 Chao Niu1,2
Chao Niu1,2 Min Li2
Min Li2 Shan Zhu1Yongchong Chen2Lei Zhou2Dongsheng Xu2Wei Li2Jiuwei Cui2*Yongjun Liu1,3*
Shan Zhu1Yongchong Chen2Lei Zhou2Dongsheng Xu2Wei Li2Jiuwei Cui2*Yongjun Liu1,3* Jingtao Chen1*
Jingtao Chen1*
- 1Department of Translational Medicine, The First Hospital of Jilin University, Changchun, China
- 2Department of Cancer Center, The First Hospital of Jilin University, Changchun, China
- 3Sanofi Research and Development, Cambridge, MA, United States
Gamma delta (γδ) T cells, which possess potent cytotoxicity against a wide range of cancer cells, have become a potential avenue for adoptive immunotherapy. Decitabine (DAC) has been reported to enhance the immunogenicity of tumor cells, thereby reinstating endogenous immune recognition and tumor lysis. However, DAC has also been demonstrated to have direct effects on immune cells. In this study, we report that DAC inhibits γδ T cell proliferation. In addition, DAC increases the number of KIR2DL2/3-positive γδ T cells, which are less cytotoxic than the KIR2DL2/3-negative γδ T cells. We found that DAC upregulated KIR2DL2/3 expression in KIR2DL2/3-negative γδ T cells by inhibiting KIR2DL2/3 promoter methylation, which enhances the binding of KIR2DL2/3 promoter to Sp-1 and activates KIR2DL2/3 gene expression. Our data demonstrated that DAC can inhibit the function of human γδ T cells at both cellular and molecular levels, which confirms and extrapolates the results of previous studies showing that DAC can negatively regulate the function of NK cells and αβ T cells of the immune system.
Introduction
Over the last decade, adoptive cell immunotherapy using gamma delta (γδ) T cells, which can secrete abundant cytokines and exert potent cytotoxicity against different cancer cells, including solid tumor cells (1–3) and hematological tumor cells, has shown high therapeutic potential in cancer treatment (4, 5). γδ T cells are a unique subset of lymphocytes that express T cell receptors composed of γ and δ chains. Although γδ T cells constitute a very small percentage of human lymphocytes, they are widely distributed in the peripheral blood and mucosal tissues (6, 7). γδ T cells rapidly recognize exogenous pathogens and endogenous stress-induced ligands in a major histocompatibility complex (MHC)-unrestricted manner and initiate adaptive immunity, thereby acting as a first line of immune defense (8–10). Many clinical trials have been conducted in an effort to harness the abilities of γδ T cells and test the efficacy of γδ T cells in adoptive immunotherapy (11–13). γδ T cells have become a potential avenue for adoptive cell immunotherapy (14, 15).
The epigenetic drug decitabine (DAC) is approved for the treatment of myelodysplastic syndrome (MDS) and has been introduced for the treatment of acute myeloid leukemia (AML). In the past two decades, the clinical benefits of DAC in solid tumors have been reported (16, 17). DAC displays a dual mechanism of action depending on the dose, i.e., reactivation of silenced genes as well as differentiation at low doses, and cytotoxicity at high doses (18–20). DAC is a DNA methyltransferase inhibitor that induces the re-expression of silenced genes such as tumor suppressor and cell cycle-regulating genes, thereby resulting in increased apoptosis or decreased proliferation of tumor cells (21–23). Recently, preclinical studies have indicated that DAC enhanced the expression of cancer-testis antigens, the death receptor FAS, MHC class I molecules, and co-stimulatory molecules, and boosted the cytolytic activity and proliferation of T cells (24–28). DAC plays an immune stimulatory role in cancer therapy by sensitizing tumor cells to immune responses. Therefore, recent clinical studies are utilizing DAC prior to or in combination with immune therapies to improve clinical outcomes (29). However, DAC can inhibit the antitumor functions of immune cells, including lysis of tumors by NK cells, naïve αβ T cell proliferation, and increasing the number of regulatory T cells (20, 30, 31).
In this study, we show that DAC inhibits γδ T cell proliferation. At the molecular levels, DAC increased the number of KIR2DL2/3-positive γδ T cells, which are less cytotoxic than KIR2DL2/3-negative γδ T cells. We found that DAC upregulated KIR2DL2/3 expression in KIR2DL2/3-negative γδ T cells by inhibiting KIR2DL2/3 promoter methylation, which enhances the binding of KIR2DL2/3 promoter to Sp-1 and activates KIR2DL2/3 gene expression. Therefore, we suggest that DAC may represent a double edged sword in the immune system that stimulates antitumor immunity by promoting tumor antigen presentation and costimulation, and inhibits antitumor immunity by blocking the function of NK cells, αβ T cells, and γδ T cells.
Materials and Methods
Patient Enrollment
Seven newly diagnosed MDS and AML patients who did not receive radiation therapy and chemotherapy before blood collection were enrolled in this study, all of whom provided written informed consent for the use of biospecimens for research purposes in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of the First Hospital of Jilin University and carried out in accordance with the approved guideline “Use of experimental animals and human subjects.” The patient information is shown in Table 1.
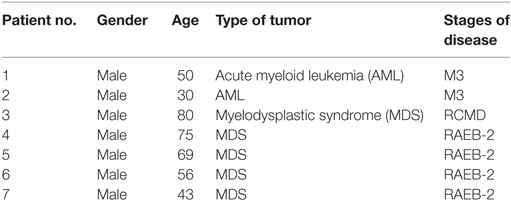
Table 1. Patient clinical characteristics.
Protections for Lab Personnel
To protect our laboratory personnel from infections, we did not collect blood samples from patients infected with HIV, HBV, or HCV. Blood sample processing was performed in a biosafety cabinet. Operators had to wear sterile gloves and masks while handling blood samples.
Pharmacological Agents
DAC was kindly provided by China Tai Tianqing Pharmaceutical Group Co., Ltd. (CTTQ) and dissolved in saline to a concentration of 10 mM. Zoledronate, which serves as an antigen to stimulate and expand γδ T cells, was purchased from Jilin Province Xidian Pharmaceutical Sci-Tech Development Co., Ltd.
Cell Culture
The human MDS cell line, SKM-1, Burkitt’s lymphoma cell line, Raji (NK-resistant), and AML cell line, K562 (NK-sensitive) were kindly provided by Dr. Jifan Hu at Stanford University Medical School, Palo Alto Veterans Institute for Research, Palo Alto, CA, USA. All cell lines were cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C in a humidified 5% CO2 incubator.
Expansion of γδ T Cells
γδ T cells were expanded as previously reported (32, 33). Briefly, heparinized peripheral blood samples were obtained from seven MDS and AML patients. Blood samples were centrifuged at 1,800 × g for 10 min, and the plasma was transferred to new tubes. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll (Nycomed Pharma AS, Oslo, Norway) at 800 × g for 30 min. To expand γδ T cells, PBMCs were cultured in AIM-V medium CTS™ (Gibco) with 1 µM zoledronate, 5% auto-plasma, and 500 U/mL human IL-2 (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) for 9 days. Fresh complete medium with IL-2 supplement (500 U/mL) was added every 2 or 3 days. The cultured cells were expanded γδ T cells and treated with DAC without sorting. KIR2DL2/3+ and KIR2DL2/3− γδ T cells were sorted from these cultured cells using a flexible BD Influx™ cell sorter (BD Biosciences, San Jose, CA, USA).
Proliferation Assay
Expanded γδ T cells (1 × 106 cells/mL) were incubated and stained with 1 µM carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR, USA) according to the manufacturer’s recommended protocol. The labeled cells were then washed, suspended (1 × 106 cells/mL), and incubated with increasing doses (0, 0.25, 0.5, 1, 2, 3, 4, and 5 µM) of DAC at 37°C in 5% CO2. After incubation for 5 days, the cells were collected and stained with Vγ9-PE (BD Biosciences). After staining, the cells were analyzed using a BD FACS Calibur (BD Biosciences) with Cell Quest Pro software, and the final analysis was performed using FlowJo software (Tree Star, Ashland, OR, USA).
Cell Viability Assay
Expanded γδ T cells (1 × 106 cells/mL) were incubated with various concentrations (0, 0.25, 0.5, 1, 2, 3, 4, and 5 µM) of DAC for 48 h. The proportions of living, dead, and apoptotic cells were determined using an Annexin V and 7-AAD staining kit (eBioscience, San Diego, CA, USA) according to the manufacturer’s protocol. After staining, the cells were analyzed using the BD FACS Calibur.
Cell Cycle Assay
After treatment with various concentrations of DAC, expanded γδ T cells were fixed with cold 70% ethanol overnight at −20°C, followed by washing once with cold phosphate-buffered saline (PBS). The fixed cells were treated with RNase and stained with propidium iodide (Sigma-Aldrich, St. Louis, MO, USA). The stained cells were analyzed by flow cytometry using ModFit LT software (Verity Software House, Topsham, ME, USA).
Surface Marker Detection
Expanded γδ T cells treated with 0.5 µM DAC for 48 h were stained with DNAM-1-PE (559789), NKG2D-APC (558071), Vγ9-FITC (555732), KIR2DL2/3 (CD158b)-PE (559785), CD3-PerCP (347344), KIR2DL1 (CD158a)-PE (556063) (BD Biosciences), CD279-APC (329908) (BioLegend, San Diego, CA, USA), KIR2DS4 (CD158i)-APC (FAB1847A), and KIR3DL1 (CD158e1)-APC (FAB1225A) (R&D Systems, Minneapolis, MN, USA). Appropriate isotype-matched antibodies (Abs) were used as controls. Data were analyzed by flow cytometry.
Cytotoxicity Assay
A calcein-AM release assay was used as previously described to assess cytotoxicity (34). Tumor cells were labeled with 3.5 µM of calcein-AM (Dojindo Laboratories, Kumamoto, Japan)/106 cells/mL and incubated in a humidified incubator at 37°C with 5% CO2 for 30 min. After washing twice with PBS, the target cells were adjusted to a concentration of 5 × 104 cells/mL with 5% FBS RPMI-1640 medium and seeded in 96-well plates. The ratio of effector to target cells was set at 10:1. DAC (0.5 μM)-treated γδ T effector cells (100 µL) and untreated cells were added to the target cells in a 96-well plate at the indicated ratio and incubated at 37°C for 4 h. For the KIR2DL2/3 Ab-blocking experiment, the effector cells were first treated with or without Abs to KIR2DL2/3 (312602) (BioLegend, San Diego, CA, USA) for 30 min and then incubated with target cells. After incubation, the supernatant was harvested and transferred to a new plate. Absorbance at 485 nm of excitation light wavelength and 528 nm of emission wavelength was assessed using a BioTek Synergy HT Microplate Reader (BioTek Instruments, Winooski, VT, USA). The specific lysis was calculated according to the following formula: [(experimental release − spontaneous release)/(maximum release − spontaneous release)] × 100%. Spontaneous release was obtained by incubating the target cells in medium alone, and maximum release was obtained after treatment with 1% Triton X-100. All experiments were performed in triplicate wells and at least three independent experiments were completed.
DNA Methylation by Bisulfite Sequencing
Genomic DNA from γδ T cells treated with and without DAC was isolated using a DNeasy® Blood and Tissue kit (Qiagen GmbH, Hilden, Germany) and modified with bisulfate using a EZ DNA Methylation-Gold™ kit (Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. DNA was amplified using specific primers (KIR2DL2/3 forward: TTGGGTTTTATGTAAGGTAGAAAGAGT; KIR2DL2/3 reverse: CCAAACCTATATCTCCAACTCTAAAC; NKG2D forward: GTGGAGAGGTTAGGTTATTTTTTAA; NKG2D reverse: TTACCTCACTCTAAACTTTCACAAAAC). The PCR products were cloned using a pJET PCR Cloning kit (K1231, Thermo Scientific, Waltham, MA, USA), and 12 independent clones from each sample were sequenced to determine DNA methylation status (34).
Chromatin Immunoprecipitation (ChIP) qPCR Assays
Chromatin immunoprecipitation assays were performed using a Pierce Agarose Chip Kit (Thermo Scientific) according to the manufacturer’s instructions. Specific anti-trimethyl-H3K4 (Cell Signaling Technology, 9727) and anti-Sp-1 Abs (Invitrogen, PA5-29165) were used to determine the promoter methylation profile of KIR2DL2/3. Normal rabbit IgG was used as a negative control. DNA was extracted and analyzed by quantitative real-time PCR (qPCR) with specific primers (Site I forward: ACGTGCTATTCCACCTTTCCT; Site I reverse: CCGGAAGCCTTAGGCAAGAA; Site II forward: GAGACAGTCTCACTCTCTCAC; Site II reverse: AGTTCAAGACCAGCTGGTCCA; Site III forward: GCAGGGCGCCAAATAACATC; Site III reverse: GATGCCCTTCCAGGACTCAC). Enrichment was calculated using the following formula: Relative Enrichment Fold = 2−[(Ct (DAC group) − (Ct (Input of DAC group) − Log2 (Input Dilution Factor of DAC group))) − (Ct (Control group) − (Ct (Input of Control group) − Log2 (Input Dilution Factor of Control group)))].
Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
Total RNA was extracted from γδ T cells treated with DAC using an EasyPure RNA kit (Beijing TransGen Biotech Co., Ltd., Beijing, China) according to the manufacturer’s instructions. After removing genomic DNA contamination with DNase I (Sigma-Aldrich), M-MLV Reverse Transcriptase (Invitrogen) was used to synthesize cDNA. KIR2DL2 and KIR2DL3 mRNA expression was quantified by qRT-PCR using a CFX384TM Real Time system (Bio-Rad, Jurong East, Singapore). The PCR reactions were set up in a final volume of 10 µL with 5 µL SYBR Green qPCR Mix (Roche, Indianapolis, IN, USA) and 10 pmol of each sense and antisense primer. The primer sequences were KIR2DL2 forward: ACCCACTGAACCAAGCTCTA; KIR2DL2 reverse: AGACTCTTGGTCCATTACCG; KIR2DL3 forward: CTCATGGTCGTCAGCATGGT; KIR2DL3 reverse: CTGTGCAGAAGGAAGTGCTG; β-actin forward: AAGATCATTGCTCCTCCTG; β-actin reverse: CGTCATACTCCTGCTTGCTG. The PCR amplification procedure was as follows: 10 s at 95°C followed by 40 cycles of 5 s at 95°C and 30 s at 64°C. Each standard and sample value was determined in three independent experiments. In each experiment, KIR2DL2 and KIR2DL3 expression under each experimental condition was calculated using threshold cycle (Ct) values standardized to β-actin (housekeeping gene), applying the 2−(ΔCt) method (35).
Western Blot Analysis
Protein samples from γδ T cells treated with DAC and mithramycin (Sigma-Aldrich) were homogenized using RIPA lysis buffer. Equal amounts of protein per sample were separated using SDS-PAGE gel and transferred to polyvinylidene fluoride membranes. The membranes were incubated for 1 h with blocking buffer containing 5% skimmed milk, and then incubated with the primary Abs for KIR2DL2, KIR2DL3, and rabbit anti-goat or goat anti-mouse IgG Ab (R&D Systems). An ECL detection system was used to visualize the proteins.
Statistical Analysis
Data were analyzed by paired t-test or one-way ANOVA using GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA). The comparative CT method was applied in the quantitative real-time RT-PCR assay according to the delta-delta CT method (36). Results are presented as means ± SD and considered significant at a p < 0.05.
Results
DAC Induced Negligible Apoptosis of γδ T Cells, but Significantly Inhibited γδ T Cell Proliferation and Cytotoxicity
γδ T cells were expanded from the PBMCs of seven MDS and AML patients. The percentage of γδ T cells in PBMCs before expansion was 2.65% (1.46–5.45%), whereas the percentage of γδ T cells after expansion was 95.2% (90–98.7%) (Figures 1A,B). After expansion, the mean fluorescence intensity (MFI) of Vγ9 increased from 10.5 (5.61–20.7) to 438 (389–569), which indicated that zoledronate plus IL-2 could increase the expression of Vγ9.
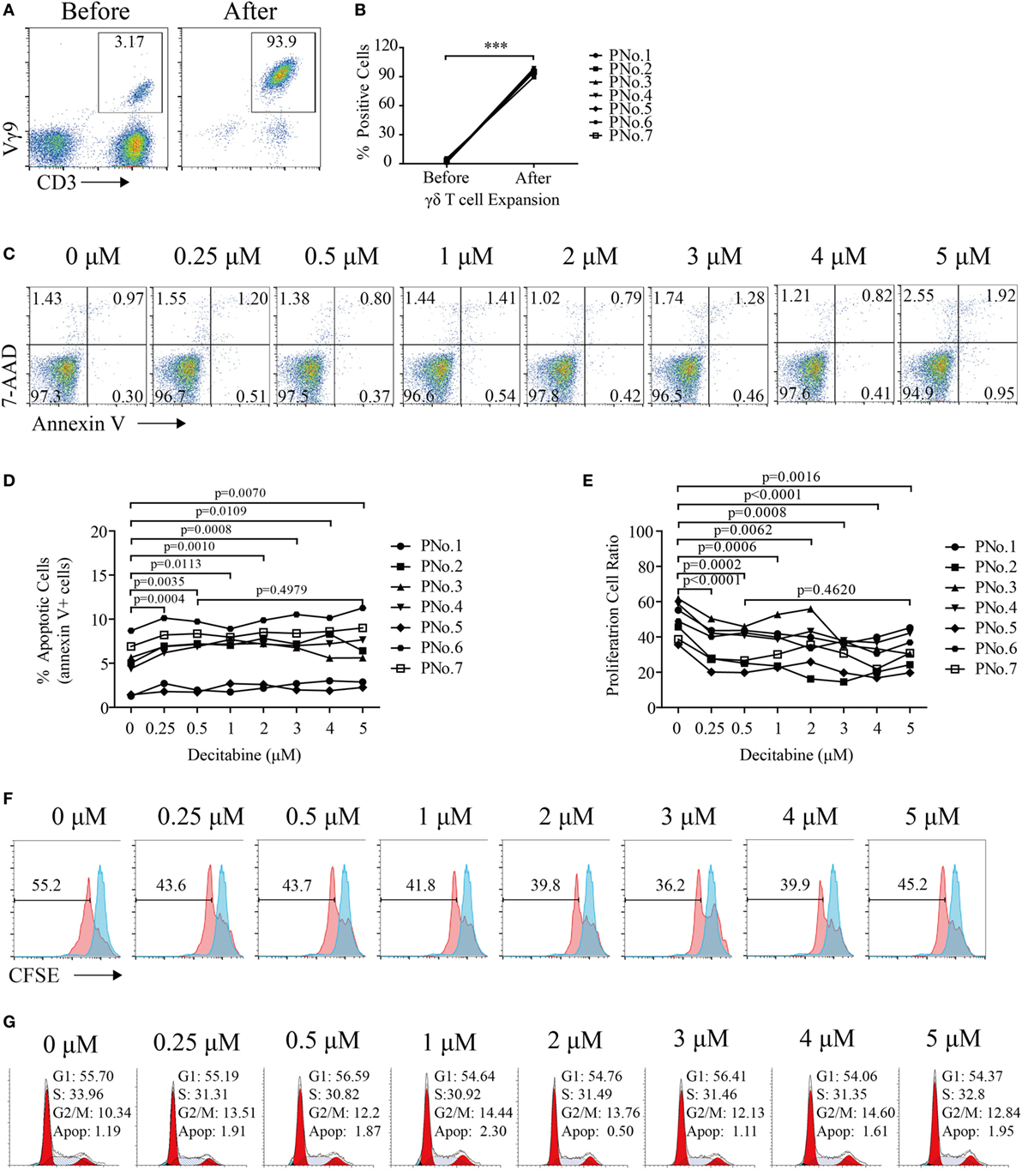
Figure 1. Decitabine inhibited the proliferation of gamma delta (γδ) T cells. (A) Representative flow cytometry analysis of CD3+Vγ9+ γδ T cells before and after expansion. (B) Percentage of γδ T cells before and after expansion (***p < 0.001; n = 7). (C) Viability of expanded γδ T cells after exposure to various DAC concentrations for 48 h. One representative experiment is shown. (D) Graph showing the apoptosis of expanded γδ T cells from seven patients after DAC treatment. The apoptosis percentages are shown as the percentage of cells that were annexin-V-positive. (E) Graph showing the proliferation of γδ T cells treated with increasing doses of DAC for 5 days. (F) Proliferation of γδ T cells treated with increasing doses of DAC. The red peaks represent the results of carboxyfluorescein succinimidyl ester (CFSE)-labeled γδ T cells after treatment with increasing concentrations of DAC for 5 days. The blue peak represents CFSE-labeled γδ T cells before DAC treatment. (G) Cell cycle of γδ T cells treated with increasing doses of DAC for 48 h.
To assess the direct impact of DAC on γδ T cell viability, expanded γδ T cells were treated with various concentrations (from 0 to 5 µM) of DAC for 48 h. DAC can induce γδ T cell apoptosis (p < 0.05), although this effect is very weak. In addition, no difference in apoptosis between γδ T cells treated with 0.5 and 5 µM DAC was observed (p > 0.05) (Figures 1C,D).
To investigate the cytostatic effects of DAC on γδ T cells, CFSE dilution was detected after 5 days of DAC treatment in CFSE-labeled γδ T cells. We observed that DAC significantly inhibited γδ T cell proliferation. However, this effect did not change with increasing drug concentrations (Figures 1E,F). DAC had no effect on the cell cycle of γδ T cells (Figure 1G).
To evaluate whether DAC treatment weakened γδ T cell-mediated cytolysis of tumor cells, three kinds of tumor cells (MDS SKM-1 cells, NK-resistant Raji cells, and NK-sensitive K562 cells) were selected as target cells. The cytotoxicity assay was performed as shown in Figure 2A. The results indicated that γδ T cells treated with 0.5 µM DAC were significantly less cytotoxic to SKM-1 and Raji cells than untreated γδ T cells (0 µM) (p < 0.05). However, these effects did not change with increasing drug concentrations (p > 0.05). DAC treatment did not affect γδ T cell lysis of K562 cells (p > 0.05) (Figure 2B).
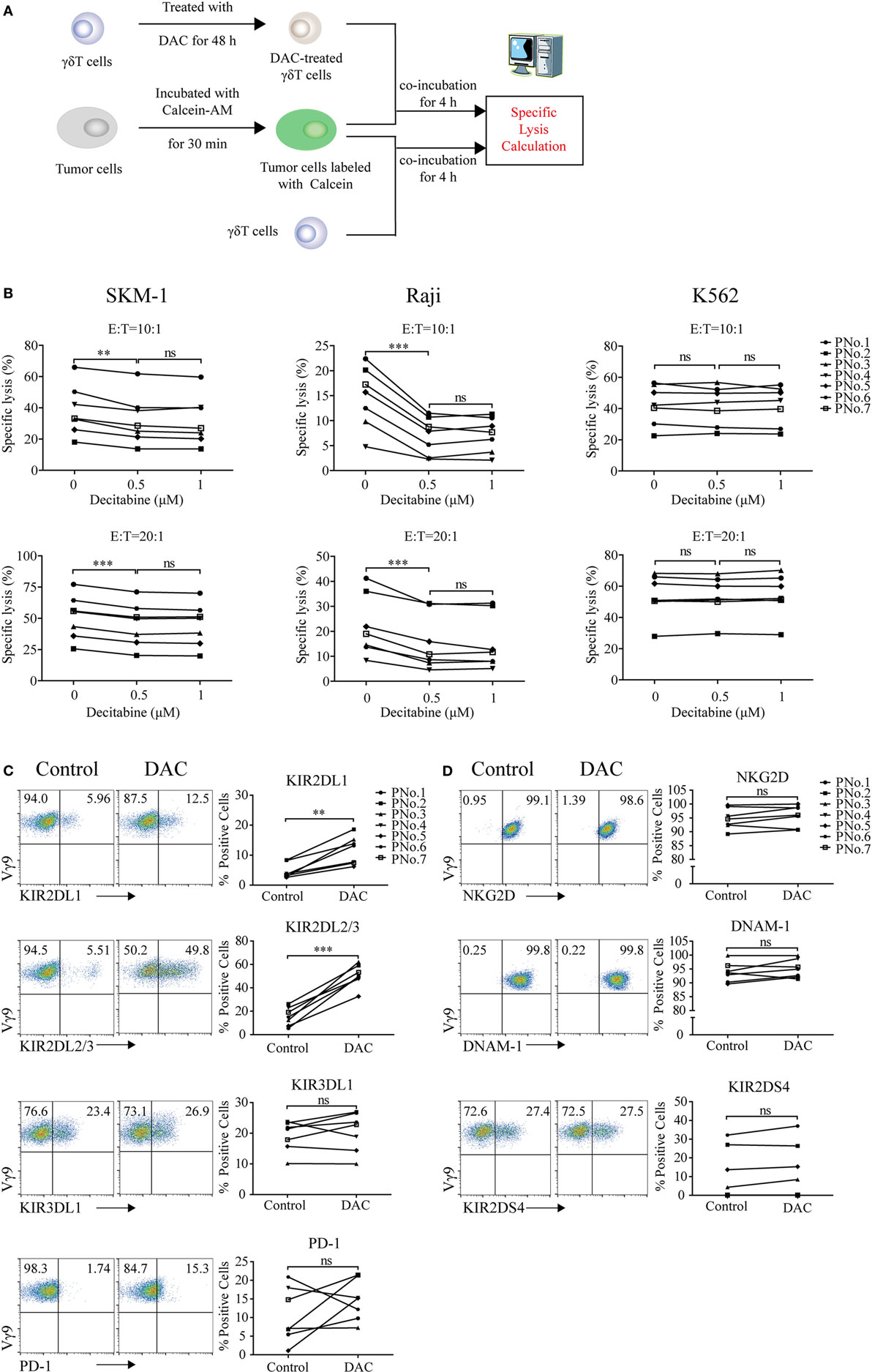
Figure 2. Decitabine treatment reduced gamma delta (γδ) T cell cytotoxicity to tumor cells and upregulated inhibitory receptor expression on γδ T cells. (A) Cytotoxicity assay protocol. (B) Cytotoxicity of expanded γδ T cells treated with increasing doses of DAC against Raji, K562, and SKM-1 cells in two effector to target cell (E:T) ratios (**p < 0.01; ***p < 0.001; ns, not significant; n = 7). (C,D) The effect of DAC on inhibitory and activating receptors expressed on γδ T cells. γδ T cells were incubated with 0.5 µM DAC for 48 h. Annexin-V-negative cells were gated and analyzed for surface marker expression (**p < 0.01; ***p < 0.001; ns, not significant; n = 7).
DAC Inhibited γδ T Cell Cytotoxicity to Tumor Cells Through Upregulation of KIR2DL2/3 Expression on γδ T Cells
To investigate whether DAC affected the expression of inhibitory and activating receptors further, γδ T cells were treated with 0.5 µM DAC for 48 h. We found that DAC increased the expression of KIR2DL1 (CD158a) and KIR2DL2/3 (CD158b) on γδ T cells (p < 0.05). In some patients, DAC upregulated the expression of KIR3DL1 (CD158e1) and PD-1 on γδ T cells (Figure 2C). However, DAC did not affect the expression of activating receptor, including that of NKG2D, DNAM-1, and KIR2DS4 (p > 0.05) (Figure 2D).
We next evaluated whether KIR2DL2/3 upregulation was relevant to the decreased cytotoxicity of DAC-treated γδ T cells. As can be seen in Figure 3A, DAC-treated γδ T cells showed a lower cytotoxicity to SKM-1 and Raji cells than γδ T cells (45.83 ± 8.01 vs. 54.65 ± 6. 97% and 10.86 ± 3.65 vs. 16.86 ± 5.05%; n = 3). However, there was no difference in cytotoxicity between γδ T cells and DAC-treated γδ T cells incubated with KIR2DL2/3-blocking Ab to SKM-1 (54.65 ± 6. 97 vs. 55.94 ± 6.71%; n = 3) and Raji (16.86 ± 5.05 vs. 16.34 ± 7.31%; n = 3) cells. These data indicated that KIR2DL2/3-blocking Ab completely restored the cytotoxicity of DAC-treated γδ T cells to SKM-1 and Raji cells. However, KIR2DL2/3-blocking Ab had no effect on the cytotoxicity of γδ T cells against K562 cells. These findings indicated that the decreased cytotoxicity of DAC-treated γδ T cells was dependent on the interaction between KIR2DL2/3 and their ligands (Figure 3A). In the blocking experiment, many γδ T cells are needed. Owing to insufficient number of γδ T cells, we performed the blocking experiment only at one effector to target cell ratio (10:1). Although only one effector to target cell ratio was measured in the blocking assay, the effector γδ T cells were obtained from three patients. We believe that the above data can support our statement.
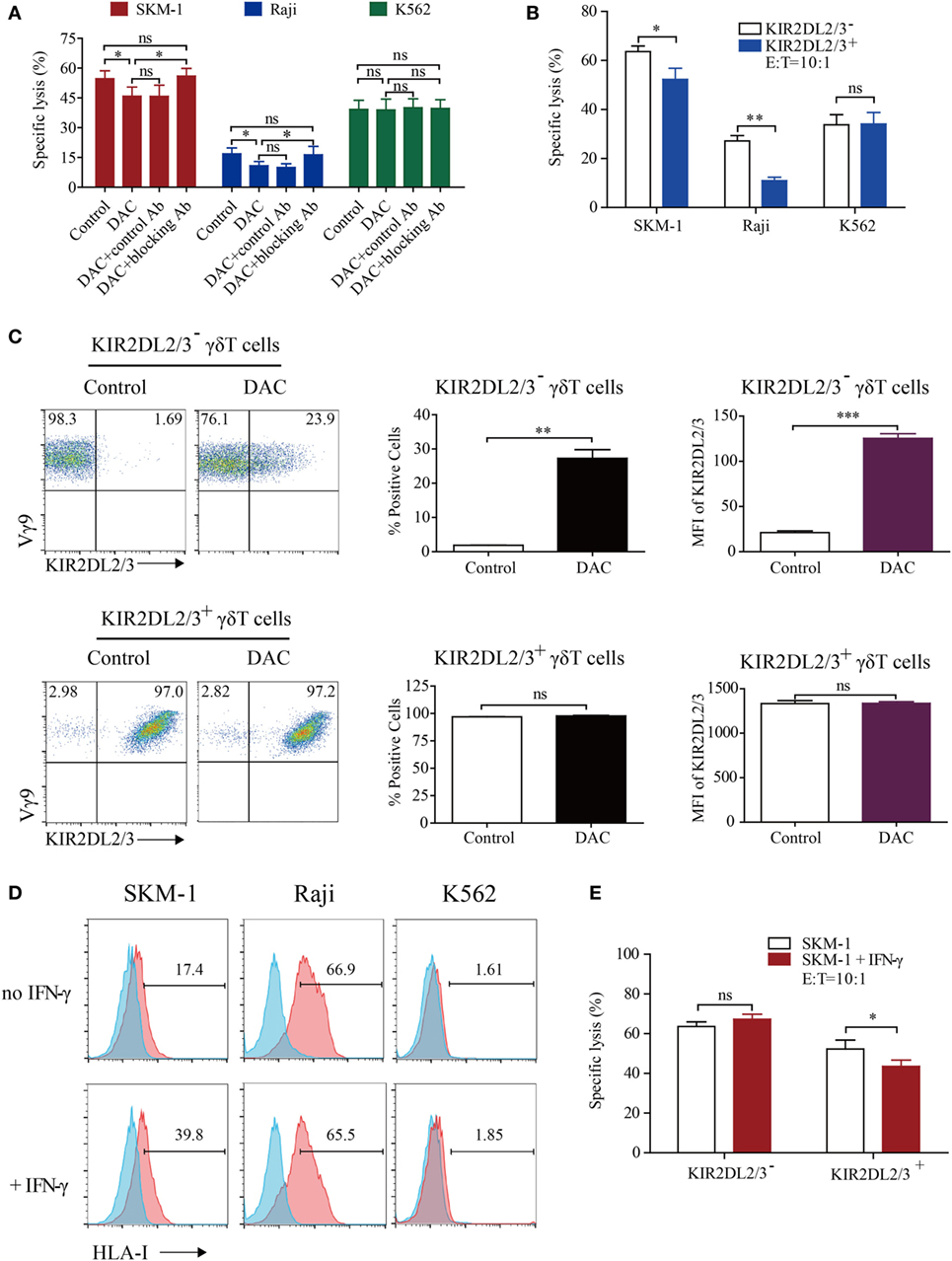
Figure 3. Decitabine induced polarization of KIR2DL2/3-negative gamma delta (γδ) T cells to KIR2DL2/3-positive cells, which are less cytotoxic to tumor cells expressing HLA-I molecules than KIR2DL2/3-negative γδ T cells. (A) The decrease in γδ T cell killing was inhibited by the KIR2DL2/3-blocking antibody (Ab). The 0.5 µM DAC-treated γδ T effector cells were first treated with or without Abs to KIR2DL2/3 for 30 min and then incubated with target cells at 37°C for 4 h to test the cytotoxicity (*p < 0.05; ns, not significant; n = 3). (B) The cytotoxicity of KIR2DL2/3-positive and -negative γδ T cells to tumor cells. KIR2DL2/3-positive and -negative γδ T cells were incubated with Raji, SKM-1, and K562 cells at 37°C for 4 h to test the cytotoxicity (n = 3; *p < 0.05; **p < 0.01; ns, not significant). (C) Effect of DAC on KIR2DL2/3 expression on KIR2DL2/3-positive and -negative γδ T cells. KIR2DL2/3-positive and -negative γδ T cells were sorted from cultured γδ T cells and treated with 0.5 µM DAC for 48 h. FACS dot plots showing the changes of KIR2DL2/3 on KIR2DL2/3-positive and -negative γδ T cells after treatment with DAC. Graph showing the percentage and mean fluorescence intensity (MFI) of KIR2DL2/3 on KIR2DL2/3-positive and -negative γδ T cells after treatment with DAC (**p < 0.01; ***p < 0.001; ns, not significant; n = 3). (D) HLA-I molecule expression on tumor cells and interferon (IFN)-γ-treated tumor cells. Tumor cells were first treated with 200 IU/mL IFN-γ for 48 h, and then harvested and stained with mouse anti-human HLA-I molecular Ab. (E) The cytotoxicity of KIR2DL2/3-positive and -negative γδ T cells to SKM-1 cells and IFN-γ-treated SKM-1 cells (*p < 0.05; ns, not significant; n = 3).
To investigate whether the expression of KIR2DL2/3 was associated with the decreased cytotoxicity of DAC-treated γδ T cells further, we determined the cytotoxicity of KIR2DL2/3+ and KIR2DL2/3− γδ T cells. γδ T cells were sorted by FACS. The purity of KIR2DL2/3-positive and -negative γδ T cells was above 95%. KIR2DL2/3+ γδ T cells were significantly less cytotoxic to SKM-1 and Raji cells than KIR2DL2/3− γδ T cells (p < 0.05). In terms of cytotoxicity to K562 cells, there was no difference between KIR2DL2/3+ and KIR2DL2/3− γδ T cells (p > 0.05) (Figure 3B). After incubation with 0.5 µM DAC for 48 h, some KIR2DL2/3− γδ T cells expressed KIR2DL2/3. However, DAC did not affect KIR2DL2/3 expression on KIR2DL2/3+ γδ T cells (Figure 3C).
To investigate whether the decreased cytotoxicity of KIR2DL2/3+ γδ T cells to SKM-1 and Raji cells was associated with the HLA-C allotypes in tumor cells, we analyzed the HLA-C allotypes in SKM-1 and Raji cell lines through genotyping by Search Biotech Co., Ltd. (Beijing, China). The HLA-C alleles of SKM-1 cells are HLA-C*04:01 homozygotes and the HLA-C alleles of Raji cells are HLA-C*03:04 and C*04:01. Both HLA-C*03:04 and HLA-C*04:01 are ligands of KIR2DL2/3 (37, 38). We further determined the expression of HLA-I molecules on target tumor cells and found that HLA-I molecular expression level was low on SKM-1 cells, high on Raji cells, and very low on K562 cells (Figure 3D). Interferon (IFN)-γ has been reported to enhance HLA-I molecular expression on tumor cells. We found that IFN-γ only increased HLA-I molecular expression on SKM-1 cells, but not on Raji and K562 cells (Figure 3D). Meanwhile, compared to SKM-1 cells, KIR2DL2/3+ γδ T cells were less cytotoxic to IFN-γ-treated SKM-1 cells (p < 0.05). However, the cytotoxicity of KIR2DL2/3− γδ T cells to IFN-γ-treated and untreated SKM-1 cells showed no difference (p > 0.05) (Figure 3E).
Epigenetic Regulation of KIR2DL2/3 Expression by DAC
To examine whether DAC treatment influenced the expression of KIR2DL2/3 at the transcriptional level, we examined the gene expression patterns of KIR2DL2/3 in γδ T cells. Real-time RT-PCR results indicated that DAC increased the expression of KIR2DL2 and KIR2DL3 (p < 0.05) (Figure 4A). The upregulation of KIR2DL2/3 expression was primarily due to the enhanced transcription of KIR2DL2/3 gene. Therefore, we determined whether epigenetic mechanisms, such as DNA methylation, were involved in regulating KIR2DL2 and KIR2DL3 genes. We found that the genes around the transcriptional star sites (TSS) of KIR2DL2 and KIR2DL3 were highly consistent (Figure 4B), and there was a CpG island and 12 CpG sites around the TSS of KIR2DL2/3 gene (between −123 and +91 positions), as predicted by the website of Li Lab (http://www.urogene.org/methprimer/) (Figure 4B). We analyzed the status of DNA methylation in the CpG islands of KIR2DL2/3 in γδ T cells treated with and without DAC from three cancer patients by using bisulfite sequencing. As shown in Figures 4C,E, there was a significant enhancement of DNA demethylation in the KIR2DL2/3 promoter of DAC-treated γδ T cells. However, DAC did not demethylate the NKG2D promoter (Figures 4D,E).
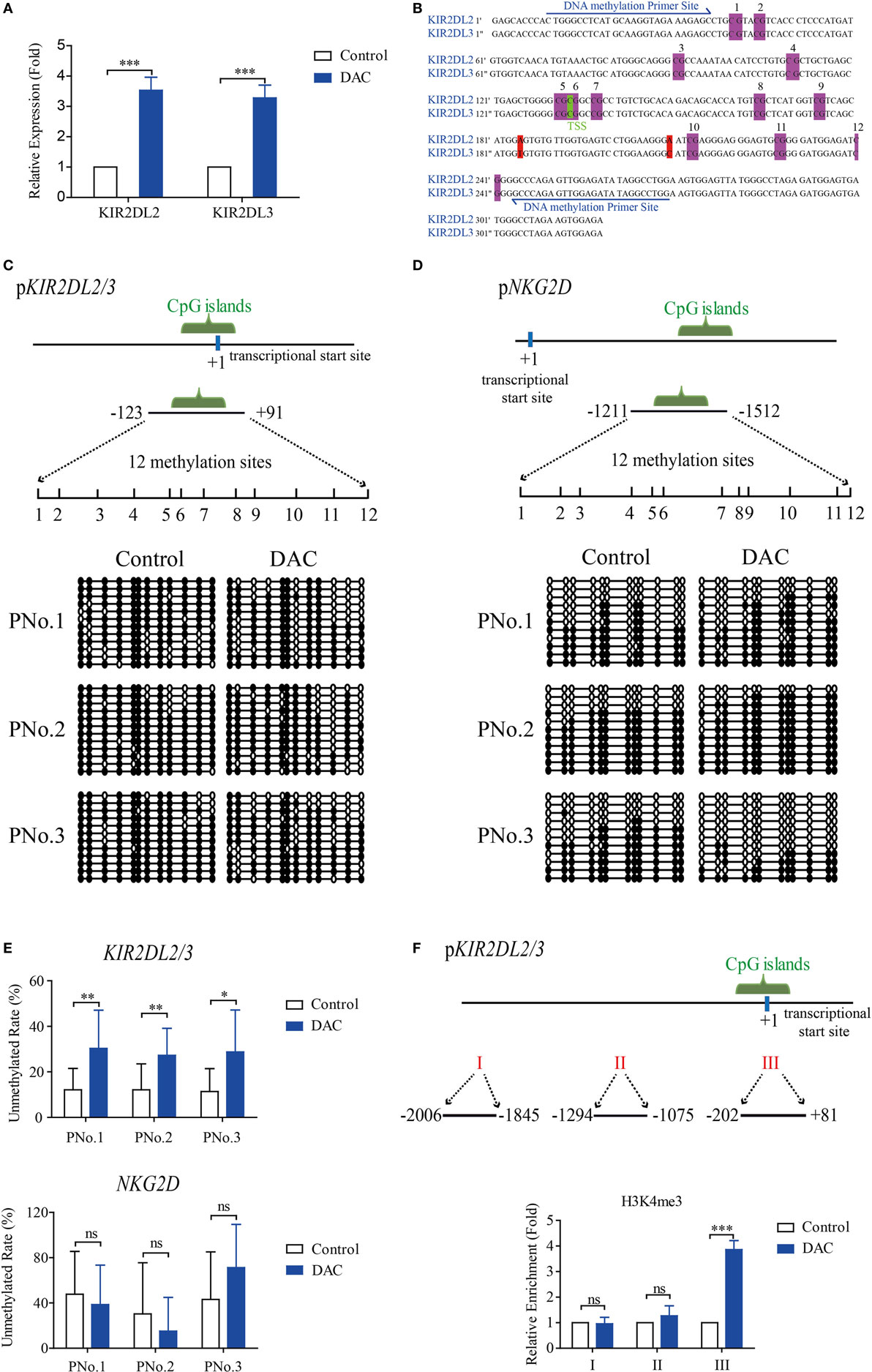
Figure 4. Decitabine increased demethylation of KIR2DL2/3 promoter, which was beneficial to the transcription of KIR2DL2/3 in gamma delta (γδ) T cells. (A) qRT-PCR analysis of KIR2DL2 and KIR2DL3 expression on γδ T cells exposed to 0.5 µM DAC for 48 h. Data are expressed as the expression of KIR2DL2 and KIR2DL3 mRNA on DAC-treated γδ T cells relative to that on γδ T cells (***p < 0.001; n = 3). (B) The genes around the transcriptional star sites (TSS) (marked green) of KIR2DL2 and KIR2DL3 are nearly the same, except for two bases (marked red). There are 12 CpG sites (marked purple) around the TSS of KIR2DL2 and KIR2DL3 genes. (C,D) DNA methylation of the promoters of both KIR2DL2/3 and NKG2D in γδ T cells. The upper images show the schematic diagram of CpG islands in KIR2DL2/3 and NKG2D promoters. Vertical lines: methylation sites. The lower images show DNA methylation of KIR2DL2/3 and NKG2D promoters in γδ T cells treated with and without (control) DAC for 48 h from three patients. Open circles: unmethylated CpGs; solid circles: methylated CpGs. (E) Statistical analysis of the demethylation states of KIR2DL2/3 and NKG2D promoters in γδ T cells from three patients (*p < 0.05; **p < 0.01; ns, not significant). (F) Three regions (I, II, and III) including TSS (+1) of KIR2DL2/3 were analyzed by chromatin immunoprecipitation qPCR for H3K4me3 occupancy in the KIR2DL2/3 promoter of γδ T cells treated with and without (control) DAC for 48 h. Enrichment of KIR2DL2/3 promoter-specific DNA sequences was measured using quantitative PCR. Data are shown from three independent experiments and each PCR was performed in triplicate (***p < 0.001; ns, not significant).
Histone H3K4 tri-methylation (H3K4me3) is positively correlated with transcriptional activity (39). We observed a significantly increased level of H3K4me3 around the CpG islands in DAC-treated γδ T cells compared with DAC-untreated γδ T cells (p < 0.05) (Figure 4F).
Altogether, the results indicated that demethylation in the KIR2DL2/3 gene promoter of DAC-treated γδ T cells correlated with the enhanced expression of KIR2DL2/3.
DAC Increased KIR2DL2/3 Expression Through Sp-1-Binding Activity
There were two Sp-1 transcription factor potential binding sites in the CpG islands of the KIR2DL2/3 promoter (Figure 5A). Therefore, we proceeded to investigate whether DAC affected the binding of Sp-1 to its potential binding sites, to upregulate the expression of KIR2DL2/3 in γδ T cells. The chromatin from γδ T cells treated with and without DAC was immunoprecipitated with anti-Sp-1 Ab. The immunoprecipitated samples were subjected to qPCR using primers to specifically amplify the CpG islands. Another two pairs of primers were also used to amplify the regions in the KIR2DL2/3 promoter, except the CpG islands. The qPCR results indicated that DAC substantially increased the binding of Sp-1 to its potential binding sites in the CpG islands of the KIR2DL2/3 promoter (p < 0.05) (Figure 5A).
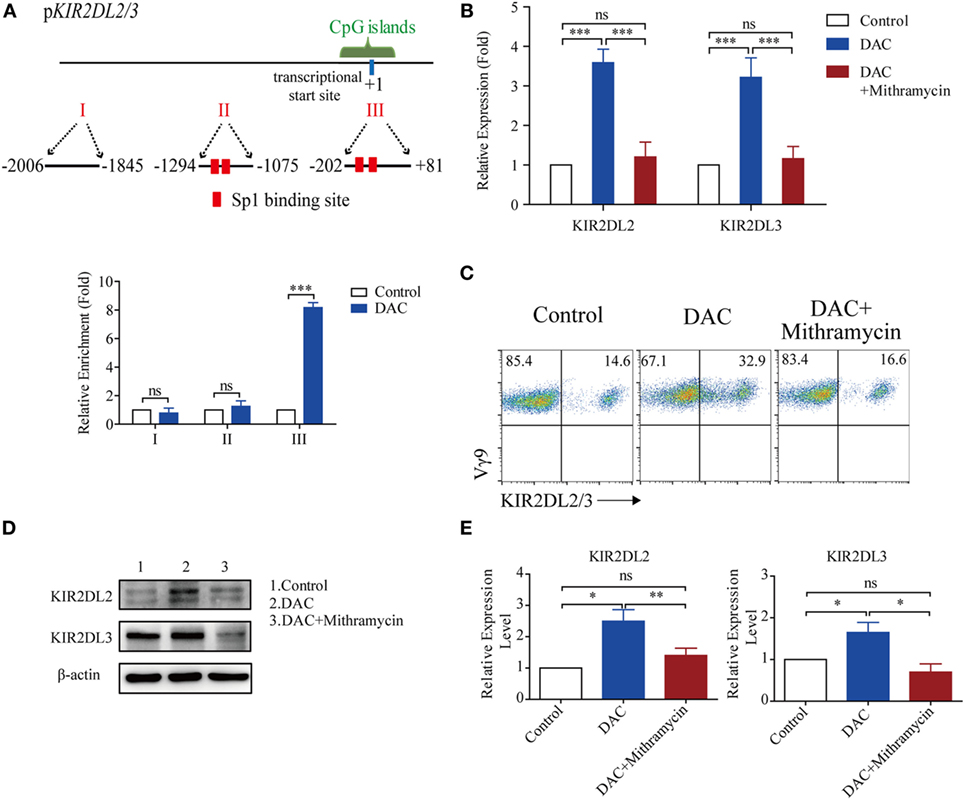
Figure 5. Decitabine increased KIR2DL2/3 expression by ligation of Sp-1 and its binding sites. (A) Three regions (I, II, and III) including transcriptional star site (+1) of KIR2DL2/3 were analyzed by chromatin immunoprecipitation qPCR for Sp-1 occupancy in KIR2DL2/3 promoter of gamma delta (γδ) T cells treated with and without (control) DAC for 48 h. Data are shown from three independent experiments and each PCR was performed in triplicate (***p < 0.001; ns, not significant). (B) The effect of the Sp-1-inhibitor mithramycin on KIR2DL2 and KIR2DL3 expression in DAC-treated γδ T cells. qRT-PCR analysis of KIR2DL2 and KIR2DL3 expression in γδ T cells, γδ T cells treated with 0.5 µM DAC and γδ T cells treated with 0.5 µM DAC and 100 µM mithramycin for 48 h. Data are expressed as the relative expression of KIR2DL2 and KIR2DL3 mRNA in different groups (***p < 0.001; ns, not significant; n = 3). (C) Flow cytometry analysis of the effect of the Sp-1-inhibitor mithramycin on DAC-treated γδ T cells. γδ T cells were treated with 0.5 µM DAC and 0.5 µM DAC plusing 100 µM mithramycin for 48 h, respectively. Representative FACS results illustrate KIR2DL2 and KIR2DL3 expression on γδ T cells, DAC-treated γδ T cells, and γδ T cells treated with both DAC and mithramycin. (D) Western blot analysis of the effect of mithramycin on DAC-treated γδ T cells. γδ T cells were treated with 0.5 µM DAC and 0.5 µM DAC plusing 100 µM mithramycin for 48 h, respectively. A representative western blot illustrates KIR2DL2 and KIR2DL3 expression on γδ T cells, DAC-treated γδ T cells, and γδ T cells treated with both DAC and mithramycin. β-Actin was used as a loading control of the cell lysates. (E) Bands of KIR2DL2 and KIR2DL3 were quantified by densitometric analysis and plotted after normalization against β-actin. The histogram shows means ± SD for three independent sets of experiments (*p < 0.05; **p < 0.01; ns, not significant).
To ascertain whether DAC increased KIR2DL2/3 expression through the ligation of Sp-1 and its binding sites in the CpG islands of the KIR2DL2/3 promoter, the Sp-1 inhibitor mithramycin was added to γδ T cells treated with DAC. Mithramycin prevented KIR2DL2 and KIR2DL3 upregulation mediated by DAC in γδ T cells (Figure 5B). Flow cytometry and western blot assay indicated that mithramycin substantially inhibited the upregulated KIR2DL2/3 expression induced by DAC (Figures 5C–E). These findings indicated that DAC enhanced KIR2DL2/3 expression in γδ T cells by increasing the binding of Sp-1 to its binding sites in the CpG islands of the KIR2DL2/3 promoter.
Discussion
DAC has been approved for the treatment of hematological malignancies, and its clinical effects on solid tumors have gained attention (17). It also affects immune cells. In a previous study, DAC induced FOXP3 hypomethylation and increased the number of regulatory T cells (30). Another study indicated that DAC inhibited naïve T cell proliferation by increasing the gene expression of the DNA dioxygenase TET2, which facilitated the expression of several cell cycle inhibitors (31). The effects of DAC on NK cells are controversial. Benjamin et al. reported that DAC augmented IFN-γ secretion by NK cells as well as cytotoxicity to tumor cells (40). However, another report claimed that the same dose of DAC treatment resulted in the inhibition of proliferation and decreased cytotoxicity of NK cells (20). It has been reported that DAC treatment resulted in a rapid and stable induction of transcription and cell surface expression of KIR2DL2/3 in NK cell lines, NK cell clones, and freshly isolated NK cells, but not in the T cell line, Jurkat, or the B cell line RPMI 8866 (41). In this study, we demonstrated that the proliferation of γδ T cells was inhibited by DAC. However, DAC had no effect on the cell cycle of γδ T cells. DAC significantly upregulated the expression of KIR2DL2/3, thereby reducing γδ T cell cytotoxicity to tumor cells expressing the ligand of KIR2DL2/3.
KIR2DL2/3 is a member of the inhibitory killer Ig-like receptors (KIRs), which recognize a defined group of polymorphic HLA-I molecules. KIR2DL2/3 inhibits NK cell-mediated lysis of target cells bearing the appropriate HLA-I allotypes through an immune tyrosine-based inhibitory motif by recruiting protein tyrosine phosphatases (SHP-1 and SHP-2) (42–45). Furthermore, the combination of KIR2DL2/3 and their ligands seems to favor NK cell inhibition and malignant melanoma dissemination (46). γδ T cells display MHC-independent cytotoxicity against various tumors (2). However, we observed that KIR2DL2/3+ γδ T cells were less cytotoxic to tumor cells expressing HLA-I molecules than KIR2DL2/3− γδ T cells. This result suggests that the lytic activity of γδ T cells against tumor cells was influenced by signaling via KIR2DL2/3.
KIR2DL2/3 protein contains KIR2DL2 and KIR2DL3. We found that the promoter regions of KIR2DL2 and KIR2DL3 are highly similar, with more than 99% DNA sequence identity. CpG islands were present in areas surrounding the transcription initiation region of both KIR2DL2 and KIR2DL3, and the DNA sequence was highly consistent. It has been reported that CpG islands surrounding the transcriptional start site of each KIR gene are consistently demethylated in expressed KIR and methylated in unexpressed KIR (41). Our data indicated that DAC treatment demethylated KIR2DL2/3 genes in γδ T cells and enriched H3K4me3 in the CpG islands of KIR2DL2/3 gene. The enrichment of H3K4me3 near the transcription start site defined an active state of gene expression (47, 48). Therefore, our data suggested that DAC increased KIR2DL2/3 expression in γδ T cells through an epigenetic mechanism.
Previous studies have shown that transcription factors such as Sp-1 play an important role in KIR gene expression (49–51). They can act as both negative and positive regulators of gene expression depending on their relative concentration and binding partners (52, 53). Through the prediction of transcription factor-binding sites using the AliBaba 2.1 website, we found that there were two Sp-1-binding sites in the CpG islands of KIR2DL2/3 gene. We noticed a significant enrichment of Sp-1 in the CpG islands of KIR2DL2/3 gene in DAC-treated γδ T cells compared with DAC-untreated γδ T cells. Mithramycin A, which inhibits the interaction of Sp-1 with its binding site (54–56), significantly attenuated KIR2DL2/3 expression in DAC-treated γδ T cells. These results indicated that DAC enhanced KIR2DL2/3 expression through the enrichment of Sp-1 binding to its binding sites.
We suggest that in activated γδ T cells, the activation signal was much stronger than the inhibitory signal when γδ T cells encountered tumor cells expressing HLA-I molecules because of the methylation of KIR2DL2/3 as well as low expression level of KIR2DL2/3. However, DAC can demethylate KIR2DL2/3 genes of γδ T cells and promote the binding of Sp-1 to KIR2DL2/3 gene promoter, which consequently promotes KIR2DL2/3 gene transcription and KIR2DL2/3 protein expression. When encountering tumor cells expressing HLA-I molecules, the cytotoxicity of DAC-treated γδ T cells, which have a higher level of KIR2DL2/3 expression, is decreased (Figure 6).
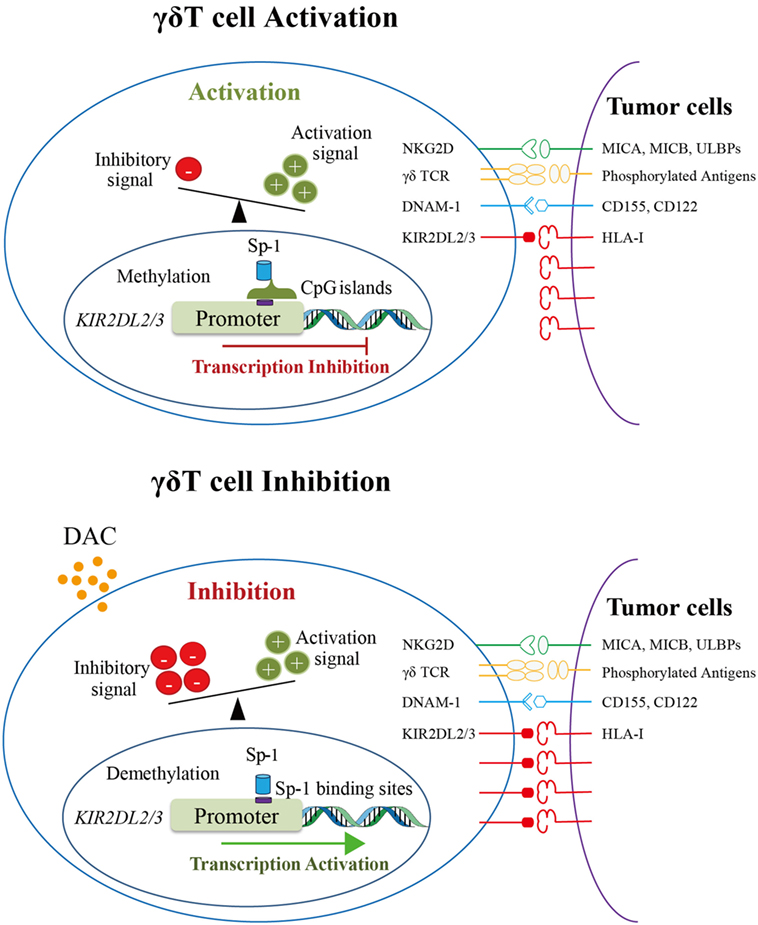
Figure 6. The proposed mechanism underlying the inhibition of gamma delta (γδ) T cell cytotoxicity by decitabine via promoting KIR2DL2/3 expression. The upper image shows the schematic diagram of γδ T cell activation. KIR2DL2/3 expression on γδ T cells was very low because the promoter of KIR2DL2/3 gene was methylated. When encountering tumor cells, γδ T cells were activated by the recognition of activation receptors and their ligands on tumors cells. The lower image shows the schematic diagram of γδ T cell inhibition by DAC. DAC demethylates the promoter of KIR2DL2/3, which increases the binding of Sp-1 to its potential binding sites in the KIR2DL2/3 promoter, and then activates KIR2DL2/3 gene and protein expression. The upregulation of KIR2DL2/3 protein expression reduces the cytotoxicity of γδ T cells against tumor cells expressing HLA-I molecules.
Our study reveals that DAC-treated γδ T cells from all patients have a high level of KIR2DL2/3 expression and are less cytotoxic than DAC-untreated γδ T cells. Furthermore, we elucidated the underlying mechanism. These observations broadened our knowledge of the effect of DAC on γδ T cells. Because of the inhibitory effect of DAC on γδ T cell cytotoxicity, it is necessary to consider this side effect of DAC before using it in the clinical setting. Therefore, we recommend the monitoring of DAC concentration in patients’ blood after DAC treatment. After DAC clearance by the human body, adoptive γδ T cell infusion might be a good method for replenishing γδ T cells affected by DAC in the human body.
Ethics Statement
Seven newly diagnosed MDS and AML patients who did not receive radiation therapy and chemotherapy before blood collection were enrolled in this study, all of whom provided with written informed consent for the use of biospecimens for research purposes in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of the First Hospital of Jilin University and carried out in accordance with the approved guideline “Use of experimental animals and human subjects.”
Author Contributions
JWC, JTC, YL, and WL conceived and designed the research. CN, ML, SZ, YC, and LZ performed the experiments and analyzed the data. DX contributed to sample collection. CN and SZ wrote the paper. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare no potential conflicts of interest.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This work was supported in part by grants from the National Key R&D Program of China (2016YFC1303800), National Key Technologies R&D Program (2015BAI12B12), National Natural Science Foundation of China (81571534, 81672275, 81402436, and 31700764), Provincial Science Fund of Jilin Province Development and Reform Commission (2014N147 and 2017C022), Provincial Science Fund of Jilin Provincial Department of Finance (20170622011JC and 20150101176JH), Youth Fund of the First Hospital of Jilin University (JDYY72016003), and JLU Science and Technology Innovative Research Team.
References
1. Kabelitz D, Wesch D, Pitters E, Zoller M. Characterization of tumor reactivity of human V gamma 9V delta 2 gamma delta T cells in vitro and in SCID mice in vivo. J Immunol (2004) 173(11):6767–76. doi:10.4049/jimmunol.173.11.6767
2. Kobayashi H, Tanaka Y, Yagi J, Osaka Y, Nakazawa H, Uchiyama T, et al. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: a pilot study. Cancer Immunol Immunother (2007) 56(4):469–76. doi:10.1007/s00262-006-0199-6
3. Kobayashi H, Tanaka Y, Yagi J, Toma H, Uchiyama T. Gamma/delta T cells provide innate immunity against renal cell carcinoma. Cancer Immunol Immunother (2001) 50(3):115–24. doi:10.1007/s002620100173
4. Sicard H, Al Saati T, Delsol G, Fournie JJ. Synthetic phosphoantigens enhance human Vgamma9Vdelta2 T lymphocytes killing of non-Hodgkin’s B lymphoma. Mol Med (2001) 7(10):711–22.
5. Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood (2000) 96(2):384–92.
6. Bonneville M, O’Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol (2010) 10(7):467–78. doi:10.1038/nri2781
7. Dieli F, Poccia F, Lipp M, Sireci G, Caccamo N, Di Sano C, et al. Differentiation of effector/memory Vdelta2 T cells and migratory routes in lymph nodes or inflammatory sites. J Exp Med (2003) 198(3):391–7. doi:10.1084/jem.20030235
8. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature (1995) 375(6527):155–8. doi:10.1038/375155a0
9. Braza MS, Klein B. Anti-tumour immunotherapy with Vgamma9Vdelta2 T lymphocytes: from the bench to the bedside. Br J Haematol (2013) 160(2):123–32. doi:10.1111/bjh.12090
10. Tyler CJ, Doherty DG, Moser B, Eberl M. Human Vgamma9/Vdelta2 T cells: innate adaptors of the immune system. Cell Immunol (2015) 296(1):10–21. doi:10.1016/j.cellimm.2015.01.008
11. Fowler DW, Bodman-Smith MD. Harnessing the power of Vdelta2 cells in cancer immunotherapy. Clin Exp Immunol (2015) 180(1):1–10. doi:10.1111/cei.12564
12. Hannani D, Ma Y, Yamazaki T, Dechanet-Merville J, Kroemer G, Zitvogel L. Harnessing gammadelta T cells in anticancer immunotherapy. Trends Immunol (2012) 33(5):199–206. doi:10.1016/j.it.2012.01.006
13. Kabelitz D, Dechanet-Merville J. Editorial: “recent advances in gamma/delta T cell biology: new ligands, new functions, and new translational perspectives”. Front Immunol (2015) 6:371. doi:10.3389/fimmu.2015.00371
14. Zou C, Zhao P, Xiao Z, Han X, Fu F, Fu L. Gammadelta T cells in cancer immunotherapy. Oncotarget (2017) 8(5):8900–9. doi:10.18632/oncotarget.13051
15. Kabelitz D. Human gammadelta T cells: from a neglected lymphocyte population to cellular immunotherapy: a personal reflection of 30 years of gammadelta T cell research. Clin Immunol (2016) 172:90–7. doi:10.1016/j.clim.2016.07.012
16. Fan H, Lu X, Wang X, Liu Y, Guo B, Zhang Y, et al. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: a phase I/II report. J Immunol Res (2014) 2014:371087. doi:10.1155/2014/371087
17. Nie J, Liu L, Li X, Han W. Decitabine, a new star in epigenetic therapy: the clinical application and biological mechanism in solid tumors. Cancer Lett (2014) 354(1):12–20. doi:10.1016/j.canlet.2014.08.010
18. Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of decitabine development: accomplishments, ongoing investigations, and future strategies. Cancer (2008) 112(11):2341–51. doi:10.1002/cncr.23463
19. Atallah E, Kantarjian H, Garcia-Manero G. The role of decitabine in the treatment of myelodysplastic syndromes. Expert Opin Pharmacother (2007) 8(1):65–73. doi:10.1517/14656566.8.1.65
20. Kopp LM, Ray A, Denman CJ, Senyukov VS, Somanchi SS, Zhu S, et al. Decitabine has a biphasic effect on natural killer cell viability, phenotype, and function under proliferative conditions. Mol Immunol (2013) 54(3–4):296–301. doi:10.1016/j.molimm.2012.12.012
21. Christman JK. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene (2002) 21(35):5483–95. doi:10.1038/sj.onc.1205699
22. Vijayaraghavalu S, Dermawan JK, Cheriyath V, Labhasetwar V. Highly synergistic effect of sequential treatment with epigenetic and anticancer drugs to overcome drug resistance in breast cancer cells is mediated via activation of p21 gene expression leading to G2/M cycle arrest. Mol Pharm (2013) 10(1):337–52. doi:10.1021/mp3004622
23. Nakamura M, Nishikawa J, Saito M, Sakai K, Sasaki S, Hashimoto S, et al. Decitabine inhibits tumor cell proliferation and up-regulates e-cadherin expression in Epstein-Barr virus-associated gastric cancer. J Med Virol (2017) 89(3):508–17. doi:10.1002/jmv.24634
24. Zhang Z, He Q, Tao Y, Guo J, Xu F, Wu LY, et al. Decitabine treatment sensitizes tumor cells to T-cell-mediated cytotoxicity in patients with myelodysplastic syndromes. Am J Transl Res (2017) 9(2):454–65.
25. Son CH, Lee HR, Koh EK, Shin DY, Bae JH, Yang K, et al. Combination treatment with decitabine and ionizing radiation enhances tumor cells susceptibility of T cells. Sci Rep (2016) 6:32470. doi:10.1038/srep32470
26. Li B, Zhu X, Sun L, Yuan L, Zhang J, Li H, et al. Induction of a specific CD8+ T-cell response to cancer/testis antigens by demethylating pre-treatment against osteosarcoma. Oncotarget (2014) 5(21):10791–802. doi:10.18632/oncotarget.2505
27. Riccadonna C, Yacoub Maroun C, Vuillefroy de Silly R, Boehler M, Calvo Tardon M, Jueliger S, et al. Decitabine treatment of glioma-initiating cells enhances immune recognition and killing. PLoS One (2016) 11(8):e0162105. doi:10.1371/journal.pone.0162105
28. Lucarini V, Buccione C, Ziccheddu G, Peschiaroli F, Sestili P, Puglisi R, et al. Combining type I interferons and 5-Aza-2’-deoxycitidine to improve anti-tumor response against melanoma. J Invest Dermatol (2017) 137(1):159–69. doi:10.1016/j.jid.2016.08.024
29. Heninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol (2015) 6:29. doi:10.3389/fimmu.2015.00029
30. Kehrmann J, Tatura R, Zeschnigk M, Probst-Kepper M, Geffers R, Steinmann J, et al. Impact of 5-aza-2’-deoxycytidine and epigallocatechin-3-gallate for induction of human regulatory T cells. Immunology (2014) 142(3):384–95. doi:10.1111/imm.12261
31. Wang X, Wang J, Yu Y, Ma T, Chen P, Zhou B, et al. Decitabine inhibits T cell proliferation via a novel TET2-dependent mechanism and exerts potent protective effect in mouse auto- and allo-immunity models. Oncotarget (2017) 8(34):56802–15. doi:10.18632/oncotarget.18063
32. Niu C, Jin H, Li M, Xu J, Xu D, Hu J, et al. In vitro analysis of the proliferative capacity and cytotoxic effects of ex vivo induced natural killer cells, cytokine-induced killer cells, and gamma-delta T cells. BMC Immunol (2015) 16:61. doi:10.1186/s12865-015-0124-x
33. Niu C, Jin H, Li M, Zhu S, Zhou L, Jin F, et al. Low-dose bortezomib increases the expression of NKG2D and DNAM-1 ligands and enhances induced NK and gammadelta T cell-mediated lysis in multiple myeloma. Oncotarget (2017) 8(4):5954–64. doi:10.18632/oncotarget.13979
34. Shi X, Li M, Cui M, Niu C, Xu J, Zhou L, et al. Epigenetic suppression of the antitumor cytotoxicity of NK cells by histone deacetylase inhibitor valproic acid. Am J Cancer Res (2016) 6(3):600–14.
35. Kang L, Sun J, Wen X, Cui J, Wang G, Hoffman AR, et al. Aberrant allele-switch imprinting of a novel IGF1R intragenic antisense non-coding RNA in breast cancers. Eur J Cancer (2015) 51(2):260–70. doi:10.1016/j.ejca.2014.10.031
36. Li T, Chen H, Li W, Cui J, Wang G, Hu X, et al. Promoter histone H3K27 methylation in the control of IGF2 imprinting in human tumor cell lines. Hum Mol Genet (2014) 23(1):117–28. doi:10.1093/hmg/ddt405
37. Frazier WR, Steiner N, Hou L, Dakshanamurthy S, Hurley CK. Allelic variation in KIR2DL3 generates a KIR2DL2-like receptor with increased binding to its HLA-C ligand. J Immunol (2013) 190(12):6198–208. doi:10.4049/jimmunol.1300464
38. Moesta AK, Parham P. Diverse functionality among human NK cell receptors for the C1 epitope of HLA-C: KIR2DS2, KIR2DL2, and KIR2DL3. Front Immunol (2012) 3:336. doi:10.3389/fimmu.2012.00336
39. Mulder KW, Brenkman AB, Inagaki A, van den Broek NJ, Timmers HT. Regulation of histone H3K4 tri-methylation and PAF complex recruitment by the Ccr4-Not complex. Nucleic Acids Res (2007) 35(7):2428–39. doi:10.1093/nar/gkm175
40. Schmiedel BJ, Arelin V, Gruenebach F, Krusch M, Schmidt SM, Salih HR. Azacytidine impairs NK cell reactivity while decitabine augments NK cell responsiveness toward stimulation. Int J Cancer (2011) 128(12):2911–22. doi:10.1002/ijc.25635
41. Santourlidis S, Trompeter HI, Weinhold S, Eisermann B, Meyer KL, Wernet P, et al. Crucial role of DNA methylation in determination of clonally distributed killer cell Ig-like receptor expression patterns in NK cells. J Immunol (2002) 169(8):4253–61. doi:10.4049/jimmunol.169.8.4253
42. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol (2013) 132(3):536–44. doi:10.1016/j.jaci.2013.07.006
43. Bryceson YT, Chiang SC, Darmanin S, Fauriat C, Schlums H, Theorell J, et al. Molecular mechanisms of natural killer cell activation. J Innate Immun (2011) 3(3):216–26. doi:10.1159/000325265
44. Chan CJ, Smyth MJ, Martinet L. Molecular mechanisms of natural killer cell activation in response to cellular stress. Cell Death Differ (2014) 21(1):5–14. doi:10.1038/cdd.2013.26
45. Bakker AB, Phillips JH, Figdor CG, Lanier LL. Killer cell inhibitory receptors for MHC class I molecules regulate lysis of melanoma cells mediated by NK cells, gamma delta T cells, and antigen-specific CTL. J Immunol (1998) 160(11):5239–45.
46. Naumova E, Mihaylova A, Ivanova M, Mihailova S. Impact of KIR/HLA ligand combinations on immune responses in malignant melanoma. Cancer Immunol Immunother (2007) 56(1):95–100. doi:10.1007/s00262-006-0151-9
47. Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, et al. Active genes are tri-methylated at K4 of histone H3. Nature (2002) 419(6905):407–11. doi:10.1038/nature01080
48. Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell (2007) 25(1):15–30. doi:10.1016/j.molcel.2006.12.014
49. Davies GE, Locke SM, Wright PW, Li H, Hanson RJ, Miller JS, et al. Identification of bidirectional promoters in the human KIR genes. Genes Immun (2007) 8(3):245–53. doi:10.1038/sj.gene.6364381
50. Liu Y, Kuick R, Hanash S, Richardson B. DNA methylation inhibition increases T cell KIR expression through effects on both promoter methylation and transcription factors. Clin Immunol (2009) 130(2):213–24. doi:10.1016/j.clim.2008.08.009
51. Presnell SR, Zhang L, Ramilo CA, Chan HW, Lutz CT. Functional redundancy of transcription factor-binding sites in the killer cell Ig-like receptor (KIR) gene promoter. Int Immunol (2006) 18(8):1221–32. doi:10.1093/intimm/dxl043
52. van Bergen J, Stewart CA, van den Elsen PJ, Trowsdale J. Structural and functional differences between the promoters of independently expressed killer cell Ig-like receptors. Eur J Immunol (2005) 35(7):2191–9. doi:10.1002/eji.200526201
53. Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol (1999) 19(8):5504–11. doi:10.1128/MCB.19.8.5504
54. Miller DM, Polansky DA, Thomas SD, Ray R, Campbell VW, Sanchez J, et al. Mithramycin selectively inhibits transcription of G-C containing DNA. Am J Med Sci (1987) 294(5):388–94. doi:10.1097/00000441-198711000-00015
55. Ray R, Snyder RC, Thomas S, Koller CA, Miller DM. Mithramycin blocks protein binding and function of the SV40 early promoter. J Clin Invest (1989) 83(6):2003–7. doi:10.1172/JCI114110
Keywords: DNA methyltransferase inhibitor, adoptive cell immunotherapy, DNA methylation, killer Ig-like receptors, myelodysplastic syndrome
Citation: Niu C, Li M, Zhu S, Chen Y, Zhou L, Xu D, Li W, Cui J, Liu Y and Chen J (2018) Decitabine Inhibits Gamma Delta T Cell Cytotoxicity by Promoting KIR2DL2/3 Expression. Front. Immunol. 9:617. doi: 10.3389/fimmu.2018.00617
Received: 31 October 2017; Accepted: 12 March 2018;
Published: 26 March 2018
Edited by:
Anahid Jewett, University of California, Los Angeles, United StatesReviewed by:
Kerry S. Campbell, Fox Chase Cancer Center, United StatesZong Sheng Guo, Harvard University, United States
Copyright: © 2018 Niu, Li, Zhu, Chen, Zhou, Xu, Li, Cui, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiuwei Cui, Y3VpandAamx1LmVkdS5jbg==;
Yongjun Liu, eW9uZy1qdW4ubGl1QHNhbm9maS5jb20=;
Jingtao Chen, anRjaGVuQGpsdS5lZHUuY24=