Sigma-Aldrich), and 1 mM sodium pyruvate (Sigma-Aldrich) and left to grow in 5% CO2 at 37°C for approximately 3 days before passaging. Cells were resuspended in RPMI and adjusted to 1 × 106 cells/well (in 1.8 ml) in a 24-well plate. To induce adherence onto the wells, THP-1 cells were treated with 50 ng/ml of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) into RPMI-1640 without FBS, penicillin or streptomycin and left to settle for at least 30 min before adding 200 µl of bacterial culture (1 × 109 bacteria/ml).
M. bovis BCG bacteria were pelleted at mid-exponential phase, at an OD600nm = 0.6–1.0 by centrifugation at 1,000 × g for 10 min at 4°C. The mycobacterial pellet was resuspended in the buffer 1. This mycobacterial culture was then separated into different microfuge tubes and treated with varying concentrations of properdin (2 or 20 µg/ml) or MBP-TSR4+5 (1 or 5 µg/ml). Control samples were left untreated, and all were incubated for 2 h at 37°C for binding to occur. The mycobacterial suspension was washed once in growth medium before resuspending in RPMI medium without FBS, penicillin, or streptomycin. 200 µl of the mycobacterial suspension was added to each well of THP-1 cells. Mycobacterial concentration was adjusted to give approximate multiplicity of infection (MOI) ratio of 10:1.
Plates were gently swirled and incubated at 37°C, 5% CO2 for up to 48 h to allow mycobacterial uptake. THP-1 cells were sampled at 15, 30, and 45 min and 1, 2, and 6 h. Supernatants were collected after 24 and 48 h of incubation for multiplex analysis. Plates were washed three times with PBS to remove extracellular bacteria. THP-1 cells were then lifted by adding 1 ml of 0.25% trypsin to the wells and incubated for 10 min at 37°C, 5% CO2. THP-1 cells were collected by centrifugation at 1,000 × g for 10 min at 4°C.
To recover and count the ingested mycobacteria, THP-1 cells were lysed by resuspending the cell pellets in 1 ml of sterile water, followed by a series of vortex mixing for 10 min at room temperature. 24-well plates containing 2 ml Middlebrook 7H10 agar with 10% Oleic Acid+ADC (OADC) (BD, BBL, Becton Dickinson) were prepared. Four serial 1/10 dilutions were made, and 10 µl of the concentrated mycobacterial suspension and diluted suspension from each time point was spotted onto the 7H10 agar wells. The 24-well plates were secured with parafilm and wrapped in aluminum foil, inverted and incubated at 37°C for 10–14 days. Wells were photographed, and the colony-forming unit (CFU) count determined. The same procedure was used to quantify the initial input number of bacteria incubated with THP-1 cells.
Fluorescence Microscopy for Phagocytosis Assay
THP-1 cells were cultured as described above and seeded at 1 × 105 cells per 13 mm coverslip and differentiated with PMA as described above. GFP-expressing M. bovis BCG was incubated with 0, 1, 10 µg/ml of TSR4+5 for 2 h at 37°C in buffer 1. Cells were also incubated with 10 µg/ml of BSA as a negative control. Cells were washed twice in PBS and then resuspended in plain RPMI media. 1 × 106 GFP-M. bovis BCG was added to the THP-1 cells (MOI of 10:1) and incubated for phagocytosis for 2 h at 37°C. THP-1 cells were then washed three times in PBS to remove extracellular bacteria and then fixed in 4% paraformaldehyde for 5 min. After washing three times in PBS, THP-1 cells were incubated with 2 µg/ml of AlexaFluor546-conjugated wheat germ agglutinin (Invitrogen) to reveal the plasma membrane. Cells were then washed three times and mounted using Vectashield antifade with DAPI (Vector Labs) to reveal nucleus. Slides were observed under a Leica DM4000 fluorescence microscope at 40× magnification. Images were processed using Image J (see text footnote 1).
Quantitative Real-Time PCR (qPCR) Analysis of mRNA Expression of Cytokines
THP-1 cell pellets were collected from each time point as described above. RNA extraction was performed using the GenElute Mammalian Total RNA Purification Kit (Sigma-Aldrich) according to the manufacturer’s protocol. Samples were then treated with DNase I (Sigma-Aldrich) to remove any contaminating DNA according to the manufacturer’s protocol. The amount of RNA was measured using the NanoDrop 2000/2000c spectrophotometer (Thermo Fisher Scientific) at 260 nm, and the ratio of absorbance at 260 and 280 nm was used to assess the purity of the RNA. Complementary DNA (cDNA) was synthesized using High Capacity RNA to cDNA Kit (Applied Biosystems, UK) according to the manufacturer’s protocol. Primer sequences (Table 1) were designed and analyzed for specificity using the nucleotide Basic Local Alignment Search Tool and Primer-BLAST.2
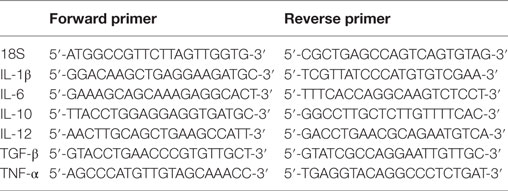
Table 1. Primers used for quantitative real-time PCR.
PCR was performed on all cDNA samples to assess the quality of the cDNA. The qPCR assays were performed for the expression of pro- and anti-inflammatory cytokines. The qPCR reaction consisted of 5 µl Power SYBR Green MasterMix (Applied Biosystems), 75 nM of forward and reverse primer, 500 ng template cDNA in a 10 µl final reaction volume. qPCR was performed in a 7900HT Fast Real-Time PCR System (Applied Biosystems). The initial steps were 2 min incubation at 50°C followed by 10 min incubation at 95°C, the template was then amplified for 40 cycles under these conditions: 15 s incubation at 95°C and 1 min incubation at 60°C. Samples were normalized using the expression of human 18S rRNA. Data were analyzed using the relative quantification (RQ) Manager Version 1.2.1 (Applied Biosystems). Cycle threshold (Ct) values for each cytokine target gene were calculated, and the relative expression of each cytokine target gene was calculated using the RQ value, using the formula: RQ = 2−ΔΔCt for each cytokine target gene, and comparing relative expression with that of the 18S rRNA constitutive gene product. Assays were conducted twice in triplicate.
Multiplex Analysis
Supernatants were collected from the phagocytosis assay at 24 and 48 h to determine the levels of secreted cytokines (IL-6, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, TNF-α, IL-13, IL-15, IL-17A, IL-9, and TNF-β), chemokines (MCP-3, MDC, Eotaxin, Fractalkine, GRO, IL-8, IP-10, MCP-1, and MIP-1α), growth factors (IL-2, EGF, FGF-2, G-CSF, GM-CSF, IL-3, IL-4, IL-5, IL-7, and VEGF), and other related ligands and receptors (IFN-α2, IFN-ϒ, FLT-3L, IL-1RA, and sCD40L). MagPix Milliplex kit (EMD Millipore) was used to measure immune response following the manufacturer’s protocol. 25 µl of assay buffer was added to each well of a 96-well plate, followed by the addition of 25 µl of standard, controls or supernatants of cells treated with M. bovis BCG in the presence or absence of properdin and MBP-TSR4+5. 25 µl of magnetic beads coupled to analytes of interest was added in each well and incubated for 18 h at 4°C. The 96-well plate was washed with the assay buffer, and 25 µl of detection antibodies was incubated with the beads for 1 h at room temperature. 25 µl of streptavidin–phycoerythrin was then added to each well and incubated for 30 min at room temperature with shaking at 750 rpm. Following a washing step, 150 µl of sheath fluid was added to each well, and the plate was read using the Luminex Magpix instrument. Assays were conducted in duplicate.
Statistical Analysis
Analysis of data for statistical significance was conducted using GraphPad Prism 6 for Windows (GraphPad Software, Inc.). Statistical analyses were made using two-way ANOVA for mRNA expression data and a one-way ANOVA for the multiplex data. p Values < 0.05 were considered statistically significant, unless otherwise stated (non-significant).
Results
Human Properdin and TSR4+5 Bind to Mycobacteria
Human properdin was purified from human plasma. SDS-PAGE, followed by western blotting using antihuman properdin polyclonal antibodies, showed a distinct band at 55 kDa (Figure 1A), which was the expected molecular weight of the glycosylated monomer. The two biologically active modules of properdin TSR4 and TSR5 were expressed together in tandem as previously described, fused to MBP (7), and is also shown on an SDS-PAGE gel, which has a molecular weight of 55 kDa (Figure 1B). Using gel filtration chromatography, we found that human properdin eluted as a mixture of monomer, dimer and trimer; a negligible amount probably formed aggregates. Nearly 60% of MBP-TSR4+5 appeared as a monomer while nearly 40% was found to migrate as a dimer (data not shown).
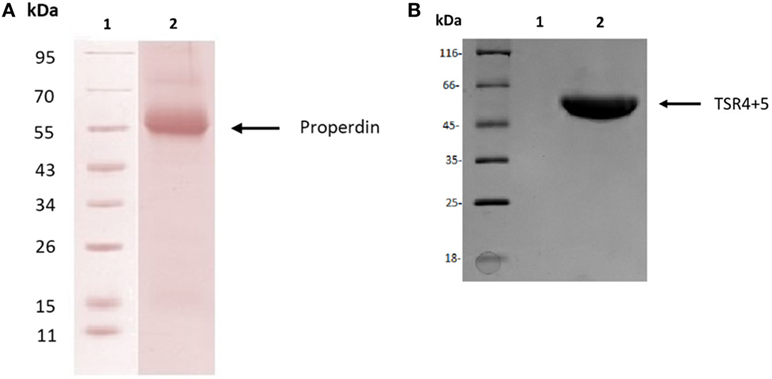
Figure 1. Purified human properdin and recombinant maltose-binding protein (MBP)-thrombospondin repeats (TSR) 4+5. (A) Properdin was purified from human plasma. Filtered plasma was applied to a non-immune IgG-Sepharose column, then to a mouse monoclonal anti-properdin Sepharose column; properdin was eluted with 3 M MgCl2. The eluted samples were dialyzed against HEPES buffer (10 mM HEPES, 140 mM NaCl, 0.5 mM EDTA, pH 7.4) overnight at 4°C. Contaminants were removed by applying the protein to a Q Sepharose column, and the product appears as a single band on SDS-PAGE and western blot at about 55 kDa. (B) MBP-TSR4+5 was purified via an amylose resin column, and the purified fusion protein also appears on SDS-PAGE as a band of about 55 kDa.
The binding of properdin to M. bovis BCG was observed to be in a dose-dependent manner; BSA was used as a negative control that showed almost no binding (Figure 2A). TSR4+5 binding was also observed to be in a dose-dependent manner. MBP was used as a negative control (Figure 2B). The two binding curves cannot be compared quantitatively, as different detection antibodies were used. Because the MBP-TSR4+5 recombinant protein and properdin monomer have about the same molecular weight, 5 µg of TSR4+5 corresponds in molar terms to about 5 µg of properdin monomer (Figures 1A,B). The binding of a mixture of the two separately expressed TSR4, and TSR5 is much lower than that of the combined expressed TSR4+5 (Figure 2B). For this comparison, the same detection antibody was used. These results suggest that both TSR4 and TSR5 modules contribute to the interaction with M. bovis BCG, and that TSR4+5 binds with similar characteristics to that of whole properdin on M. bovis BCG surface. These results were further confirmed using microscopy where TSR4+5 specifically bound to M. bovis BCG in a dose-dependent manner (Figure 2C).
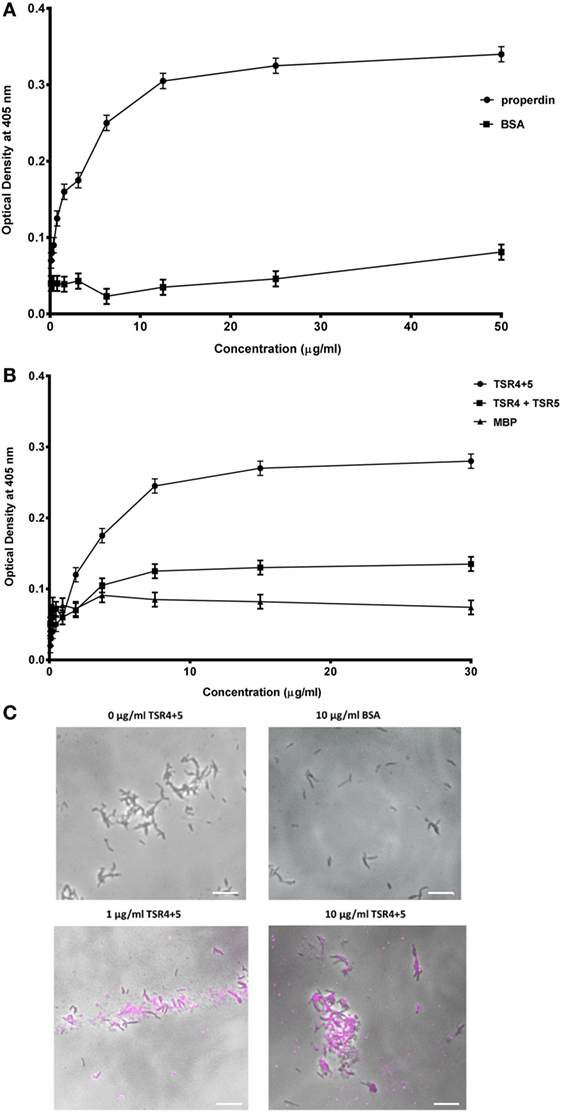
Figure 2. Human properdin binds mycobacteria via thrombospondin repeats (TSR) 4+5. (A) Properdin binding to mycobacteria; BSA was used as a negative control protein. (B) Comparison between TSR4+5 and individual TSR4 and TSR5 binding to mycobacteria; maltose-binding protein (MBP) as negative control. Assays were conducted in 10 mM HEPES, 140 mM NaCl, 0.5 mM CaCl2 + 0.5 mM MgCl2, 100 µg/ml hen ovalbumin, and pH 7.5. Serial dilutions of properdin were incubated in mycobacteria coated wells followed by incubation with mouse anti-properdin monoclonal antibody and mouse anti-BSA monoclonal antibody, respectively; serial dilutions of TSR4+5, TSR4 or TSR5 were incubated in another set of mycobacteria coated wells followed by incubation with mouse anti-MBP monoclonal antibody. Anti-mouse IgG conjugated with alkaline phosphatase and substrate p-nitrophenol phosphate were incubated in both sets of wells, and the color was measured at 405 nm using a plate reader. Assay was conducted in quadruplicate. Error bars represent SD. (C) Differential direct binding of 0, 1, and 10 µg/ml of TSR4+5 to Mycobacterium bovis BCG. 10 µg/ml of BSA was used as a negative control. Cells were incubated for 2 h with either TSR4+5 or BSA. Cells were washed, fixed, and stained with mouse anti-MBP monoclonal antibody followed by goat anti-mouse 1gG-conjugated with AlexaFluor488. Images are shown as single sections taken using a Leica DM4000 microscope; bar scale 10 µm.
Properdin Inhibits Uptake of M. bovis BCG by THP-1 Cells
Properdin inhibited the uptake of M. bovis BCG by THP-1 cells. At a concentration of 20 µg/ml, uptake of M. bovis BCG was significantly reduced by properdin (Figure 3A). TSR4+5 was also able to substantially inhibit uptake of M. bovis BCG by THP-1 cells (Figure 3B). The effect of properdin and TSR4+5 on the phagocytosis of M. bovis BCG was dose dependent. The input number of M. bovis BCG was about 7.8 × 106 CFU/ml, which was the total number of bacteria added to THP-1 cells. Without properdin or TSR4+5, 5.0 × 106 CFU/ml of M. bovis BCG was phagocytosed which was approximately 66% efficiency of phagocytosis compared with the input number. At the highest concentration tested, properdin showed an inhibition of uptake of approximately 60% compared with M. bovis BCG with no properdin (Figure 3A). For TSR4+5, the effect on M. bovis BCG was slightly lower at approximately 40% inhibition (Figure 3B). These results were also confirmed by microscopy, with TSR4+5 having a suppressive effect on the uptake of GFP-expressing M. bovis BCG by THP-1 cells (Figure 3C). PMA stimulation was used to induce differentiation of THP-1 cells before incubation with M. bovis BCG. PMA has been shown to activate protein kinase C and increase cell adherence and expression of surface markers associated with macrophage differentiation (30). These data show that (i) properdin has an anti-opsonic effect on M. bovis BCG, inhibiting phagocytosis; and (ii) TSR4+5 modules play a major role in this interaction of M. bovis BCG and macrophages. These observations demonstrate, for the first time, a novel, non-complement-related role for properdin in host–pathogen interactions in tuberculosis.
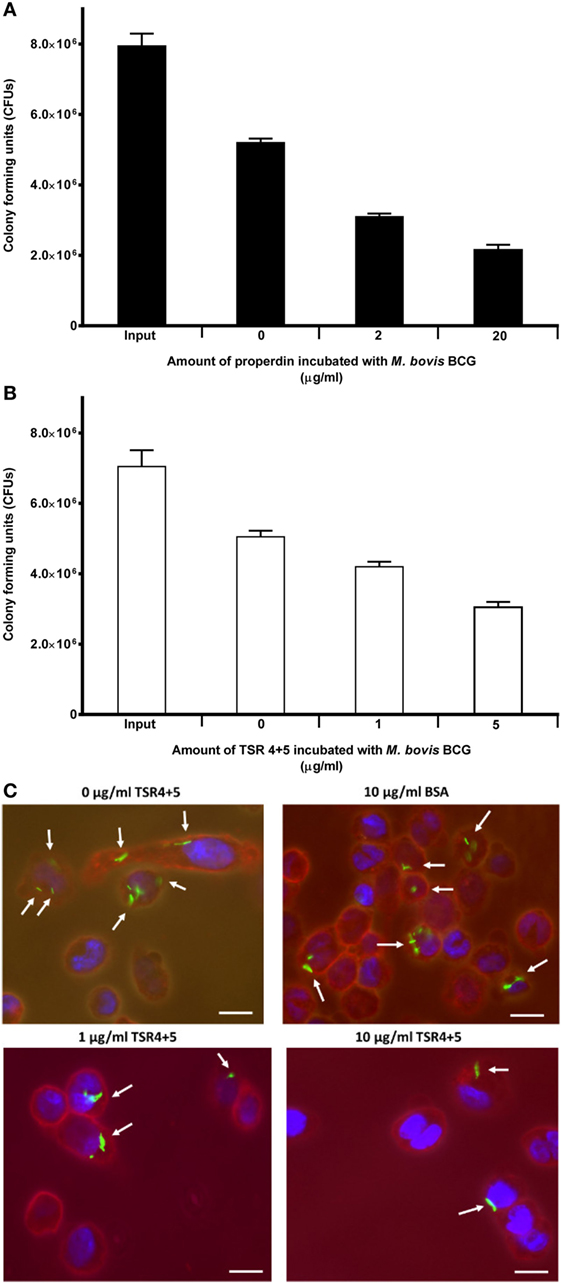
Figure 3. Effect of properdin and thrombospondin repeats (TSR) 4+5 on the phagocytosis of Mycobacterium bovis BCG by THP-1 cells. (A) M. bovis BCG was treated with properdin at concentrations of 0, 2, and 20 µg/ml or with (B) TSR4+5 at concentrations 0, 1, and 5 µg/ml. The mycobacteria were incubated with macrophage for 2 h. After THP-1 cell lysis, surviving internalized M. bovis BCG were measured by plating lysates on 7H10 media to obtain colony-forming units (CFUs). The input value is the starting number of M. bovis BCG added to the THP-1 cells, before phagocytosis. A one-way ANOVA test was performed on the data to determine significant differences in CFU count by properdin or TSR4+5. All comparisons were significant (p < 0.05), unless where shown (ns, not significant, p > 0.05). Samples were analyzed in triplicate. (C) Differential uptake of GFP-M. bovis BCG by THP-1 macrophages after treatment with 0, 1, and 10 µg/ml of TSR4+5, or 10 µg/ml of BSA, used as a negative control. Cells were incubated for 2 h. Cells were then washed, fixed, and stained with AlexaFluor546-conjugated wheat germ agglutinin to reveal the plasma membrane (red), and the nucleus was stained with DAPI (blue). Images are shown as single sections, taken using a Leica DM4000 microscope; bar scale 10 µm.
Properdin Induces a Pro-Inflammatory Response During the Early Phase of Phagocytosis of M. bovis BCG by THP-1 Cells
The effect of properdin on the inflammatory response during the phagocytosis of M. bovis BCG was measured. The gene expression of key pro- and anti-inflammatory cytokines in tuberculosis was determined using quantitative real-time PCR. Our data showed that properdin significantly enhanced the upregulation of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 from THP-1 cells challenged by M. bovis BCG (Figure 4A), particularly at the initial stage of uptake (within the first hour of phagocytosis), which decreased gradually toward the later stages of phagocytosis. The increase in TNF-α transcript was particularly striking as TNF-α is well known for activating macrophages for killing of intracellular mycobacteria. In addition, TNF-α is a key mediator in the early stages of granuloma formation. By contrast, the anti-inflammatory cytokines measured from THP-1 cells (IL-10 and TGF-β) were shown to be downregulated in the presence of properdin, when challenged by M. bovis BCG (Figure 4B). IL-12 also appeared to be downregulated (Figure 4A). Properdin, therefore, appears to play an important role in pro-inflammatory cytokine production by macrophages infected by M. bovis BCG, which may have significant implications in shaping the adaptive immune response during M. tuberculosis infection.
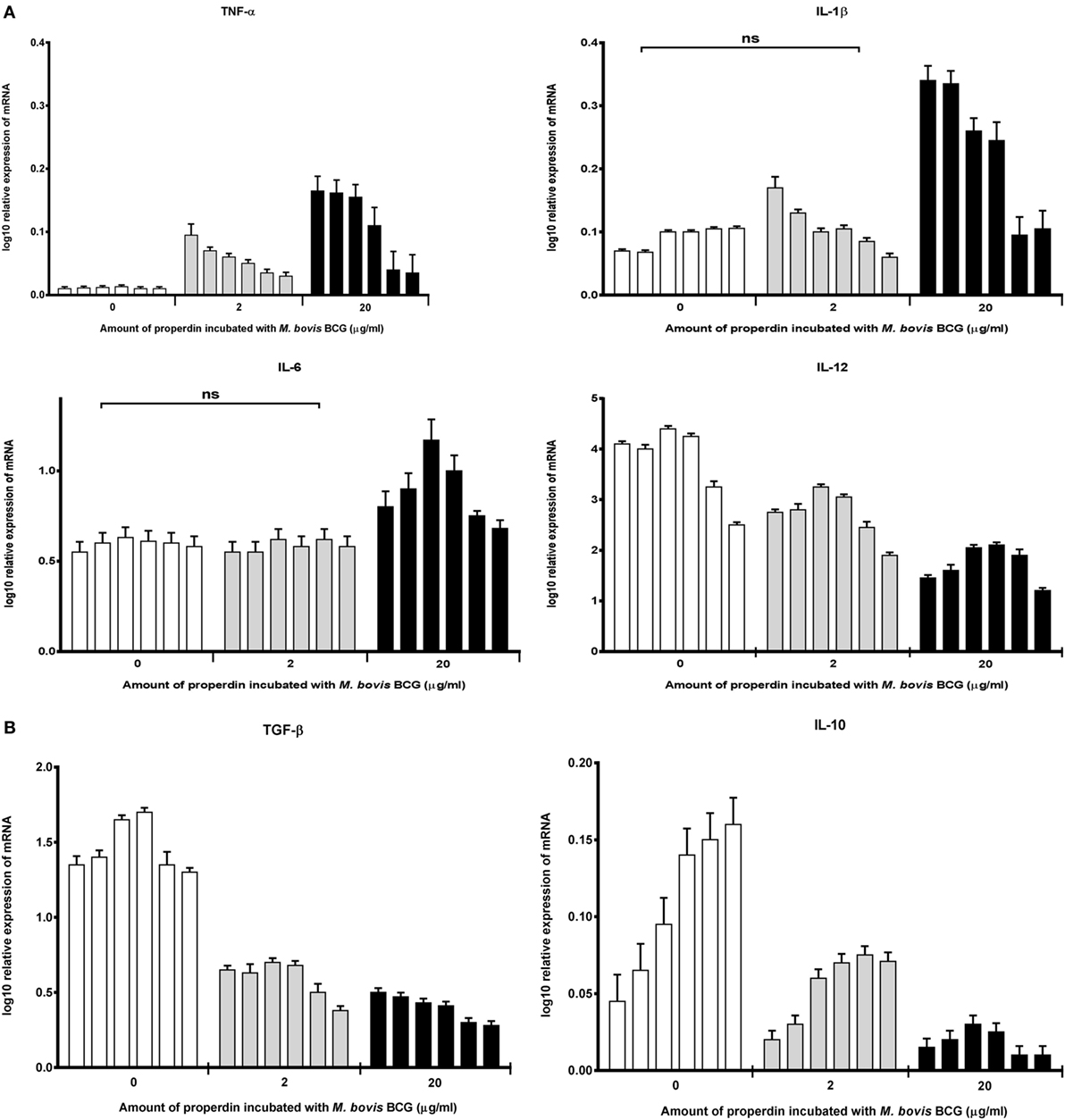
Figure 4. Temporal mRNA expression profile of cytokines produced by THP-1 cells incubated with different concentrations of properdin and Mycobacterium bovis BCG. (A) Pro-inflammatory cytokines: TNF-α, IL-1β, IL-6, and IL-12; (B) anti-inflammatory cytokines: TGF-β and IL-10. The expression of each cytokine was measured using qPCR, and the relative expression [relative quantification (RQ)] calculated by normalizing the data using human 18S rRNA expression as a control. The RQ value was calculated using the formula: RQ = 2−ΔΔCt. Assays were conducted twice in triplicates. Error bars represent SD. A two-way ANOVA test was performed on the data to determine significant differences in expression of cytokine production by properdin. All comparisons were significant (p < 0.05), unless where shown (ns, not significant, p > 0.05).
Cytokine gene expression by THP-1 cells infected with M. bovis BCG were also studied in the presence of TSR4+5, which revealed that TSR4+5 also has a significant effect on the pro-inflammatory response. TNF-α was upregulated (Figure 5A), while IL-10 was found to be downregulated (Figure 5B), during the first hour of phagocytosis. IL-12 was also shown to be significantly downregulated (Figure 5A). These data mirror the observations for properdin, and hence, validate the importance of TSR4+5 in the binding of properdin to M. bovis BCG and in its modulation of the inflammatory response. These data are also similar to recent published observations of another complement regulatory protein, factor H (19), thus offering potentially novel insights into the involvement of these proteins in host–pathogen interactions in tuberculosis.
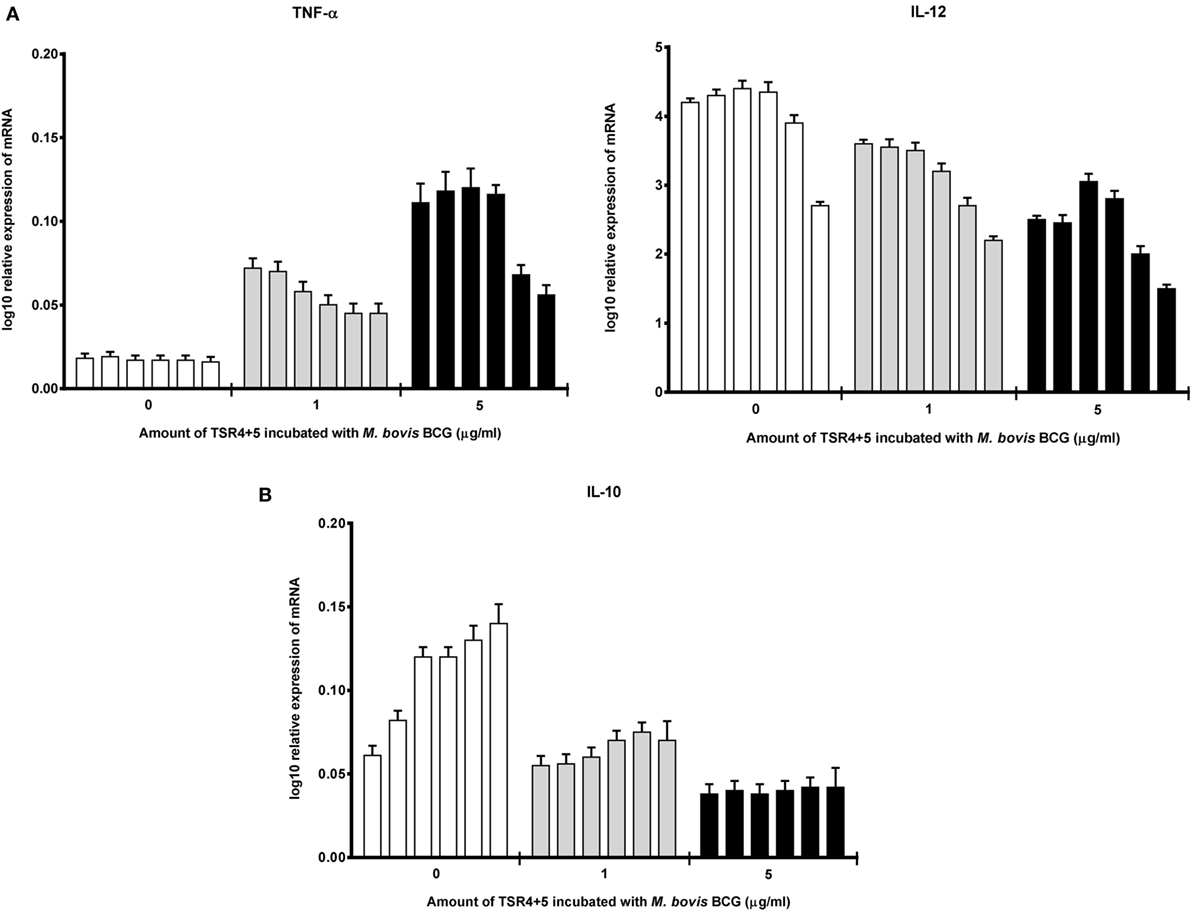
Figure 5. Temporal mRNA expression profile of cytokines produced by THP-1 cells incubated with different concentrations of thrombospondin repeats (TSR) 4+5 and Mycobacterium bovis BCG. (A) Pro-inflammatory cytokines: TNF-α and IL-12; (B) anti-inflammatory cytokine: IL-10. The expression of each cytokine was measured using qPCR, and the relative expression [relative quantification (RQ)] calculated by normalizing the data using human 18S rRNA expression as a control. The RQ value was calculated using the formula: RQ = 2−ΔΔCt. Assays were conducted twice in triplicates. Error bars represent SD. A two-way ANOVA test was performed on the data to determine significant differences in expression of cytokine production by TSR4+5. All comparisons were significant (p < 0.05), unless where shown (ns, not significant, p > 0.05).
Multiplex Analysis of Cytokine Secretion
The inflammatory response during the phagocytosis of M. bovis BCG by THP-1 cells was further determined by measuring the secretion of cytokines, chemokines, and other growth factors using the Multiplex analysis of supernatants collected at 24 and 48 h post phagocytosis (Figures 6A–D). The secretion of pro-inflammatory cytokines TNF-α, IL-1β, and IL-1α was significantly enhanced by treatment with properdin or TSR4+5 at the 24 h time point (Figure 6A). The enhancement of these pro-inflammatory cytokines can be critical for controlling mycobacterial infection, particularly in the formation of the granuloma. However, by 48 h, there was a decrease in the production of pro-inflammatory cytokines (IL-6, IL-12p40, IL-12p70, IL-1α, IL-1β, TNF-α, IL-13, IL-15, and IL-9) in the presence of properdin- and TSR4+5-treated M. bovis BCG (Figure 6A). Properdin and TSR4+5 also downregulated the anti-inflammatory response such as IL-10 after 24 and 48 h of phagocytosis, although this was less pronounced for IL-12 at 48 h (Figure 6A). These observations again mirror the initial responses observed in cytokine gene expression of during the first few hours of phagocytosis, in the presence of properdin or TSR4+5 (Figures 5A,B). The effect of properdin and TSR4+5 also led to marked downregulation of a number of growth factors MCP-3 (24 h), MDC, Eotaxin, Fractalkine (24 h), GRO (24 h), IP-10, MCP-1, MIP-1, VEGF, G-CSF (48 h), GM-CSF (48 h), and VEGF (24 h) (Figures 6B,C). Additional ligands and receptors (IFN-α2, IFN-γ, FLT-3L, IL-1RA, and sCD40L) did not show any significant changes (Figure 6D).
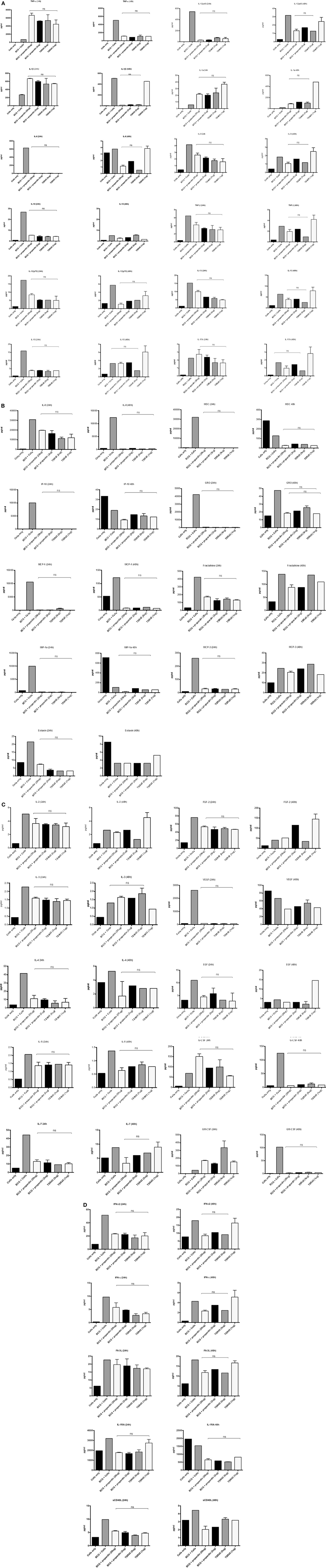
Figure 6. Multiplex cytokine analysis of supernatants collected at 24 and 48 h phagocytosis of Mycobacterium bovis BCG by THP-1 cells incubated with or without properdin and thrombospondin repeats (TSR) 4+5. The supernatants were collected from phagocytosis assay of M. bovis BCG in the presence or absence of properdin and TSR4+5 at 24 and 48 h time point. The levels of cytokine production were measured for (A) (IL-6, IL-10, IL-12p40, IL-12p70, IL-1α, IL-1β, TNF-α, IL-13, IL-15, IL-17A, IL-9, and TNF-β), (B) chemokines (MCP-3, MDC, Eotaxin, Fractalkine, GRO, IL-8, IP-10, MCP-1, and MIP-1α), (C) growth factors (IL-9, IL-2, EGF, FGF-2, G-CSF, GM-CSF, IL-3, IL-4, IL-5, IL-7, and VEGF), and (D) related ligands and receptors (IFN-α2, IFN-γ, FLT-3L, IL-1RA, and sCD40L) using multiplex analysis. Error bars represent SD. A one-way ANOVA test was performed on the data to determine significant differences in expression of cytokine production by properdin or TSR4+5. All comparisons were significant (p < 0.05), unless where shown (ns, not significant, p > 0.05). Supernatants were analyzed in duplicate.
Discussion
We have previously shown that a complement regulatory protein, factor H, can bind to M. bovis BCG and inhibit its uptake by THP-1 macrophages (19). Factor H can also enhance the pro-inflammatory response during this host–pathogen interaction (19). This study highlighted a novel complement-independent property of factor H as an anti-opsonin and in the modulation of the inflammatory response against a pathogen. With the goal of further elucidating the role of complement control proteins in the early stages of mycobacterial infection, this study looked at the role of properdin, an upregulator of the alternative complement pathway. Properdin and thrombospondin repeat (TSR) modules TSR4+5 were shown to bind to M. bovis BCG and inhibit bacterial uptake by THP-1 cells, augmenting the inflammatory response. These observations are similar to what has been observed previously with factor H (19), which is intriguing, since properdin and factor H have opposing effects on the regulation of complement activation (3). These findings were also consistent with previous reports, which have demonstrated that properdin deficient mice have a reduced M1 (IL-1β) and increased M2 (arginase-1, MCP-1, IL-10) profile, crucial for the tumor microenvironment (31). This suggests that the production of IL-1β and reduction in IL-10 mediated by properdin may be required for protection against M. tuberculosis in the initial phase of infection.
The functions of properdin have been extensively investigated within the remit of the complement alternative pathway, and its involvement with pathogens has largely been characterized as complement dependent. In this study, we aimed to look at the complement-independent interaction of properdin with mycobacteria (i.e., effects in the absence of other complement proteins), with a view to examining its possible role in the pathogenesis of tuberculosis. The role of complement in tuberculosis has been examined, but little is understood about the role of the individual complement proteins in tuberculosis infection, especially complement control proteins. Properdin has been shown to play a role in a number of pathogenic infections such as those by C. pneumoniae, in which properdin promotes complement C3b deposition and opsonization (12). A recent study also demonstrated that a low dose of properdin, which is highly polymerized, is able to protect against N. meningitidis and Streptococcus pneumoniae, by assembling the alternative complement pathway (16). The central premise in these recent studies is that properdin is an upregulator of the complement alternative pathway, and thus, when in contact with pathogens, the alternative pathway is triggered and stabilized by properdin.
Properdin has also been shown to enhance the uptake of apoptotic T cells by dendritic cells (DCs) and macrophages, thus promoting phagocytosis (27). Properdin may also bind to the DNA found to be exposed on apoptotic and necrotic cells, suggesting that this may also be a crucial site for alternative pathway activation (32). There is recent evidence to suggest that properdin, locally produced by tolerogenic DCs, binds to necrotic cells, confirming its role a pattern recognition molecule of the innate immunity. In addition, properdin is also involved in the interaction of DC and T cell responses. Interestingly, silencing of properdin by treating DCs with siRNA in the presence/absence of IFN-γ reduced the proliferation of allogenic T cells (33). Properdin binds to early apoptotic cells via sulfated GAGs, resulting in C3b deposition and uptake by phagocytes. Activation of neutrophils drives the deposition of properdin, which binds apoptotic T cells. Since properdin has been shown to bind apoptotic cells via GAGs (27) or DNA, it remains unclear what other ligands and receptors are involved in properdin–apoptotic cell interaction. It is likely that properdin as a soluble factor is acting as an adaptor molecule. Furthermore, properdin also binds to NKp46 expressed on natural killer cells, innate lymphoid cells (ILC)1 and ILC3. This study demonstrated that the control of meningococcal infection was dependent on NKp46 and group 1 ILCs, further elucidating the role of properdin as an independent pattern recognition molecule (34).
In our study, we show that purified native properdin and TSR4+5 bind to M. bovis BCG in a dose-dependent manner, suggesting that the binding of properdin to M. bovis BCG may be via TSR4+5. The physiological concentration of properdin in serum is about 25 µg/ml (35). We also demonstrate that coating of M. bovis BCG with properdin inhibits the uptake of the bacterium by THP-1 cells; however, only about 60% inhibition was achieved at the highest dose of native properdin. TSR4+5 was also able to mirror the effects of properdin, in inhibiting the uptake of M. bovis BCG by macrophages by up to 40%.
The recruitment of properdin by mycobacteria may be particularly crucial in the initial stages of tuberculosis infection, when after inhalation, the first host cell M. tuberculosis encounters is the alveolar macrophage.
In the lungs, mycobacteria are phagocytosed by alveolar macrophages, which are unable to completely eliminate them, and so produce crucial chemoattractants (36), which recruit inflammatory cells such as neutrophils, macrophages, γδ-T cells, and natural killer cells that stimulate inflammation and tissue remodeling (36–38). Our findings may be indicative of the early inflammatory processes in vivo, involved in granuloma formation, which are nodular-type lesions that cordon-off M. tuberculosis infection, and provide an environment for the bacilli to persist and survive as a latent infection. TNF-α and IFN-γ are involved in recruitment of cells in the granuloma (39, 40). Thus, properdin may play a role in granuloma formation by promoting pro-inflammatory cytokines. Inflammatory balance is essential particularly of Th2/Th1 cytokines, which are required to maintain a protective granuloma (41). This is determined by the balance in IFN-γ/TNF-α versus IL-4/IL-10/TGF-β within the granuloma. Properdin and TSR4+5 may be implicated in maintaining this balance. It is not known whether complement proteins reside in the granuloma; however, during infection, complement proteins may be produced locally at sites of infection. Properdin may be secreted by neutrophils, monocytes and T cells locally at the site of infection (3). Thus, innate immune molecules residing in or being recruited at sites of infection may play a role in the balance of Th1/Th2, which may cause granuloma necrosis and replication of M. tuberculosis (41–43).
TNF-α was dramatically increased in the first 24 h of phagocytosis in the presence of properdin and TSR4+5, compared to non-treated mycobacteria. TNF-α plays a major role in granuloma formation and our results suggest that properdin may have a role in potentiating the pro-inflammatory response that results in granuloma formation. These observations are further strengthened by the concurrent increase in IL-1α levels over 24 h which have been shown to be key in macrophage proliferation and maturation during granuloma formation (44).
During phagocytosis, pro- and anti-inflammatory cytokines were produced by THP-1 cells when treated with properdin or TSR4+5. In the initial stages of infection by M. bovis BCG, pretreated with properdin, during phagocytosis, the expression of TNF-α was significantly enhanced. Other pro-inflammatory responses that were also elevated in the presence of properdin at initial stages of infection are IL-1β and IL-6. IL-1β is a mediator of inflammation and is required for host resistance to M. tuberculosis infection (45). IL-6 is a biomarker for tuberculosis, as increased levels are observed in patients with tuberculosis (46) that is required for a T cell response against M. tuberculosis infection (47, 48). Conversely, IL-10, IL-12, and TGF-β were downregulated by properdin, thus suppressing the anti-inflammatory response. The downregulation of IL-12 by properdin in vivo may suppress the Th1 response. TSR4+5 was also able to mimic the cytokine response like properdin, suggesting that the modules responsible for the major part of the interaction with M. bovis BCG may be TSR4+5.
Macrophages play a significant role in the innate immune response to pathogens and so are also crucial for an adaptive immune response (49). However, M. tuberculosis can evade the innate immune defense, inhibiting phagosome maturation (36), resisting anti-microbial agents damaging the bacterial cell wall and facilitating replication within the host and escaping early immune recognition. Thus, these pathogens interfere with the early immune response and the induction of pro-inflammatory cytokines (49).
IL-10 and TGF-β suppression by properdin may enhance the clearance of mycobacteria by the host during the early stages of M. tuberculosis infection. After phagocystosis of M. tuberculosis, IL-10 has been shown to block phagolysosome maturation and antigen presentation by macrophages, thus aiding the survival of the pathogen (50, 51). Furthermore, IL-10 can inhibit the generation of reactive oxygen and nitrogen intermediates in IFN-γ activated macrophages, which are required for intracellular killing (52, 53). The enhanced levels of IL-10 and TGF-β in the lungs of active tuberculosis patients demonstrate a weakened immune response to M. tuberculosis, and hence, a role in the pathogenesis and disease progression (54, 55). VEGF was also found to be at a significantly higher level in tuberculosis patients with extrapulmonary tuberculosis (EPTB) than those with pulmonary disease (56). In our study, properdin and TSR4+5 seems to result in a marked elevation of VEGF after 48 h. Since our data also shows that mycobacteria have a reduced phagocytosis by macrophages, the resulting extracellular bacteria may be encouraged by VEGF to disseminate. The beneficial effect of properdin may be to inhibit the mechanisms involved in evasion and, thus, facilitate a protective response against mycobacterial infection.
The downregulation of IL-12 by properdin or TSR4+5 may be due to the reduced phagocytosis of M. bovis BCG, thus, downregulating the Th1 response. This may be necessary for the Th1/Th2 homeostasis in the protective granuloma (41, 57, 58). Both IL-10 and TGF-β levels were supressed, whilst TNF-α was elevated during the first 24 h after phagocytosis.
Although M. bovis BCG shares 99% genome homology to M. tuberculosis, there are some genetic differences which lead to its avirulence. The major difference between M. bovis BCG and M. tuberculosis is the large genomic deletion RD1, which causes the loss of various virulent genes coding for proteins such as ESAT-6, CFP-10 and also a bacterial secretion system (59, 60). Therefore the findings in our study will need to be validated using virulent strains of M. tuberculosis.
Properdin deficiency renders the host susceptible to a range of bacterial infections, especially Neisseria species. Three types of properdin deficiency have been reported: type I (absence of the properdin protein), type 2 (low level of properdin about 1–10% found in the serum), and type 3 deficiency (normal levels of protein being produced, but functionally defective). The most commonly reported deficiency is the type I properdin deficiency that exhibits fulminant infections. The incidence of tuberculosis has not been reported in properdin deficient subjects, possibly due to the majority of studies being in Scandinavia or western Europe, in populations where there is a low incidence of tuberculosis.
The data in this study suggest that properdin, via TSR4+5, may help in the clearance of mycobacterial infection by circumventing pathogen immune evasion strategies by upregulating the pro-inflammatory response. Properdin may also promote the formation and maintenance of the protective granuloma. The data in this study give further insights into the involvement of complement regulatory proteins in shaping the cellular immune response against mycobacteria in a complement activation-independent manner. Further studies are needed to fully characterize the nature and extent of involvement of properdin in tuberculosis pathogenesis, particularly in the early stages of infection.
The complement-independent interaction between human properdin and mycobacteria is a novel observation, which is independent of C3b deposition and aggregation of properdin (61). This is consistent with our recent study where we have shown that properdin can recognize chemical patterns on nanoparticles via TSR4+5 and modulate immune response by THP-1 cells (26) without involving complement activation/deposition. In conclusion, properdin may be involved in modulating host-pathogen interactions in tuberculosis. However, further studies are needed on pathogenic M. tuberculosis and in vivo, to understand the precise role of this complement regulatory protein in pathogenesis, which may give new insights into therapies against this formidable disease.
Author Contributions
MA-M, AT, MA-A, and LK carried out crucial experiments. SA, MNA-A, AAP, VM, EMG, AK, and RBS provided crucial reagents and expertise. UK, AT and LK wrote the manuscript in addition to designing the experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MA-A has been supported by the Ministry of Higher Education, Malaysia and the Universiti Sains Malaysia.
Footnotes
References
1. Pangburn MK, Muller-Eberhard HJ. The C3 convertase of the alternative pathway of human complement. Enzymic properties of the bimolecular proteinase. Biochem J (1986) 235:723–30. doi:10.1042/bj2350723
2. Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science (1954) 120:279–85. doi:10.1126/science.120.3112.279
3. Kouser L, Abdul-Aziz M, Nayak A, Stover CM, Sim RB, Kishore U. Properdin and factor h: opposing players on the alternative complement pathway “see-saw”. Front Immunol (2013) 4:93. doi:10.3389/fimmu.2013.00093
4. Smith CA, Pangburn MK, Vogel CW, Muller-Eberhard HJ. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. J Biol Chem (1984) 259:4582–8.
5. Higgins JM, Wiedemann H, Timpl R, Reid KB. Characterization of mutant forms of recombinant human properdin lacking single thrombospondin type I repeats identification of modules important for function. J Immunol (1995) 155:5777–85.
6. Goundis D, Reid KB. Properdin, the terminal complement components, thrombospondin and the circumsporozoite protein of malaria parasites contain similar sequence motifs. Nature (1988) 335:82–5.
7. Kouser L, Abdul-Aziz M, Tsolaki AG, Singhal D, Schwaeble WJ, Urban BC, et al. A recombinant two-module form of human properdin is an inhibitor of the complement alternative pathway. Mol Immunol (2016) 73:76–87. doi:10.1016/j.molimm.2016.03.005
8. Pangburn MK. Analysis of the natural polymeric forms of human properdin and their functions in complement activation. J Immunol (1989) 142:202–7.
9. Alcorlo M, Tortajada A, Rodriguez de Cordoba S, Llorca O. Structural basis for the stabilization of the complement alternative pathway C3 convertase by properdin. Proc Natl Acad Sci U S A (2013) 110:13504–9. doi:10.1073/pnas.1309618110
10. Gulati S, Agarwal S, Vasudhev S, Rice PA, Ram S. Properdin is critical for antibody-dependent bactericidal activity against Neisseria gonorrhoeae that recruit C4b-binding protein. J Immunol (2012) 188:3416–25. doi:10.4049/jimmunol.1102746
11. Kimura Y, Miwa T, Zhou L, Song WC. Activator-specific requirement of properdin in the initiation and amplification of the alternative pathway complement. Blood (2008) 111:732–40. doi:10.1182/blood-2007-05-089821
12. Cortes C, Ferreira VP, Pangburn MK. Native properdin binds to Chlamydia pneumoniae and promotes complement activation. Infect Immun (2011) 79:724–31. doi:10.1128/IAI.00980-10
13. Ferreira VP, Cortes C, Pangburn MK. Native polymeric forms of properdin selectively bind to targets and promote activation of the alternative pathway of complement. Immunobiology (2010) 215:932–40. doi:10.1016/j.imbio.2010.02.002
14. Saggu G, Cortes C, Emch HN, Ramirez G, Worth RG, Ferreira VP. Identification of a novel mode of complement activation on stimulated platelets mediated by properdin and C3(H2O). J Immunol (2013) 190:6457–67. doi:10.4049/jimmunol.1300610
15. Carroll MV, Lack N, Sim E, Krarup A, Sim RB. Multiple routes of complement activation by Mycobacterium bovis BCG. Mol Immunol (2009) 46:3367–78. doi:10.1016/j.molimm.2009.07.015
16. Ali YM, Hayat A, Saeed BM, Haleem KS, Alshamrani S, Kenawy HI, et al. Low-dose recombinant properdin provides substantial protection against Streptococcus pneumoniae and Neisseria meningitidis infection. Proc Natl Acad Sci U S A (2014) 111:5301–6. doi:10.1073/pnas.1401011111
17. Tsolaki AG. Innate immune recognition in tuberculosis infection. Adv Exp Med Biol (2009) 653:185–97. doi:10.1007/978-1-4419-0901-5_13
18. Rooijakkers SH, van Strijp JA. Bacterial complement evasion. Mol Immunol (2007) 44:23–32. doi:10.1016/j.molimm.2006.06.011
19. Abdul-Aziz M, Tsolaki AG, Kouser L, Carroll MV, Al-Ahdal MN, Sim RB, et al. Complement factor H interferes with Mycobacterium bovis BCG entry into macrophages and modulates the pro-inflammatory cytokine response. Immunobiology (2016) 221:944–52. doi:10.1016/j.imbio.2016.05.011
20. Ferguson JS, Weis JJ, Martin JL, Schlesinger LS. Complement protein C3 binding to Mycobacterium tuberculosis is initiated by the classical pathway in human bronchoalveolar lavage fluid. Infect Immun (2004) 72:2564–73. doi:10.1128/IAI.72.5.2564-2573.2004
21. Ramanathan VD, Curtis J, Turk JL. Activation of the alternative pathway of complement by mycobacteria and cord factor. Infect Immun (1980) 29:30–5.
22. Cywes C, Godenir NL, Hoppe HC, Scholle RR, Steyn LM, Kirsch RE, et al. Nonopsonic binding of Mycobacterium tuberculosis to human complement receptor type 3 expressed in Chinese hamster ovary cells. Infect Immun (1996) 64:5373–83.
23. Hu C, Mayadas-Norton T, Tanaka K, Chan J, Salgame P. Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J Immunol (2000) 165:2596–602. doi:10.4049/jimmunol.165.5.2596
24. Schlesinger LS, Bellinger-Kawahara CG, Payne NR, Horwitz MA. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J Immunol (1990) 144:2771–80.
25. Cywes C, Hoppe HC, Daffé M, Ehlers MR. Nonopsonic binding of Mycobacterium tuberculosis to complement receptor type 3 is mediated by capsular polysaccharides and is strain dependent. Infect Immun (1997) 65:4258–66.
26. Kouser L, Paudyal B, Kaur A, Stenbeck G, Jones LA, Abozaid SM, et al. Human properdin opsonizes nanoparticles and triggers a potent pro-inflammatory response by macrophages without involving complement activation. Front Immunol (2018) 9:131. doi:10.3389/fimmu.2018.00131
27. Kemper C, Hourcade DE. Properdin: new roles in pattern recognition and target clearance. Mol Immunol (2008) 45:4048–56. doi:10.1016/j.molimm.2008.06.034
28. Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol (2010) 28:131–55. doi:10.1146/annurev-immunol-030409-101250
29. Perdikoulis MV, Kishore U, Reid KB. Expression and characterisation of the thrombospondin type I repeats of human properdin. Biochim Biophys Acta (2001) 1548:265–77. doi:10.1016/S0167-4838(01)00238-2
30. Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One (2010) 5:e8668. doi:10.1371/journal.pone.0008668
31. Al-Rayahi IA, Browning MJ, Stover C. Tumour cell conditioned medium reveals greater M2 skewing of macrophages in the absence of properdin. Immun Inflamm Dis (2017) 5:68–77. doi:10.1002/iid3.142
32. Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, et al. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol (2008) 180:7613–21. doi:10.4049/jimmunol.180.11.7613
33. Dixon KO, O’Flynn J, Klar-Mohamad N, Daha MR, van Kooten C. Properdin and factor H production by human dendritic cells modulates their T-cell stimulatory capacity and is regulated by IFN-gamma. Eur J Immunol (2017) 47:470–80. doi:10.1002/eji.201646703
34. Narni-Mancinelli E, Gauthier L, Baratin M, Guia S, Fenis A, Deghmane AE, et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol (2017) 2(10):eaam9628. doi:10.1126/sciimmunol.aam9628
35. Agarwal S, Specht CA, Haibin H, Ostroff GR, Ram S, Rice PA, et al. Linkage specificity and role of properdin in activation of the alternative complement pathway by fungal glycans. MBio (2011) 2(5):e00178-11. doi:10.1128/mBio.00178-11
36. Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol (2001) 2:569–77. doi:10.1038/35085034
37. Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, et al. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J Immunol (2006) 177:7086–93. doi:10.4049/jimmunol.177.10.7086
38. Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary tuberculosis. Chest (2010) 137:122–8. doi:10.1378/chest.09-0903
39. Smith D, Hansch H, Bancroft G, Ehlers S. T-cell-independent granuloma formation in response to Mycobacterium avium: role of tumour necrosis factor-alpha and interferon-gamma. Immunology (1997) 92:413–21. doi:10.1046/j.1365-2567.1997.00384.x
40. Algood HM, Chan J, Flynn JL. Chemokines and tuberculosis. Cytokine Growth Factor Rev (2003) 14:467–77. doi:10.1016/S1359-6101(03)00054-6
41. Ehlers S, Schaible UE. The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front Immunol (2012) 3:411. doi:10.3389/fimmu.2012.00411
42. Dannenberg AM Jr. Delayed-type hypersensitivity and cell-mediated immunity in the pathogenesis of tuberculosis. Immunol Today (1991) 12:228–33. doi:10.1016/0167-5699(91)90035-R
43. Sanghi S, Grewal RS, Vasudevan B, Lodha N. Immune reconstitution inflammatory syndrome in leprosy. Indian J Lepr (2011) 83:61–70.
44. Huaux F, Lo Re S, Giordano G, Uwambayinema F, Devosse R, Yakoub Y, et al. IL-1α induces CD11b(low) alveolar macrophage proliferation and maturation during granuloma formation. J Pathol (2015) 235(5):698–709. doi:10.1002/path.4487
45. Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol (2010) 184:3326–30. doi:10.4049/jimmunol.0904189
46. Correia JW, Freitas MV, Queiroz JA, PereiraPerrin M, Cavadas B. Interleukin-6 blood levels in sensitive and multiresistant tuberculosis. Infection (2009) 37:138–41. doi:10.1007/s15010-008-7398-3
47. Leal IS, Smedegard B, Andersen P, Appelberg R. Interleukin-6 and interleukin-12 participate in induction of a type 1 protective T-cell response during vaccination with a tuberculosis subunit vaccine. Infect Immun (1999) 67:5747–54.
48. Appelberg R, Castro AG, Pedrosa J, Minoprio P. Role of interleukin-6 in the induction of protective T cells during mycobacterial infections in mice. Immunology (1994) 82:361–4.
49. Bhatt K, Salgame P. Host innate immune response to Mycobacterium tuberculosis. J Clin Immunol (2007) 27:347–62. doi:10.1007/s10875-007-9084-0
50. Shaw TC, Thomas LH, Friedland JS. Regulation of IL-10 secretion after phagocytosis of Mycobacterium tuberculosis by human monocytic cells. Cytokine (2000) 12:483–6. doi:10.1006/cyto.1999.0586
51. O’Leary S, O’Sullivan MP, Keane J. IL-10 blocks phagosome maturation in Mycobacterium tuberculosis-infected human macrophages. Am J Respir Cell Mol Biol (2011) 45:172–80. doi:10.1165/rcmb.2010-0319OC
52. Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol (2001) 19:683–765. doi:10.1146/annurev.immunol.19.1.683
53. Gazzinelli RT, Oswald IP, James SL, Sher A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-gamma-activated macrophages. J Immunol (1992) 148:1792–6.
54. Almeida AS, Lago PM, Boechat N, Huard RC, Lazzarini LC, Santos AR, et al. Tuberculosis is associated with a down-modulatory lung immune response that impairs Th1-type immunity. J Immunol (2009) 183:718–31. doi:10.4049/jimmunol.0801212
55. Barnes PF, Lu S, Abrams JS, Wang E, Yamamura M, Modlin RL. Cytokine production at the site of disease in human tuberculosis. Infect Immun (1993) 61:3482–9.
56. Ranaivomanana P, Raberahona M, Rabarioelina S, Borella Y, Machado A, Randria MJD. Cytokine biomarkers associated with human extra-pulmonary tuberculosis clinical strains and symptoms. Front Microbiol (2018) 9:275. doi:10.3389/fmicb.2018.00275
57. Aly S, Mages J, Reiling N, Kalinke U, Decker T, Lang R, et al. Mycobacteria-induced granuloma necrosis depends on IRF-1. J Cell Mol Med (2009) 13:2069–82. doi:10.1111/j.1582-4934.2008.00470.x
58. Rook GA. Th2 cytokines in susceptibility to tuberculosis. Curr Mol Med (2007) 7:327–37. doi:10.2174/156652407780598557
59. Gordon SV, Brosch R, Billault A, Garnier T, Eiglmeier K, Cole ST. Identification of variable regions in the genomes of tubercle bacilli using bacterial artificial chromosome arrays. Mol Microbiol (1999) 32:643–55. doi:10.1046/j.1365-2958.1999.01383.x
60. Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, et al. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun (2006) 74:88–98. doi:10.1128/IAI.74.1.88-98.2006
Keywords: complement, cytokine, properdin, macrophage, Mycobacterium tuberculosis, Mycobacterium bovis BCG, phagocytosis, thrombospondin repeats
Citation: Al-Mozaini MA, Tsolaki AG, Abdul-Aziz M, Abozaid SM, Al-Ahdal MN, Pathan AA, Murugaiah V, Makarov EM, Kaur A, Sim RB, Kishore U and Kouser L (2018) Human Properdin Modulates Macrophage: Mycobacterium bovis BCG Interaction via Thrombospondin Repeats 4 and 5. Front. Immunol. 9:533. doi: 10.3389/fimmu.2018.00533
Received: 03 July 2017; Accepted: 01 March 2018;
Published: 08 May 2018
Edited by:
Cees Van Kooten, Leiden University, NetherlandsReviewed by:
Michael Kirschfink, Universität Heidelberg, GermanyLubka T. Roumenina, INSERM UMRS 1138, France
Copyright: © 2018 Al-Mozaini, Tsolaki, Abdul-Aziz, Abozaid, Al-Ahdal, Pathan, Murugaiah, Makarov, Kaur, Sim, Kishore and Kouser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Uday Kishore, dWRheS5raXNob3JlQGJydW5lbC5hYy51aw==, dWtpc2hvcmVAaG90bWFpbC5jb20=;
Lubna Kouser, bHVibmFfa0Bob3RtYWlsLmNvLnVr