 Defne Bayik
Defne Bayik Debra Tross
Debra Tross Dennis M. Klinman
Dennis M. Klinman- Cancer and Inflammation Program, National Cancer Institute at Frederick, Frederick, MD, United States
Monocytic myeloid-derived suppressor cells (mMDSC) accumulate within tumors where they create an immunosuppressive milieu that inhibits the activity of cytotoxic T and NK cells thereby allowing cancers to evade immune elimination. The toll-like receptors 7/8 agonist R848 induces human mMDSC to mature into inflammatory macrophage (MACinflam). This work demonstrates that TNFα, IL-6, and IL-10 produced by maturing mMDSC are critical to the generation of MACinflam. Neutralizing any one of these cytokines significantly inhibits R848-dependent mMDSC differentiation. mMDSC cultured in pro-inflammatory cytokine IFNγ or the combination of TNFα plus IL-6 differentiate into MACinflam more efficiently than those treated with R848. These mMDSC-derived macrophages exert anti-tumor activity by killing cancer cells. RNA-Seq analysis of the genes expressed when mMDSC differentiate into MACinflam indicates that TNFα and the transcription factors NF-κB and STAT4 are major hubs regulating this process. These findings support the clinical evaluation of R848, IFNγ, and/or TNFα plus IL-6 for intratumoral therapy of established cancers.
Introduction
Infiltration of the tumor microenvironment by immunosuppressive leukocytes protects cancers from immune elimination (1). Myeloid-derived suppressor cells (MDSC) are key contributors to this immunosuppressive milieu. MDSC are classified into monocytic or granulocytic subsets based on their phenotype, morphology, and function. Both subsets are present at very low frequencies in the peripheral blood of healthy donors but are much more prevalent in cancer patients (2–5). MDSC migrate from the peripheral blood into the tumor bed, where they inhibit the activity of tumoricidal NK and cytotoxic T cells (5, 6). As monocytic myeloid-derived suppressor cells (mMDSC) have the greatest immunosuppressive activity per cell, efforts to enhance the efficacy of immunotherapy have focused on blocking the recruitment/activation of that cell type (2, 4, 5).
Toll-like receptors (TLRs) comprise a family of highly conserved germline-encoded pattern recognition receptors (7). TLR engagement stimulates elements of the innate immune system to produce pro-inflammatory cytokines, such as IL-6 and IL-12 that help bolster adaptive immunity (7, 8). Our group previously showed that injecting TLR7 agonists into murine tumors induced resident mMDSC to differentiate into tumoricidal M1-like macrophages (MACinflam) and led to the elimination of established cancers (9, 10). Human mMDSC cultured with the TLR7/8 agonist R848 also differentiate primarily into MACinflam (11). By comparison, human mMDSC treated with the TLR2/1 agonist Pam3CSK4 (hereafter PAM3) mature primarily into immunosuppressive M2-like macrophages (MACsuppress) (11). This study seeks to identify the factors and gene networks that influence the generation of MACinflam from mMDSC by comparing the effects of R848 treatment with that of other stimulants.
Materials and Methods
Reagents
R848, Pam3CSK4, Celastrol, and Ruxolitinib were purchased from InvivoGen (San Diego, CA, USA) and all human recombinant cytokines were obtained from Miltenyi Biotec (Auburn, CA, USA). CD163 (Clone #GHI/61), CD206 (Clone #15-2), and CD14 (Clone #M5E2) antibodies used to purify or stain human mMDSC, and anti-IL-6 (Clone #MQ2-13A5), anti-IL-10 (Clone #JES3-19F1), anti-IL-12 (Clone #C11.5), anti-TNFα (Clone #Mab1), and anti-IFNγ (Clone #NIB42) utilized to neutralize secreted cytokines were purchased from BioLegend (San Diego, CA, USA) with the exception of CD14 (Clone #MφP9), EGFR (Clone #EGFR.1), and HLA-DR (Clone #G46-6) antibodies (BD Biosciences, Franklin Lakes, NJ, USA) and the marker of active macrophage 25F9 (Clone #eBio25F9, Thermo Scientific, Waltham, MA, USA).
Preparation of Human mMDSC
Elutriated mononuclear cells and apheresis collections were obtained from healthy donors on NCI IRB-approved NIH protocol 99-CC-0168. Research blood donors provided written informed consent and blood samples were de-identified prior to distribution (NCT00001846) (National Institutes of Health, Bethesda, MD, USA). Peripheral blood mononuclear cells (PBMC) and elutriated monocytes were separated by gradient centrifugation over Histopaque (Sigma-Aldrich, St. Louis, MO, USA) and cultured overnight in RPMI 1640 medium supplemented with 2% FCS (both from Lonza, Walkersville, MD, USA), 2 mM glutamine, and 25 mM HEPES buffer (both from Invitrogen, Carlsbad, CA, USA). Elutriated mononuclear cells or PBMC in suspension were stained with fluorescence-conjugated antibodies against CD14 and HLA-DR. mMDSC represented by CD14bright HLA-DR−/low population was FACS sorted using a FACSAria II (BD Biosciences, Franklin Lakes, NJ, USA) with >95% purity.
In Vitro Stimulation of mMDSC
FACS-purified mMDSC were stimulated with 1 µg/ml PAM3, 3 µg/ml R848 [previously defined to be the optimal concentration to drive mMDSC maturation (11)], and/or 250 ng/ml of IL-6, IL-10, IL-12, TNFα, IFNγ, or M-CSF in RPMI supplemented with 2% FCS. Where indicated, cytokine neutralizing Abs (25 µg/ml), the IκB kinase (IKK) inhibitor Celastrol (1 µM), and/or the Janus kinase1/2 (JAK1/2) inhibitor Ruxolitinib (1 µM) were added throughout the duration of MDSC culture (3–5 days).
Analysis of Surface Marker Expression by mMDSC
Stimulated mMDSC were incubated with Fc Block for 15 min on ice and stained with fluorochrome-conjugated antibodies against 25F9, CD206, and CD163 on ice for 20 min. Cells were washed with PBS/2% BSA followed by Fix & Perm Medium A (Invitrogen, Carlsbad, CA, USA). Cells were washed again, re-suspended in PBS, and analyzed using an LSRFortessa (BD Biosciences, Franklin Lakes, NJ, USA).
Cytotoxicity Assay
FACS-purified mMDSC were cultured with R848, IL-6 plus TNFα, M-CSF, or IFNγ for 5 days. Cells were then collected through scraping, counted, and incubated with A549 tumor cells at a 1:40 ratio in fresh media for 6 h. Samples were trypsinized and stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Life Technologies, Carlsbad, CA, USA) followed by fluorescein-conjugated anti-EGFR and anti-CD14 Ab for 30° on ice. After washing, cells were re-suspended in PBS/2% BSA and analyzed using the LSRFortessa.
ELISAs
Cell supernatants were collected on day 3 and frozen until further use. Immunol 2HB microtiter plates (Thermo Scientific) were coated with anti-cytokine antibodies anti-IL-6 (Clone #6708), anti-IL-10 (Clone #127107), anti-TNFα (Clone #28401), and anti-M-CSF (Clone #21113) (R&D Systems, Minneapolis, MN, USA) and then blocked with PBS/2% BSA. Serially diluted standards and culture supernatants were added to these plates overnight. Plates were incubated with biotinylated anti-cytokine Ab (R&D Systems), followed by phosphatase-streptavidin (BD Biosciences) and K-Gold PNPP Substrate (Neogen Corporation, Lexington, KY, USA). Human IL-12p70 Quantikine, IL-4 Quantikine, and TGFβ1 Quantikine ELISAs were performed based on manufacturer’s instructions (R&D Systems). ELISAs were read using a SpectraMax M5 Microplate Reader and SoftMax Pro Acquisition and Analysis Software (both Molecular Devices, Sunnyvale, CA, USA).
RNA-Seq Analysis
After 4 h stimulation [a duration previously found to be optimal for monitoring changes in gene expression in differentiating mMDSC (11)], stimulated mMDSC were stored in RNA Protect (Qiagen, Frederick, MD, USA). Total RNA was isolated using the RNeasy micro kit (Qiagen) and RNA quality was assessed using an Agilent 2200 TapeStation. mRNA libraries were generated using the Smart-Seq ultra-low input kit (Clontech) and sequenced using a HiSeq2500 sequencer using IlluminaTruSeq v4 chemistry with 125 bp paired-end reads. Sequences were aligned to the human (hg19) reference genome. Genes that were differentially expressed compared to untreated samples were identified using CLC genomics workbench (version 10). Genes that were significantly upregulated (FDR p < 0.01) were imported into Ingenuity Pathway Analysis (Qiagen, version 10). Networks involving genes that interacted with more than two other genes were used to build networks. Accession code in GEO repository: GSE105142.
Statistical Analysis
A two-sided unpaired Student’s t-test was used for analysis, and p-value less than 0.05 was considered statistically significant (GraphPad Software Inc., La Jolla, CA, USA).
Results
R848 Induces mMDSC to Differentiate Into MACinflam
Macrophage has historically been categorized into two subsets: MACinflam and MACsuppress (12). These subsets differ both phenotypically and functionally. While all human macrophages express the 25F9 surface marker, only MACsuppress upregulate the CD163 scavenger receptor and the CD206 C-type mannose receptor (13, 14). Our lab previously demonstrated that the TLR7/8 agonist R848 induced human mMDSC to differentiate into MACinflam, while the TLR2/1 agonist PAM3 supported their preferential generation of MACsuppress (11). To clarify the mechanism underlying the generation of MACinflam, normal healthy volunteers were leukapheresed and mMDSC were isolated by FACS sorting based on the absence of HLA-DR and presence of CD14 (3, 11). As previously documented, mMDSC constitute 0.4 + 0.3% of PBMC in normal donors (11). Consistent with the earlier report, TLR stimulation induced a majority of CD14+/HLA-DR− mMDSC to differentiate into 25F9+ macrophage (Figures 1A,B). Preliminary studies further showed that increasing the duration of culture from 3 to 5 days increased the generation of MACinflam following R848 stimulation without altering the generation of MACsuppress by PAM3 (Figure S1 in Supplementary Material, p < 0.05). In the absence of stimulation, less than 1% of mMDSC survived 5 days in culture (yielding too few cells for further study). By contrast, viability was high in cultures stimulated with R848 or PAM3 (80.2 + 11.3%).
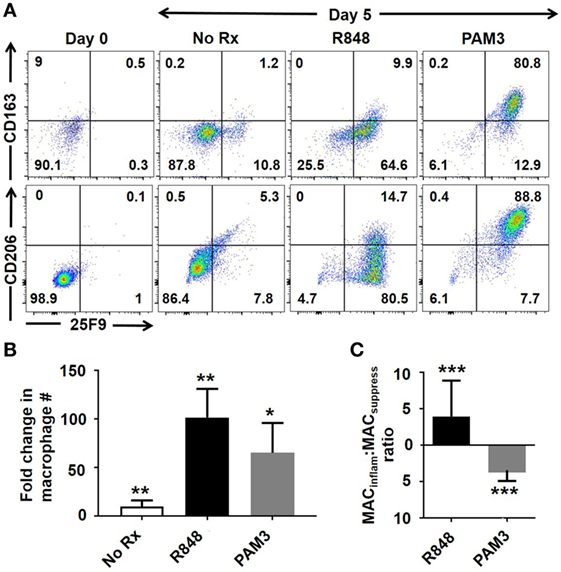
Figure 1. Effect of Toll-like receptor stimulation on mMDSC polarization. FACS-purified human mMDSC were stimulated with R848 (3 µg/ml) or PAM3 (1 µg/ml) for 5 days. (A) Representative dot plots depicting changes in 25F9, CD163, and CD206 expression. (B) Fold change in the number of macrophage present at the end of culture over the baseline of freshly isolated mMDSC (mean ± SD of six independently studied donors/data point). (C) Ratio of CD163+ to CD163− 25F9+ macrophage in the samples described in (B). *p < 0.05, **p < 0.01, ***p < 0.001 versus unstimulated cells.
A majority of the macrophage generated by R848 treatment expressed only 25F9 and thus were phenotypically MACinflam (Figure 1C). This contrasted to PAM3 treated cells that typically expressed CD163 and CD206 in addition 25F9 (Figures 1A,C). On average, R848 treatment generated a four-fold excess of MACinflam compared to MACsuppress, while PAM3 generated fourfold more MACsuppress than MACinflam (Figure 1C), consistent with previous findings (11).
Contribution of Cytokines to R848-Induced Generation of MACinflam
To better understand the factors that influence the generation of MACinflam rather than MACsuppress, the production of cytokines by human mMDSC stimulated with R848 versus PAM3 was compared. Our lab previously used intracytoplasmic cytokine staining to show that mMDSC cultured for 1–3 days with R848 accumulated cells containing IL-6 and IL-12, while those treated with PAM3 accumulated cells containing IL-6 and IL-10 (11). To measure secreted cytokine levels in the surrounding environment, culture supernatants (representing the balance of cytokines produced and metabolized over 3 days) were examined. Levels of IL-6 and IL-10 rose significantly after stimulation with either R848 or PAM3, with concentrations being higher after R848 stimulation (Figure 2). By contrast, the pro-inflammatory cytokines TNFα and IL-12 were significantly elevated only after R848 treatment (Figure 2). Levels of IFNγ, M-CSF, IL-4, and TGFβ1 did not change in response to either TLR agonist. These results suggested that TNFα and/or IL-12 might contribute to the preferential generation of MACinflam mediated by R848.
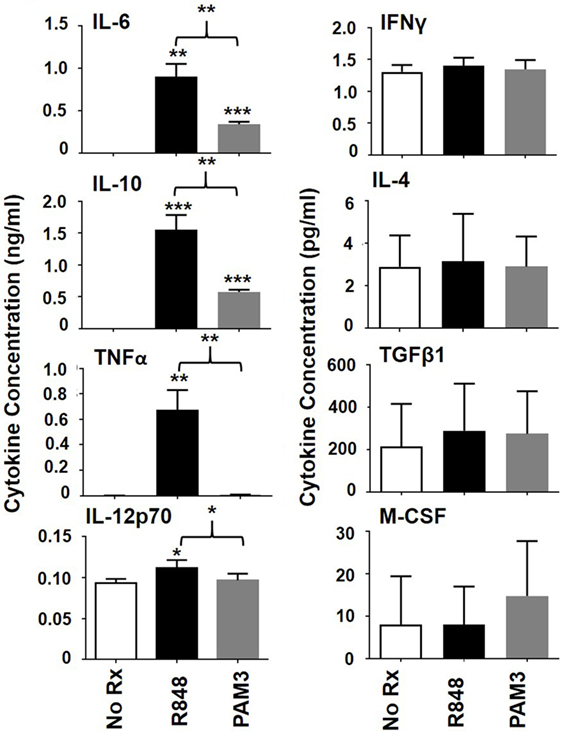
Figure 2. Effect of Toll-like receptor stimulation on cytokine production by mMDSC. FACS-purified mMDSC were stimulated as described in Figure 1. Cytokine and chemokine levels in culture supernatants were determined on day 3 by ELISA (mean ± SD of samples from four to eight independently analyzed donors). *p < 0.05; **p < 0.01, ***p < 0.001 versus unstimulated cells or between PAM3 and R848 stimulated mMDSC.
Cytokine neutralization experiments were conducted to examine these possibilities. Blocking TNFα resulted in a dramatic reduction in the generation of MACinflam but had no effect on the number of MACsuppress. This finding suggests that TNFα may play an important role in determining the type of macrophage generated following TLR stimulation of mMDSC (Figure 3). Blocking IL-6 or IL-10 reduced the generation of both MACsuppress and MACinflam (p < 0.01), consistent with those cytokines contributing to the general process by which mMDSC mature into macrophage (Figure 3). Blocking IL-12 led to a modest reduction in MACinflam, while having no effect on MACsuppress. The addition of neutralizing Ab against cytokines that were not detected in stimulated cultures (such as IFNγ) had no effect on macrophage generation (Figure 3).
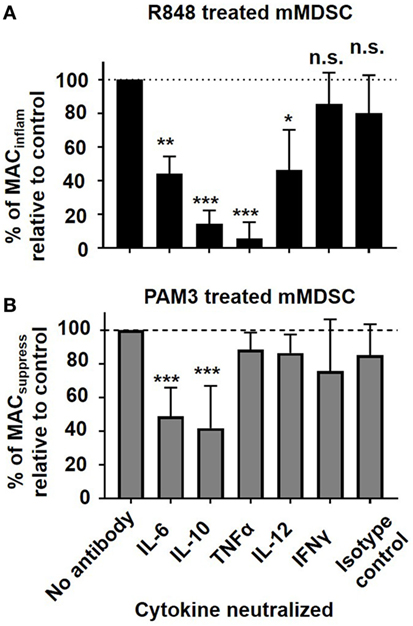
Figure 3. Effect of cytokine neutralization on Toll-like receptor-induced macrophage generation. FACS-purified mMDSC were stimulated with R848 or PAM3 for 3 days in the presence of 25 µg/ml neutralizing anti-cytokine or isotype control antibodies. Data show the change in frequency of (A) MACinflam generated by R848 versus (B) MACsuppress generated by PAM3 (mean ± SD of samples from 4 to 11 independently analyzed donors/group). *p < 0.05; **p < 0.01, ***p < 0.001 versus isotype control antibody group.
Cytokines Can Directly Induce mMDSC Differentiation
To further examine whether TNFα and to a lesser extent IL-6, IL-10, and/or IL-12 could influence the differentiation of mMDSC, their effect was compared to that of R848. The addition of IL-6 and/or IL-10 significantly increased the generation of macrophages from mMDSC but did not preferentially support the generation of MACinflam (Figure 4). By comparison, TNFα mirrored the ability of R848 to generate MACinflam rather than MACsuppress (Figure 4B). IL-12 had no significant effect on the maturation of mMDSC into MACinflam (Figures 4A,C).
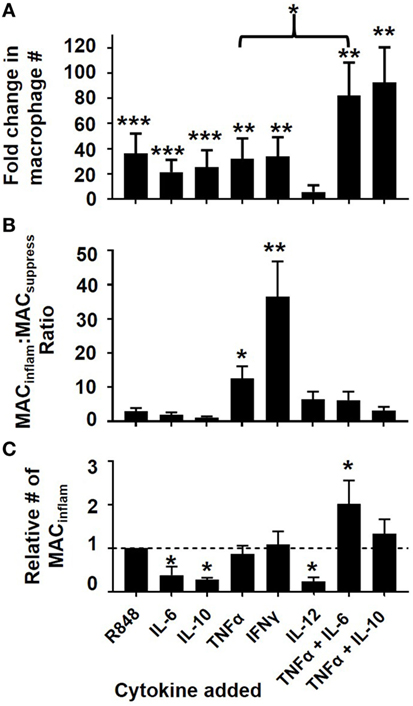
Figure 4. Effect of cytokines on mMDSC polarization. FACS-purified mMDSC were stimulated with 3 µg/ml of R848 or various cytokines (250 ng/ml) for 5 days. (A) Fold change in the fraction of macrophage present at the end of culture (mean ± SD of five to eight independently studied donors/data point), (B) ratio of CD163+ to CD163− macrophage present at the end of culture, (C) relative number of MACinflam present in culture versus a baseline of R848 for each donor. *p < 0.05, **p < 0.01, ***p < 0.001 versus unstimulated cells.
Based on the observation that IL-6 and IL-10 supported the general process of mMDSC differentiation, the effect of co-administering them with TNFα was examined. The combination of TNFα plus IL-6 generated a greater number of MACinflam than any other treatment (p < 0.05; Figure 4C). IFNγ was also evaluated in these studies. While IFNγ played no role in R848-driven mMDSC maturation (Figures 2 and 3), previous reports indicated that IFNγ could induce classical monocytes to differentiate into MACinflam (15, 16). Current results show that IFNγ also supports the generation of MACinflam from mMDSC (Figure 4). These findings suggest that multiple distinct stimuli can play a role in the generation of MACinflam. To examine that possibility, subsequent experiments focusing on the mechanism underlying the generation of 25F9+CD163− macrophage compared the effect of TNFα plus IL-6 to IFNγ as well as to R848.
Functional Activity of MACinflam Generated From mMDSC
We previously established that MACinflam but not MACsuppress could lyse A549 tumor cells. The functional activity of MACinflam generated by treating mMDSC with R848, IL-6 plus TNFα, or IFNγ was therefore evaluated using this assay. As expected, MACinflam generated by R848 treatment lysed tumor targets (p < 0.05; Figure 5). MACinflam produced in cultures containing IL-6 plus TNFα or IFNγ also mediated significant tumor cell lysis (Figure 5). There was no statistically significant difference in the activity of MACinflam generated by any of these treatments, suggesting the MACinflam generated by distinct stimuli were not only phenotypically alike but also shared functional characteristics.
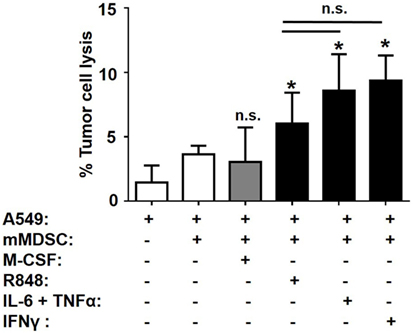
Figure 5. Tumoricidal activity of MACinflam. Purified mMDSC were stimulated with 3 µg/ml R848 or 250 ng/ml IL-6, TNFα, IFNγ, and/or M-CSF for 5 days. Their ability to lyse A549 tumor targets during 6 h of incubation at an E:T ratio of 40:1 is shown. Data shown mean ± SD of four to eight independently analyzed donors per group. *p < 0.05 versus unstimulated mMDSC.
Regulatory Networks Underlying the Differentiation of mMDSC Into MACinflam
The above findings established that mMDSC treated with R848, IFNγ, or the combination of TNFα plus IL-6 matured into MACinflam based on both phenotypic and functional metrics. Previous studies examined the gene expression signatures of mMDSC stimulated with PAM3 versus R848 (9). While differences in mRNA levels were detected and the optimal timepoint for analyzing shifts in gene expression identified (4 h) that study was unable to identify the regulatory networks underlying the generation of MACinflam (11). We reasoned that comparing the effects of R848, IFNγ, and TNFα plus IL-6 treatments might clarify whether there was a common pathway underlying the differentiation of mMDSC into MACinflam. To test that possibility, mRNA libraries generated from mMDSC cultured for 4 h with each stimulant were sequenced. Significantly upregulated genes (p < 0.01) that formed network connections with at least two other genes were identified. TNFα plus IL-6 upregulated 820 genes, whereas R848 upregulated ~2.3 times that many (Figure 6A). Consistent with the observation that blocking either IL-6 or TNFα significantly inhibited R848-driven mMDSC differentiation, 82% of the genes upregulated by IL-6 plus TNFα were also upregulated by R848 (Figure 6A). By contrast, only 37% of the genes upregulated by IFNγ were shared with R848 (Figure 6A).
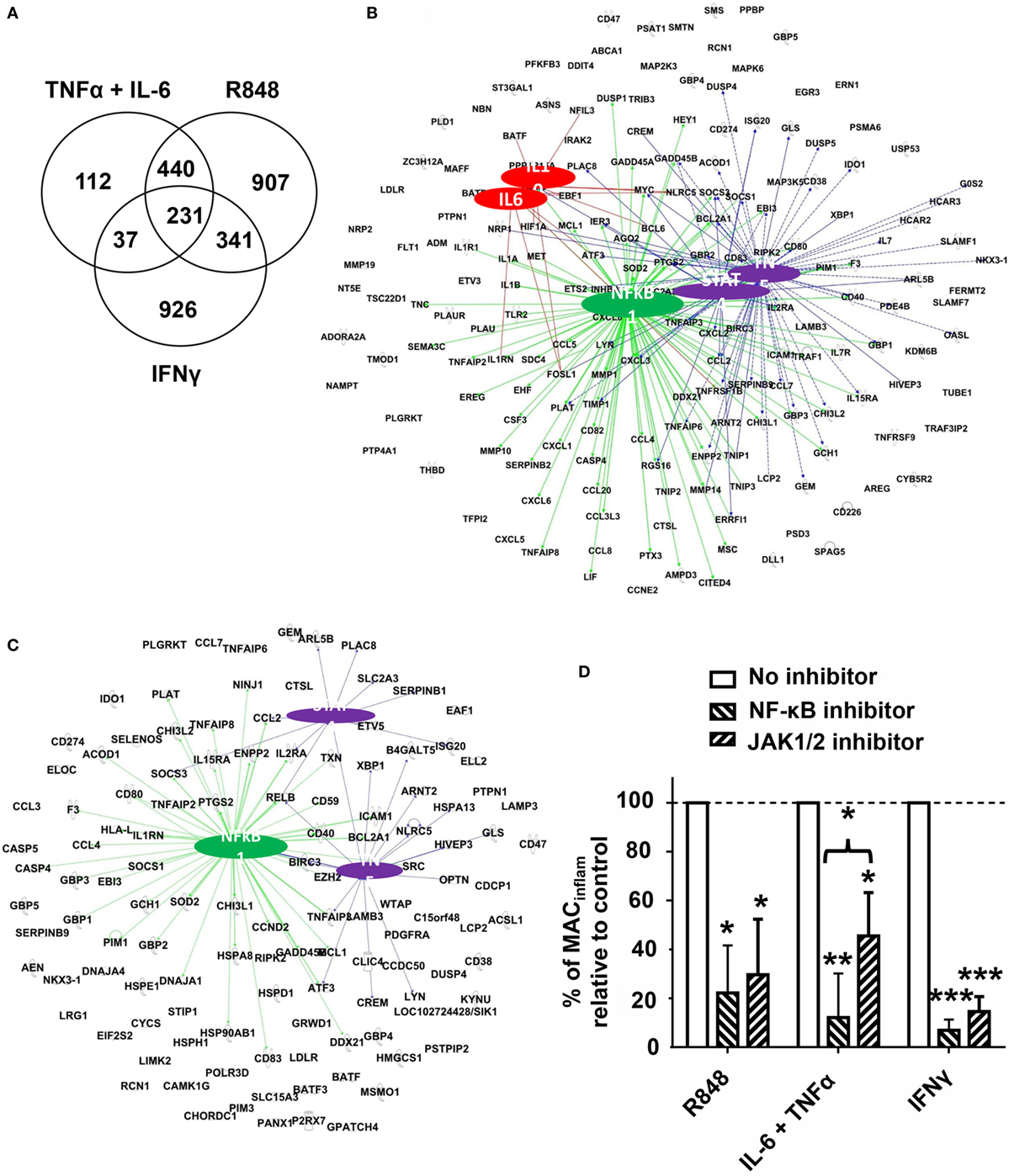
Figure 6. Regulatory networks associated with differentiation of mMDSC into MACinflam. Purified mMDSC were stimulated with R848, IFNγ, or the combination of TNFα plus IL-6 for 4 h. Changes in gene expression were detected using RNA-Seq by comparison to unstimulated samples from the same donors. (A) Venn diagram showing the number of genes upregulated by each treatment. (B) Genes activated by both R848 and IL-6 plus TNFα whose regulatory interactions could be mapped by IPA. Colored lines identify regulatory gene interactions involving NF-κB (green), TNFα or STAT4 (purple), and IL-6 or IL-10 (red). (C) Genes activated by all three methods of MACinflam generation (R848, IFNγ, and TNFα plus IL-6). Colored lines identify regulatory gene interactions involving NF-κB (green) and TNFα or STAT4 (purple). (D) Purified mMDSC were stimulated with R848, IFNγ, or TNFα plus IL-6 for 5 days in the continuous presence of the IκB kinase inhibitor Celastrol or the Janus kinase1/2 (JAK1/2) inhibitor Ruxolitinib (mean ± SD of three to four independently studied donors/data point). Inhibitors were added on day 0. *p < 0.05, **p < 0.01 versus unstimulated cells.
Ingenuity Pathway Analysis was used to identify the regulatory networks involved in the activation of these shared genes. Initial studies compared R848 with IL-6 plus TNFα, given the close relationship already established between those two forms of mMDSC activation. Consistent with results from the cytokine neutralization studies (Figure 3), TNFα and to a lesser extent IL-6 and IL-10 were found to be regulatory hubs associated with the maturation of mMDSC into MACinflam (Figure 6). TNFα influenced the expression of a large set of genes associated with the generation of MACinflam that was largely distinct from those regulated by IL-6 or IL-10 (Figure 6B). IPA also predicted that regulatory networks mediated via the NF-κB complex and STAT4 would be relevant to the generation of MACinflam (Figure 6B). To determine whether the same regulatory pathways contributed to the generation of MACinflam mediated by an unrelated stimulant, we performed IPA analysis of the genes upregulated by IFNγ as well R848 and IL-6 plus TNFα. This analysis confirmed that NF-κB, STAT4, and TNFα were major hubs regulating the differentiation of mMDSC into MACinflam (Figure 6C). Of interest, mMDSC stimulated with IFNγ did not upregulate expression of IL-6/IL-10 but instead triggered genes encoding IFNγ-induced regulatory factors (Figure 6C and data not shown).
To validate the IPA prediction that the transcription factors NF-κB and STAT4 drove MACinflam differentiation and inhibitors of IKK (an upstream regulator of the NF-κB complex) and JAK1/2 (an upstream regulator of STAT4) were added throughout the period of mMDSC culture. Inhibition of either the NF-κB complex or JAK-STAT4 axis blocked the polarizing activity of R848, IFNγ, and IL-6 plus TNFα by 55–90%, consistent with the conclusion that these pathways are essential for the generation of MACinflam from mMDSC (Figure 6D).
Discussion
Myeloid-derived suppressor cells contribute to the immunosuppressive microenvironment that protects cancers from elimination by tumoricidal NK and T cells (5). In animal models, survival is significantly prolonged by interventions that block the generation, expansion, and/or trafficking of MDSC to the tumor bed (17–19). Epidemiologic studies show that the presence of large numbers of MDSC correlates with a worse prognosis and poorer response to therapy in cancer patients (20–22). Targeting MDSC has thus become an important component of cancer immunotherapy as they are highly immunosuppressive and accumulate in the tumor microenvironment (2, 4, 5, 19, 23). Clinical trials designed to eliminate mMDSC and/or induce them to differentiation into tumoricidal macrophages are underway (17–19). In support of that goal, this work examined the mechanisms regulating the maturation of human mMDSC into MACinflam.
Previous studies showed that TLR7/8 agonists, including R848, preferentially induced mMDSC isolated from healthy donors or cancer patients to mature into tumoricidal macrophage with the ability to produce pro-inflammatory cytokines (11). Studies in mice verified that intratumoral delivery of TLR7 agonists facilitated the elimination of large tumors, an effect accompanied by the differentiation of mMDSC into MACinflam (9, 10, 24). Yet, not all TLR agonists have this effect on human mMDSCs: the TLR2/1 agonist PAM3 induces them to differentiate into MACsuppress that interfere with tumor elimination (11). Yet mMDSC differentiation has a stochastic component in that a minority of the macrophage generated by R848 were suppressive, while a minority of those generated by PAM3 were inflammatory (Figure 1) (11). This work sought to clarify the processes that support the generation of MACinflam with the goal of improving their generation for anti-tumor therapy.
As previously reported, (1) R848 preferentially supported the generation of 25F9+CD163− MACinflam, while PAM3 supported the generation primarily of 25F9+CD163+ MACsuppress (Figure 1) and (2) R848 elicited the production of a different pattern of cytokines than PAM3 (Figure 2) (11). Expanding on those findings, this work examined additional cytokines and found that TNFα production was significantly elevated in R848 but not PAM3 stimulated cultures (Figure 2). Given the limited number of cytokines detected in R848-treated mMDSC cultures, we explored whether they might play a role in the generation of MACinflam. Consistent with that possibility, neutralizing TNFα (and to a much lesser extent IL-12) significantly reduced the generation of MACinflam by R848, while leaving the generation of MACsuppress by PAM3 intact (Figure 3). Neutralizing IL-6 or IL-10 blocked the development of both MACinflam and MACsuppress, indicating that those cytokines contribute to the general process of mMDSC maturation. TNFα, IL-6, and IL-10 (but not IL-12) also supported the differentiation of freshly isolated mMDSC into macrophages yet only TNFα selectively supported the generation of MACinflam (Figure 4). Of interest, although IFNγ was not present in TLR-stimulated cultures and had no effect on R848-dependent generation of MACinflam, that cytokine was able to stimulate human mMDSC to differentiate into MACinflam. This finding builds on earlier studies showing that IFNγ supports the generation of antigen presenting cells from classical monocytes (15, 16).
Several earlier studies examined the effect of TNFα and IFNγ on murine rather than human MDSC. While the accumulation of MDSC in inflammatory states was initially associated with increased TNFα and IL-6 levels (25–28), more recent data suggest that increased TNFα expression in the tumor microenvironment reduces MDSC infiltration and supports tumor regression (29). Similarly, IFNγ was shown to augment the suppressive activity of murine MDSC by triggering nitric oxide production, a mediator used by MDSCs to suppress T cell activity (30, 31). However, human myeloid cells differ from mice in terms of their ability to produce inducible nitric oxide synthase and respond to some stimulants (32). Given the importance of mMDSC in tumor immunology, this work examined how the differentiation of these cells was regulated in humans. As seen in Figure 4, our findings indicate that TNFα and IFNγ induce human mMDSC to differentiate into MACinflam.
Consistent with the cytokine neutralization data, adding IL-6 or IL-10 to cultures of human mMDSC increased total macrophage yield but did not selectively generate MACinflam rather than MACsuppress (Figures 4A,B). By contrast, TNFα and IFNγ induced mMDSC to preferentially differentiate into MACinflam. The yield and relative frequency of MACinflam was maximized by treating mMDSC with a combination of TNFα plus IL-6 (Figure 4C). Coupled with the observation that neutralizing TNFα or IL-6 significantly inhibited the activity of R848 (Figure 3), these findings identify TNFα as a central driver of R848-induced mMDSC polarization and suggest that the general process by which mMDSC differentiate into macrophage is supported by IL-6 and perhaps IL-10. Inflammatory macrophages contribute to cancer immunotherapy by killing tumor targets via the secretion of various mediators including TNFα (33, 34). Thus, the functional activity of the macrophages identified as being MACinflam based on phenotypic markers was verified by their ability to lyse tumor targets (Figure 5). Consistent with earlier findings, R848-treated mMDSCs lysed tumor targets as did cells cultured with IFNγ or the combination of IL-6 plus TNFα (Figure 5).
RNA-Seq was used to identify the genes and regulatory networks critical to the differentiation of mMDSC to MACinflam. Analysis focused on those genes whose expression was significantly increased by all three forms of stimulation: R848, TNFα plus IL-6, and IFNγ. 82% of the genes upregulated by the cytokine combination were also activated by R848 as opposed to 37% common genes between IFNγ and R848 (Figure 6A). IPA analysis revealed that a majority of the genes whose expression was increased by all three stimulants were linked via networks involving TNFα, NF-κB, and the STAT4 pathways. Inhibiting either NF-κB or STAT4 transcription factors significantly reduced the differentiation of human mMDSC into MACinflam (Figure 6D). While consistent with evidence that NF-κB influences the differentiation of human monocytes, these findings are at odds with studies in mice showing that NF-κB activation causes MDSC to accumulate at sites of inflammation (supporting the importance of evaluating the activity of human mMDSC) (35, 36). Less is known of the role of STAT4 in the differentiation of myeloid cells. Originally identified as a transcription factor supporting the maturation of Th1 and NK cells, it is expressed by activated blood monocytes (37, 38). Unfortunately, no STAT4-specific inhibitor has been described, so Ruxolitinib was used in this work to monitor inhibition. Ruxolitinib blocks signal transduction mediated by multiple STATs (39). As STAT4 was the only member of the STAT family significantly upregulated by R848, IFNγ, and TNFα plus IL-6, this combination of findings suggest that STAT4 plays a role in the differentiation of human mMDSC into MACinflam. Importantly, one of the four targets jointly regulated by NF-κB and STAT4 was inhibitor of STAT3 (SOCS3, Figure 6C) (40). STAT3 has been implicated in the maintenance and function of MDSCs in cancer patients (41, 42). Our findings are consistent with the possibility that inflammatory stimuli drive mMDSC to differentiate into MACinflam by limiting STAT3 activity, a conclusion supportive of further development of STAT3 inhibitors for clinical use. However, the role of STAT3 was not analyzed in this study given that currently available inhibitors lack specificity and RNA interference is accompanied by technical challenges related to primary myeloid cultures (43, 44). By contrast, upregulation of IL-6 and IL-10 was present only when mMDSC were stimulated with R848 or IL-6 plus TNFα, while expression of multiple IRFs was found only in IFNγ stimulated cultures (Figures 6B,C and data not shown). These findings suggest that IL-6, IL-10, and IRFs can support but are not central to the generation of MACinflam.
The therapeutic utility of R848 is limited by the development of lymphopenia (45, 46). Systemic administration of TNFα can lead to hypotension and hepatotoxicity while IL-6 is known to be present in the tumor microenvironment, where it supports the survival and proliferation of cancer cells (47, 48). Our findings indicate that the behavior of IL-6 is altered when combined with TNFα and that it augments TNFα-mediated conversion of mMDSC into tumoricidal MACinflam (Figure 5). Although IFNγ is a potent anti-tumoral agent, it is reported to support tumor growth by increasing the proliferative capacity of the cancer cells and upregulating immune checkpoint inhibitors via a negative feedback loop (49, 50). Current findings suggest that tumor growth might be inhibited by targeting the immunosuppressive milieu through local delivery of R848, IFNγ, or TNFα.
Author Contributions
DB and DT performed the experiments and analyzed results; DB and DK designed the research, analyzed results, and wrote the paper; DK supervised the research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Kathleen Noer and Roberta Matthai for their help in fluorescence-activated cell sorter purification of human mMDSCs. This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government. The funders had no role in study design, data collection or analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00608/full#supplementary-material.
Figure S1. Effect of longer incubation period on the percentage of MACinflam and MACsuppress. FACS-purified human monocytic myeloid-derived suppressor cells were stimulated with R848 (3 µg/ml) or PAM3 (1 µg/ml) for 3–5 days. The percentage of cells bearing MACinflam (25F9+, CD206−) versus MACsuppress (25F9+, CD206+) phenotype is shown (mean ± SD of three independently studied donors per group). *p < 0.05.
References
1. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
2. Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111:4233–44. doi:10.1182/blood-2007-07-099226
3. Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside TL. Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples. J Immunol Methods (2012) 381:14–22. doi:10.1016/j.jim.2012.04.004
4. Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol (2013) 14:211–20. doi:10.1038/ni.2526
5. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol (2016) 37:208–20. doi:10.1016/j.it.2016.01.004
6. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology (2013) 138:105–15. doi:10.1111/imm.12036
7. Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature (1997) 388:394–7. doi:10.1038/41131
8. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4:499–511. doi:10.1038/nri1391
9. Shirota Y, Shirota H, Klinman DM. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J Immunol (2012) 188:1592–9. doi:10.4049/jimmunol.1101304
10. Zhao BG, Vasilakos JP, Tross D, Smirnov D, Klinman DM. Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer (2014) 2:12. doi:10.1186/2051-1426-2-12
11. Wang J, Shirota Y, Bayik D, Shirota H, Tross D, Gulley JL, et al. Effect of TLR agonists on the differentiation and function of human monocytic myeloid-derived suppressor cells. J Immunol (2015) 194:4215–21. doi:10.4049/jimmunol.1402004
12. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6:13. doi:10.12703/P6-13
13. Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol (2000) 67:97–103. doi:10.1002/jlb.67.1.97
14. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm (2015) 2015:816460. doi:10.1155/2015/816460
15. Becker S. Interferons as modulators of human monocyte-macrophage differentiation. I. Interferon-gamma increases HLA-DR expression and inhibits phagocytosis of zymosan. J Immunol (1984) 132:1249–54.
16. Geng YJ, Hansson GK. Interferon-gamma inhibits scavenger receptor expression and foam cell formation in human monocyte-derived macrophages. J Clin Invest (1992) 89:1322–30. doi:10.1172/JCI115718
17. Albeituni SH, Ding C, Yan J. Hampering immune suppressors: therapeutic targeting of myeloid-derived suppressor cells in cancer. Cancer J (2013) 19:490–501. doi:10.1097/PPO.0000000000000006
18. Wesolowski R, Markowitz J, Carson WE III. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi:10.1186/2051-1426-1-10
19. Draghiciu O, Lubbers J, Nijman HW, Daemen T. Myeloid derived suppressor cells – an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology (2015) 4:e954829. doi:10.4161/21624011.2014.954829
20. Montero AJ, Diaz-Montero CM, Kyriakopoulos CE, Bronte V, Mandruzzato S. Myeloid-derived suppressor cells in cancer patients: a clinical perspective. J Immunother (2012) 35:107–15. doi:10.1097/CJI.0b013e318242169f
21. Idorn M, Kollgaard T, Kongsted P, Sengelov L, Thor Straten P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol Immunother (2014) 63:1177–87. doi:10.1007/s00262-014-1591-2
22. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63:247–57. doi:10.1007/s00262-013-1508-5
23. Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol (2009) 182:4499–506. doi:10.4049/jimmunol.0802740
24. Lee M, Park CS, Lee YR, Im SA, Song S, Lee CK. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch Pharm Res (2014) 37:1234–40. doi:10.1007/s12272-014-0379-4
25. Sumida K, Wakita D, Narita Y, Masuko K, Terada S, Watanabe K, et al. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur J Immunol (2012) 42:2060–72. doi:10.1002/eji.201142335
26. Wu CT, Hsieh CC, Lin CC, Chen WC, Hong JH, Chen MF. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J Mol Med (Berl) (2012) 90:1343–55. doi:10.1007/s00109-012-0916-x
27. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest (2012) 122:4094–104. doi:10.1172/JCI64115
28. Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity (2013) 38:541–54. doi:10.1016/j.immuni.2013.02.007
29. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-alpha promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol (2014) 192:1320–31. doi:10.4049/jimmunol.1203195
30. Greifenberg V, Ribechini E, Rossner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol (2009) 39:2865–76. doi:10.1002/eji.200939486
31. Medina-Echeverz J, Haile LA, Zhao F, Gamrekelashvili J, Ma C, Metais JY, et al. IFN-gamma regulates survival and function of tumor-induced CD11b+ Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur J Immunol (2014) 44:2457–67. doi:10.1002/eji.201444497
32. Rehli M. Of mice and men: species variations of toll-like receptor expression. Trends Immunol (2002) 23:375–8. doi:10.1016/S1471-4906(02)02259-7
33. Keller R, Keist R, Wechsler A, Leist TP, Van Der Meide PH. Mechanisms of macrophage-mediated tumor cell killing: a comparative analysis of the roles of reactive nitrogen intermediates and tumor necrosis factor. Int J Cancer (1990) 46:682–6. doi:10.1002/ijc.2910460422
34. Howard AD, Erickson KL. The induction and augmentation of macrophage tumoricidal responses by platelet-activating factor. Cell Immunol (1995) 164:105–12. doi:10.1006/cimm.1995.1148
35. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell (2008) 14:408–19. doi:10.1016/j.ccr.2008.10.011
36. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol (2014) 5:614. doi:10.3389/fimmu.2014.00614
37. Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, et al. STAT4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J Immunol (2000) 164:4659–64. doi:10.4049/jimmunol.164.9.4659
38. Fukao T, Frucht DM, Yap G, Gadina M, O’shea JJ, Koyasu S. Inducible expression of STAT4 in dendritic cells and macrophages and its critical role in innate and adaptive immune responses. J Immunol (2001) 166:4446–55. doi:10.4049/jimmunol.166.7.4446
39. Bottos A, Gotthardt D, Gill JW, Gattelli A, Frei A, Tzankov A, et al. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat Commun (2016) 7:12258. doi:10.1038/ncomms12258
40. Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol (2014) 5:58. doi:10.3389/fimmu.2014.00058
41. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123:1580–9. doi:10.1172/JCI60083
42. Bu LL, Yu GT, Deng WW, Mao L, Liu JF, Ma SR, et al. Targeting STAT3 signaling reduces immunosuppressive myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology (2016) 5:e1130206. doi:10.1080/2162402X.2015.1130206
43. Zhang X, Edwards JP, Mosser DM. The expression of exogenous genes in macrophages: obstacles and opportunities. Methods Mol Biol (2009) 531:123–43. doi:10.1007/978-1-59745-396-7_9
44. Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem Biol (2016) 11:308–18. doi:10.1021/acschembio.5b00945
45. Engel AL, Holt GE, Lu H. The pharmacokinetics of toll-like receptor agonists and the impact on the immune system. Expert Rev Clin Pharmacol (2011) 4:275–89. doi:10.1586/ecp.11.5
46. Perkins H, Khodai T, Mechiche H, Colman P, Burden F, Laxton C, et al. Therapy with TLR7 agonists induces lymphopenia: correlating pharmacology to mechanism in a mouse model. J Clin Immunol (2012) 32:1082–92. doi:10.1007/s10875-012-9687-y
47. Roberts NJ, Zhou S, Diaz LA Jr, Holdhoff M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget (2011) 2:739–51. doi:10.18632/oncotarget.344
48. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol (2014) 26:38–47. doi:10.1016/j.smim.2014.01.008
49. Zaidi MR, Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res (2011) 17:6118–24. doi:10.1158/1078-0432.CCR-11-0482
Keywords: myeloid-derived suppressor cells, inflammatory macrophage, TNFα, STAT4, NF-κB, IFNγ
Citation: Bayik D, Tross D and Klinman DM (2018) Factors Influencing the Differentiation of Human Monocytic Myeloid-Derived Suppressor Cells Into Inflammatory Macrophages. Front. Immunol. 9:608. doi: 10.3389/fimmu.2018.00608
Received: 12 January 2018; Accepted: 12 March 2018;
Published: 26 March 2018
Edited by:
Rudolf Lucas, Augusta University, United StatesReviewed by:
Christine E. Cutucache, University of Nebraska Omaha, United StatesNathan Karin, Technion – Israel Institute of Technology, Israel
Rachael Zoe Murray, Queensland University of Technology, Australia
Copyright: © 2018 Bayik, Tross and Klinman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dennis M. Klinman, a2xpbm1hbmRAbWFpbC5uaWguZ292