 Yakir Guri
Yakir Guri Thierry M. Nordmann
Thierry M. Nordmann Jason Roszik
Jason Roszik- 1Biozentrum, University of Basel, Basel, Switzerland
- 2Department of Dermatology, University Hospital Zurich, Zurich, Switzerland
- 3Department of Melanoma Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 4Department of Genomic Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
Cancer is a complex disease and a leading cause of death worldwide. Immunity is critical for cancer control. Cancer cells exhibit high mutational rates and therefore altered self or neo-antigens, eliciting an immune response to promote tumor eradication. Failure to mount a proper immune response leads to cancer progression. mTOR signaling controls cellular metabolism, immune cell differentiation, and effector function. Deregulated mTOR signaling in cancer cells modulates the tumor microenvironment, thereby affecting tumor immunity and possibly promoting carcinogenesis.
Introduction
Tumor bulk is a mass containing heterogeneous cell populations including malignant cancer cells, non-malignant cells, and supporting stroma (1). In addition to tumor cells, non-malignant cells and the supporting stroma play a dynamic and possibly tumor promoting role (2). Non-malignant cells in the tumor microenvironment include cells of the lymphoid and myeloid immune system (3). The supporting stroma is largely composed of cancer-associated fibroblasts (CAFs), vascular and lymphatic endothelial cells, and pericytes. Cells within the tumor “communicate” by secretion of various factors to the tumor microenvironment, including matrix remodeling enzymes, cytokines, chemokines, growth factors, and metabolites (4, 5). This interplay between malignant, non-malignant, and stromal cells has functional consequences on tumor progression.
Target Of Rapamycin (TOR) is an evolutionarily conserved serine/threonine protein kinase. TOR controls cellular metabolism and growth and functions in two complexes: TOR Complex 1 (TORC1) and TORC2 (6, 7) (Figure 1). Mammalian TORC1 (mTORC1) comprises mTOR, mammalian lethal with sec-13 protein 8 (mLST8), and regulatory-associated protein of mammalian target of rapamycin (RAPTOR). mTORC1 is activated by growth factors, nutrients (amino acids), and cellular energy (8, 9), and is allosterically inhibited by rapamycin (10). Various growth factors regulate mTORC1 via a heterotrimeric tuberous sclerosis complex (TSC) complex. Growth factors bind receptor tyrosine kinases (RTKs) and activate Phosphatidylinositol-4,5-Bisphosphate 3-Kinase (PI3K), which generates Phosphatidylinositol-3,4,5-Trisphosphate (PIP3) (11). PI3K activity is counteracted by the tumor suppressor, phosphatase, and Tensin Homolog Deleted on Chromosome 10 (PTEN). mTORC1 promotes anabolic processes, such as protein and nucleotide synthesis and inhibits catabolic processes, such as autophagy (12–14). mTORC2 contains mTOR, mLST8, mammalian stress-activated map kinase-interacting protein 1 (mSIN1), and Rapamycin-Insensitive Companion of mTOR (RICTOR), and is activated by growth factors in association with ribosomes (15) (Figure 1). mTORC1 and mTORC2 are frequently activated in human cancers and, as discussed below, reported to modulate the tumor microenvironment or respond to its changes.
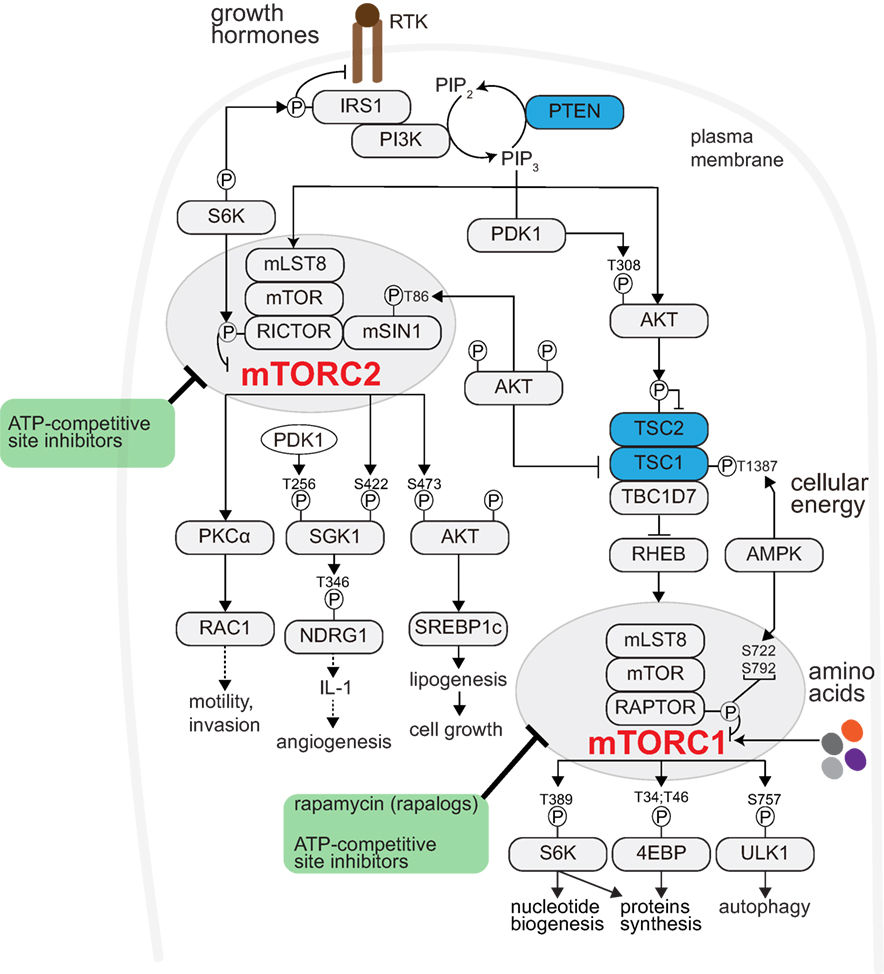
Figure 1. mTOR signaling promotes anabolism. Receptor Tyrosine Kinases (RTKs)- Phosphatidyl-Inositol-4,5-bisphosphate 3-Kinase (PI3K) activated by growth factor (like insulin). PI3K generates phosphatidylinositol-3,4,5-trisphosphate (PIP3) from the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2). Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN) counteracts PI3K activity (restoring PIP3 to PIP2). PIP3 recruits to the plasma membrane and activates phosphoinositide-dependent kinase 1 (PDK1) and AKT. PDK1 phosphorylates and activates AKT (pAKT-Thr308). pAKT-Thr308 phosphorylates and inhibits the TSC complex. The TSC complex, composed of tuberous sclerosis complex 1 (TSC1) and TSC2 and TRE2-BUB2-CDC16 domain family member 7 (TBC1D7), activates the lysosomal RAS homolog enriched in brain (RHEB). RHEB interacts with and activates mTORC1. mTORC1 comprises mTOR, mammalian lethal with sec-13 protein 8 (mLST8), and regulatory-associated protein of mammalian target of rapamycin (RAPTOR). mTORC1 can also be activated by nutrients (such as amino acids). Cellular energy status also regulates mTORC1 through AMPK-mediated TSC or RAPTOR phosphorylation. mTORC1 promotes anabolism, among others, through ribosomal protein S6 kinase (S6K), eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1), and blocks cellular catabolism through Unc-51-like kinase 1 (ULK1). Through S6K-mediated IRS1 phosphorylation, mTORC1 negatively regulates mTORC2-AKT signaling. Rapamycin and its analogs (so-called rapalogues) acutely inhibit mTORC1 allosterically. The ATP-site competitive inhibitor(s) potently block both mTORC1 and mTORC2 signaling. mTORC2 is also activated by RTKs, and consists of mTOR, mLST8, mammalian stress-activated map kinase-interacting protein 1 (mSIN1), and rapamycin-insensitive companion of mTOR (RICTOR). mTORC2 regulates the AGC kinase family members AKT, serum/glucocorticoid-regulated kinase (SGK), and protein kinase C (PKC). Prolonged rapamycin administration may block mTORC2 activity.
Cancer Cell-Intrinsic mTOR Activation Modulating the Tumor Microenvironment
Oncogenic mutations drive tumorigenesis by activating various growth controlling signaling pathways (16). The PI3K–mTOR–AKT signaling pathway is activated in the majority of tumors, due to upstream oncogenic mutation(s). Alternatively, parallel growth controlling (oncogenic) pathways, such as the MEK–ERK, may also activate PI3K–mTOR–AKT signaling (12). Either way, PI3K–mTOR–AKT activation promotes cell growth and proliferation (Figure 1). In addition to the cell-intrinsic growth-promoting effect, PI3K–mTOR–AKT activation appears to alter the tumor microenvironment.
T Lymphocytes
T cells play a critical role in adaptive and innate immunity. Antigen recognition and adaptive immunity involves, among others, CD4 + and CD8 + T cells. While tumor eradication is largely mediated by cytotoxic CD8 + T lymphocytes (CTL), CD4 + T cells are critical in regulating and propagating the immune response, hence referred to as T helper cells (Th) (17). In solid tumors, the extent of T-cell infiltration is an important prognostic determinate. Increased CD4 + and CD8 + T-cell levels are associated with an improved clinical outcome (18). In colorectal tumors, increased density of T cells (i.e., Th1 adaptive immunity) correlated with reduced tumor recurrence, and provided a better prognostic tool than conventional histopathological methods (19). Conversely, tumors with a higher density of immune-suppressive cells (such T regulatory cells, as discussed below) exhibit a worse prognosis, in colorectal (19) and other tumor types (20). Thus, adaptive immunity plays a critical role in tumor progression and prognosis.
Various cytokines and chemokines attract immune cells to the site of inflammation (21). In addition to cytokines and chemokines, also metabolites in the tumor microenvironment (some of which are secreted by cancer cells) activate immune cells (22). Non-Alcoholic Fatty Liver Disease (NAFLD) is a metabolic disorder and a risk factor for hepatocellular carcinoma (HCC) (23). In NAFLD, increased linoleic acid levels disrupt adaptive immunity, specifically by depleting CD4 + T cells, which in turn promotes HCC (24). These data indicate that a metabolite accumulating in the tumor microenvironment may affect neighboring T cells, disturb their function, and promote cancer. It is not fully understood what regulates linoleic acid accumulation, but hepatic fatty acid (FA) synthesis (including linoleic acid) is controlled by mTORC2 (25). Importantly, constitutively active hepatic mTORC2 signaling is oncogenic and promotes HCC (26), and is particularly important in case of NAFLD to HCC transition (27). Thus, it is likely that mTORC2-mediated FA (and perhaps lipid) synthesis in cancer cells modulates immunity.
mTORC2 mediates various cellular processes via AGC kinase family members AKT, serum/glucocorticoid-regulated kinase (SGK), and protein kinase C (PKC) (28, 29) (Figure 1). In a mammary gland tumor model, Rictor deletion disrupted secondary mammary ductal branching, cell motility, and survival. This effect was mediated by PKCα-Rac1, but not AKT (30), suggesting an AKT-independent role of mTORC2 in motility and metastasis. mTORC2 phosphorylates and activates AKT (pAKT-Ser473). Melanoma with increased pAKT-Ser473 correlated with reduced T-cell infiltration, possibly due to increased secretion of inhibitory cytokines by cancer cells, and exhibit resistance to immune checkpoint inhibitors (31). The mTORC2 target SGK is frequently expressed in tumors (32). In gastric tumors, increased expression of the SGK1 target, NDRG1, is suggested to stimulate IL-1 expression and promote angiogenesis (33). Taken together, these data suggest that increased PI3K–mTORC2–AKT signaling in cancer cells may affect T cells and thereby tumorigenesis. It is possible that other immune cells in the tumor microenvironment are also modulated by PI3K–mTORC2–AKT, as described further below.
Regulatory T Cells (Tregs)
Regulatory T cells suppress inflammation and are detrimental in tumor immunity. Genetic and pharmacological (rapamycin) abrogation of mTOR signaling induce Treg expansion via Foxp3 expression (34, 35). Furthermore, Treg-specific conditional TSC deletion in mice (constitutively active mTORC1) propelled Treg differentiation and a strong effector-like phenotype, reversed by S6K1 knockdown (36), suggesting that mTORC1 is an important checkpoint in Treg homeostasis.
Programmed Death 1 (PD-1) and Cytotoxic T-Lymphocyte-associated Antigen 4 (CTLA-4) immune checkpoints negatively regulate T-cell immune function. Immune suppression in the tumor microenvironment through PD-1 or CTLA-4 occurs in various tumors, and immune checkpoint inhibitors (anti-PD-1, anti-PD-L1, or anti-CTLA-4) amplify antitumor T-cell response (37). The surface protein PD-L1 is widely expressed in various tumors. PD-L1 binds to either the T-cell-expressed PD-1 or CD80 receptors thereby inhibiting their effector responses. PD-L1 and PD-1 interaction induces differentiation of naïve CD4 + T cells into Tregs, leading to an immune suppressive environment. In addition to inhibiting T-cell effector function, cancer cell-intrinsic PD-1 expression may promote tumor growth (38). Thus, PD-1 axis has a twofold effect in tumorigenesis: first by inhibiting cancer cell clearance by T cells, and second, promoting cancer cell growth. In a lung carcinoma mouse model, mTORC1 increased PD-L1 expression, allowing cancer cells to escape killing by immune cells (39, 40). Within the tumor, PD-L1 seems to be enriched in Tumor Initiating Cells (TICs) (also referred to as Cancer Stem Cells) (41–43). TICs are tumor cells with self-renewal capacity and considered to be more resistant to targeted cancer therapies. In syngeneic ovarian mouse model experiments, PD-L1 appeared to control the expression of canonical “stemness” genes, such as Oct4 and Nanog (44, 45). PD-L1 expression correlated with mTOR activation in human lung adenocarcinomas and squamous cell carcinomas (39), suggesting that oncogenic AKT-mTOR activation promotes immune escape through PD-L1 upregulation. Furthermore, anti-PD-1 therapy inhibited human melanoma xenograft growth and reduced S6 phosphorylation, suggesting that PD-1 in tumor cells activates mTORC1. Importantly, cells expressing high levels of PD-L1 appear to be more sensitive to the mTORC1 inhibitor rapamycin, further suggesting that some of the PD-L1 growth-controlling mechanisms are via mTOR signaling. Collectively, these data suggest a functional relationship between mTOR signaling, PD-L1 expression, and resistance to targeted therapies (i.e., TICs). However, the mechanism(s) by which mTORC1 signaling regulates PD-L1 expression remains to be elucidated. We note that in addition to Treg and Th1, other T-cell subsets, such as Th17, may be involved in cancer immune response.
Tumor-Associated Macrophages (TAMs)
Tumor-associated macrophages originate from expansion of tissue-resident macrophages or are recruited to tumor site (by chemotactic factors), and are present at multiple stages of tumor progression (2). Macrophages are not a homogenous population and can be subdivided into M1 and M2. M1 macrophages produce Th1 cytokines, promoting phagocyte-dependent inflammation and thereby an antitumor response. M2 macrophages enforce antibody response, but inhibit several phagocytic functions, therefore seemingly enabling a growth-tolerant tumor microenvironment. TAMs predominantly exhibit M2 phenotypes, therefore considered tumor-promoting. Several factors can promote polarization of TAMs to M2 during cancer progression, including IL-4, IL-10, TGF-β, and M-CSF (46). TAMs promote tumorigenesis by modulating lymph- and angiogenesis (47), but more recently, TAMs were shown to express PD-1. The presence of TAM expressing PD-1 steadily increases with cancer progression and results in an overall reduction in cancer cell phagocytosis (48). Because macrophages activation and function is, at least in part, controlled by PI3K–mTOR–AKT (49), it would be valuable to examine whether the observed reduction in phagocytosis is related to mTOR signaling. Furthermore, mTOR regulates macrophage polarization (50), and M1 and M2 macrophages exhibit dependency on distinct metabolic pathways. While M1 macrophages upregulate glycolysis and lipogenesis, M2 macrophages upregulate beta-oxidation. This is important because metabolic shifts are coupled to macrophage function (51, 52). For instance, IL-4 activate AKT and thereby inducing M2 gene transcription, possibly via ACLY expression and regulation of histone acetylation (53), indicating that mTOR signaling couple metabolic inputs to modulate immune response. Moreover, PI3K–AKT appears to recruit immune-suppressive monocytes to tumors via monocyte chemoattractant protein-1 (MCP-1) expression, in a mechanism that potentially involves TGFβ1 (54). MCP1 plays a similar role in other tumors (55), but whether PI3K–AKT induced MCP1 expression can be generalized to other tumors remains to be investigated. mTORC2 appears to be particularly important for differentiation of M2 macrophages (as opposed to M1), as not only monocytes recruitment but also monocyte polarization is involved in tumor progression (56); therefore, mTORC2 plays a dual immunosuppressive role.
Antigen-presenting cells (APCs), especially dendritic cells (DCs), are crucial in mounting antitumor immune response (57). Indeed, abrogation of mTORC2 signaling in the professional APCs, DCs, led to enhanced tumor eradication possibly via engagement of CTLs (58). Rapamycin administration augmented the expression of costimulatory molecules and enhanced DC life span, via modulation of glucose metabolism (59). These data suggest that mTOR signaling in APC cells imposes an immunosuppressive environment.
Myeloid-Derived Suppressor Cells (MDSCs)
Myeloid-derived suppressor cells are a heterogeneous population defined as CD11b + Gr1 + cells. Based on Ly6G and Ly6C expression, MDSCs can be further classified as granulocytic or monocytic subsets, respectively. Both CD11b + Ly6G + and CD11b + Ly6C + cells play immunosuppressive roles. The allosteric mTORC1 inhibitor, rapamycin, inhibits MDSC accumulation in tumors and skin allografts (60). In breast cancer, accumulation of MDSCs in tumors occurred via G-CSF. Rapamycin administration or Raptor deletion (a core-component of mTORC1) reduced G-CSF levels (61), suggesting that mTORC1 in tumor cells attracts MDSCs by upregulating G-CSF. Increased G-CSF levels also correlated with elevated mTOR activity in human tumors. Interestingly, there is correlation between presence of TICs, elevated mTORC1 signaling, and G-CSF production. Moreover, rapamycin administration leads to reduced TIC levels (61). These data suggest that mTOR activity in a subset of cells within the tumor mass (i.e., intra-tumoral heterogeneity) mediates MDSC accumulation.
Other Cells of the Tumor Microenvironment: CAFs
Fibroblasts are not only involved in the deposition of stromal extra-cellular matrix (ECM) but also in the secretion of growth factors. CAFs seem to play a role in cancer progression and initiation, particularly in stroma-rich tumors like pancreatic cancers (62, 63). In pancreatic tumors, CAFs are also involved in resistance to anticancer drugs (64). Interleukin-6 (IL-6) is linked to resistance-to-cancer drug therapies (65), possibly via its downstream effector pSTAT3 (66). In pancreatic CAFs, the somatostatin receptor sst1 inhibits mTOR-mediated IL-6 protein synthesis, thereby counteracting mTOR/IL-6-driven resistance to anticancer drugs (67). How mTOR regulates IL-6 expression in stromal cells remains to be investigated, but this mechanism seems to involve the quintessential mTORC1 target, 4E-BP1 (67). In lung carcinoma, paracrine IGF-II secretion by CAFs activated insulin growth factor receptor 1 (IGF1R) signaling in cancer cells, possibly activating a TICs (stemness)-like phenotype (68). Conversely, in irradiated tumors, IGF-II secreted from CAFs appears to block mTORC1 signaling in neighboring cancer cells. mTORC1 inhibition allowed autophagy initiation and thereby tumor regrowth (69). It seems counterintuitive that mTOR inhibition allows tumor growth, but possibly under stress or nutrient-poor conditions autophagy initiation provide the required nutrients. Nevertheless, this hypothesis needs to be examined in other cancer models. Yet, liver specific Raptor knockout mice (abrogated mTORC1 signaling) developed more HCC when challenged with the hepato-carcinogen diethyl-nitrosamine, as compared with wild-type mice (70). These data suggest that “too low” mTORC1 activity may also be oncogenic. Taken together, it is likely that the response to drug therapies is not only dependent on stromal cells and their secretome but also on the conditions in which therapies are given.
mTOR on the Receiving End of Cancer Immunity
mTOR signaling is also on the receiving end of cues coming from the tumor microenvironment. For example, non-tumorigenic (stromal) cells of the tumor microenvironment secrete MCP1 to activate the mTOR pathway in neighboring breast cancer cells (71). Moreover, metabolic activation of natural killer (NK) cells is dependent on IL-15 stimulation to prompt intracellular mTOR signaling (72). NK cells are suggested to play a pivotal role in cancer control and are increased in metastatic melanoma (73). Conversely, TGF-β represses mTOR signaling, both in mice and humans, to inhibit NK cell activation (74), suggesting an mTOR-dependent immune suppressive role for TGF-β in tumor microenvironment. Additionally, genetic activation of mTORC1 (mutated TSC) causes impairment of NK cell development (75). Notably, mTOR also regulates Th1 and Th2 differentiation; and while mTORC1 is distinctly critical for Th1 and Th17 differentiation, mTORC2 seems to promote Th2 differentiation (76). Furthermore, mTORC1 regulates CD8 + T-cell effector function (77), thereby allowing better clearance of tumor cells. Although mTORC2 seems to be dispensable for the effector function of CD8 + T cells, it is critical for generation of CD8 + memory cells (77). Further studies are required to examine how extracellular signals affect mTOR in T cells; nonetheless, the data demonstrate that mTOR signaling differentially regulates T cells.
Clinical Implications
Various mTOR inhibitors are in ongoing clinical trials and the FDA-approved rapalog everolimus is used in various cancer cell types (10). Because mTOR signaling plays a key role in cancer and immune cell function (78), it is possible that some of the anticancer effect of mTOR inhibitors is via immune modulation (Figure 2). Indeed, rapamycin is clinically used for prevention of renal graft rejection and is traditionally considered as a “pure” immunosuppressant, possibly by blocking T-cell activation. However, as discussed above, mTOR seems to play a more complex role in immunity. Under certain conditions, mTOR inhibition poses an immune-activating function, such as induction of memory CD8 + T cell (77) that may in turn increase the durability of antitumor effector T-cell function. It is also likely that ATP kinase mTOR inhibitors, blocking robustly mTORC1, and mTORC2 signaling (79) act on cancer cells, as well as on the tumor microenvironment.
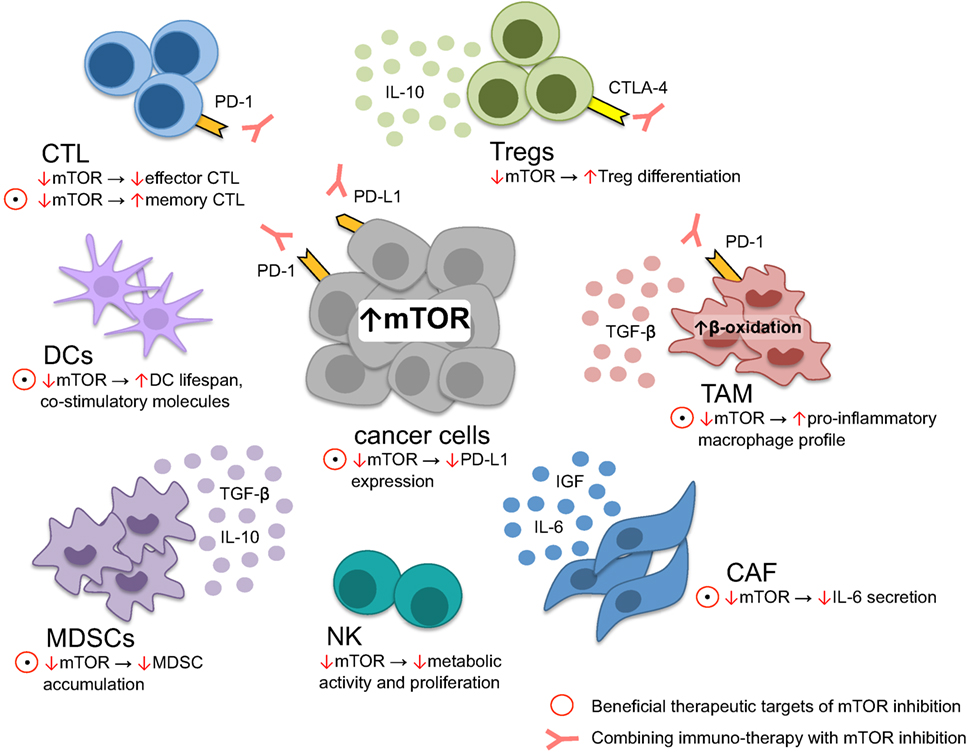
Figure 2. Tumor microenvironment modulation through mTOR. mTOR inhibition induces memory cytotoxic CD8 + T lymphocytes (CTL) formation while reducing effector CTL function, critical for cellular antitumor response. In dendritic cells, lifespan and the expression of costimulatory molecules is increased upon mTOR suppression, leading to improved foreign-antigen recognition. On the other hand, metabolic NK-cell function, essential for antitumor response, is diminished upon mTOR inhibition. Myeloid derived stem cells (MDSC), regulatory T cells (Tregs), tumor-associated macrophages (TAM), and cancer-associated fibroblasts (CAF) contribute to tumor immune-evasion and tumor growth. The immune-suppressive environment generated through MDSC is limited through mTOR blockage by restraining MDSC accumulation. Similarly, anti-inflammatory TAM may be skewed toward a more pro-inflammatory profile upon mTOR inhibition. CAF secrete various cytokines promoting tumor growth and therapy resistance, counteracted by mTOR blockage. In contrast, Tregs are preferentially differentiated upon mTOR downregulation. Within the majority of cancer cells, the PI3K–mTOR–AKT pathway is upregulated, driving PD-L1 expression maintaining an immune-suppressive state within the tumor microenvironment: a process that may be interrupted through mTOR inhibition. However, not all therapeutic targets of mTOR inhibition seem to be beneficial, such as reducing effector CTL function and T-Reg differentiation. Accordingly, rationale exists to combine anti-PD-1/PD-L1 or anti-CTLA-4 and mTOR inhibitors, alleviating reduced CTL effector function and Treg differentiation.
While checkpoint and mTOR inhibitors have revolutionized cancer treatment, as monotherapies these drugs seem to be insufficient to fully block cancer progression. Oncogenic PI3K–mTOR–AKT pathway reduces T-cell tumor infiltration and causes inferior outcome after PD-1 inhibition (31), providing a rationale for the design of combination therapies of mTOR and immune checkpoint inhibitors, as recently shown for HCC (80) (Figure 2). Nonetheless, the specific oncogenic mechanism downstream of mTOR remains unknown. Understanding these pathways is critical for the rational design of selective inhibitors. The combination of checkpoint and mTOR inhibitors might be limited by its side effects because: (i) PI3K–mTOR–AKT signaling plays a critical role in physiological cell homeostasis, (ii) rapamycin administration reduces the effector CD8 + T-cell function (that are otherwise required for execution of anticancer effect), and (iii) possibly relieve negative feedback loops that may induce compensatory pathway activation.
Conclusion
Collectively, the above suggests that mTOR signaling has both tumor-intrinsic and tumor-extrinsic (i.e., tumor microenvironment) activities. mTOR-kinase quickly responds to stimuli in the tumor microenvironment and executes various (possibly opposing) effects on immune cells. Thus, a prime challenge is to dissect the role of mTOR in the different cell types in the tumor microenvironment and to assess the overall “net effect” of mTOR blockade.
Author Contributions
YG wrote the original draft. YG, TMN, and JR wrote and approved the final version of the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dale Braunschweig for helpful suggestions on our manuscript. This work was supported by generous philanthropic contributions to The University of Texas MD Anderson Moon Shots Program, the Swiss National Science Foundation and the Swiss Cancer Research foundation (KFS).
References
1. Allen E, Mieville P, Warren CM, Saghafinia S, Li L, Peng MW, et al. Metabolic symbiosis enables adaptive resistance to anti-angiogenic therapy that is dependent on mTOR signaling. Cell Rep (2016) 15:1144–60. doi:10.1016/j.celrep.2016.04.029
2. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21:309–22. doi:10.1016/j.ccr.2012.02.022
3. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol (2013) 14:1014–22. doi:10.1038/ni.2703
4. Wendler F, Favicchio R, Simon T, Alifrangis C, Stebbing J, Giamas G. Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene (2017) 36:877–84. doi:10.1038/onc.2016.253
5. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med (2013) 19:1423–37. doi:10.1038/nm.3394
6. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124:471–84. doi:10.1016/j.cell.2006.01.016
7. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149:274–93. doi:10.1016/j.cell.2012.03.017
8. Shimobayashi M, Hall MN. Multiple amino acid sensing inputs to mTORC1. Cell Res (2016) 26:7–20. doi:10.1038/cr.2015.146
9. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol (2013) 15:555–64. doi:10.1038/ncb2763
10. Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov (2011) 10:868–80. doi:10.1038/nrd3531
11. Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol (2015) 25:545–55. doi:10.1016/j.tcb.2015.06.002
12. Guri Y, Hall MN. mTOR signaling confers resistance to targeted cancer drugs. Trends Cancer (2016) 2:688–97. doi:10.1016/j.trecan.2016.10.006
13. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene (2006) 25:6416–22. doi:10.1038/sj.onc.1209888
14. Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol (2009) 10:307–18. doi:10.1038/nrm2672
15. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell (2011) 144:757–68. doi:10.1016/j.cell.2011.02.014
16. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
17. Disis ML. Immune regulation of cancer. J Clin Oncol (2010) 28:4531–8. doi:10.1200/JCO.2009.27.2146
18. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer (2012) 12:298–306. doi:10.1038/nrc3245
19. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
20. Kitano Y, Okabe H, Yamashita YI, Nakagawa S, Saito Y, Umezaki N, et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br J Cancer (2018) 118(2):171–80. doi:10.1038/bjc.2017.401
21. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12:269–81. doi:10.1038/nri3191
22. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol (2018) 18(1):19–34. doi:10.1038/nri.2017.99
23. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol (2013) 10:656–65. doi:10.1038/nrgastro.2013.183
24. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature (2016) 531:253–7. doi:10.1038/nature16969
25. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab (2012) 15:725–38. doi:10.1016/j.cmet.2012.03.015
26. Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell (2006) 11:859–71. doi:10.1016/j.devcel.2006.10.007
27. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell (2017) 32:807–23.e12. doi:10.1016/j.ccell.2017.11.011
28. Cybulski N, Polak P, Auwerx J, Ruegg MA, Hall MN. mTOR complex 2 in adipose tissue negatively controls whole-body growth. Proc Natl Acad Sci U S A (2009) 106:9902–7. doi:10.1073/pnas.0811321106
29. Sparks CA, Guertin DA. Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene (2010) 29:3733–44. doi:10.1038/onc.2010.139
30. Morrison MM, Young CD, Wang S, Sobolik T, Sanchez VM, Hicks DJ, et al. mTOR directs breast morphogenesis through the PKC-alpha-Rac1 signaling axis. PLoS Genet (2015) 11:e1005291. doi:10.1371/journal.pgen.1005291
31. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov (2016) 6:202–16. doi:10.1158/2159-8290.CD-15-0283
32. Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J (2013) 452:499–508. doi:10.1042/BJ20130342
33. Murakami Y, Watari K, Shibata T, Uba M, Ureshino H, Kawahara A, et al. N-myc downstream-regulated gene 1 promotes tumor inflammatory angiogenesis through JNK activation and autocrine loop of interleukin-1alpha by human gastric cancer cells. J Biol Chem (2013) 288:25025–37. doi:10.1074/jbc.M113.472068
34. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol (2006) 177:8338–47. doi:10.4049/jimmunol.177.12.8338
35. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
36. Park Y, Jin HS, Lopez J, Elly C, Kim G, Murai M, et al. TSC1 regulates the balance between effector and regulatory T cells. J Clin Invest (2013) 123:5165–78. doi:10.1172/JCI69751
37. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell (2017) 31:311–25. doi:10.1016/j.ccell.2017.02.008
38. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell (2015) 162:1242–56. doi:10.1016/j.cell.2015.08.052
39. Lastwika KJ, Wilson W III, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res (2016) 76:227–38. doi:10.1158/0008-5472.CAN-14-3362
40. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res (2016) 76:6964–74. doi:10.1158/0008-5472.CAN-16-0258
41. Yao Y, Tao R, Wang X, Wang Y, Mao Y, Zhou LF. B7-H1 is correlated with malignancy-grade gliomas but is not expressed exclusively on tumor stem-like cells. Neuro Oncol (2009) 11:757–66. doi:10.1215/15228517-2009-014
42. Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X, et al. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS One (2015) 10:e0135528. doi:10.1371/journal.pone.0135528
43. Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, et al. CD44+ cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin Cancer Res (2016) 22:3571–81. doi:10.1158/1078-0432.CCR-15-2665
44. Clark CA, Gupta HB, Curiel TJ. Tumor cell-intrinsic CD274/PD-L1: a novel metabolic balancing act with clinical potential. Autophagy (2017) 13:987–8. doi:10.1080/15548627.2017.1280223
45. Gupta HB, Clark CA, Yuan B, Sareddy G, Pandeswara S, Padron AS, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther (2016) 1. doi:10.1038/sigtrans.2016.30
46. Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol (2011) 55:861–7. doi:10.1387/ijdb.113371dl
47. Riabov V, Gudima A, Wang N, Mickley A, Orekhov A, Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front Physiol (2014) 5:75. doi:10.3389/fphys.2014.00075
48. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature (2017) 545:495–9. doi:10.1038/nature22396
49. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol (2015) 27:286–96. doi:10.1016/j.smim.2015.08.001
50. Zhu L, Yang T, Li L, Sun L, Hou Y, Hu X, et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat Commun (2014) 5:4696. doi:10.1038/ncomms5696
51. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity (2016) 45:817–30. doi:10.1016/j.immuni.2016.09.016
52. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42:419–30. doi:10.1016/j.immuni.2015.02.005
53. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife (2016) 5. doi:10.7554/eLife.11612
54. Diaz-Valdes N, Basagoiti M, Dotor J, Aranda F, Monreal I, Riezu-Boj JI, et al. Induction of monocyte chemoattractant protein-1 and interleukin-10 by TGFbeta1 in melanoma enhances tumor infiltration and immunosuppression. Cancer Res (2011) 71:812–21. doi:10.1158/0008-5472.CAN-10-2698
55. Furukawa S, Soeda S, Kiko Y, Suzuki O, Hashimoto Y, Watanabe T, et al. MCP-1 promotes invasion and adhesion of human ovarian cancer cells. Anticancer Res (2013) 33:4785–90.
56. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol (2002) 23:549–55. doi:10.1016/S1471-4906(02)02302-5
57. Martin K, Schreiner J, Zippelius A. Modulation of APC function and anti-tumor immunity by anti-cancer drugs. Front Immunol (2015) 6:501. doi:10.3389/fimmu.2015.00501
58. Raich-Regue D, Fabian KP, Watson AR, Fecek RJ, Storkus WJ, Thomson AW. Intratumoral delivery of mTORC2-deficient dendritic cells inhibits B16 melanoma growth by promoting CD8(+) effector T cell responses. Oncoimmunology (2016) 5:e1146841. doi:10.1080/2162402X.2016.1146841
59. Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, et al. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J Immunol (2012) 189:2151–8. doi:10.4049/jimmunol.1103741
60. Wu T, Zhao Y, Wang H, Li Y, Shao L, Wang R, et al. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci Rep (2016) 6:20250. doi:10.1038/srep20250
61. Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol (2016) 18:632–44. doi:10.1038/ncb3355
62. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell (2010) 17:135–47. doi:10.1016/j.ccr.2009.12.041
63. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res (2012) 18:4266–76. doi:10.1158/1078-0432.CCR-11-3114
64. Erkan M. Understanding the stroma of pancreatic cancer: co-evolution of the microenvironment with epithelial carcinogenesis. J Pathol (2013) 231:4–7. doi:10.1002/path.4213
65. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol (2016) 37:11553–72. doi:10.1007/s13277-016-5098-7
66. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell (2009) 15:103–13. doi:10.1016/j.ccr.2009.01.001
67. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F, et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med (2015) 7:735–53. doi:10.15252/emmm.201404346
68. Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun (2014) 5:3472. doi:10.1038/ncomms4472
69. Wang Y, Gan G, Wang B, Wu J, Cao Y, Zhu D, et al. Cancer-associated fibroblasts promote irradiated cancer cell recovery through autophagy. EBioMedicine (2017) 17:45–56. doi:10.1016/j.ebiom.2017.02.019
70. Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab (2014) 20:133–44. doi:10.1016/j.cmet.2014.05.001
71. Riverso M, Kortenkamp A, Silva E. Non-tumorigenic epithelial cells secrete MCP-1 and other cytokines that promote cell division in breast cancer cells by activating ERalpha via PI3K/Akt/mTOR signaling. Int J Biochem Cell Biol (2014) 53:281–94. doi:10.1016/j.biocel.2014.05.023
72. Marcais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol (2014) 15:749–57. doi:10.1038/ni.2936
73. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell (2017) 171:934–49.e15. doi:10.1016/j.cell.2017.09.028
74. Viel S, Marcais A, Guimaraes FS-F, Loftus R, Rabilloud J, Grau M, et al. TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal (2016) 9:ra19. doi:10.1126/scisignal.aad1884
75. Yang M, Chen S, Du J, He J, Wang Y, Li Z, et al. NK cell development requires Tsc1-dependent negative regulation of IL-15-triggered mTORC1 activation. Nat Commun (2016) 7:12730. doi:10.1038/ncomms12730
76. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
77. Pollizzi KN, Patel CH, Sun IH, Oh MH, Waickman AT, Wen J, et al. mTORC1 and mTORC2 selectively regulate CD8(+) T cell differentiation. J Clin Invest (2015) 125:2090–108. doi:10.1172/JCI77746
78. O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol (2018) 48:91–103. doi:10.1016/j.semcancer.2017.04.015
79. Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature (2016) 534:272–6. doi:10.1038/nature17963
80. Li H, Li X, Liu S, Guo L, Zhang B, Zhang J, et al. Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology (2017) 66:1920–33. doi:10.1002/hep.29360
Keywords: signaling, tumorigenesis, metabolism, immunity, immunotherapy, rapamycin
Citation: Guri Y, Nordmann TM and Roszik J (2018) mTOR at the Transmitting and Receiving Ends in Tumor Immunity. Front. Immunol. 9:578. doi: 10.3389/fimmu.2018.00578
Received: 14 December 2017; Accepted: 07 March 2018;
Published: 27 March 2018
Edited by:
Sherven Sharma, VA Greater Los Angeles Healthcare System (VHA), United StatesReviewed by:
Kawaljit Kaur, University of California, Los Angeles, United StatesAmedeo Amedei, Università degli Studi di Firenze, Italy
Copyright: © 2018 Guri, Nordmann and Roszik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yakir Guri, eWFraXIuZ3VyaUB1bmliYXMuY2g=;
Jason Roszik, anJvc3ppa0BtZGFuZGVyc29uLm9yZw==