 Brianne Cruickshank1†
Brianne Cruickshank1† Michael Giacomantonio1†
Michael Giacomantonio1† Paola Marcato1,2
Paola Marcato1,2 Sherri McFarland3,4
Sherri McFarland3,4 Jonathan Pol5,6,7,8,9*
Jonathan Pol5,6,7,8,9* Shashi Gujar1,2,10,11*
Shashi Gujar1,2,10,11*
- 1Department of Pathology, Dalhousie University, Halifax, NS, Canada
- 2Department of Microbiology and Immunology, Dalhousie University, Halifax, NS, Canada
- 3Department of Chemistry and Biochemistry, The University of North Carolina at Greensboro, Greensboro, NC, United States
- 4Department of Chemistry, Acadia University, Wolfville, NS, Canada
- 5Gustave Roussy Comprehensive Cancer Institute, Villejuif, France
- 6INSERM, U1138, Paris, France
- 7Equipe 11 labellisée par la Ligue Nationale contre le Cancer, Centre de Recherche des Cordeliers, Paris, France
- 8Université Paris Descartes, Université Sorbonne Paris Cité, Paris, France
- 9Université Pierre et Marie Curie, Paris, France
- 10Department of Biology, Dalhousie University, Halifax, NS, Canada
- 11Centre for Innovative and Collaborative Health Services Research, IWK Health Centre, Halifax, NS, Canada
Immunogenic cell death (ICD) activates both innate and adaptive arms of the immune system during apoptotic cancer cell death. With respect to cancer immunotherapy, the process of ICD elicits enhanced adjuvanticity and antigenicity from dying cancer cells and consequently, promotes the development of clinically desired antitumor immunity. Cancer ICD requires the presentation of various “hallmarks” of immunomodulation, which include the cell-surface translocation of calreticulin, production of type I interferons, and release of high-mobility group box-1 and ATP, which through their compatible actions induce an immune response against cancer cells. Interestingly, recent reports investigating the use of epigenetic modifying drugs as anticancer therapeutics have identified several connections to ICD hallmarks. Epigenetic modifiers have a direct effect on cell viability and appear to fundamentally change the immunogenic properties of cancer cells, by actively subverting tumor microenvironment-associated immunoevasion and aiding in the development of an antitumor immune response. In this review, we critically discuss the current evidence that identifies direct links between epigenetic modifications and ICD hallmarks, and put forward an otherwise poorly understood role for epigenetic drugs as ICD inducers. We further discuss potential therapeutic innovations that aim to induce ICD during epigenetic drug therapy, generating highly efficacious cancer immunotherapies.
Introduction
Antitumor T cells can detect and eliminate cancer cells in a highly precise, antigen-specific fashion. Appropriately activated antitumor T cells can target cancer cells at both local and metastatic sites and, most importantly, can kill existing as well as possibly relapsing cancerous cells. Numerous patient cohort studies thus far have reported a clear positive correlation between the activities of antitumor T cells and better patient outcomes (1–3). Therapeutic interventions promoting antitumor T cell immunity are at the forefront of next-generation cancer therapeutic strategies and as such are highly desired in clinics.
Promoting antitumor T cell responses in cancer-bearing hosts is challenging (4). This is largely because cancers employ numerous evasion strategies that are non-conducive toward T cell activation and function. In particular, cancer-associated immune evasion is supported through the plastic nature of the tumor microenvironment (TME), which harbors the processes that actively suppress the development of antitumor T cells. Some prominent examples of such evasion mechanisms include the presence of immunosuppressive cytokines like transforming growth factor beta 1 (TGF-β) and immune checkpoint molecules such as programmed death-ligand 1 (PD-L1). In addition, immune cells such as myeloid-derived suppressor cells and regulatory T cells contribute to the ability of cancers to evade the immune system (5, 6). Moreover, decreased tumor antigen presentation in the TME further contributes to the impaired functions of antigen-presenting cells (APCs). Consequently, antitumor T cells remain impaired or absent in the immunosuppressive TME, and the tumor persists. Not surprisingly, many modern-day immunotherapies focus on correcting the underlying TME-associated immune evasion strategies, with the goal of facilitating the initiation of an antitumor T cell response (7).
Functional activation of antitumor T cells requires three signals: (#1) tumor antigen presentation in the context of major histocompatibility complex (MHC), (#2) co-stimulatory signals such as cluster of differentiation 28 signaling, and (#3) the presence of cytokines like interferons (IFNs) (8, 9). Although the TME actively discourages the presence of one or more of these essential signals, therapeutic interventions can be used to overcome these immunosuppressive effects. One such strategy is to induce immunogenic cell death (ICD) during cancer therapy (10). As the name suggests, ICD is a process where apoptotic cells elicit an immunogenic response through the induction of damage-associated molecular patterns (DAMPs) that can be recognized by various immune cells (11, 12). More specifically, through the release of DAMPs, ICD increases the adjuvanticity, facilitating the signals # 2 and 3, within TME (10). This occurs through the production of chemoattracting agents such as chemokine C–X–C motif ligand (CXCL) 1 and chemokine (C–C motif) ligand 2 (CCL2) by dying cancer cells and subsequent recruitment of innate immune cells such as neutrophils and DCs to the TME (13). These events, in combination with both the release of nucleic acids from dying cancer cells and a cascade of other DAMPs, enable neo-epitope presentation of the cancer cell (10). This increased antigenicity, facilitating the signal # 1, is reflected through the ICD-enhanced antigen presentation (capture, processing, and presentation via MHC) from recruited APCs (10, 13, 14). Consequently, this leads to the activation of T cell response. Importantly, ICD-induced T cell immunity can establish immunological memory capable ensuring the longevity of remission, as opposed to non-regulated cell death. Such processes have been linked to tumor cell death in in vitro as well as in vivo mouse models (15). Taken together, ICD enhances the adjuvanticity and antigenicity of the cancer cells in the TME and facilitates the development of the three essential signals discussed earlier that are necessary for the activation of antitumor T cell responses (10).
For ICD to be successfully induced, the onset of a specific combination of DAMPs is required. The exact combination of DAMPs needed to induce ICD lies outside of the scope of this review and has been described elsewhere (10, 16). It is important to note, however, that the DAMPs that drive the induction of ICD are dependent on the treatment modality which is being used. For example, while chemotherapy-induced ICD requires the induction of autophagy, pathogen-induced ICD does not (10). Regardless, in the context of ICD, the initiation of an immune response begins with the release of lymphocyte chemoattracting agents, and the presentation of early apoptotic surface markers that tag dying cells for phagocytosis by APCs. In this process, the unfolded protein response (UPR) causes the translocation and expression of endoplasmic reticulum (ER) chaperones, such as calreticulin (CALR), to the cell surface. The induction of autophagy enables the cell to attract APCs to the TME via the release of intracellular ATP stores. This further functions to activate both inflammasome signaling and the APCs themselves (17). The secretion of annexin A1 (ANXA1) helps guide the APCs to the dying cancer cells where they become activated. In addition, the extracellular release of high-mobility group box-1 (HMGB1) stimulates an inflammatory response via toll-like receptor (TLR)-4 signaling (18). This involves the induction of the type 1 IFN response, resulting in CXCL10 release that enables neutrophil, APC, and T cell recruitment (10, 19, 20). Cumulatively, these ICD hallmarks activate APCs, which then stimulate antitumor T cells, leading to tumor eradication.
Interestingly, the expression of many of these ICD-associated DAMPs is governed by small heritable changes to the genome called epigenetic modifications. Epigenetic modifications result in changes to gene expression through chromatin remodeling mechanisms that include DNA methylation, histone modification, and non-coding RNA (ncRNA) (21, 22). Epigenetic modifications can silence or activate genes involved in tumor suppression or oncogenesis, respectively. In relation to immunity, epigenetic modifying drugs have the potential to boost the immune response by increasing antigen presentation, the expression of co-stimulatory molecules, and the display of MHC molecules; all paving the way for more efficient antigen presentation to T cells (23). In particular, DNA methylation has been investigated in many immune-related studies, where it silences genes such as TLR-3 and mitochondrial–antiviral signaling protein (24–26). Therefore, it is plausible that epigenetic modifications have a regulatory role to play when considering the induction of antitumor immunity.
In this review, we propose that various epigenetic events are actively involved in the regulation of ICD-associated DAMP expression. By recognizing that epigenetic modifications are involved in the induction of individual DAMPs, the efficacy of many cancer immunotherapies can be improved. Herein, we extensively discuss the current evidence that identifies direct links between epigenetic modifications and ICD in the context of TME and cancer immunotherapy.
Epigenetic Regulation of ICD Hallmarks
In the context of cancer therapy, ICD occurs when a therapeutic treatment induces the expression of a specific combination of “hallmarks” during cancer cell death. These hallmarks are a set of premortem stress responses that promote the expression of “danger signals” from the dying cancer cell, which can then be recognized by immune cells to trigger antitumor T cell activation. As shown in Figure 1A, major ICD hallmarks consist of various DAMPs that inevitably result in the development of T cell immunity.
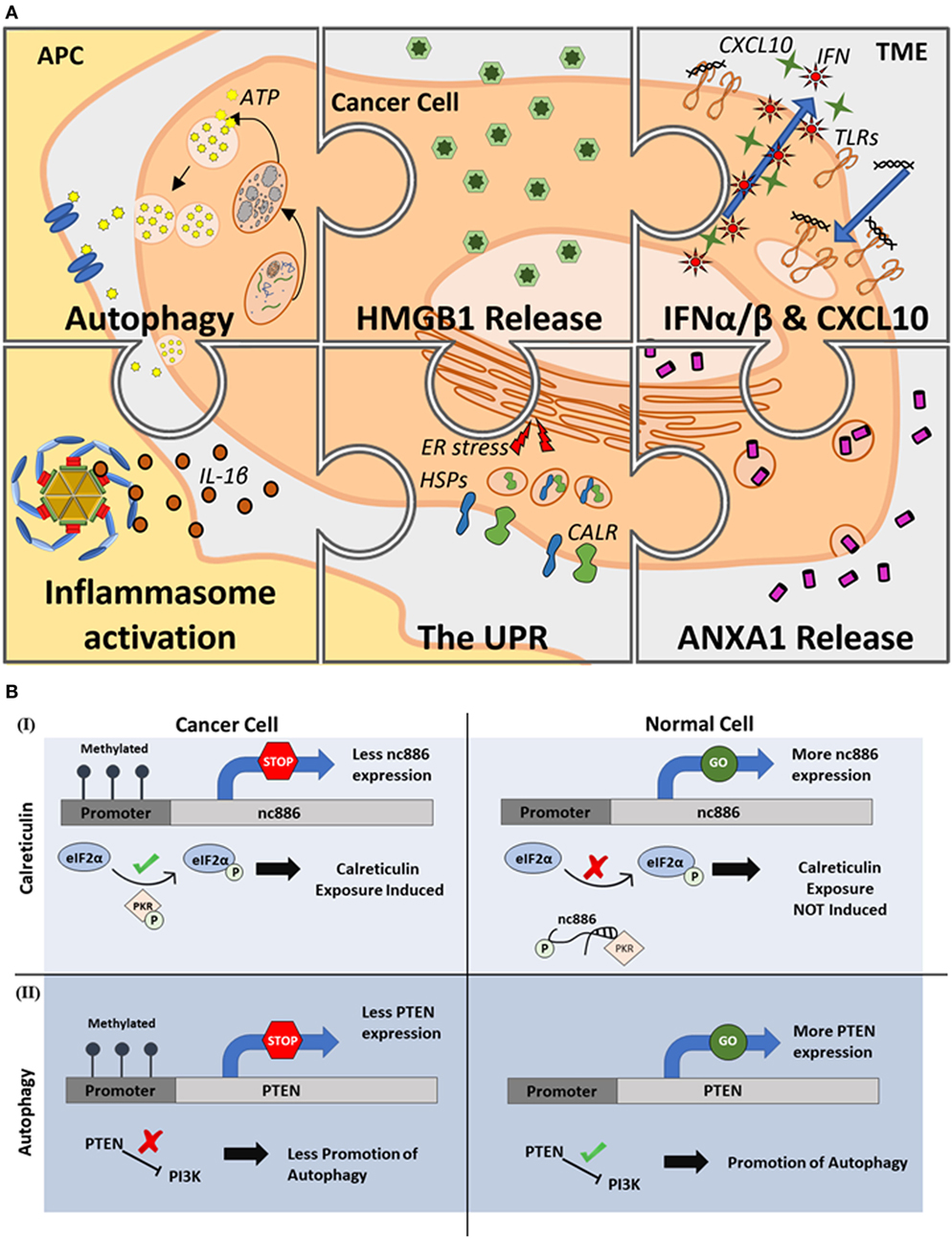
Figure 1. Epigenetic regulation of immunogenic cell death (ICD). (A) Major hallmarks of ICD. Induction of ICD has been shown to be associated with six major hallmark processes including the induction of autophagy and release of ATP, high-mobility group box-1 (HMGB1) and annexin A1 (ANXA1) release, toll-like receptor (TLR) signaling that leads to interferon (IFN) α/β and CXCL10 release, inflammasome activation and interleukin-1β (IL-1β) secretion, and endoplasmic reticulum (ER) stress causing the unfolded protein response (UPR) that induces ER chaperones, especially calreticulin (CALR), expression on the cell surface. (B) Positive and negative regulation of ICD through epigenetic mechanisms. As illustrated through two distinct examples, activatory (I) or suppressive (II) effects of DNA methylation can either promote or suppress the molecular events leading to ICD. [(B), I], DNA methylation events positively influence the induction of ICD by suppressing the expression of a non-coding RNA (nc886) whose function prevents the successful phosphorylation of eukaryotic transcription initiation factor 2 (eIF2α) inhibiting CALR exposure. [(B), II] DNA methylation events negatively influence the induction of ICD by suppressing the expression of phosphatase and tensin homolog (PTEN) whose expression is needed to initiate pathways leading to autophagy initiation. Abbreviations: APC, antigen-presenting cell; TME, tumor microenvironment.
What is becoming increasingly clear is that most of the ICD hallmarks are directly or indirectly regulated through epigenetic mechanisms. In addition, many currently investigated therapeutic epigenetic modulators (e.g., HDAC inhibitors) are being recognized for their actions in dendritic cell activation, antigen uptake, and T cell activation (27, 28) (Table 1). Thus, the epigenome, through its inherent or therapeutically modified activities, can be exploited to harness the antitumor benefits of ICD.
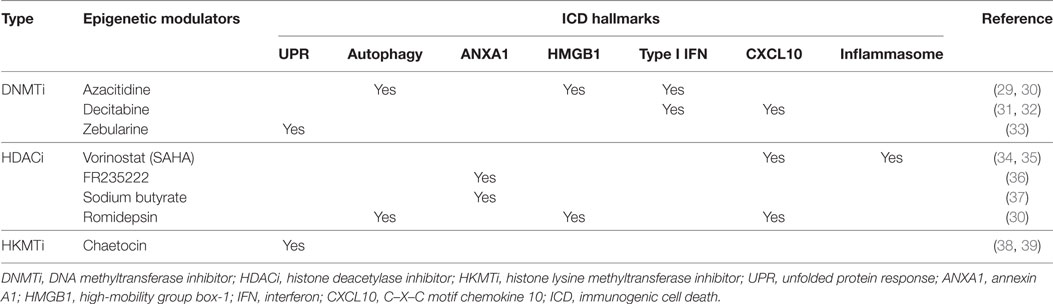
Table 1. Epigenetic modulators shown to induce the expression of various ICD hallmarks, studied outside the context of ICD.
The UPR and ER Chaperones
The ER is critically important in the synthesis, modification, and transport of proteins (40, 41). When under physiological stress, the ER initiates the UPR, an evolutionarily conserved mechanism which, in the context of ICD, is characterized by the translocation of ER chaperones to the cell surface (42). Herein, ER chaperones function as “eat me” signals that mark the cell for uptake by APCs (10). Some ER chaperones that have been implicated in ICD include heat shock proteins (HSPs, e.g., HSP70 and HSP90) as well as CALR (43). The UPR is initiated by the activation of three main stress sensors; inositol-requiring enzyme-1 (IRE1), protein kinase RNA-like ER kinase (PERK), and transcription factor 6 (44). The ER chaperone immunoglobin protein (BiP) binds to IRE1 and PERK, suppressing their activity (45). Under ER stress conditions, BiP binds to misfolded proteins and no longer suppresses the sensor’s activity, triggering the UPR (44).
One of the implicated sensors, IRE1, increases in expression upon treatment with an inhibitor of histone lysine methyltransferase (HKMTi). HKMT enzymes work by transferring methyl groups to lysine residues of histone proteins, and in this case, result in the transcriptional silencing of IRE1 (46, 47). Specifically, treatment of lung cancer cells with Chaetocin (Table 1), a non-specific HKMTi, increases the expression of IRE1, suggesting that not only is IRE1 regulated via BiP but it may also be regulated via histone methylation (38, 39).
In relation to the UPR, HSP expression increases in response to stress stimuli as an effort to cope with the denaturation of proteins (48). Two types of HSPs (HSP70 and HSP90) have been shown to be directly regulated via promoter methylation in mammalian cells (49, 50).
The distal portion of the promoter region of HSP70 is aberrantly methylated during thermal stress, restricting the binding of POU class 2 homeobox-1 (POU2F1) to the HSP70 promoter (51). In T cells, the role of POU2F1 has been shown to contribute to the timing of cytokine expression in CD4 T cells and has also been shown to promote the development of effector T cell lineages (52). In addition, the expression of HSP90 is inhibited by DNA methylation in both pancreatic and colon cancer cell lines as a consequence of enhanced DNA methyltransferase (DNMT) expression (53). HSP90 regulates the transcription of DNMT enzymes, where HSP90s decrease in expression has been shown to result in an increased expression of DNMTs. DNMT-mediated hypermethylation events then result in the silencing of known tumor suppressor genes (TSGs). For aberrantly methylated HSPs, epigenetic drugs such as Zebularine may correct detrimental hypermethylation events in addition to inducing the UPR helping to induce a more robust immune response (Table 1). Other ER chaperones, such as CALR, are regulated by the presence of ncRNAs whose promoters are hypermethylated in some cancer models (54, 55). In this case, methylation events can work in favor of inducing CALR exposure.
Calreticulin is the most studied “eat me” signal in regards to ICD and is a main player in cultivating the ICD-induced antitumor response (56–58). Although their roles have not been fully elucidated, ncRNA such as RB1 and miR-27a are beginning to be recognized as key players in regulating CALR exposure (54, 55). Within the recent years, however, the roles of a few ncRNA have indeed been more thoroughly analyzed. For example, nc886 has been shown to regulate phosphorylation events that are necessary for proper CALR exposure (Figure 1B) (59). Specifically, eukaryotic transcription initiation factor 2 (eIF2α) is an important protein involved in the exposure of CALR and must be phosphorylated for CALR exposure to be initiated (60). In cholangiocarcinoma, the downregulation nc886 leads to the induction of apoptosis through the phosphorylation of eIF2α (59). When compared with normal tissues, gastric cancer cell lines were found to have hypermethylated CpG islands in the nc886 gene (61). Hypermethylation of nc886 prevents its typical function of discouraging the activation of protein kinase R (PKR), allowing proper phosphorylation of eIF2α and subsequent CALR exposure (Figure 1B). Conversely, expression of nc886 discourages CALR exposure; its expression prevents PKR from catalyzing the phosphorylation of eIF2α, revealing an adverse effect that may be observed with the use of an epigenetic modifying drug. These drugs could further have adverse effects by co-upregulating counterbalancing molecules of ICD hallmarks, such as CD47, a molecule that offsets the pro-phagocytic functions of CALR (62).
Autophagy Induction
Autophagy is an evolutionarily conserved mechanism that functions to maintain cellular homeostasis during times of starvation and stress (63). The induction of autophagy enables harmful or damaged cellular components to be sequestered into autophagosomes, and then broken down via lysosomal degradation (64). While the role of autophagy in cancer is still being fully elucidated, it appears to be context dependent (65, 66).
During the process of ICD, the induction of autophagy results in vesicular ATP pools to be transported and secreted from the cell (10). The secretion of ATP activates signaling pathways via purinergic receptors P2Y2 (P2RY2) and P2RX7 acting as a “find me” signals that promote maturation as well as TME recruitment of APCs (19, 20, 67). In APCs, the interaction of ATP with P2RY2 induces a robust chemotactic effect, while its interaction with P2RX7 results in the release of immunostimulatory cytokines (67). The expression of the P2RX7 receptor has been shown to be controlled via promoter methylation in submandibular carcinomas, where aberrant methylation events decrease its expression, which presumably would prevent proper P2RX7 signaling during ICD (68).
It is important to note that while other mechanisms are capable of triggering ATP release (69–71), a successful autophagic response is required for the optimal levels of ATP to be released for an immunogenic response (10, 64, 67). Autophagy induction requires multiple cellular processes to occur in tandem, such as the expression of TSGs phosphatase, tensin homolog (PTEN), and autophagy-related protein 5 (ATG5) (10). PTEN promotes autophagy by inhibiting the activation of phosphoinositide 3-kinase (PI3K) signaling (67) (Figure 1B), while ATG5 mediates autophagosome formation (72). Interestingly, PTEN is one of the most commonly mutated or inactivated genes during cancer development (73). The PTEN promoter is also known to be hypermethylated in breast and gastric cancers, as well as in melanoma and soft tissue sarcomas (74–77). During the development of many cancers, including colorectal cancer and melanoma, ATG5 is often downregulated (78, 79). Interestingly, it has been demonstrated in melanoma that ATG5 downregulation is a consequence of a hypermethylation of the promoter site (79). The hypermethylation status of these genes in cancers represents an ideal target for demethylating agents (e.g., Azacitidine) to promote autophagy (Table 1).
It is possible to induce autophagy through many mechanisms. Caloric restriction mimetics (CRMs), which induce autophagy by mimicking biochemical effects of nutrient deprivation, have been shown to stimulate ATP release in a protein deacetylation-dependent manner. Specifically, CRMs influence the acetylation of histone proteins, ultimately influencing gene expression and displaying a potential epigenetic mechanism that influences whether or not autophagy is induced (32, 80). Related, autophagy can also be induced via photodynamic therapy. Following exposure to photosensitizers, multiple human cancer cell lines showed the surface expression of CALR and released ATP before the signatures of apoptosis could be detected. In fact, both of these processes seem to have overlapping regulatory mechanisms, operating through PERK signaling and PI3K pathways, suggesting that the interplay between ICD and DAMP induction requires further elucidation (58).
ANXA1 Release
Annexin A1, known for its immunosuppressive functions (81), has recently been found to contribute to DC function during ICD. Here, ANXA1 released from the apoptotic cells can bind to formyl peptide receptor 1 receptor on APCs, enabling the stable interaction between the APC and dying cancer cell (82–84). As such, ANXA1 functions to enable antigen uptake and cross presentation of tumor antigens (84). Interestingly, ANXA1 is silenced by methylation in nasopharyngeal cancer cell lines and aberrantly methylated in breast and non-small cell lung cancer (85–88). Here, the use of a DNMTi may be an attractive tool, allowing the restoration of ANXA1 expression and secretion in the context of ICD (Table 1).
In head and neck squamous carcinoma, the expression of ANXA1 is inversely correlated with the expression of a specific microRNA (miRNA-196). This miRNA directly targets ANXA1 by binding to the untranslated region on the ANXA1 mRNA transcript (82). The expression of this miRNA is controlled by DNA methylation in many different cancer cell lines including breast, colon, liver, lung, brain, and oral (89). Without the expression of miRNA-196, ANXA1 would no longer be silenced, allowing proper ANXA1 release during ICD induction. As mentioned, epigenetic modifiers (either hypo- or hypermethylating) can be employed in the context of cancers and TME. In this case, induction of de novo DNA hypermethylation by inserting CpG-free DNA may control the miRNA-196-regulated release of ANXA1 for ICD induction (90).
HMGB1 Release
High-mobility group box-1 is found in nearly all eukaryotic cells and is highly conserved and abundant (91). Much like autophagy, it is important to note that HMGB1 has both a positive and negative correlation in regard to cancer progression (92). HMGB1 has multiple roles: within the nucleus it facilitates the transcription of many genes by modulating nucleosomes, while when secreted, it functions as a DAMP (91, 93). The mechanisms that regulate this secretion, however, remain unclear (10, 94). Extracellular HMGB1 signaling is facilitated by numerous receptors, where its binding is heavily dependent on its redox form (94). An important signaling pathway in the context of ICD is the extracellular binding of HMGB1 to TLR4 on APCs, initiating a signal transduction through the adaptor protein MyD88 (10, 18, 94). This pathway has been shown to be required to evoke ICD and subsequent T cell immunity, as Tlr4−/− and Myd88−/− mice do not develop antitumor immunological memory (18).
The link between HMGB1 and epigenetic regulation has already been hypothesized, and it is postulated that HMGB1 itself acts as an epigenetic modifier that leads to the silencing of tumor necrosis factor alpha and interleukin-1 beta (IL-1β) (91). Like the miRNA-196-based regulatory mechanism discussed in relation to ANXA1 release, miRNA-129-2, a tumor suppressor in glioma and hepatocellular carcinoma (95, 96), directly targets HMGB1 and inhibits its release. The regulatory region of this miRNA is heavily methylated in portions of its promoter region, resulting in its suppression and subsequent expression of HMGB1 (97, 98). In relation to gliomas, this methylation occurs more frequently in cancerous tissues when compared with normal tissues (95). As with some of the previously discussed ICD hallmarks, this implication is positive in relation to ICD. Interestingly, inducing expression of this miRNA would not be beneficial in this case. In fact, similar to ANXA1, the induction of de novo methylation in models where this miRNA is expressed may be a better choice of treatment (90).
Type I IFN Production and CXCL10 Secretion
Type I IFNs are secreted as DAMPs from infected cells to both signal and activate antimicrobial responses and initiate the innate and adaptive immune system (10). Characteristic type I IFNs (IFNα and IFNβ) primarily signal through the heterodimeric IFNα receptor (10, 99), eliciting a vast range of responses that are dependent on environmental factors, the extent of infection, and the hosts’ physiological status (99). In the context of ICD, a major role for type I IFNs is to activate signaling cascades to produce more IFNs that act in both an autocrine and paracrine fashion (10). Moreover, like ATP secretion, type I IFNs also act as chemokines to attract APCs to the TME and play a pivotal role in APC maturation and T cell activation (100, 101). Thus, type I IFNs are important mediators of the signals # 2 (co-stimulatory signals) and 3 (cytokine presence) that are required for the induction of T cell immunity.
A clear link exists between epigenetic regulation and IFNs. First, expression of HDAC3 (a histone deacetylase) has been found to be necessary for IFN-β expression showing the regulatory role of HDAC3 in controlling IFN-β expression (99). Second, CXCL10 secretion is a subsequent result of IFN signaling (102). Its expression has been found to increase upon treatment with demethylating agents in ovarian cancer cells, suggesting that promoter methylation controls CXCL10 expression (103). The epigenetic regulation of CXCL10 in ovarian cancer suggests that treatment with a demethylating agent such as Decitabine (which is also known to induce type I IFN signaling) could aid in the induction of ICD (Table 1).
NLRP3 Inflammasome Signaling
During ICD induction, DAMPs are able to trigger pro-inflammatory events by activating inflammasomes (104). Inflammasomes are large multi-protein complexes, often consisting of caspase 1, within which the maturation of pro-inflammatory cytokines such as IL-1β and IL-18 takes place (104). One of the most well-characterized inflammasome complexes is called NLR family pyrin domain containing 3 inflammasome (NLPR3), which consists of a caspase recruiting domain (ASC), a cytosolic pattern recognition receptor, and a pro-caspase 1 (105). In 2014, Salminen et al. found that the ASC domain is identical to the domain that was termed as methylation-induced silencing-1 (TMS1) (105). The same study outlined that the promoter of the TSM1 gene is aberrantly methylated in many cancer cell lines, suggesting that this process regulates the expression of inflammasomes and the induction of apoptosis. Assuming methylation events alter the function of the ASC domain, aberrant events may prevent the successful induction of ICD by preventing proper inflammasome formation.
Another layer of complexity is added when the production of IL-1β is considered. Many interleukin genes have been shown to be methylated in cancers (106–108). In addition, interleukins have also been shown to have powerful antitumor roles by inhibiting the growth of lung tumors, and by stimulating the immune system to engage antiangiogenic mechanisms (109). Interestingly, the promoter of IL-1β has the highest methylation status out of all interleukin genes studied in lung cancer (29). In this model, aberrant promoter methylation of this important ICD-related interleukin would prevent the successful induction of ICD, revealing a potential therapeutic target using a demethylating drug (Table 1).
Conclusion and Future Directions
The immunogenic response initiated through ICD can overcome the immunosuppressive nature of the TME. This leads to the restoration of the three signals required for proper T cell activation, including increased antigen presentation following cancer cell apoptosis and phagocytosis (signal # 1), co-stimulation from matured and recruited APCs (signal # 2), and the production of cytokines from both the cancer (e.g., IFNs) and APCs (e.g., IL-1β) (signal # 3). Therefore, the successful induction of ICD leads to the activation of antitumor T cells, which in turn can kill cancer cells and prevent recurring disease. Therefore, understanding how epigenetic modifications contribute to ICD is important when aiming to improve the efficacy of current cancer immunotherapies.
As highlighted above, many of the processes that govern ICD are regulated through epigenetic modifications. Interestingly, the initiation of individual ICD hallmarks, upon treatment with epigenetic modifying drugs, has been observed in studies that may not have been directly evaluating ICD induction (Table 1). As a result, the combination of epigenetic modifiers and immunotherapies offer an attractive avenue to elicit more robust antitumor T cell immunity. In fact, this concept is already being applied. The combination of Azacitidine and Romidepsin with IFNα elicits bona fide ICD in colorectal cancer cells (30). Further, treatment with Decitabine triggers a “viral” or “altered-self” mimicry state in these cells that leads to ICD hallmark expression through the retinoic acid inducible gene-I (RIG-I) pathway (31). This pathway has been shown to evoke ICD in melanoma, acute-promyelocytic leukemia, and pancreatic cancer models (31). Most recently, this concept has been shown to be important in neutrophil-based anticancer activity, where apoptotic cancer cells release epigenetically regulated cytokines such as CXCL1, CXCL10, and CCL2, driving nucleic acid-elicited phagocytosis of dying cancer cells by neutrophils (13, 110, 111).
However, it is still important to consider the possibility that epigenetic modifications could negatively affect the ability of CD8 T cells to recognize a cancer cell through ICD. It has already been established that epigenetic mechanisms tightly regulate the expression of MHC molecules, cytokines and other co-stimulatory molecules (112). Therefore, it cannot be ignored that adjusting these regulatory pathways using epigenetic modifiers may reduce the successful activation of specific CD8 T cells. In addition, increasing the secretion of a desired DAMP using epigenetic modulators may affect the expression of checkpoint molecules such as PD-L1 or suppressive metabolites such as indoleamine 2,3-dioxygenase (IDO1) in the cancer cells, causing them to respond to immunotherapeutic strategies differently. It has also been established that the miRNA-regulated mechanisms that control the expression of PD-L1 are also involved in the repression of IDO1 in cancer cells (113, 114). These points stress the complex relationship that exists between using epigenetic modifiers and their effect on ICD DAMPs.
Finally, the context-dependent roles of DAMPs must also be noted while considering ICD-based implications. For example, while HMGB1 excretion is involved in DC-based nucleic acid-sensing systems in ICD (115), it has also been shown to silence the expression of IL-1β in severe systemic inflammation by binding with histone H1, causing a change from euchromatin to heterochromatin at the IL-1β promoter (116). Therefore, the induction of one process (e.g., autophagy) that regulates a hallmark may suppress another (e.g., CALR exposure). Thus, when aiming to improve cancer therapy using epigenetic modifiers to induce hallmarks of ICD, the methylation status of ICD-related genes should be analyzed in each cancer model. This will enable an evaluation of both the benefits and adverse events that could result from the treatment modality of interest. Nonetheless, there is an undeniable link between the regulation of ICD hallmarks and epigenetics that cannot be ignored when evaluating the efficacy of novel cancer treatments.
Author Contributions
BC and MG: conception, research, writing, editing. SG: conception, research, writing, editing, funding. PM, JP, and SM: research, writing, editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the Canadian Institute of Health Research (CIHR) and Beatrice Hunter Cancer Research Institute (BHCRI). BC is supported by Nova Scotia Graduate Scholarship (NSGS), BHCRI scholarship, and Killam Predoctoral Scholarship.
References
1. Cassetta L, Pollard JW. Repolarizing macrophages improves breast cancer therapy. Cell Res (2017) 27(8):963. doi:10.1038/cr.2017.63
2. Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol (2017) 18(8):940–50. doi:10.1038/ni.3775
3. Srivastava RM, Trivedi S, Concha-Benavente F, Gibson SP, Reeder C, Ferrone S, et al. CD137 stimulation enhances cetuximab-induced natural killer: dendritic cell priming of antitumor T-cell immunity in patients with head and neck cancer. Clin Cancer Res (2017) 23(3):707–16. doi:10.1158/1078-0432.CCR-16-0879
4. Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer (2016) 4:51. doi:10.1186/s40425-016-0156-7
5. Rodríguez PC, Zea AH, Ochoa AC. Mechanisms of tumor evasion from the immune response. Cancer Chemother Biol Response Modif (2003) 21:351–64. doi:10.1016/S0921-4410(03)21018-8
6. Wang M, Zhang C, Song Y, Wang Z, Wang Y, Luo F, et al. Mechanism of immune evasion in breast cancer. Onco Targets Ther (2017) 10:1561–73. doi:10.2147/OTT.S126424
7. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi:10.1016/j.cell.2017.01.017
8. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol (2013) 13(4):257. doi:10.1038/nri3403
9. Shissler SC, Lee MS, Webb TJ. Mixed signals: co-stimulation in invariant natural killer T cell-mediated cancer immunotherapy. Front Immunol (2017) 8:1447. doi:10.3389/fimmu.2017.01447
10. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol (2016) 17(2):97–111. doi:10.1038/nri.2016.107
11. Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X, et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials (2016) 102:187–97. doi:10.1016/j.biomaterials.2016.06.032
12. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer (2012) 12(12):860–75. doi:10.1038/nrc3380
13. Garg AD, Vandenberk L, Fang S, Fasche T, Van Eygen S, Maes J, et al. Pathogen response-like recruitment and activation of neutrophils by sterile immunogenic dying cells drives neutrophil-mediated residual cell killing. Cell Death Differ (2017) 24(5):832–43. doi:10.1038/cdd.2017.15
14. Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev (2017) 280(1):126–48. doi:10.1111/imr.12574
15. Fucikova J, Kralikova P, Fialova A, Brtnicky T, Rob L, Bartunkova J, et al. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res (2011) 71(14):4821–33. doi:10.1158/0008-5472.CAN-11-0950
16. Gebremeskel S, Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget (2015) 6(39):41600–19. doi:10.18632/oncotarget.6113
17. Garg AD, Martin S, Golab J, Agostinis P. Danger signalling during cancer cell death: origins, plasticity and regulation. Cell Death Differ (2014) 21(1):26. doi:10.1038/cdd.2013.48
18. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med (2007) 13(9):1050–9. doi:10.1038/nm1622
19. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ (2014) 21(1):79–91. doi:10.1038/cdd.2013.75
20. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31(1):51–72. doi:10.1146/annurev-immunol-032712-100008
21. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol (2010) 28(10):1057. doi:10.1038/nbt.1685
22. Baylin SB. DNA methylation and gene silencing in cancer. Nat Rev Clin Oncol (2005) 2(S1):S4–11. doi:10.1038/ncponc0354
23. Héninger E, Krueger TEG, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol (2015) 6:29. doi:10.3389/fimmu.2015.00029
24. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell (2015) 162(5):974–86. doi:10.1016/j.cell.2015.07.011
25. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol (2007) 37(9):2378–89. doi:10.1002/eji.200737594
26. Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov (2006) 5(1):37–50. doi:10.1038/nrd1930
27. McCaw TR, Randall TD, Forero A, Buchsbaum DJ. Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy (2017) 9(16):1359–72. doi:10.2217/imt-2017-0134
28. Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol (2017) 87:227–39. doi:10.1016/j.molimm.2017.04.012
29. Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediators Inflamm (2015) 2015:201703. doi:10.1155/2015/201703
30. Buoncervello M, Romagnoli G, Buccarelli M, Fragale A, Toschi E, Parlato S, et al. IFN-α potentiates the direct and immune-mediated antitumor effects of epigenetic drugs on both metastatic and stem cells of colorectal cancer. Oncotarget (2016) 7(18):26361–73. doi:10.18632/oncotarget.8379
31. Roulois D, Yau HL, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell (2015) 162(5):961–73. doi:10.1016/j.cell.2015.07.056
32. Mariño G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell (2014) 53(5):710–25. doi:10.1016/j.molcel.2014.01.016
33. Yang P-M, Lin Y-T, Shun C-T, Lin S-H, Wei T-T, Chuang S-H, et al. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci Rep (2013) 3:3219. doi:10.1038/srep03219
34. Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martínez Leal JF, de La Cueva P, et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica (2010) 95(4):613–21. doi:10.3324/haematol.2009.013870
35. Abujamra AL, dos Santos MP, Roesler R, Schwartsmann G, Brunetto AL. Histone deacetylase inhibitors: a new perspective for the treatment of leukemia. Leuk Res (2010) 34(6):687–95. doi:10.1016/j.leukres.2009.08.021
36. Petrella A, D’Acunto CW, Rodriquez M, Festa M, Tosco A, Bruno I, et al. Effects of FR235222, a novel HDAC inhibitor, in proliferation and apoptosis of human leukaemia cell lines: role of annexin A1. Eur J Cancer (2008) 44(5):740–9. doi:10.1016/j.ejca.2008.01.023
37. Mu D, Gao Z, Guo H, Zhou G, Sun B. Sodium butyrate induces growth inhibition and apoptosis in human prostate cancer DU145 cells by up-regulation of the expression of annexin A1. PLoS One (2013) 8(9):e74922. doi:10.1371/journal.pone.0074922
38. Liu X, Guo S, Liu X, Su L. Chaetocin induces endoplasmic reticulum stress response and leads to death receptor 5-dependent apoptosis in human non-small cell lung cancer cells. Apoptosis (2015) 20(11):1499–507. doi:10.1007/s10495-015-1167-4
39. Cherblanc FL, Chapman KL, Brown R, Fuchter MJ. Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat Chem Biol (2013) 9(3):136–7. doi:10.1038/nchembio.1187
40. Shen X, Zhang K, Kaufman RJ. The unfolded protein response – a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat (2004) 28(1):79–92. doi:10.1016/j.jchemneu.2004.02.006
41. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest (2005) 115(10):2656–64. doi:10.1172/JCI26373
42. Rufo N, Garg AD, Agostinis P. The unfolded protein response in immunogenic cell death and cancer immunotherapy. Trends Cancer (2017) 3(9):643–58. doi:10.1016/j.trecan.2017.07.002
43. Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. J Chem Neuroanat (2004) 28(1–2):51–65. doi:10.1016/j.jchemneu.2003.08.007
44. Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol (2015) 17(7):829. doi:10.1038/ncb3184
45. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol (2000) 2(6):326–32. doi:10.1038/35014014
46. Li J, Zhu S, Ke X-X, Cui H. Role of several histone lysine methyltransferases in tumor development. Biomed Rep (2016) 4(3):293–9. doi:10.3892/br.2016.574
47. Zindl CL, Chaplin DD. Immunology. Tumor immune evasion. Science (2010) 328(5979):697–8. doi:10.1126/science.1190310
48. Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol (1999) 61:243–82. doi:10.1146/annurev.physiol.61.1.243
49. Yang Q, Liu S, Tian Y, Hasan C, Kersey D, Salwen HR, et al. Methylation-associated silencing of the heat shock protein 47 gene in human neuroblastoma. Cancer Res (2004) 64(13):4531–8. doi:10.1158/0008-5472.CAN-04-0956
50. Qi W, White MC, Choi W, Guo C, Dinney C, McConkey DJ, et al. Inhibition of inducible heat shock protein-70 (Hsp72) enhances bortezomib-induced cell death in human bladder cancer cells. PLoS One (2013) 8(7):e69509. doi:10.1371/journal.pone.0069509
51. Kisliouk T, Cramer T, Meiri N. Methyl CpG level at distal part of heat-shock protein promoter HSP70 exhibits epigenetic memory for heat stress by modulating recruitment of POU2F1-associated nucleosome-remodeling deacetylase (NuRD) complex. J Neurochem (2017) 141(3):358–72. doi:10.1111/jnc.14014
52. Hwang SS, Kim LK, Lee GR, Flavell RA. Role of OCT-1 and partner proteins in T cell differentiation. Biochim Biophys Acta (2016) 1859(6):825–31. doi:10.1016/j.bbagrm.2016.04.006
53. Nagaraju GP, Wu C, Merchant N, Chen Z, Lesinski GB, El-Rayes BF. Epigenetic effects of inhibition of heat shock protein 90 (HSP90) in human pancreatic and colon cancer. Cancer Lett (2017) 402:110–6. doi:10.1016/j.canlet.2017.05.021
54. Musahl A-S, Huang X, Rusakiewicz S, Ntini E, Marsico A, Kroemer G, et al. A long non-coding RNA links calreticulin-mediated immunogenic cell removal to RB1 transcription. Oncogene (2015) 34(39):5046–54. doi:10.1038/onc.2014.424
55. Colangelo T, Polcaro G, Ziccardi P, Pucci B, Muccillo L, Galgani M, et al. Proteomic screening identifies calreticulin as a miR-27a direct target repressing MHC class I cell surface exposure in colorectal cancer. Cell Death Dis (2016) 7:e2120. doi:10.1038/cddis.2016.28
56. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
57. Yang Y, Li XJ, Chen Z, Zhu XX, Wang J, Zhang LB, et al. Wogonin induced calreticulin/annexin A1 exposure dictates the immunogenicity of cancer cells in a PERK/AKT dependent manner. PLoS One (2012) 7(12):e50811. doi:10.1371/journal.pone.0050811
58. Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, et al. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J (2012) 31(5):1062–79. doi:10.1038/emboj.2011.497
59. Kunkeaw N, Jeon SH, Lee K, Johnson BH, Tanasanvimon S, Javle M, et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene (2013) 32(32):3722–31. doi:10.1038/onc.2012.382
60. Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X, et al. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol (2015) 33:86–92. doi:10.1016/j.semcancer.2015.02.004
61. Lee K-S, Park J-L, Lee K, Richardson LE, Johnson BH, Lee H-S, et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget (2014) 5(11):3944–55. doi:10.18632/oncotarget.2047
62. Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med (2010) 2(63):63ra94. doi:10.1126/scitranslmed.3001375
63. Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med (2017) 6:24. doi:10.1186/s40169-017-0154-5
64. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science (2011) 334(6062):1573–7. doi:10.1126/science.1208347
65. Ayna G, Petrovski G, Fésüs L. Chapter 14 – immunogenicity of dying cancer cells – the inflammasome connection: autophagic death arrives on the scene. In: Hayat MA, editor. Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Amsterdam: Academic Press (2014). p. 203–19. Available from: https://www.sciencedirect.com/science/article/pii/B9780124055308000145 (Accessed: October 26, 2017)
66. Rosenfeldt MT, O’Prey J, Flossbach L, Nixon C, Morton JP, Sansom OJ, et al. PTEN deficiency permits the formation of pancreatic cancer in the absence of autophagy. Cell Death Differ (2017) 24(7):1303. doi:10.1038/cdd.2016.120
67. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, et al. Autophagy in malignant transformation and cancer progression. EMBO J (2015) 34(7):856–80. doi:10.15252/embj.201490784
68. Shin Y-H, Kim M, Kim N, Choi S-K, Namkoong E, Choi S-Y, et al. Epigenetic alteration of the purinergic type 7 receptor in salivary epithelial cells. Biochem Biophys Res Commun (2015) 466(4):704–10. doi:10.1016/j.bbrc.2015.09.095
69. Pellegatti P, Falzoni S, Pinton P, Rizzuto R, Di Virgilio F. A novel recombinant plasma membrane-targeted luciferase reveals a new pathway for ATP secretion. Mol Biol Cell (2005) 16(8):3659–65. doi:10.1091/mbc.E05-03-0222
70. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle (2009) 8(22):3723–8. doi:10.4161/cc.8.22.10026
71. Increasing ATP release to increase metastasis. Sci Signal (2018). Available from: http://stke.sciencemag.org/content/8/385/ec188. (accessed Feb 19, 2018).
72. Liu Y, Shoji-Kawata S, Sumpter RM, Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A (2013) 110(51):20364–71. doi:10.1073/pnas.1319661110
73. Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature (2005) 436(7051):725–30. doi:10.1038/nature03918
74. García JM, Silva J, Peña C, Garcia V, Rodríguez R, Cruz MA, et al. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer (2004) 41(2):117–24. doi:10.1002/gcc.20062
75. Kang Y-H, Lee HS, Kim WH. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Invest (2002) 82(3):285–91. doi:10.1038/labinvest.3780422
76. Roh MR, Gupta S, Park K-H, Chung KY, Lauss M, Flaherty KT, et al. Promoter methylation of PTEN is a significant prognostic factor in melanoma survival. J Invest Dermatol (2016) 136(5):1002–11. doi:10.1016/j.jid.2016.01.024
77. Yin L, Cai W-J, Liu C-X, Chen Y-Z, Hu J-M, Jiang J-F, et al. Analysis of PTEN methylation patterns in soft tissue sarcomas by MassARRAY spectrometry. PLoS One (2013) 8(5):e62971. doi:10.1371/journal.pone.0062971
78. Cho D-H, Jo YK, Kim SC, Park IJ, Kim JC. Down-regulated expression of ATG5 in colorectal cancer. Anticancer Res (2012) 32(9):4091–6.
79. Liu H, He Z, von Rütte T, Yousefi S, Hunger RE, Simon H-U. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med (2013) 5(202):202ra123. doi:10.1126/scitranslmed.3005864
80. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell (2016) 30(1):147–60. doi:10.1016/j.ccell.2016.05.016
81. Huang P, Zhou Y, Liu Z, Zhang P. Interaction between ANXA1 and GATA-3 in immunosuppression of CD4. Mediators Inflamm (2016) 2016:9. doi:10.1155/2016/1701059
82. Álvarez-Teijeiro S, Menéndez ST, Villaronga MÁ, Pena-Alonso E, Rodrigo JP, Morgan RO, et al. Annexin A1 down-regulation in head and neck squamous cell carcinoma is mediated via transcriptional control with direct involvement of miR-196a/b. Sci Rep (2017) 7(1):6790. doi:10.1038/s41598-017-07169-w
83. Pupjalis D, Goetsch J, Kottas DJ, Gerke V, Rescher U. Annexin A1 released from apoptotic cells acts through formyl peptide receptors to dampen inflammatory monocyte activation via JAK/STAT/SOCS signalling. EMBO Mol Med (2011) 3(2):102–14. doi:10.1002/emmm.201000113
84. Cucolo L, Minn AJ. Getting tumor dendritic cells to engage the dead. Cancer Cell (2015) 28(6):685–7. doi:10.1016/j.ccell.2015.11.009
85. Tan S-X, Hu R, Dai A-G, Tang C-E, Yi H, Cheng A-L, et al. DNA methylation inhibits ANXA1 gene expression in nasopharyngeal carcinoma cell lines. Prog Biochem Biophys (2009) 36:1319–26. doi:10.3724/SP.J.1206.2009.00170
86. Wang W, Zhong W, Chen C, Meng Q, Wei J. Circulating antibodies to linear peptide antigens derived from ANXA1 and FOXP3 in lung cancer. Anticancer Res (2017) 37(6):3151–5. doi:10.21873/anticanres.11673
87. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (2015) 350(6263):972–8. doi:10.1126/science.aad0779
88. Palomeras S, Díaz Lagares Á, Blancafort A, Sarrats A, Crujeiras Martinez AB, Sandoval J, et al. 33PDNA methylation signature to identify trastuzumab response in HER2 breast cancer models. Ann Oncol (2017) 28(Suppl_1):mdx138.010. doi:10.1093/annonc/mdx138.010
89. Tsai K-W, Hu L-Y, Wu C-W, Li S-C, Lai C-H, Kao H-W, et al. Epigenetic regulation of miR-196b expression in gastric cancer. Genes Chromosomes Cancer (2010) 49(11):969–80. doi:10.1002/gcc.20804
90. Takahashi Y, Wu J, Suzuki K, Martinez-Redondo P, Li M, Liao H-K, et al. Integration of CpG-free DNA induces de novo methylation of CpG islands in pluripotent stem cells. Science (2017) 356(6337):503–8. doi:10.1126/science.aag3260
91. Guo ZS, Liu Z, Bartlett DL, Tang D, Lotze MT. Life after death: targeting high mobility group box 1 in emergent cancer therapies. Am J Cancer Res (2013) 3(1):1–20.
92. Ulloa L, Messmer D. High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev (2006) 17(3):189–201. doi:10.1016/j.cytogfr.2006.01.003
93. Bell CW, Jiang W, Reich CF, Pisetsky DS. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol (2006) 291(6):C1318–25. doi:10.1152/ajpcell.00616.2005
94. Bianchi ME, Crippa MP, Manfredi AA, Mezzapelle R, Rovere Querini P, Venereau E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol Rev (2017) 280(1):74–82. doi:10.1111/imr.12601
95. Yang Y, Huang J-Q, Zhang X, Shen L-F. MiR-129-2 functions as a tumor suppressor in glioma cells by targeting HMGB1 and is down-regulated by DNA methylation. Mol Cell Biochem (2015) 404(1–2):229–39. doi:10.1007/s11010-015-2382-6
96. Zhai J, Qu S, Li X, Zhong J, Chen X, Qu Z, et al. MiR-129 suppresses tumor cell growth and invasion by targeting PAK5 in hepatocellular carcinoma. Biochem Biophys Res Commun (2015) 464(1):161–7. doi:10.1016/j.bbrc.2015.06.108
97. He DX, Gu F, Gao F, Hao JJ, Gong D, Gu XT, et al. Genome-wide profiles of methylation, microRNAs, and gene expression in chemoresistant breast cancer. Sci Rep (2016) 6:24706–24706. doi:10.1038/srep24706
98. Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A (2008) 105(36):13556–61. doi:10.1073/pnas.0803055105
99. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14(1):36–49. doi:10.1038/nri3581
100. Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:187. doi:10.3389/fimmu.2015.00187
101. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol (2015) 15(2):87–103. doi:10.1038/nri3787
102. Trinchieri G. Type I interferon: friend or foe? J Exp Med (2010) 207(10):2053–63. doi:10.1084/jem.20101664
103. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of Th1 type chemokines shapes tumor immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi:10.1038/nature15520
104. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol (2016) 16(7):407–20. doi:10.1038/nri.2016.58
105. Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Epigenetic regulation of ASC/TMS1 expression: potential role in apoptosis and inflammasome function. Cell Mol Life Sci (2014) 71(10):1855–64. doi:10.1007/s00018-013-1524-9
106. Gasche JA, Hoffmann J, Boland CR, Goel A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer (2011) 129(5):1053–63. doi:10.1002/ijc.25764
107. Tekpli X, Landvik NE, Anmarkud KH, Skaug V, Haugen A, Zienolddiny S. DNA methylation at promoter regions of interleukin 1B, interleukin 6, and interleukin 8 in non-small cell lung cancer. Cancer Immunol Immunother (2013) 62(2):337–45. doi:10.1007/s00262-012-1340-3
108. Larco JED, Wuertz BRK, Yee D, Rickert BL, Furcht LT. Atypical methylation of the interleukin-8 gene correlates strongly with the metastatic potential of breast carcinoma cells. Proc Natl Acad Sci U S A (2003) 100(24):13988–93. doi:10.1073/pnas.2335921100
109. Kolb R, Liu G-H, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell (2014) 5(1):12–20. doi:10.1007/s13238-013-0001-4
110. Kiguchi N, Saika F, Kobayashi Y, Kishioka S. Epigenetic regulation of CC-chemokine ligand 2 in nonresolving inflammation. Biomol Concepts (2014) 5(4):265–73. doi:10.1515/bmc-2014-0022
111. Baird A-M, Gray SG, O’Byrne KJ. Epigenetics underpinning the regulation of the CXC (ELR+) chemokines in non-small cell lung cancer. PLoS One (2011) 6(1):e14593. doi:10.1371/journal.pone.0014593
112. van den Elsen PJ. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol (2011) 2:48. doi:10.3389/fimmu.2011.00048
113. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol (2016) 27(3):409–16. doi:10.1093/annonc/mdv615
114. Dewi DL, Mohapatra SR, Blanco Cabañes S, Adam I, Somarribas Patterson LF, Berdel B, et al. Suppression of indoleamine-2,3-dioxygenase 1 expression by promoter hypermethylation in ER-positive breast cancer. Oncoimmunology (2017) 6(2):e1274477. doi:10.1080/2162402X.2016.1274477
115. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi:10.1038/ni.2376
Keywords: tumor microenvironment, immunogenic cell death, epigenetics, T cell immunity, cancer immunotherapy, immune evasion
Citation: Cruickshank B, Giacomantonio M, Marcato P, McFarland S, Pol J and Gujar S (2018) Dying to Be Noticed: Epigenetic Regulation of Immunogenic Cell Death for Cancer Immunotherapy. Front. Immunol. 9:654. doi: 10.3389/fimmu.2018.00654
Received: 19 December 2017; Accepted: 16 March 2018;
Published: 03 April 2018
Edited by:
Andreas Behren, Olivia Newton-John Cancer Research Institute, AustraliaReviewed by:
Abhishek D. Garg, KU Leuven, BelgiumMarc Diederich, Seoul National University, South Korea
Copyright: © 2018 Cruickshank, Giacomantonio, Marcato, McFarland, Pol and Gujar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Pol, cG9sX2pvbmF0aGFuQHlhaG9vLmZy;
Shashi Gujar, c2hhc2hpLmd1amFyQGRhbC5jYQ==
†These authors have contributed equally to this work.